

Abstract
The immunoglobulin heavy chain (IgH) 3′ regulatory region modulates IgH locus transcription, upon induction by specific trans-acting factors, and plays a significant role in class switch DNA recombination (CSR) and, perhaps, somatic hypermutation (SHM). CSR and SHM are central to the maturation of the antibody response. In contrast to the single 5′-hs3a-hs1,2-hs3b-hs4-3′ mouse IgH 3′ regulatory region, the human IgH 3′ regulatory region exists as a 5′-hs3-hs1,2-hs4–3′ cluster duplicated 3′ of Ca1 and Cα2. We show here that the human hs1,2 element is the strongest enhancer of transcription, as directed by a VH1 or the ECS-Iγ3 promoter, thereby suggesting a dominant role for hs1,2 over hs3 and hs4 in the overall activity of the 3′ regulatory region. Within hs1,2, we identified three regions (1, 2, and 3) that are all necessary, but individually not sufficient, for enhancement of transcription. In region 2, a HoxC4 site and a HoxC4/embedded octamer (HoxC4/Oct) site are conserved across human, mouse, rat, and rabbit. These two sites recruit HoxC4 and Oct-1/Oct-2, which act synergistically with the Oca-B coactivator to effect the full hs1,2-enhancing activity. HoxC4, Oct-1/Oct-2, and Oca-B recruitment is negligible in pro-B cells, moderate in pre-B cells, and maximal in germinal center B cells and plasma cells, where HoxC4, Oct-2, and Oca-B expression correlates with hs1,2 activation and ongoing CSR. The hs1,2-mediated enhancement of VH and CH promoter-driven transcription as induced by HoxC4 and Oct-1/Oct-2 suggests an important role of these homeodomain proteins in the overall regulation of the IgH locus expression.
Gene transcription of the Ig heavy (H) and light (L) chain locus proceeds in a lymphoid-restricted and developmental stage-specific fashion, leading to Ig V(D)J recombination and the emergence of mature B cells expressing unique receptors for antigen. Upon encounter with antigen, B cells undergo somatic hypermutation (SHM)1 and class switch DNA recombination (CSR) to produce affinity-mature, isotype-switched antibodies. Like V(D)J recombination, SHM and CSR are critically dependent on transcription, as driven by three main cis-regulatory regions: the promoter upstream of each V gene (V), the IgH evolutionarily conserved sequence (ECS)-intervening (I) region promoter, which is located upstream of each constant region (CH) exon cluster, and the IgH intronic enhancer (iEμ) (1–4). The identification of an additional cis-regulatory region was suspected after mouse cell lines lacking the iEμ enhancer were found to still effectively transcribe IgH genes, and a mouse cell line containing an intact iEμ enhancer but showing a large deletion of sequence downstream of the Cα gene showed decreased IgH transcription (5, 6).
Indeed, a second B cell-specific regulatory region was identified ~25 kb downstream of the rat Cα gene and 16 kb downstream of the mouse Cα gene, with 82% sequence identity (7–9). The murine 3′ regulatory region consists of five B cell-specific DNase I hypersensitivity sites, each characterized as an IgH 3′ enhancer (3′EH): hs3a, hs1,2, hs3b, and hs4, with hs1,2 lying at the center of a region of symmetry flanked by inverted repeat sequences (6). hs1–4 collectively function as a locus control region (LCR) (5, 6), as suggested by the position-independent and copy number-dependent deregulation of c-MYC expression in plasmacytoma cells transfected with a hs1,2-hs3b-hs4-linked c-MYC construct (10). Additional sequences may be required to allow the murine hs1–4 enhancers to act as a classical LCR, as in transgenic mice harboring a VH promoter-β-globin reporter gene linked to the Ig 3′EH regulatory region, transgene expression was strictly confined to B cells, and reporter gene activity was integration-independent but not copy number-dependent (11).
A role for the mouse hs1,2 and hs3a enhancers in CSR to IgG2b and IgE has been suggested by Cre/loxP gene targeting experiments (12). These extended previous findings obtained by replacement of DNA encompassing the mouse hs1,2 element with the neomycin (neo) gene (13). Further experiments involving hs3b and hs4 and Cre/loxP knockouts showed severe impairment of germ-line Ih-Ch transcription and CSR to IgG2a, IgG2b, IgG3, IgE, and IgA, indicating that the distal portion of the regulatory region is required for CSR to most isotypes (14). The role of the 3′ regulatory region in SHM awaits better definition. The use of a transgenic construct containing murine hs1,2 suggested that this element does not play a role in SHM, even when coupled with the iEμ enhancer (15). In contrast, the use of a transgene construct containing the distal hs3b and hs4 elements, but not hs1,2, resulted in an increase in the SHM level of transgenes driven by a VH promoter, pointing to a role for hs3b and hs4 in SHM (15, 16). However, more recent experiments have indicated that hs3b and hs4 are dispensable for SHM and VHDJH gene assembly (17).
The human IgH 3′ regulatory region comprises the B cell-specific DNase I hypersensitivity sites hs1,2, hs3, and hs4, which are arranged in the 5′ hs3-hs1,2-hs4 3′ sequence and duplicated as discrete enhancer clusters 3′ of Ca1 and 3′ of Cα2 (supplementary Fig. S1), with the Cα1 and Cα2 hs1,2 sequences inverted with respect to each other (18, 19). Like their murine counterparts, the human 3′EH hs1,2 and hs4 elements are induced by the Oct-2 transcription factor (trans-factor) and its interacting coactivator Oca-B (6). In contrast to the mouse, negative regulatory mechanisms would not be conserved, as binding sites for B cell-specific activator protein (BSAP), a repressor of the mouse 3′EH hs1,2, are lacking in human hs1,2 (6). Human 3′EH elements have been shown to enhance transcription as driven by a mouse Vκ or Vλ promoter or a human ECS-Iα or ECS-Iγ3 promoter (18–21). Like its murine counterpart, the human 3′EH regulatory region likely functions as an LCR and may play a role in VH gene rearrangement, CSR, and SHM (2, 6).
To identify the elements involved in the induction of the human IgH 3′ regulatory region, we created hs1,2, hs3, and hs4 enhancer-DNA constructs and 5′, 3′, and internal deletion hs1,2 mutants, and inserted them into luciferase (luc) reporter gene vectors driven by a human VH or ECS-IH promoter. By bearing the luc gene downstream of the promoter and upstream of hs1,2, hs3, or hs4, these vectors mimicked the physiological promoter-gene-enhancer structure found in the IgH locus. In addition, we generated hs1,2 enhancer constructs containing mutations of selected cis-elements and enforced expression of respective trans-factors. Finally, to determine the stages of B cell ontogeny at which the human hs1,2 is activated, we transfected pro-B cells, pre-B cells, early and late germinal center B cells, and plasma cells with a reporter gene vector containing a fully inducible hs1,2 enhancer construct, as driven by the human β-globin promoter. We then measured the enhancement of such a promoter activity and analyzed it with the levels of endogenous germ-line IH-Ch and mature VHDJH-CH transcripts, the expression of trans-factors targeting hs1,2, and the formation of specific nucleoprotein complexes.
EXPERIMENTAL PROCEDURES
Human IgH 3′ Regulatory Region
The human hs1,2, hs3, and hs4 DNAs were from the 3′EH cluster lying 3′ of the Cα2 exon (19). The 1079-bp hs1,2 DNA spans residues 322–1400 according to the numeration of this genomic clone (GenBank™ accession nos. AF013724 and U84574); the 695-bp hs3 DNA spans residues 526–1221 (GenBank™ accession nos. AF013719 and Y14406); and the 426-bp hs4 DNA spans residues 1–426 (GenBank™ accession no. AF013726). The hs1,2 region 1 G-rich repeats were identified by Pustell DNA matrix analysis (22), enabling the search for regions of high similarity between two nucleic acid sequences using a dot matrix plot. The human, mouse, rat, and rabbit hs1,2 region 2 sequences were compared using the ClustalW algorithm (23) allowing for multiple alignments of nucleotide sequences. All sequence comparisons were implemented by MacVector, version 6.5.3 (Accelrys Inc., San Diego, CA). The identification of putative cis-regulatory binding sites was performed using Matlnspector (www.genomatix.de/cgibin/matinspector/matinspector.pl), which utilizes the TRANSFAC library of matrices (www.gene-regulation.de/) to locate consensus matches in nucleotide sequences (24).
Vectors
The pGL3-Basic luc-reporter gene vector (Promega Corp., Madison, WI) was modified by inserting three different promoter sequences between the SacI and BglII restriction sites: −449/+265 ECS-Iγ3 promoter DNA (GenBank™ accession no. S79588) (25), VH1 promoter DNA isolated from the IgG1 mAb57-producing cell line (26, 27), or the human β-globin promoter DNA (GenBank™ accession no. U01317). The pGL3 vectors containing the VH1, ECS-Iγ3 or β-globin promoter were further modified by inserting between the BamHI/SalI restriction sites the hs3, hs1,2, and hs4 elements, resulting in the positioning of each 3′EH ~2 kb downstream of the respective promoter and immediately flanking the firefly luc gene. To generate sequential 5′- and 3′-end truncation mutants, hs1,2 was PCR-amplified using appropriate primers with BamHI/SalI overhangs. Internal hs1,2 deletion mutants were generated by first amplifying 5′- and 3′-halves, minus the targeted sequence, ligating the two fragments, and re-amplifying the “complete” DNA. Ligation of the 5′ and 3′ PCR fragments flanking the targeted motifs generated site-targeted mutations of the 5′ HoxC4 and 3′ HoxC4/Oct-binding sites. The 5′ HoxC4 site (ATTT, residues 715–718) was replaced by cggg. The HoxC4/Oct site (ATTTGCAT, residues 773–780) was replaced with a KpnI restriction site. The double mutant hs1,2 DNA was generated using the single 5′ HoxC4 and 3′ HoxC4/Oct mutants as templates. All digested PCR products were gel-purified and subcloned into the pGL3 vectors driven by the VH1 or ECS-Iγ3 promoter.
