

Abstract
The ability to characterize bacterial cell-wall composition and structure is crucial to understanding the function of the bacterial cell wall, determining drug modes of action and developing new-generation therapeutics. Solid-state NMR has emerged as a powerful tool to quantify chemical composition and to map cell-wall architecture in bacteria and plants, even in the context of unperturbed intact whole cells. In this review, we discuss solid-state NMR approaches to define peptidoglycan composition and to characterize the modes of action of old and new antibiotics, focusing on examples in Staphylococcus aureus. We provide perspectives regarding the selected NMR strategies as we describe the exciting and still-developing cell-wall and whole-cell NMR toolkit. We also discuss specific discoveries regarding the modes of action of vancomycin analogues, including oritavancin, and briefly address the reconsideration of the killing action of β-lactam antibiotics. In such chemical genetics approaches, there is still much to be learned from perturbations enacted by cell-wall assembly inhibitors, and solid-state NMR approaches are poised to address questions of cell-wall composition and assembly in S. aureus and other organisms.
Keywords: bacterial cell wall, peptidoglycan, S. aureus, antibiotics, solid-state NMR, whole-cell NMR
1. Overview
Gram-positive bacteria, including Staphylococcus aureus, surround themselves with a thick cell wall that is essential to cell survival and growth, and is a major target of antibiotics [1]. Penicillin and vancomycin are among the antibiotics that interfere with synthesis of the bacterial cell wall, yet patients are succumbing to infections caused by bacteria that have emerged resistant even to the drugs of last resort, such as vancomycin. S. aureus is commonly found on the skin and mucosal surfaces in healthy humans, but it can serve as a devastating pathogen when it colonizes undesired niches. Infection can result in pneumonia, endocarditis, bacteraemia and sepsis [2,3]. As many as 60% of clinically isolated strains of S. aureus are resistant to methicillin and other β-lactam antibiotics, and multi-drug resistant organisms or ‘superbugs' are wreaking havoc in the clinic [4–6].
The ability to characterize cell-wall composition and structure in a non-perturbative manner is crucial to understanding the structure and function of the bacterial cell wall, determining drug modes of action and developing new-generation therapeutics. The modes of action of penicillin and many classic antibiotics have been examined extensively during and after the golden age of antibiotics in which most antibiotics were discovered [7]. However, the Gram-positive cell wall is a heterogeneous insoluble macromolecular polymeric matrix that surrounds the cell and poses a challenge to non-perturbative and quantitative compositional analysis. The primary component and structural scaffold of the cell wall is the peptidoglycan, composed of repeating units of a disaccharide-multi-peptide building block that are polymerized and cross-linked to create a continuous network that envelops the cell (figure 1). Peptidoglycan biosynthesis is coordinated through the action of more than 10 proteins [8]. Different cell-wall inhibitors target distinct steps in peptidoglycan biosynthesis, ranging from inhibiting the production of the disaccharide inside the cell (fosfomycin) [9] to preventing the cross-linking of peptide stems outside the cell (penicillin) [10,11]. Wall teichoic acids and proteins are also covalently attached to the peptidoglycan to generate a complete cell wall that protects the cell from turgor pressure and external stress, and confers functional benefits in adhesion and host interactions during infection [12,13].
Figure 1.

Peptidoglycan assembly and the chemical composition of the primary constituents of the S. aureus cell wall. (a) A scanning electron micrograph of whole cells reveals the thick cell-wall layer surrounding S. aureus, alongside a TEM image of an isolated cell-wall sacculus. (b) The peptidoglycan precursor lipid II is assembled inside the cell with the pentaglycine bridge attached and is transported outside the cell to be incorporated into the peptidoglycan network through transglycosylation (to connect the glycan strands) and transpeptidation (to cross-link the peptides). (c) Chemical structure of the S. aureus peptidoglycan and wall teichoic acid. (Online version in colour.)