For enforced expression studies, cDNA encoding human HoxC4, Oct-1, Oct-2, or Oca-B was subcloned into pcDNA3.1 vectors using the pcDNA3.1/V5-His TOPO TA expression kit (Invitrogen, Carlsbad, CA). The pcDNA3.1 expression vector contains a cytomegalovirus promoter for high level expression and a T7 promoter for in vitro translation using the TnT Quick Coupled Transcription/Translation System (Promega Corp.). The expression vector encoding the dominant negative HoxC4 lacking the homeodomain was described (28). The glutathione S-transferase (GST) fusion proteins were generated by subcloning human HoxC4, Oct-1, Oct-2, and Oca-B cDNAs into the pGEX-6P-1 vector as described previously (28). GST fusion proteins were expressed in BL21 bacteria, purified using GSH-agarose beads according to the manufacturer’s protocol (Sigma–Aldrich) and analyzed for homogeneity by SDS-PAGE and silver staining. Proteins were eluted in 15 mM reduced glutathione in 50 mM Tris buffer, pH 8.0. Restriction enzyme mapping and DNA sequencing were used to verify all of the correct plasmids.
Cell Lines
RS4;11 is a human cytoplasmic and surface IgM− pro-B cell line (29). Nalm-6 is a human cytoplasmic IgM+ and surface IgM− pre-B cell line (30). 4B6 is an IgM+, IgDλ+ cell line with an early germinal center phenotype and undergoes spontaneous switching to IgG, IgE, and IgA (28). It was derived from our inducible CL-01 IgM+, IgDλ+ B cell line (31–33) by sequential subcloning and selection for IgG, IgA, and IgE secretion. HS Sultan is a human IgGκ+ Burkitt’s lymphoma with a late germinal center phenotype (34). U266 is a human myeloma IgEλ+ cell line (35). All cell lines were cultured in RPMI 1640 medium (Invitrogen) supplemented with 10% heat-inactivated fetal calf serum (Sigma–Aldrich), 2 mML-glutamine, and 1× antibiotic-antimy-cotic mixture (100 units/ml penicillin, 100 μg/ml streptomycin, 0.25 μg/ml, amphotericin B fungizone, Invitrogen).
Transfection Assays
B cells (5 × 106) in 0.5 ml of medium in an 0.4-cm cuvette were electroporated using Gene Pulser II (Bio-Rad). All electroporations were performed in duplicate, adhering to the following parameters: 4B6 cells, 210 V, 950 μF; HS Sultan cells, 200 V, 900 μF; and U266 cells, 250 V, 1050 μF. For luc -reporter transient transfec-tions, 10 μg of pGL3-promoter-only and/or pGL3-enhancer-promoter plasmid DNA and 10 ng Renilla pRL-TK control vector (as an internal control for transfection efficiency) were used. For enforced expression studies, pcDNA3.1 expression vector encoding Oct-1, Oct-2, and/or Oca-B (2 μg) were cotransfected with pGL3-promoter-only and/or pGL3-enhancer-promoter plasmid DNA (5 μg), with salmon sperm DNA added to normalize total DNA at 20 μg. RS4;11 and Nalm-6 cells were transfected using Cellfectin following the manufacturer’s instructions (Invitrogen). Briefly, plasmid DNA (2 μg) was gently mixed with Cellfectin (15 μl) diluted in Opti-MEM I (Invitrogen) reduced serum medium. The ensuing DNA-lipid complex solution was used to resuspend 2 × 106 cells. Transfected cells were cultured for 24–48 h. Luc extracts were prepared in 1× passive lysis buffer (Promega). Firefly and Renilla Luc activities were measured using an MLX microtiter plate luminometer (Thermo Labsystems, Beverly, MA).
Antibodies
mAbs to Ku70 (Ab-5), Ku86 (Ab-2), and Ku70/Ku86 (Ab-3) were from Lab Vision/NeoMarkers (Fremont, CA). The anti-Oct-1 (YL 15) mAb was from Upstate Biotech (Charlottesville, VA) and the anti-HoxC4 mAb from Covance (Princeton, NJ). The rabbit antibodies to Oct-1 (C-21), Oct-2 (C-20), and Oca-B (C-20) as well as rabbit IgG were from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). Horseradish peroxidase-conjugated goat IgG to mouse IgG and rabbit IgG were also from Santa Cruz Biotechnology, Inc. The rabbit IgG to Stat-1 was from Transduction Laboratories (San Diego, CA). The mouse IgG and mAb to β-actin were from Sigma–Aldrich.
EMSAs
Nuclear and cytoplasmic extracts from mammalian cells were prepared using a micro-procedure involving hypotonic lysis followed by high salt nuclei extraction (28). Whole-cell extracts were prepared using extraction buffer (20 mM HEPES, 0.5% Nonidet P-40, 2 mM EDTA, 0.3 M KCl, 1 mM dithiothreitol, 5% glycerol, pH 7.5) and a mixture of protease inhibitors (1 μg/ml aprotinin, 1 μg/ml leupeptin, 1 μg/ml pepstatin, 1 mM phenylmethylsulfonyl fluoride, 1 mM Na3VO4, and 10 mM NaF) (25). Oligonucleotides encompassing the hs1,2 5′ HoxC4 (residues 704–729) and 3′ HoxC4/Oct (residues 764–789) cis-elements were as follows: 5′ HoxC4 wild type (wt), TCGTGCAAGCTATTTCTGGAAACAGC; 5′ HoxC4 mt4, TCGTGCAAGCTcgggCTGGAAACAGC; 3′ HoxC4/Oct wt, CAAACAACCATTTGCATGTGCCCTGA; 3′ HoxC4/Oct mt5, CAAACAACCcgggtaccGTGCCCTGA, 3′ HoxC4/Oct mt6, CAAACAACCcgggGCATGTGCCCTGA; 3′ HoxC4/Oct mt7, CAAACAACCccTTGCATGTGCCCTGA; and 3′ HoxC4/Oct mt8, CAAACAACCATTTGCcgGTGCCCTGA (underlines denote HoxC4/Oct sites). All reactions were performed as reported (25, 28). For EMSAs involving GST fusion proteins, 0.25 μg of the GST fusion protein (or GST moiety alone) was incubated, in place of nuclear extracted proteins, with the specified oligonucleotide probe for 20 min. For supershifting EMSA reactions, antibodies were pre-incubated with nuclear extracts for 30 min prior to addition of probe (25,000 CPM). All EMSA gels were 7% polyacrylamide and subjected to electrophoresis in 0.25× Tris borate/EDTA. Gels were dried and exposed for autoradiography.
Reverse Transcriptase PCRs
Total RNA was extracted from human B cells using the RNeasy Mini Kit (Qiagen, Valencia, CA). First strand cDNA syntheses were performed using SuperScript II reverse transcriptase (Invitrogen) with oligo(dT)12−18 as primer. HoxC4, Oct-1, Oct-2, Oca-B, and β-actin cDNAs were amplified using the following primers: HoxC4 forward, 5′-ATGGGATCATGAGCTCGTATTTG-3′; HoxC4 reverse, 5′-CTATAACCTGGTAATGTCCTCTGC-3′; Oct-1 forward, 5′-ATGGGGAACAATCCGTCAGAAACCAGTAAA-3′; Oct-1 reverse, 5′-CTACTGTGCCTTGGAGGCGGTGGT-3′; Oct-2 forward, 5′-CAAAATAAGACCTCCCCATTCTCC-3′; Oct-2 reverse, 5′-TTGATGCGGCGTTGCTTGAAGG-3′; Oca-B forward, 5′-CCTCAGTGTTGACCTATGCCTCTC-3′; Oca-B reverse, 5′-TCCTCTTCCTCCAAAAGCAGC-3′; β-actin forward, 5′-GTACCACTGGCATCGTGATGGACT-3′; and β-actin reverse, 5′-ATCCACACGGAGTACTTGCGCTCA-3′. Germ-line IH-CH and mature VHDJ-CH transcripts were detected using specific primers, as reported (28). Before each reverse transcriptase PCR, cDNAs were denatured for 5 min at 94 °C. PCRs involved denaturation (1 min at 94 °C), annealing (1 min at 68 °C), and extension (1 min at 72 °C).
Western Blot Analysis
Whole human B cell extracts (25 μg) were fractionated through 10% SDS-PAGE. Proteins were blotted onto polyvinylidene difluoride membranes (Bio-Rad) overnight (30 V) at 4 °C and then detected by primary (1:250 to 1:1000) and secondary (1:2500) antibodies. After washing with phosphate-buffered saline-Tween (0.05%), bound horseradish peroxidase-conjugated antibodies were detected using Enhanced Chemiluminescence Plus reagents (Amersham Biosciences).
Chromatin Immunoprecipitation (ChIP) Assays
B cells (2.5 × 107) were treated with 1% formaldehyde for 10 min at room temperature to cross-link chromatin (36). After washing with cold phosphate-buffered saline containing protease inhibitors, chromatin was separated using nuclei-lysis buffer (10 mM Tris-HCl, 0.5 M NaCl, 1 mM EDTA, 1% Trition-X-100, 0.5% sodium deoxycholate, 0.5% sarkosyl, pH 8.0), resuspended in IP-1 buffer (20 mM Tris-HCl, 0.2 M NaCl, 2 mM EDTA, 0.1% sodium deoxycholate, 0.1% SDS, protease inhibitors), and sonicated to yield 500–1000-bp DNA fragments. These were pre-cleared with agarose beads bearing protein A or G (Santa Cruz Biotechnology) and then incubated with mAb to HoxC4, rabbit antibody to Oct-1, Oct-2, or Oca-B, or mAb to Ku70/Ku86 or rabbit antibody to Stat-1 overnight at 4 °C. The immune complexes were isolated using beads bearing protein A or G, eluted with elution buffer (50 mM Tris-HCl, 0.5% SDS, 200 mM NaCl, 100 μg/ml proteinase K, pH 8.0) and then heated at 65 °C overnight to reverse the cross-links. DNA was recovered by phenol extraction followed by ethanol precipitation and then solubilized in Tris/EDTA buffer. The recovered DNA was specified using the following primers: forward ChIP 670–689 (5′-TGGGTGGGTGGCGGCTGCAG3′) and reverse ChIP 812–793 (5′-CTCATTCTGGGCAGACTTGG-3′). DNA was then transferred to Hybond-N+ membrane (Amersham Bio-sciences) for Southern blotting using the 753–770 5′-TCGGTGTGGAACAAACAA-3′ oligonucleotide as probe.
RESULTS
hs1,2 Is the Strongest Enhancer in the Human Cα2 3′Regulatory Region
To address the function of the human IgH 3′ regulatory region, we analyzed the ability of the Cα2 hs1,2, hs3 and hs4 elements to enhance transcription of a luciferase (luc)-reporter gene vector, as driven by a human VH1 promoter, the human ECS-Iγ3 promoter, or the Ig-irrelevant human β-globin promoter, upon transfection of human 4B6 B cells. These cells are IgM+IgD+ and spontaneously switch to IgG, IgA, and IgE. They are the neoplastic equivalent of centroblasts/centrocytes (28), a B cell differentiation stage at which the 3′ regulatory region has been suggested to be active (6). In 4B6 cells, the hs1,2 element was the strongest enhancer of transcription, yielding a 172-fold (VH1 promoter), 100-fold (ECS-Iγ3 promoter), and 77-fold (β-globin promoter) increase in Luc activity; hs4 was second, yielding a 46-fold (VH1 promoter), 10-fold (ECS-Iγ3 promoter), and 15-fold (β-globin promoter) increase in Luc activity, whereas hs3 was the weakest of the three elements, yielding 6-fold (VH1 promoter), 2-fold (ECS-Iγ3 promoter), and 2-fold (β-globin promoter) increase in Luc activity (Fig. 1).
Fig. 1. The hs1,2 element is the strongest IgH 3′ enhancer.
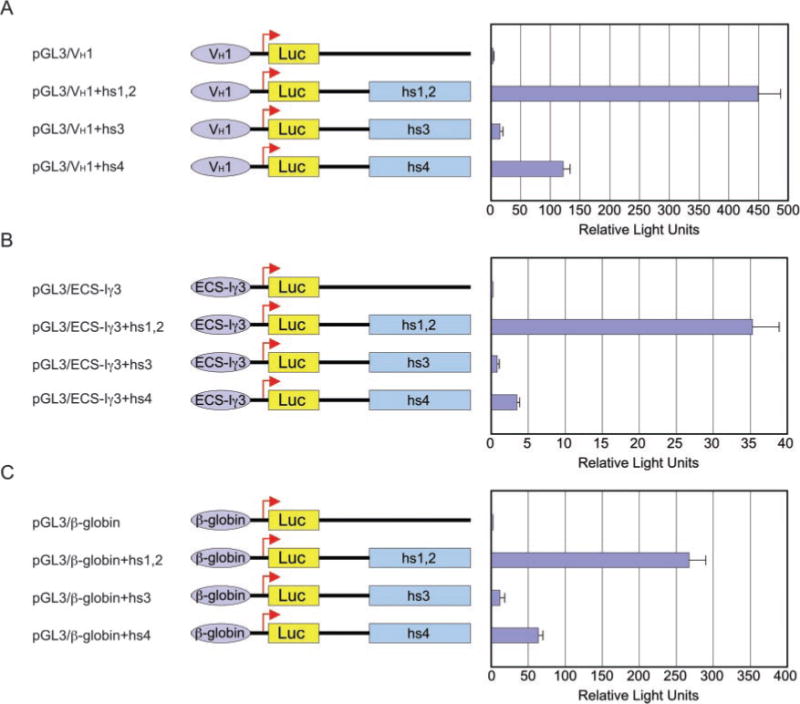
Human 4B6 B cells were transfected with the indicated enhancer-promoter or promoter-only pGL3 luc-reporter gene vector to assess the enhancing activity of each Cα2 3′EH element. Three separate human promoters were used: VH1 (A), ECS-Iγ3 (B), and β-globin (C). Luc activity is expressed as relative light units. Data are the mean values of three independent experiments plus standard deviation.
hs1,2 Contains Three Regions with Multiple cis-Elements
Because of its dominant activity, we analyzed hs1,2 for putative trans-factor-binding motifs and segregated it into three regions (1, 2, and 3) based on the identified cis-elements (Fig. 2). Region 1 encompasses residues 452–669 and comprises four highly G-rich ~53-bp tandem arrayed segments (G-rich repeats 1–4 (GRR)), which have been suggested to display some polymorphism (19, 37). In each GRR, we identified a putative κB (GGGGYNNCCY consensus) site. Region 2, originally classified as an ECS core (19), encompasses residues 684–818. In this region, we identified two Hox-binding sites (ATTT, 5′ at residues 715–718 and 3′ at residues 773–780), in addition to the previously identified E-Box (characterized as a μE5 site, CANNTG, residues 687–698)-, Oct (ATTTGCAT, residues 773–780)-, and AP-1 (TGAGTCA, residues 809–815)-binding sites (19). We found that these ATTT sites recruit HoxC4 (not shown). We determined that the 3′ HoxC4-binding site is embedded within the 5′ portion of the Oct site and therefore designated it as HoxC4/Oct site. Region 3 encompasses residues 819–1133 and contains eight putative cis-elements: a GATA (WGATAR consensus) site, an E-Box site, two Myb (YAACKG consensus) sites, two Ikaros (GGGAA consensus), and two AP sites (an AP-4 (CAGCTG consensus) and an AP-2 (GSSWGSCC consensus)). Regions 1 and 3 are only 47 and 39% identical, respectively, in human, mouse, rat, and rabbit (Fig. 3). In contrast, region 2 is 87% identical overall and 100% identical in the identified HoxC4 and HoxC4/Oct elements across human, mouse, rat, and rabbit.
Fig. 2. Identification of putative cis-binding sites within the hs1,2 and segregation of regions 1, 2, and 3.
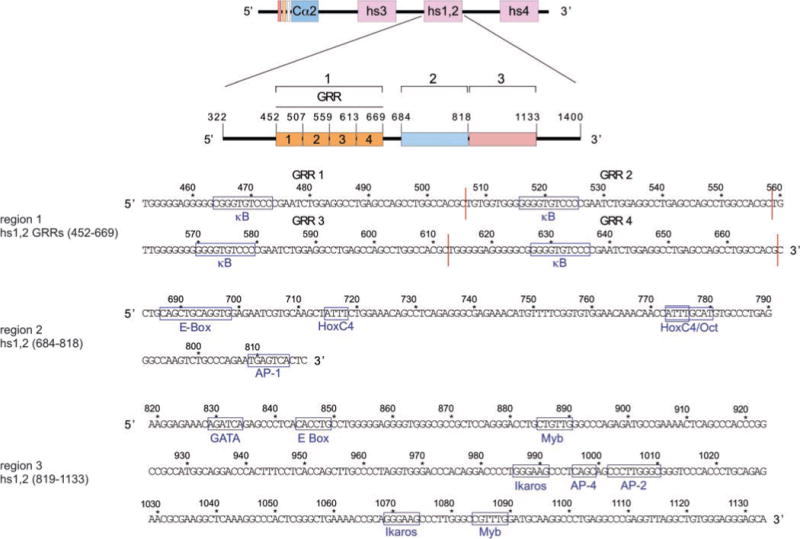
The Cα2 hs1,2 sequence (originally obtained as a genomic clone spanning residues 322–1400) was segregated into three regions based on the nature and the distribution of the identified motifs: region 1, consisting of four GRRs, each ~53 bp in length and spanning residues 452–669; region 2, spanning residues 684–818; and region 3, spanning residues 819–1133. Putative cis-regulatory motifs are indicated and boxed within each region. The 3′ HoxC4/Oct-, Myb-, and AP-2-binding sites match the respective consensus in the reverse orientation. See “Results” for consensus sites.
Fig. 3. hs1,2 region 2 and the 5′ HoxC4 and 3′ HoxC4/Oct cis-elements are conserved across species.