Radiolabelling studies during the 1950s and 1960s showed that many antibiotics leave signatures of their mode of action in the depletion and accumulation of specific cell-wall precursors or components [14–16]. Complementary but elaborate cell digestions and radiochemical assays or chromatographic and mass spectrometry-based detection of muropeptides attempt to quantify solubilized components (often using muramidase to cleave the MurNAc–GlcNAc disaccharide units), although complete digestion is not always possible, e.g. S. aureus [17–20]. Other creative biochemical strategies have been used to infer antibiotic modes of action, yet care should be taken as these can lead to conflicting conclusions owing to differences in the model organism or the in vitro assays being employed. Naturally, complementary methods are invaluable. Solid-state NMR is of great value in observing and quantifying compositional changes in intact cell walls and whole cells, and in mapping the placement of antibiotics to help understand antibiotic modes of action.
Solid-state NMR is well suited to defining composition and structural detail in complex and insoluble macromolecular systems including cell walls, intact cells, biofilms and other multicellular assemblies. Indeed, solid-state NMR has a rich history as an analytical tool to study the composition, structure, dynamics and function of solid materials, ranging from coal and earth materials, industrial polymers and catalysts to biomaterials including spider silk, insect exoskeletons, amyloids, membrane proteins, cell walls, whole cells, biofilms and intact tissues [21–27]. In this contribution, we discuss solid-state NMR approaches to define peptidoglycan composition and to characterize the modes of action of old and new antibiotics in cell walls and whole cells, focusing on examples in S. aureus. The specific examples are meant to provide a review as well as an experimental primer of how to examine fundamental cell-wall composition and changes owing to antibiotics or growth conditions using well-established solid-state NMR approaches. We provide perspectives regarding NMR strategy and the selection of specific NMR experiments as we describe the exciting and still-developing NMR toolkit that can be recruited to investigate bacterial cell walls and whole cells in different organisms. Through comparisons with biochemical methods and insights, we strive to make the NMR analyses conceptually accessible for non-NMR experts. We also discuss the discoveries regarding the modes of action of vancomycin analogues including oritavancin, and briefly address the reconsideration of the killing action of β-lactam antibiotics. Although not covered in this review, solid-state NMR has also been employed to investigate peptidoglycan architecture [28] as well as dynamics [29]. In addition, solid-state NMR approaches have helped to dissect the influences of antimicrobial peptides in disrupting membrane integrity in different lipid systems [30–33]. There are many future avenues to be explored and important problems to be addressed in understanding the composition, structure and function of cell-surface assemblies using solid-state NMR.
2. Solid-state NMR for quantifying composition in biological systems
(a). CPMAS
Simple one-dimensional solid-state NMR spectra of intact materials can provide us with important parameters of chemical composition. By way of background, most 13C and 15N solid-state NMR studies of biological solids employ cross-polarization magic-angle spinning (CPMAS) coupled with high-power proton decoupling to obtain NMR spectra. Introduced 40 years ago, CPMAS combines cross polarization (CP) and magic-angle spinning (MAS) [34]. MAS involves mechanically spinning the NMR sample rotor at the magic angle (54.74°) to average over the spatial coordinates of the dipolar coupling interactions and chemical shift anisotropy, something which is achieved without external spinning in solution-state NMR of smaller assemblies owing to rapid tumbling of molecules and proteins [35]. CP involves the transfer of magnetization typically from 1H to the observe nucleus of interest (13C or 15N in this review) yielding signal enhancements and also decreasing the experimental acquisition time owing to an aspect of CP that results in the spin–lattice relaxation time being determined by that of 1H rather than by the carbons, which in a solid material is much longer than that of 1H.
CPMAS is the platform for most biological solid-state NMR experiments today. Alternatively, one can also obtain spectra by direct excitation, where carbons are directly observed without CP. Some carbons, however, in biological solids can have very long T1 relaxation times, requiring tens of seconds or even minutes of time in between scans, and 10 000–100 000 scans can sometimes be required for natural abundance 13C spectra. Thus, direct excitation experiments can require considerably more time and may, ultimately, miss some signals from the solid material for spins that do not return to equilibrium upon subsequent NMR scans. CPMAS spectra can also be used quantitatively to compare absolute peak intensities across a spectrum by performing control experiments where the CP time is varied (typically arrayed between 100 µs and 10 ms) to determine whether there are different efficiencies of CP for some spins or different relaxation behaviour such that they need to be taken into account to scale the measured peak heights or areas. Recent uses of quantitative CPMAS have led to the determination of bacterial biofilm matrix composition in Escherichia coli and Vibrio cholerae [36,37]. In practice, CP corrections are often not necessary for the types of carbon spin systems in cell walls, whole cells and biofilms, but this should be determined if absolute quantification is desired.
(b). Access to dipolar couplings
A powerful aspect of CPMAS NMR is the fact that solution-like high-resolution spectra are achieved by sample spinning in a coherent way, such that the distance information contained in the dipolar couplings is not lost and can be measured by manipulating the spin coordinates with pulse sequences to re-introduce the dipolar couplings, which depend on the distance (as recently reviewed in [38,39]). In contrast, in solution NMR, molecules are rapidly tumbling, and the averaging of the dipolar couplings is random. Thus, long-range dipolar couplings cannot be accessed in solution without using strategies to restrict motion or align molecules in the sample. The major solid-state NMR recoupling measurement tool covered in this review is rotational echo double resonance (REDOR), which enables the measurement of long-range heteronuclear distances [40]. In the context of this review, REDOR can be used to measure a distance between an antibiotic and a specific cell wall site, for example, and permits the spectroscopic filtering of CPMAS spectra to select and quantify only one-bond pairs of interest, such as d-Ala–Gly cross-links, based on their strong dipolar coupling. In practice, REDOR experiments are done in two parts: one spectrum is collected with rotor-synchronized dephasing pulses on the dephasing spin (S), and a second spectrum is collected without dephasing pulses and provides the full echo spectrum (S0). The difference of the two yields the REDOR difference spectrum (ΔS) and reflects the observed spins that are coupled to the dephasing spin (reviewed in reference [39]).
(c). Multi-dimensional NMR
The power of one-dimensional NMR spectra to examine composition in biological systems is complemented by many solution-state and solid-state NMR studies employing multi-dimensional two-, three- and four-dimensional NMR methods, often to determine protein structures, where every atom is defined in space. Total structure determinations require multi-dimensional methods in order to separate and assign carbons and nitrogens for each residue in the protein or complex (or for as many residues as possible). Bacterial cell walls are not crystalline and do not yield the very sharp peaks associated with crystalline proteins, although two-dimensional NMR methods are still valuable in cell-wall studies as described in §6, and two- and three-dimensional experiments have been performed to study cell-wall assembly proteins including l,d-transpeptidase to provide evidence for its interaction with peptidoglycan [41]. Yet, there is great power in obtaining and analysing the one-dimensional NMR spectra of complex assemblies including cell walls and whole cells.
(d). NMR sample preparation
Finally, we would like to highlight an additional practical consideration when preparing samples for solid-state NMR studies. Most compositional and structural studies of the S. aureus peptidoglycan have been performed on lyophilized samples of whole cells and cell walls. Structural studies have been performed with antibiotics bound to cell walls or whole cells, typically prepared in trehalose- or ethanolamine-based lyophilization buffers, wherein trehalose serves as an effective hydrogen bonding partner as bulk water is removed during lyophilization. Such lyophilized formulations have permitted structure-based studies of enzymes such as lumazine synthase [42], EPSP synthase [43], factor Xa [44] and uracil DNA glycosylase [45], and are not perturbative to structure. The enzymes can maintain their enzymatic activity even after NMR measurements upon sample rehydration and assay. This was specifically reported for uracil DNA glycosylase, where greater than 60% activity was recovered after 2 years in the lyophilized state, typical of the decrease observed for normal hydrated enzyme stocks stored at −80°C [45]. Proteins such as insulin and others of interest to the pharmaceutical industry are regularly prepared and supplied as lyophilized formulations, and bacteria are often stored and shipped as lyophilized powders. Naturally, care should always be taken to verify or optimize ideal lyophilization buffer conditions for new systems.