ClustalW-formatted alignments of human, mouse, rat, and rabbit 3′EH hs1,2 regions 1, 2, and 3. The Ca1 hs1,2 element contains only one GRR, whereas the Ca2 hs1,2 element contains four GRRs. Residue numbers are as follows: human Cα1, 452–506 (region 1), 507–641 (region 2), and 642–956 (region 3) (GenBank™ accession numbers AF013722 and U84575); human Cα2, 452–669 (region 1), 684–818 (region 2) and 819–1133 (region 3) (GenBank™ accession no. AF013724, U84574); mouse, 498–711 (region 1), 851–988 (region 2), and 1098–1394 (region 3) (GenBank™ accession no. X62778); rat, 328–526 (region 1), 645–774 (region 2), and 1059–1353 (region 3) (GenBank™ accession no. A28224); and rabbit, 372–592 (region 1), 605–733 (region 2), and 738–1018 (region 3) (GenBank™ accession no. AF314408). Gaps in the sequence for each species are as follows: human Ca1, none; human Ca2, 669–683; mouse, 712–761, 777–850, and 989–1097; rat, 527–644 and 775–1058; and rabbit, 593–604 and 734–737. All cis-elements are boxed. Note the identity of the ATTT and ATTTGTAC sites across human, mouse, rat, and rabbit. Also, note the lack of BSAP sites in the human.
Regions 1, 2, and 3 Are All Necessary for Cα2 hs1,2 Activation
To determine the minimal sequence responsible for mediating hs1,2 activation, we generated 5′ and 3′ truncation and internal deletion mutants and inserted them in luc-reporter gene vectors, which were then used to transfect 4B6 cells and to measure the enhancement of transcription, as driven by a VH1 or the ECS-Iγ3 promoter. The transcription-enhancing activity mediated by the full-length Cα2 hs1,2 construct (322–1400) was referred to as 100% and used as the term of comparison for all other constructs (Fig. 4). Analysis of the 5′ and 3′ truncation mutants showed that the sequences 322–451 (compare C6 with C2) and 1133–1400 (compare C13 with C2) do not make a significant contribution to the overall enhancement of transcription, as mediated by VH1 or ECS-Iγ3 promoter. This was confirmed by the analysis of the 5′ and 3′ truncated construct C17, which displayed 97% of the activity of C2. Analysis of sequential 5′ deletion mutants showed that each of the four GRRs critically contributes to the overall enhancing activity of region 1 (C6–C11), whereas 3′ deletion mutants suggested a significant role for GATA, E-Box, Ikaros, AP-2, AP-4, and/or Myb in the overall enhancing activity of region 3 (C14 and C15). Furthermore, deletion of region 1 (Δ452–669, C3), 2 (Δ684–818, C4), or 3 (Δ819–1133, C5) DNA resulted in 84, 96, and 93% loss, respectively, of such an activity, regardless of the promoter used. Conversely, when alone, region 1, 2, or 3 failed to significantly enhance transcription, as driven by the VH1 or ECS-I73 promoter (C12, C16, C20, and C21). Regions 1 and 2 together displayed only partial enhancing activity when disjoined from region 3 (C18), whereas regions 2 and 3 together displayed only partial activity when disjoined from region 1 (C19). Thus, regions 1, 2, and 3 are all necessary, but none is sufficient per se to effect full Cα2 hs1,2 activation.
Fig. 4. Regions 1, 2, and 3 are necessary but individually are not sufficient for transcription enhancement.

Human 4B6 B cells were transfected with the indicated pGL3 luc -reporter gene vectors to measure the contribution of different DNA regions to the overall hs1,2-enhancing activity. Sequential 5′- and 3′-end truncation, internal deletion, and subfragment mutants of the hs1,2 sequence were created and subcloned into the pGL3 vector driven by VH1 or ECS-Iγ3 promoter. The basal enhancing activity was determined and expressed as relative light units. Data are the mean values of three independent experiments plus standard deviations.
The HoxC4 and HoxC4/Oct Motifs of Region 2 Critically Mediate hs1,2 Region 2-enhancing Activity
The absolute conservation of the HoxC4 and HoxC4/Oct sites prompted us to test the hypothesis that these two cis-elements mediate the hs1,2 activity. We transfected 4B6 cells with a luc-reporter gene vector containing the Cα2 hs1,2 DNA sequence spanning regions 1, 2, and 3 (residues 452–1133), which were variously mutated in the region 2 HoxC4 and/or HoxC4/Oct sites to potentially hamper recruitment of the relevant trans-factors. Mutation of the HoxC4 element (mt1) reduced hs1,2-enhancing activity by 80% (VH1 promoter), 55% (ECS-Iγ3 promoter), and 58% (β-globin promoter) (Fig. 5). Mutation of the HoxC4/Oct element (mt2) reduced hs1,2-mediated transcription by 75% (VH1), 43% (ECS-Iγ3), and 58% (β-globin promoter). Finally, mutation of both the HoxC4 and the HoxC4/Oct sites (mt3) resulted in virtual ablation of the hs1,2-enhancing activity, regardless of the promoter used, thereby confirming that these cis-elements critically mediate the Cα2 hs1,2-enhancing activity.
FIG. 5. The HoxC4 and HoxC4/Oct cis-elements are required for full hs1,2-enhancing activity.
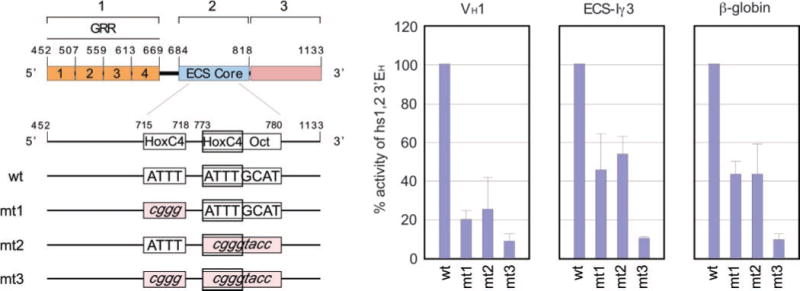
The hs1,2 HoxC4 and HoxC4/Oct-binding sites were mutagenized by site-directed PCR, resulting in single and double mutant enhancer pGL3 luc-reporter gene constructs driven by the VH1, ECS-I γ3, or β-globin promoter. Each construct was used to transfect 4B6 cells to measure Luc activity. Data are represented as percentage of wild-type hs1,2 (regions 1–3)-enhancing activity and are the mean values of three independent experiments plus standard deviations.
The HoxC4 and Oct-1/Oct-2 Homeodomain Proteins Are Specifically Recruited to Cα2 hs1,2
To identify the trans-factors that are recruited to the HoxC4 and HoxC4/Oct sites of hs1,2 region 2, we used wild-type (wt) and mutated (mt) oligonucleotide probes encompassing residues 704–729 (HoxC4 site) and 764–789 (HoxC4/Oct site) (Fig. 6) in EMSAs involving 4B6 nuclear cell extracts. These gave rise to a distinct nucleoprotein complex when incubated with the HoxC4 probe (Fig. 7A, top panel). Such a complex was specific, as its formation was inhibited by cold wild-type but not mutant hs1,2 HoxC4 (HoxC4 mt4) oligonucleotides or oligonucleotides containing an Igκ NF-κB or BSAP site. The involvement of HoxC4 in the nucleoprotein complex was demonstrated by the inhibition of complex formation by a mouse mAb to HoxC4 and the direct in vitro interaction of a GST-HoxC4 fusion protein with the radiolabeled DNA containing the wild-type but not the mutant hs1,2 HoxC4-binding site (Fig. 7A, bottom panel).
Fig. 6. hs1,2 HoxC4 and HoxC4/Oct oligonucleotides.

HoxC4- and HoxC4/Oct-binding site oligonucleotides used as radiolabeled probes in EMSA. For the HoxC4 site, the ATTT motif was mutated to cggg (HoxC4 mt4). For the HoxC4/Oct site, the ATTTGCAT motif was mutated as follows: HoxC4/Oct mt5, complete element replaced with a KpnI sequence; HoxC4/Oct mt6, 5′ HoxC4 portion (ATTT) mutated to cggg; HoxC4/Oct mt7, AT of 5′ HoxC4 portion mutated to gg; and HoxC4/Oct mt8, AT of 3′ Oct portion mutated to cg.
Fig. 7. Recruitment of HoxC4, Oct-1/Oct-2, and Oca-B to the 3′EH hs1,2.