As highlighted in §14, important structural insights have emerged regarding the modes of action of antibiotics, including oritavancin [46,47] and plusbacin [48] and how they bind to the cell wall, with data from both whole-cell and cell-wall samples. For studies of only chemical composition, samples are often lyophilized without special buffers, because any subtle or even dramatic changes in conformations would not affect chemical bonding and composition, and this also avoids natural abundance contributions of buffer components to NMR spectra. Compared with hydrated samples, NMR sensitivity is higher in lyophilized samples [49,50], and the prevention of high-frequency motions also permits the accurate determination of dipolar couplings and hence distances in antibiotic complexes, for example. Hydrated samples, on the other hand, have advantages when one aims to obtain sets of relaxation parameters to evaluate dynamics in biological systems. Studies of this type can be performed on hydrated whole cells or hydrated isolated peptidoglycan [29,51]. Yet, cell-wall dynamics should preferentially be measured in the whole-cell context. Dipolar rotational spin echo solid-state NMR experiments on actively metabolizing Aerococcus viridans showed that amide NH dipolar sideband patterns retained 50% coupling and thus, while the cell wall in whole cells is naturally ‘fully hydrated’, it does not exhibit the solution-state-like motional averaging that can be observed with isolated cell walls [49].
3. Cell-wall preparations
The study we will discuss first is actually the most recent one, but the NMR comparisons provide the most fundamental and general information that can be readily obtained from solid-state NMR of any cell wall system. The cell wall preparations were based on a carefully optimized cell wall preparation that was reported in 2012 to stringently remove unbroken cells with their unwanted cytoplasmic contributions that have been observed in some older NMR studies using conventional cell-wall preparation protocols [52]. In particular, fluorescence imaging of DAPI-stained cell walls confirmed that no DNA or proteins from unbroken cells were present in the peptidoglycan preparations (figure 2a). The protocol included several low-speed spins (5000g) after the typical procedure of: bead-beater lysis, boiling SDS treatment, protease and DNase digestion and centrifugation (38 000g). This served to remove remaining high molecular mass cell debris (mostly unbroken cells), which actually remains after these steps. It may be useful for cell-wall researchers to note the analytical yields of the cell-wall preparations using this protocol, based on growth in S. aureus synthetic medium (SASM). Cell wall yields by lyophilized dry mass of cell walls with respect to whole cells were 16% for cells harvested at OD660 0.7, 18% at OD660 2.0 and 21% at OD660 4.0. These harvests corresponded to growth times of 3.5, 8 and 12 h, respectively, growing 300 ml cells shaking in 1 l flasks, each started with a 1 : 300 inoculum of an overnight culture.
Figure 2.

Cell-wall preparation and characterization. (a) Fluorescence microscopy of DAPI-stained whole cells (top) and isolated peptidoglycan (bottom) confirms the removal of whole-cell contaminants in the stringent cell wall preparation. (b) 13C and 15N CPMAS spectra of S. aureus cell walls reveal the carbon and nitrogen composition of isolated cell walls. (c) 13C{15N} REDOR performed for 1.68 ms (12 Tr) of [u-15N]-labelled cell walls reveals directly bonded 13C–15N pairs owing to peptide-bond carbons and α carbons. The difference spectrum (ΔS) represents the exact difference of the S0 (full-echo) spectrum and the S (dephased) spectrum and shows just the carbons that were selected through their one-bond dipolar coupling to nitrogen. (d) The 15N CPMAS spectrum (black) reveals all nitrogens in the sample. Frequency-selective 15N experiments select one or the other of two amide peaks in the 15N spectrum (red and blue spectra obtained from separate frequency-selective experiments). (e) Frequency-selective 13C{15N} REDOR measurements with a 2.24 ms evolution time. Recoupling with the downfield 15N peak at 120 ppm results in dephasing of carbonyls and preferential dephasing of the non-Gly α carbons centred at 55 ppm (left, blue). Recoupling with the upfield 15N peak centred at 107 ppm results in preferential dephasing of the 42-ppm Gly α carbons (right, red). Adapted from Nygaard et al. [53] with permission from Elsevier. (Online version in colour.)