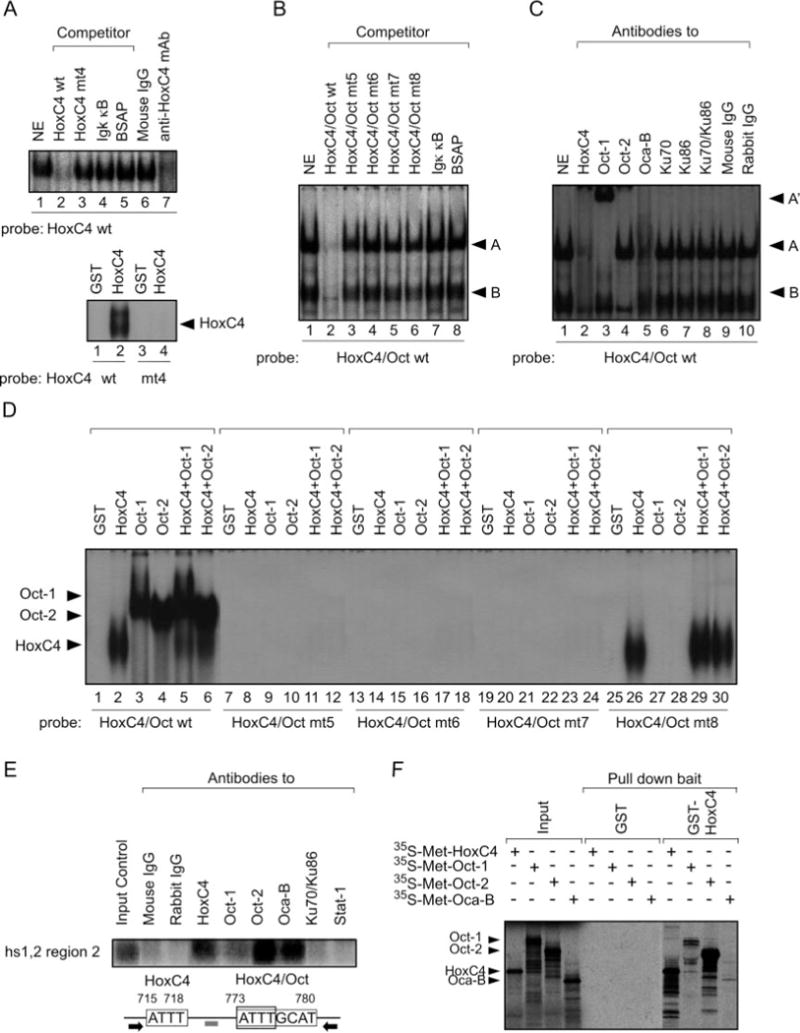
A, nuclear proteins from spontaneously switching 4B6 cells specifically bind an oligonucleotide probe containing the HoxC4-binding site of hs1,2 region 2 (top panel). Efficient competition was achieved by 50-fold molar excess of wild-type (HoxC4 wt) but not mutant (HoxC4 mt4), Igκ κB, or BSAP cold oligonucleotides. The formation of the DNA-binding complex was inhibited by a specific mAb to HoxC4. Mouse IgG served as a negative control. The specificity of the HoxC4 oligonucleotide probe was further verified by binding of recombinant GST-HoxC4 protein (250 ng) to wild-type HoxC4 but not to mutated HoxC4 (mt4) oligonucleotide probe (bottom panel). NE, nuclear extract. B, nuclear proteins from 4B6 cells bind specifically to radiolabeled hs1,2 HoxC4/Oct wt probe. Efficient competition was achieved by 50-fold molar excess of wt (HoxC4/Oct wt) but not mt (HoxC4/Oct mt5–8) or nonspecific (Igκ κB and BSAP) cold oligonucleotides. C, identity of the nuclear protein complexes as assessed by supershift EMSA. 4B6 nuclear extracts were preincubated with the indicated antibodies prior to the addition of the HoxC4/Oct oligonucleotide probe and EMSA. Arrowheads indicate complexes A and B as well as the supershifted complex A′. D, direct binding of HoxC4, Oct-1, andOct-2 tohs1,2 DNA. Recombinant GST fusion HoxC4, Oct-1, Oct-2, HoxC4, and Oct-1 or HoxC4 and Oct-2 protein (250 ng) were analyzed for direct binding to HoxC4/Oct probes with GST serving as a negative control. Sequences of HoxC4/Oct mt5, mt6, mt7, and mt8 were as described in the legend to Fig. 6. Note that mutation of the 3′-end of HoxC4/Oct motif (mt8) allowed for binding of HoxC4 only, as predicted. E, in vivo binding of HoxC4, Oct-1, and Oct-2 proteins to hs1,2 region 2 as determined by ChIP. Cross-linked chromatin from HS Sultan B cells was precipitated by mAb to human HoxC4 or antibodies specific for human Oct-1, Oct-2, or Oca-B. The precipitated DNA was specified by PCR using primers (black arrows) and detected by Southern blotting using a specific probe (gray bar) as listed under “Experimental Procedures.” mAbs that detect the interface between Ku70 and Ku86 and a rabbit antibody to Stat-1 were used as controls, along with mouse and rabbit IgG. F, GST fusion protein pull-down assays. In vitro 35S-labeled translated HoxC4, Oct-1, Oct-2, or Oca-B proteins were mixed with GST or GST-HoxC4 immobilized on glutathione-agarose resin, subjected to 12% SDS-PAGE, and exposed for autoradiography.
Incubation of nuclear extracts from 4B6 cells with the HoxC4/Oct probe gave rise to the two major nucleoprotein complexes, A and B (Fig. 7B). These were specific, as shown by the failure of mutated HoxC4/Oct oligonucleotides (HoxC4/Oct mt1–4) or Igκ NF-κB and BSAP oligonucleotides to inhibit the formation of both complexes. They included the HoxC4 and Oct-1 or Oct-2 homeodomain proteins as well as the Oca-B coactivator, as inferred from their supershifting or inhibition by specific antibodies to HoxC4, Oct-1, Oct-2, and Oca-B (Fig. 7C) and the binding of GST-HoxC4, GST-Oct-1, and/or GST-Oct-2 to the HoxC4/Oct oligonucleotide probe (Fig. 7D, lanes 1–6). Such a binding was specific, as mutation of the whole HoxC4/Oct-binding site (mt5) or its 5′-end containing the HoxC4-binding motif (mt6 and mt7) abolished binding of HoxC4, Oct-1, and Oct-2 (Fig. 7D, lanes 7–24), whereas mutation of the HoxC4/Oct 3′ portion (mt8) abrogated the binding of Oct-1 and Oct-2 but not HoxC4 (lanes 25–30). Finally, the Hox/Oct nucleoprotein complexes did not include homeodomain-interacting Ku70/Ku86 proteins, as Ku-specific antibodies did not supershift either complex A or B (Fig. 7C, lanes 6–8).
That HoxC4, Oct-1, Oct-2, and Oca-B are bound in vivo to hs1,2 region 2 was demonstrated by ChIP experiments in which the hs1,2 HoxC4-HoxC4/Oct sequence was specified in DNA precipitated from HS Sultan B cells using antibodies to HoxC4, Oct-1, Oct-2, or Oca-B but not Ku70–Ku86 or Stat-1 (Fig. 7E). In addition, the binding of in vitro translated 35S-Oct-1, 35S-Oct-2, and 35Oca-B to immobilized GST-HoxC4 indicated that Oct-1, Oct-2, and Oca-B can be recruited to hs1,2 by DNA-bound HoxC4 through direct protein-protein interaction (Fig. 7F). Thus, the hs1,2 5′ HoxC4 and 3′ HoxC4/Oct cis-elements effectively recruit HoxC4, Oct-1/Oct-2 and Oca-B as discrete components of a nucleoprotein complex that can be detected in vivo in germinal center B cells.
HoxC4, Oct-1, Oct-2, and Oca-B Activate Cα2 hs1,2
To verify that HoxC4 and Oct-1/Oct-2 indeed activate hs1,2, we co-transfected 4B6 cells with expression vectors encoding HoxC4, Oct-1, Oct-2, or Oca-B together with the hs1,2 (residues 452–1133) luc-reporter gene vector. The human β-globin promoter was used rather than the VH1 or the ECS-Iγ3 promoter to minimize the effect of these overexpressed trans-factors on promoter induction because the ß-globin promoter lacks binding sites for HoxC4 or Oct. Enforced expression of HoxC4, Oct-1, Oct-2, or Oca-B alone in 4B6 cells resulted in up to a 2.4-fold enhancement of ß-globin-driven luc transcription (Fig. 8). Further enhancement of transcription was achieved when HoxC4 was co-expressed with Oct-1 (3.8-fold), Oct-2 (5.0-fold), or Oca-B (6.4-fold). The role of HoxC4 in activating hs1,2 was confirmed by the virtual ablation of hs1,2-enhancing activity when a mutant HoxC4 lacking its homeodomain (HD mt) was expressed, even in the presence of Oct-1, Oct2, or Oca-B coexpression. Thus, HoxC4 synergizes with Oct-1/Oct-2 and Oca-B to activate hs1,2 in a homeodomain-dependent manner for maximal transcription-enhancing activity.
Fig. 8. Enforced expression of HoxC4, Oct-1, Oct-2, and/or Oca-B activate the hs1,2 3′EH.

Mammalian expression constructs of full-length HoxC4, Oct-1, Oct-2, and/or Oca-B proteins together with the luc-reporter vector containing the β-globin promoter and hs1,2 (regions 1, 2, and 3) were used to transfect 4B6 cells. A mutant HoxC4 (HD mt) was coexpressed in the presence or absence of Oct-1, Oct-2, or Oca-B expression vector. Results represent the fold induction of Luc activity expressed by the reporter gene vector containing the promoter only. Data are the mean values of three experiments plus standard deviations.
Activation of Cα2 hs1,2 Is Maximal in Germinal Center B Cells and Plasma Cells
To confirm the relevance of our findings to B cell ontogeny, we transfected human cell lines corresponding to sequential stages of B cell differentiation with the Cα2 hs1,2 (452–1133) enhancer-luc reporter gene vector, as driven by the β-globin promoter. We then measured Luc activity, the levels of endogenous germ-line IH-Ch and mature VH-DJH-CH transcripts, and the expression of HoxC4, Oct-1/Oct-2, and Oca-B, and we monitored the formation of HoxC4·Oct-1·Oct-2·Oca-B nucleoprotein complexes. Cα2 hs1,2 was not activated in pro-B cells (RS4;11), was moderately activated (4-fold) in pre-B cells (Nalm-6), and was significantly activated in early (4B6) and late (HS Sultan) germinal center B cells (12–50-fold) and plasma cells (U266) (10-fold) (Fig. 9A). Increased hs1,2-enhancing activity was associated with the appearance of germ-line IH-CH and/or mature V(D)J-CH transcripts, expression of HoxC4, Oct-1/Oct-2, and Oca-B transcripts and proteins, and the formation of related nucleoprotein complexes involving the ATTT and ATTTGCAT DNA motifs (Fig. 9, B–D). Thus, hs1,2 activation and consequent enhancement of IgH locus transcription is B cell stage-specific and occurs concomitantly with the formation of HoxC4, HoxC4/Oct-1, and HoxC4/Oct-2 nucleoprotein complexes.
Fig. 9. IgH hs1,2 activation is dependent on HoxC4, Oct-2, and Oca-B, and is B cell stage-specific.

A, human B cell lines representing different stages of B cell ontogeny were transfected with the luc -reporter vector containing the β-globin promoter and hs1,2 (containing regions 1, 2, and 3). Results represent the -fold induction of Luc activity expressed by the reporter gene vector containing the promoter only. Data are the mean values of three experiments plus standard deviations. B, expression of germ-line IH-CH and mature VHDJH-CH transcripts by pro-B cell (RS4;11), pre-B cell (Nalm-6), early germinal center B cells (4B6), late germinal center B cells (HS Sultan), and plasmacytoma cells (U266). β-Actin transcripts were used to normalize the amount of cDNA in each cell type. C, expression of HoxC4, Oct-1, Oct-2, and Oca-B in the same B cells as in A. Transcripts were detected by specific reverse transcriptase PCR analysis (RT-PCR, top panel) and proteins by immunoblotting (bottom panel). β-Actin transcripts and proteins were used as normalizing loading controls. D, EMSA involving hs1,2 region 2 HoxC4 and HoxC4/Oct DNA probes with nuclear extracts from the same B cells as in B. Arrowheads indicate each specified nucleoprotein complex(s). The amounts of proteins analyzed were normalized by calibrating Sp1 complex content using the Sp1 probe.
DISCUSSION
Together with our previous studies on the human Iγ and Iε promoters (25, 28), these findings point to HoxC4 as an important regulator of transcription in the human IgH locus and outline a critical role for this homeodomain protein in B cell differentiation. We show here that HoxC4 mediates activation of the 3′EH hs1,2 enhancer in human B cells by binding to newly identified conserved ATTT and ATTTGCAT motifs and through synergy with two other homeodomain proteins, Oct-1/Oct-2, and the Oca-B coactivator. By showing that hs1,2 is dominant over hs4 and hs3 in enhancing transcription, as driven by a human VH, ECS-IH or the ß-globin promoter, our experiments extend previous findings (18–21). Further, they show that hs1,2 can be segregated into three regions (1, 2, and 3), which are all necessary to enhance germ-line IH-CH and mature VHDJH-CH transcription. Finally, they define the minimal requirements for human hs1,2-mediated transcriptional enhancement and determine a B cell stage specificity in HoxC4-dependent activation of hs1,2 (25, 28).
The enhancement of VH1 or ECS-Iγ3 promoter-driven transcription suggests a primary role for hs1,2 in the overall function of the IgH 3′ regulatory region, as VH1 and ECS-Iγ3 are IgH locus promoters. The VH promoter is required for steady-state VH gene transcription, VHDJH gene rearrangement, and IgH SHM; the ECS-IH promoter is required for germ-line IH-Ch transcription and CSR. The enhancing activity displayed by hs1,2 in conjunction with the Ig-irrelevant β-globin promoter emphasizes the strength of hs1,2 as a bona fide enhancer (1, 5, 6, 38, 39). This dominant transcription-enhancing activity of human hs1,2 would be reflected in the SHM-enhancing activity displayed by this element, but not hs3 or hs4, when inserted downstream of a SHM “inducible” human DNA VHDJH-iEμ-Sμ-Sγ-Cγ1 construct2 (40).
Our analysis of the human Cα2 hs1,2 identified two HoxC4-binding sites, which are both critical for full hs1,2 activation. The 3′ cis-element is a HoxC4/Oct-binding site and was previously recognized as a mere Oct-binding site in the reports originally detailing the structure of the human IgH 3′ regulatory region (18–21). That Oct site was included together with Ets/AP-1 and E-Box sites in the ~0.3 kb PstI/PstI fragment P300. P300 comprised region 2, perhaps together with region 1 or 3, and was found to yield about 55% of the overall hs1,2-enhancing activity, to which neither region 1 nor region 3 was identified as a further contributor (19). In another study (18), a sequence corresponding to our region 2, but also containing one copy of a ~53-bp motif (equal to region 1 GRR4), was identified as an ECS lacking CTGCAGCTGCAGGT, which includes the E-Box site, and resulting in an overall structure/function of hs1,2 significantly different from that reported here. In that study, the cis-binding sites downstream of the ECS were not identified, nor was a “region 3” defined and shown to be required for full hs1,2 activation. An analysis of the rat hs1,2 element resulted in the dissection of the enhancer into three domains, designated as A, B, and C, which were shown to effect transcriptional enhancer activity (41). As in the human region 2, such enhancer activity was contributed mainly by the second hs1,2 DNA region, domain B. But unlike our human region 2, the main rat hs1,2 domain B cis-elements are a group of Ets-like binding sites.
The maximal activation of hs1,2 in human germinal center B cells originally went unrecognized, as IgG+ HS Sultan B cells were classified as a myeloma rather than a germinal center B cell line (18, 19, 34). It has been further confirmed here by the demonstration that hs1,2 is highly activated in 4B6 cells, which effectively express IH-CH transcripts and undergo CSR to IgG, IgA and IgE (28). The maximal activation of hs4 in Ramos B cells (42) further indicates that the 3′ regulatory region plays an important role in the IgH locus transcriptional regulation in germinal center B cells. However, significant hs1,2 activation was seen in murine Ig-secreting plasma cells but not in germinal center B cells (8–10, 43, 44), perhaps reflecting the presence of BSAP-binding motifs in this murine IgH enhancer element (5, 6, 39). By binding to hs1,2 and hs4, BSAP represses the 3′EH activity in murine germinal center B cells, and BSAP down-regulation is likely central to the full activation of the IgH 3′ regulatory region observed in plasma cells (45, 46). Because of the putative lack of BSAP-binding sites in the hs1,2 and hs4 sequences (2, 6, 19), human germinal center B cells would likely evade BSAP-mediated repression. This and the demonstration that, like hs3a, the murine hs1,2 is dispensable for germ-line IH-CH transcription and CSR (14) underscore significant differences between the mouse and human IgH 3′ regulatory regions.
Hox proteins are phylogenetically conserved helix-loop-helix homeodomain proteins that recognize the ATTT/A consensus (47, 48). They regulate embryonic pattern formation, axis specification and organogenesis, selective hematopoietic differentiation, and stem cell renewal (49). Genes belonging to the C cluster are preferentially expressed in developing and differentiated lymphoid lineages. HoxC4 is expressed in activated and/or proliferating T, B, and NK cells. No data, even from targeted deletions in the mouse (50, 51), have been available on HoxC4 function in the lymphoid system. Its early expression and nuclear localization suggest an involvement of HoxC4 in the regulation of genes controlling lymphocyte activation and/or proliferation (52). By defining critical new roles in the regulation of the IgH locus expression, our present and previous (28) findings point to HoxC4 as an important element in human lymphocyte differentiation.
HoxC4 synergizes with Oct-1/Oct-2 and the Oca-B coactivator, which it recruits to induce the human hs1,2 enhancer. Indeed, Oct-1/Oct-2 and Oca-B are components of the newly identified nucleoprotein complexes A and B, which assemble on the hs1,2 ATTTGCAT cis-element through recruitment of HoxC4 to the 5′-ATTT end (Fig. 7C). Although HoxC4/Oct-1 heterodimers can form and bind hs1,2 we propose that it is HoxC4/Oct-2 that is recruited preferentially, as suggested by the ChIP assays and the specific DNA binding by increased HoxC4 and Oct-2 proteins in germinal B cells (Figs. 7E and 9, C and D). This heterodimer assembly and recruitment further involves Oca-B. The ATTT cis-element would not recruit Oct-1/Oct-2 and Oca-B directly but through DNA-bound HoxC4, as suggested by protein-protein interaction experiments (Fig. 7F). The trans-factors recruited at the HoxC4 site would synergize with the HoxC4·Oct-1/Oct-2/Oca-B complex recruited at the HoxC4·Oct site to potentiate hs1,2 activation. This paradigm of transcriptional regulation through protein-protein cooperation has been effectively shown for other Hox homeodomain proteins in which the transcriptional activation function appears to be dependent on the nature of the target DNA sequence, implicating the importance of partner(s) or cofactor(s) and the relative properties of this interaction in mediating specific transcriptional regulation.
The HoxC4 homeodomain is important for hs1,2 activation, as expression of a mutant HoxC4 lacking this domain abolished hs1,2 activity. Overexpression of Oct-1, Oct-2, or Oca-B could not overcome this inactivation, presumably because the HoxC4 homeodomain mutant behaved as a potent “dominant negative” regulator of Oct-mediated hs1,2 activation. Oct-1 and Oct-2 are members of the POU family, a group of homeodomain-containing trans-factors that contain the DNA-binding POU domain. This comprises the “POU-homeodomain” and “POU-specific” subdomains (53). Oct-1 and Oct-2 regulate both general and cell type-specific genes (54), including VH and CH (53). Although Oct-1 is ubiquitous, Oct-2 is preferentially expressed in B cells. In the human IgH 3′ regulatory region, Oct-2 is required for not only hs1,2 (our data), but also hs4 activation (42). Accordingly, in Oct-2-deficient mice, B cell development to surface IgM expression is normal, but germinal center formation is impaired, and IgG1 and IgG3 levels are severely decreased (55, 56), indicating that Oct-2 is required for germinal center formation, CSR to secondary isotypes, and a high level of Ig transcription.