4. Carbon and nitrogen composition in Staphylococcus aureus cell walls
In this first example, a general approach to profile cell-wall composition was reported which can be performed with 13C at natural abundance (with no isotopic labelling required) or on uniformly 13C-labelled samples (which makes possible other correlation experiments that require higher enrichment levels) [53]. One-dimensional NMR spectra of isolated cell walls reveal the unique compositional signatures of cell-wall material (figure 2b). The carbon contributions include carbonyls from each peptide and from the acetyl modifications on GlcNAc, MurNAc and teichoic acid ManNAc (166–182 ppm); anomeric (100 ppm) and ring sugar carbons (55–90 ppm) from the aforementioned sugars, as well as ribitol carbons present in teichoic acid (62–75 ppm); α carbons from Gly (42 ppm); α carbons from l-Ala, l-Lys and d-Ala (48–58 ppm) and aliphatics from amino acid sidechains (10–38 ppm). A cell-wall spectrum reflects the total compositional make-up of the cell wall, with its five amino acids, pentaglycine bridge, subunit disaccharide, plus teichoic acid. REDOR NMR was also employed in this work to confirm assignments of the Gly and other peptide α-carbon signals. The cell-wall 15N spectrum reveals the highly cross-linked nature of the material (as amides between 100 and 140 ppm) with little detectable amine intensity, and no significant lysyl amine observed from non-cross-linked lysines (figure 2b), which would exhibit a peak near 32 ppm, as shown later [52]. There are no amines reporting from the wall teichoic acid d-Ala either, as the alkaline cell wall preparation protocol is known to readily de-esterify the appended d-Ala [54,55].
The spectra in figure 2c–e demonstrate how additional atomic-level specificity can be accessed for 15N-labelled samples using REDOR. For these REDOR experiments, samples have 13C present at natural abundance levels (no 13C labelling) as uniform 13C labelling and the resulting homonuclear 13C couplings interfere with the measurement of longer-range heteronuclear dipolar couplings. For a 13C{15N} REDOR experiment with an evolution time of 1.68 ms to select one-bond C–N pairs, the α carbons and the carbonyls exhibit nearly complete dephasing as expected, because all carbonyls in the cell wall and all α carbons, in general, are one bond away from a nitrogen (figure 2c). Naturally, in other types of samples, e.g. whole cells, lipid systems and bacterial extracellular matrix material, the percentage of carbonyls adjacent to a nitrogen could be reduced, and there could be other types of carbons that contribute intensity to the upfield α-carbon region without being adjacent to a nitrogen [39,40]. This can be quantified by REDOR. In S. aureus cell walls, figure 2d,e shows results corresponding to a frequency-selective REDOR experiment that highlight how the spectral contributions in figure 2c can be annotated even more specifically. In particular, by observing all carbons and dephasing with only the non-Gly amide nitrogens (those represented in the experimental frequency-selective spectrum in blue in figure 2d), only the non-Gly α carbons are dephased. When frequency-selective recoupling pulses are applied only to the Gly amide nitrogens (contributions from the nitrogens observed in the red spectrum in figure 2d), primarily Gly α carbons are dephased. Of note here, is the ability to identify the glycine carbons and nitrogens. These measurements could be used to compare cell-wall glycine content among a sample set. In addition, the ability to select these specific carbons and nitrogens would also make possible the measurement of distances from these sites to bound antibiotics, as discussed for selectively labelled samples in §13.
5. Detecting cell wall content and major alterations in whole-cell NMR spectra
Signatures of cell walls can be identified in whole-cell NMR spectra, again for either unlabelled or uniformly labelled cells. The use of selective labelling will be highlighted in §7. In particular, with a high ratio of sugar to peptide, reductions in cell wall are manifest by reductions in the polysaccharide region of a whole-cell spectrum [53]. The most straightforward demonstration of this is illustrated by the comparative 13C CPMAS spectra of whole cells and protoplasts (figure 3a). Protoplasts are osmotically delicate cells in which the majority of the cell wall has been removed by lysostaphin treatment [56,57]. Protoplasts must be stabilized by sucrose or trehalose to prevent cell lysis from the turgor pressure of the cell. The protoplast preparation in figure 3a was stabilized by the addition of trehalose, which does not contribute to the anomeric carbon peak intensity near 100 ppm as sucrose would. The anomeric carbon peak intensity was reduced in the protoplast preparation, consistent with the reduced cell-wall content. The residual anomeric peak intensity can arise from the thin cell wall that remains cell-associated as well as anomeric carbons associated with lipoteichoic acid [58]. In the whole-cell and protoplast spectra, one can observe the many other cellular carbon contributions, including nucleic acids and the full repertoire of proteins and lipids not present in the clean cell-wall spectrum in figure 2b.