Coexpression of Oca-B and HoxC4 yielded the highest level of hs1,2 activation, presumably through interactions with the endogenous pool of Oct-1/Oct-2 proteins (Fig. 8). Oca-B (Oct coactivator from B cells, or Oct-binding factor-1 (OBF-1)) functions as an important transcriptional coactivator in B cells. It increases the binding affinity of Oct-1 and Oct-2 for DNA by clamping the POUH and POUS subdomains, which can further stimulate Oct-dependent gene transcription (57). Our demonstration that Oca-B plays an important role in HoxC4/Oct-2-mediated activation of human hs1,2 further emphasizes the critical role of this coactivator in B cell differentiation. Accordingly, Oca-B interacts with Oct-2 in modulating the activity of the 3′EH and IgH transcription in murine B cells (58, 59), and Oca-B−/− B cells stimulated with anti-CD40 and interleukin-4 fail to activate a luc-reporter gene construct bearing the regulatory hs3a-hs1,2-hs3b-hs4 cluster (44). Further, mice lacking Oca-B are viable and have normal serum IgM levels but lack GCs and show a significant impairment in CSR to IgG and serum IgG levels (60 – 62). Finally, Oct-2/Oca-B double deficient mice display a similar but more pronounced phenotype with impairment of T cell-dependent antibody responses (59).
The transcription-enhancing activity induced by the binding of HoxC4 to the human hs1,2 ATTT and ATTTGCAT motifs contrasts with the repression of germ-line Iγ-Cγ and Iε-Cε transcription that, as we showed (28), is dependent on recruitment of HoxC4 to ATTT sites embedded in the human ECS-Iγ and ECS-Iε promoters. ATTT motifs exist as multiple copies in the human ECS-Iγ and ECS-Iε promoters (28). Because of the lack of ATTT sites in the ECS-Iα1/Iα2 promoters, the Cα1/Cα2 loci can undergo CSR to IgA even when HoxC4 expression is up-regulated (28). The repression exerted by HoxC4 on the ECS-Iγ and ECS-Iε promoters, germ-line Iγ-Cγ and Iε-Cε transcription, and CSR to IgG and IgE is dependent on the recruitment of the Ku70/Ku86 heterodimer, as a mutant Ku70 lacking the homeodomain interaction motif relieved all HoxC4-mediated inhibition (28). As we show here (Fig. 7), instead of recruiting Ku70/Ku86, HoxC4 bound to human hs1,2 recruits Oct-1/Oct-2 and Oca-B, consistent with the notion that Hox proteins are multifunctional transcriptional regulators that interact with different cofactors and/or components of the transcriptional machinery depending on the broader structure of their target regulatory elements (63).
The combined recruitment of HoxC4 and Oct-2 to the HoxC4 and HoxC4/Oct sites, as complemented by Oca-B, would represent a paradigm of gene regulation by homeodomain transcription factors (Fig. 10). Once the HoxC4·Oct-1·Oct-2·Oca-B complex is bound to hs1,2, long-range interactions with VH and ECS-IH promoters, presumably by looping of the 3′ regulatory region, would confer greater IgH locus accessibility. This would result, perhaps in the context of a promoter competition mechanism as proposed in the mouse (12, 13, 64), in markedly differential enhancement levels of transcription, as seen with human ECS-Iα1/Iα2 and ECS-Iγ3 promoters (21). Further studies are needed to address such possible mechanisms and the role of hs1,2, hs3 and hs4 in the human IgH 3′ regulatory region as a LCR. Such studies would require the generation of constructs containing hs1,2, hs3 and/or hs4 together with the appropriate promoters and rearrangeable, switchable, or hypermutable Ig DNA for in vitro and in vivo expression, CSR, and SHM.
Fig. 10. IgH 3′ regulatory region in B cell differentiation.

A, induction of the IgH 3′ hs1,2 enhancer at sequential stages of (human) B cell differentiation. B, schematic depiction of the structure and putative long-range activity of IgH 3′ regulatory region. Once the HoxC4′Oct-1/Oct-2/Oca-B complex is bound to hs1,2, long-range interactions with VH and ECS-IH promoters, presumably by looping of the 3′ regulatory region, would confer greater IgH locus accessibility.
Supplementary Material
Acknowledgments
We thank Dr. Edward E. Max for human Cα2 hs1,2, hs3, and hs4 DNAs, HS Sultan cells, and β-globin promoter, as well as for insightful suggestions. We thank Dr. Robert G. Roeder for Oct-1, Oct-2, and Oca-B cDNAs, Dr. Max D. Cooper for the Nalm-6 cell line, Dr. Pengbo Zhou for HoxC4 cDNA, and Fatih Uckun for the RS4;11 cell line. We thank Dr. Hong Zan and Dr. Peggy Crow for helpful discussions. Finally, P. Casali thanks Dr. Sudhir Gupta for help and encouragement.
Footnotes
This work was supported by National Institutes of Health Grants AI 45011 and AR 40908 (to P. C.).
The on-line version of this article (available at http://www.jbc.org) contains supplemental Fig. 1.
The abbreviations used are: SHM, somatic hypermutation; BSAP, B cell-specific activator protein; C, constant region; ChIP, chromatin immunoprecipitation; CSR, class switch DNA recombination; D, diversity; ECS, evolutionarily conserved sequence; EH, IgH enhancer; EMSA, electrophoresis mobility shift assay; GRR, G-rich repeat; GST, glutathione S-transferase; IgH, immunoglobulin heavy chain; hs, DNase I hypersensitivity site; HD, homeodomain; IH, IgH DNA intervening region; iEμ, IgH intronic enhancer; J, joining; L, Ig light chain; LCR, locus control region; Luc, luciferase; μF, microfarad; mAb, monoclonal antibody; mt, mutant; Oca-B, Oct-coactivator from B cells; Oct, octamer; POU, Pict, Oct, Unc (homeodomain); Stat-1, signal transducer and activator of transcription-1; trans-factor, transcription factor; V, Ig variable (region); wt, wild type
A. Komori, X. Wu, E. Kim, H. Zau, and P. Casali, manuscript in preparation.
References
- 1.Henderson A, Calame K. Annu Rev Immunol. 1998;16:163–200. doi: 10.1146/annurev.immunol.16.1.163. [DOI] [PubMed] [Google Scholar]
- 2.Max EE. In: Fundamental Immunology. 4th. Paul WE, editor. Lippincott-Raven; Philadelphia: 1999. pp. 148–163. [Google Scholar]
- 3.Stavnezer J. Curr Top Microbiol Immunol. 2000;245:127–168. doi: 10.1007/978-3-642-59641-4_6. [DOI] [PubMed] [Google Scholar]
- 4.Honjo T, Kinoshita K, Muramatsu M. Annu Rev Immunol. 2002;20:165–196. doi: 10.1146/annurev.immunol.20.090501.112049. [DOI] [PubMed] [Google Scholar]
- 5.Birshtein BK, Chen C, Saleque S, Michaelson JS, Singh M, Little RD. Curr Top Microbiol Immunol. 1997;224:73–80. doi: 10.1007/978-3-642-60801-8_7. [DOI] [PubMed] [Google Scholar]
- 6.Khamlichi AA, Pinaud E, Decourt C, Chauveau C, Cogne M. Adv Immunol. 2000;75:317–345. doi: 10.1016/s0065-2776(00)75008-5. [DOI] [PubMed] [Google Scholar]
- 7.Pettersson S, Cook GP, Bruggemann M, Williams GT, Neuberger MS. Nature. 1990;344:165–168. doi: 10.1038/344165a0. [DOI] [PubMed] [Google Scholar]
- 8.Dariavach P, Williams GT, Campbell K, Pettersson S, Neuberger MS. Eur J Immunol. 1991;21:1499–1504. doi: 10.1002/eji.1830210625. [DOI] [PubMed] [Google Scholar]
- 9.Lieberson R, Giannini SL, Birshtein BK, Eckhardt LA. Nucleic Acids Res. 1991;19:933–937. doi: 10.1093/nar/19.4.933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Madisen L, Groudine M. Genes Dev. 1994;8:2212–2226. doi: 10.1101/gad.8.18.2212. [DOI] [PubMed] [Google Scholar]
- 11.Chauveau C, Jansson EA, Muller S, Cogne M, Pettersson S. Immunol. 1999;163:4637–4641. [PubMed] [Google Scholar]
- 12.Manis JP, van der Stoep N, Tian M, Ferrini R, Davidson L, Bottaro A, Alt FW. J Exp Med. 1998;188:1421–1431. doi: 10.1084/jem.188.8.1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cogne M, Lansford R, Bottaro A, Zhang J, Gorman J, Young F, Cheng HL, Alt FW. Cell. 1994;77:737–747. doi: 10.1016/0092-8674(94)90057-4. [DOI] [PubMed] [Google Scholar]
- 14.Pinaud E, Khamlichi AA, Le Morvan C, Drouet M, Nalesso V, Le Bert M, Cogne M. Immunity. 2001;15:187–199. doi: 10.1016/s1074-7613(01)00181-9. [DOI] [PubMed] [Google Scholar]
- 15.Tumas-Brundage KM, Vora KA, Manser T. Mol Immunol. 1997;34:367–378. doi: 10.1016/s0161-5890(97)00065-5. [DOI] [PubMed] [Google Scholar]
- 16.Terauchi A, Hayashi K, Kitamura D, Kozono Y, Motoyama N, Azuma T. J Immunol. 2001;167:811–820. doi: 10.4049/jimmunol.167.2.811. [DOI] [PubMed] [Google Scholar]
- 17.Morvan CL, Pinaud E, Decourt C, Cuvillier A, Cogne M. Blood. 2003;102:1421–1427. doi: 10.1182/blood-2002-12-3827. [DOI] [PubMed] [Google Scholar]
- 18.Chen C, Birshtein BK. J Immunol. 1997;159:1310–1318. [PubMed] [Google Scholar]
- 19.Mills FC, Harindranath N, Mitchell M, Max EE. J Exp Med. 1997;186:845–858. doi: 10.1084/jem.186.6.845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pan Q, Petit-Frere C, Stavnezer J, Hammarstrom L. Eur J Immunol. 2000;30:1019–1029. doi: 10.1002/(SICI)1521-4141(200004)30:4<1019::AID-IMMU1019>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 21.Hu Y, Pan Q, Pardali E, Mills FC, Bernstein RM, Max EE, Sideras P, Hammarstrom L. J Immunol. 2000;164:6380–6386. doi: 10.4049/jimmunol.164.12.6380. [DOI] [PubMed] [Google Scholar]
- 22.Pustell JM. Nucleic Acids Res. 1988;16:1813–1820. doi: 10.1093/nar/16.5.1813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Thompson JD, Higgins DG, Gibson TJ. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Quandt K, Frech K, Karas H, Wingender E, Werner T. Nucleic Acids Res. 1995;23:4878–4884. doi: 10.1093/nar/23.23.4878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schaffer A, Cerutti A, Shah S, Zan H, Casali P. J Immunol. 1999;162:5327–5336. [PubMed] [Google Scholar]
- 26.Ueki Y, Goldfarb IS, Harindranath N, Gore M, Koprowski H, Notkins AL, Casali P. J Exp Med. 1990;171:19–34. doi: 10.1084/jem.171.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ikematsu H, Harindranath N, Ueki Y, Notkins AL, Casali P. J Immunol. 1993;150:1325–1337. [PMC free article] [PubMed] [Google Scholar]
- 28.Schaffer A, Kim EC, Wu X, Zan H, Testoni L, Salamon S, Cerutti A, Casali P. J Biol Chem. 2003;278:23141–23150. doi: 10.1074/jbc.M212952200. [DOI] [PubMed] [Google Scholar]
- 29.Stong RC, Korsmeyer SJ, Parkin JL, Arthur DC, Kersey JH. Blood. 1985;65:21–31. [PubMed] [Google Scholar]
- 30.Hurwitz R, Hozier J, LeBien T, Minowada J, Gajl-Peczalska K, Kubonishi I, Kersey J. Int J Cancer. 1979;23:174–180. doi: 10.1002/ijc.2910230206. [DOI] [PubMed] [Google Scholar]
- 31.Cerutti A, Zan H, Schaffer A, Bergsagel L, Harindranath N, Max EE, Casali P. J Immunol. 1998;160:2145–2157. [PMC free article] [PubMed] [Google Scholar]
- 32.Zan H, Cerutti A, Dramitinos P, Schaffer A, Casali P. J Immunol. 1998;161:5217–5225. [PMC free article] [PubMed] [Google Scholar]
- 33.Zan H, Komori A, Li Z, Cerutti A, Schaffer A, Flajnik MF, Diaz M, Casali P. Immunity. 2001;14:643–653. doi: 10.1016/s1074-7613(01)00142-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Drexler HG, MacLeod RA, Dirks WG. Blood. 2001;98:3495–3496. doi: 10.1182/blood.v98.12.3495. [DOI] [PubMed] [Google Scholar]
- 35.Nilsson K, Bennich H, Johansson SG, Ponten J. Clin Exp Immunol. 1970;7:477–489. [PMC free article] [PubMed] [Google Scholar]
- 36.Zan H, Wu X, Komori A, Holloman WK, Casali P. Immunity. 2003;18:727–738. doi: 10.1016/s1074-7613(03)00151-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Denizot Y, Pinaud E, Aupetit C, Le Morvan C, Magnoux E, Aldigier JC, Cogne M. Immunology. 2001;103:35–40. doi: 10.1046/j.1365-2567.2001.01217.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Manis JP, Tian M, Alt FW. Trends Immunol. 2002;23:31–39. doi: 10.1016/s1471-4906(01)02111-1. [DOI] [PubMed] [Google Scholar]
- 39.Johnson K, Calame K. Curr Opin Genet Dev. 2003;13:522–528. doi: 10.1016/j.gde.2003.08.004. [DOI] [PubMed] [Google Scholar]
- 40.Wu X, Feng J, Komori A, Kim EC, Zan H, Casali P. J Clin Immunol. 2003;23:235–246. doi: 10.1023/a:1024571714867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Grant PA, Arulampalam V, Ahrlund-Richter L, Pettersson S. Nucleic Acids Res. 1992;20:4401–4408. doi: 10.1093/nar/20.17.4401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sepulveda MA, Emelyanov AV, Birshtein BK. J Immunol. 2004;172:1054–1064. doi: 10.4049/jimmunol.172.2.1054. [DOI] [PubMed] [Google Scholar]
- 43.Saleque S, Singh M, Little RD, Giannini SL, Michaelson JS, Birshtein BK. J Immunol. 1997;158:4780–4787. [PubMed] [Google Scholar]
- 44.Ong J, Stevens S, Roeder RG, Eckhardt LA. J Immunol. 1998;160:4896–4903. [PubMed] [Google Scholar]
- 45.Neurath MF, Stuber ER, Strober W. Immunol Today. 1995;16:564–569. doi: 10.1016/0167-5699(95)80078-6. [DOI] [PubMed] [Google Scholar]
- 46.Singh M, Birshtein BK. Proc Natl Acad Sci U S A. 1996;93:4392–4397. doi: 10.1073/pnas.93.9.4392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Saleh M, Rambaldi I, Yang XJ, Featherstone MS. Mol Cell Biol. 2000;20:8623–8633. doi: 10.1128/mcb.20.22.8623-8633.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Galant R, Carroll SB. Nature. 2002;415:910–913. doi: 10.1038/nature717. [DOI] [PubMed] [Google Scholar]
- 49.Hombria JC, Lovegrove B. Differentiation. 2003;71:461–476. doi: 10.1046/j.1432-0436.2003.7108004.x. [DOI] [PubMed] [Google Scholar]
- 50.Boulet AM, Capecchi MR. Dev Biol. 1996;177:232–249. doi: 10.1006/dbio.1996.0159. [DOI] [PubMed] [Google Scholar]
- 51.Saegusa H, Takahashi N, Noguchi S, Suemori H. Dev Biol. 1996;174:55–64. doi: 10.1006/dbio.1996.0051. [DOI] [PubMed] [Google Scholar]
- 52.Meazza R, Faiella A, Corsetti MT, Airoldi I, Ferrini S, Boncinelli E, Corte G. Blood. 1995;85:2084–2090. [PubMed] [Google Scholar]
- 53.Matthias P. Semin Immunol. 1998;10:155–163. doi: 10.1006/smim.1998.0117. [DOI] [PubMed] [Google Scholar]
- 54.Phillips K, Luisi B. J Mol Biol. 2000;302:1023–1039. doi: 10.1006/jmbi.2000.4107. [DOI] [PubMed] [Google Scholar]
- 55.Corcoran LM, Karvelas M, Nossal GJ, Ye ZS, Jacks T, Baltimore D. Genes Dev. 1993;7:570–582. doi: 10.1101/gad.7.4.570. [DOI] [PubMed] [Google Scholar]
- 56.Humbert PO, Corcoran LM. J Immunol. 1997;159:5273–5284. [PubMed] [Google Scholar]
- 57.Teitell MA. Trends Immunol. 2003;24:546–553. doi: 10.1016/j.it.2003.08.002. [DOI] [PubMed] [Google Scholar]
- 58.Tang H, Sharp PA. Immunity. 1999;11:517–526. doi: 10.1016/s1074-7613(00)80127-2. [DOI] [PubMed] [Google Scholar]
- 59.Schubart K, Massa S, Schubart D, Corcoran LM, Rolink AG, Matthias P. Nat Immunol. 2001;2:69–74. doi: 10.1038/83190. [DOI] [PubMed] [Google Scholar]
- 60.Schubart DB, Rolink A, Kosco-Vilbois MH, Botteri F, Matthias P. Nature. 1996;383:538–542. doi: 10.1038/383538a0. [DOI] [PubMed] [Google Scholar]
- 61.Kim MK, Lesoon-Wood LA, Weintraub BD, Chung JH. Mol Cell Biol. 1996;16:4366–4377. doi: 10.1128/mcb.16.8.4366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nielsen PJ, Georgiev O, Lorenz B, Schaffner W. Eur J Immunol. 1996;26:3214–3218. doi: 10.1002/eji.1830261255. [DOI] [PubMed] [Google Scholar]
- 63.Vigano MA, Di Rocco G, Zappavigna V, Mavilio F. Mol Cell Biol. 1998;18:6201–6212. doi: 10.1128/mcb.18.11.6201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Seidl KJ, Manis JP, Bottaro A, Zhang J, Davidson L, Kisselgof A, Oettgen H, Alt FW. Proc Natl Acad Sci USA. 1999;96:3000–3005. doi: 10.1073/pnas.96.6.3000. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.