Figure 3.
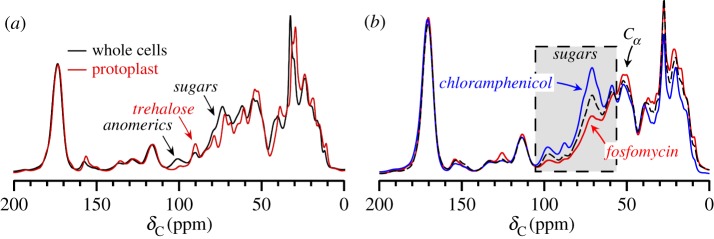
Spectral comparisons of S. aureus whole cells, protoplasts, and antibiotic-treated cells. (a) Protoplast spectra exhibit a reduced polysaccharide region owing to cell-wall removal. (b) Antibiotic-treatment with fosfomycin (a cell wall inhibitor) and chloramphenicol (a protein synthesis inhibitor) results in significantly altered carbon composition in whole cells. From Nygaard et al. [53] with permission from Elsevier. (Online version in colour.)
This general inspection of whole cell carbons at natural abundance was sensitive enough to reveal contrasting compositional changes owing to the action of either a cell-wall synthesis inhibitor or a protein synthesis inhibitor. Untreated cells were compared with cells treated with fosfomycin and chloramphenicol (figure 3b). Fosfomycin is a cell-wall biosynthesis inhibitor that inactivates the enzyme MurA, preventing the formation of muramic acid from N-acetylglucosamine, which is ultimately required inside the cell to generate cell-wall precursors [9,59,60]. Chloramphenicol targets the bacterial ribosome to generally inhibit cellular protein synthesis by interfering with peptidyl transferase activity [61]. Compared with untreated cells, fosfomycin treatment resulted in notable carbon pool decreases among polysaccharide carbons and glycine α carbons owing to the inhibition of cell-wall synthesis. In contrast, treatment with the protein synthesis inhibitor chloramphenicol resulted in an altered compositional balance where the cell-wall spectral contributions were increased relative to untreated cells. Carbons associated with proteins were decreased as cytoplasmic proteins were not replenished during antibiotic treatment. Thus, the cell-wall carbons represent a higher percentage of the whole-cell carbon pools after treatment with chloramphenicol. This example demonstrates that relevant changes in carbon composition can be captured through whole-cell NMR spectra. This approach is general, does not require selective labelling strategies, and can be implemented to examine cell-wall alterations, such as those owing to antibiotics, genetic mutations or other environmental conditions.
In addition to using traditional CPMAS methods, the use of dynamic nuclear polarization (DNP) has been used to enable sensitivity enhancements in the acquisition of spectra from biological solids, including bacterial and plant cell walls [62,63], through the transfer of polarization from electrons (with a much larger gyromagnetic ratio than nuclei) to nuclei. The use of DNP typically requires the use of an external paramagnetic polarizing agent to be added to the sample and that the polarizing agent be accessible to the nuclear spins which are to be enhanced [64]. In this way, DNP can also enable the selective observation of compositional contributions from the outside of the cell versus those from inside the cell, for example, if the polarizing agent stays on the outside and cannot penetrate the cell [62].
6. Two-dimensional NMR of cell-wall signatures in cell walls and whole cells
Two-dimensional NMR experiments have also been used to identify bacterial cell-wall nuclei, including work by Vollmer & Simorre [41,51,65], and even revealed the complexation of divalent ions with peptidoglycan [29]. Changes in chemical shifts have also been noted to occur when proteins bind to the cell wall, inferring that the protein may be binding to those residues that experienced a chemical shift change [41]. A representative set of cell wall and whole cell two-dimensional 13C spin diffusion-based spectra obtained in our laboratory is shown in figure 4 and is useful for qualitative insight and comparisons [53]. The carbohydrate spin system, reflected by 13C–13C correlations, is comparable between cell walls and whole cells. A one-dimensional slice of the two-dimensional spectrum, with correlations arising from the anomeric 13C near 100 ppm, was nearly identical in cell walls and whole cells. This is consistent with the comparative analysis using protoplasts (figure 3a) that demonstrated that a significant portion of the anomeric signal intensity in whole cells results from cell-wall material.
Figure 4.

Polysaccharide correlations in cell walls and whole cells. Comparative two-dimensional 13C–13C DARR spectra (using a spin-diffusion mixing time of 2 s) for [u-13C, u-15N]-labelled whole cells (black) and cell walls (red) allow the identification of cell-wall signals in the whole-cell sample, with clear overlap in the polysaccharide spin system. One-dimensional traces (top) identify the carbons arising from spin-diffusion starting at the anomeric carbon at 100 ppm. From Nygaard et al. [53] with permission from Elsevier. (Online version in colour.)
7. Unique lysine sidechain linkages in the cell wall and specific labelling
Lysine is a key amino acid present in the peptidoglycan of many bacterial cell walls. In S. aureus, the mature peptidoglycan precursor, lipid II, consists of the disaccharide GlcNAc–MurNAc with its attached pentapeptide stem with a pentaglycine bridge connected to the stem through the lysine sidechain [66]. This yields an amine-to-amide transformation for the lysyl sidechain nitrogen and an associated change in chemical shift (figure 5), where lysyl amines appear at 32 ppm and amides at 117 ppm when spectra are referenced to external liquid ammonia. Historically, older solid-state NMR studies used solid ammonium sulfate referenced to 0 ppm as a 15N chemical shift reference standard, although more recently and most commonly, 15N chemical shifts have been referenced to external ammonia. This results in ppm values that are similar to solution NMR, and we have also adopted this convention. Thus, in some older cell-wall papers, the amine and amide chemical shifts are approximately 7 and 92 ppm, respectively [68]. We have adjusted older figures in this review to the current convention.
Figure 5.
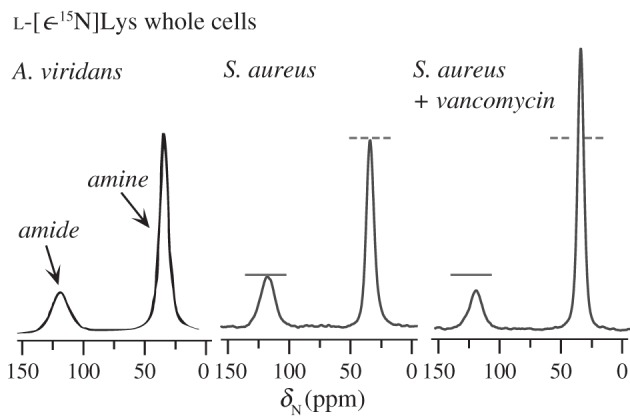
Comparison of whole-cell lysine incorporation for A. viridans and S. aureus biosynthetically labelled with l-[ε-15N]Lys and the influence of vancomycin. The 15N CPMAS spectra of l-[ε-15N]Lys labelled whole cells of A. viridans (left) and S. aureus (middle) reveal a lysyl amide attributed to peptidoglycan or peptidoglycan precursors with lysine sidechain linkages. Vancomycin treatment of S. aureus results in a reduced amide peak and an increased amine peak (right), consistent with inhibition of cell wall synthesis and the accumulation of Park's nucleotide. Reproduced with permission from Cegelski et al. [67]. Copyright © 2002 American Chemical Society.
The manifestation of the amidation of the lysine sidechain in peptidoglycan and the ability to detect it in whole-cell samples was first reported just over 30 years ago in 1983 by Jacob et al. [69] in the solid-state NMR examination of A. viridans whole cells (figure 5a). After introducing CPMAS in 1976, Schaefer was the first to implement solid-state NMR to examine whole-cell systems and bacterial cell walls, including studies with A. viridans [69], B. subtilis [70], S. aureus [50] and E. faecium [71], although the most extensive work to date has resulted from experiments with S. aureus.
In A. viridans, the lysine participates directly in a cross-link with the d-Ala of a neighbouring stem, in contrast to S. aureus where the final glycine of the pentaglycine bridge cross-links to the neighbouring d-Ala. A comparison of the lysine labelling of whole cells from A. viridans (from 1985) [72] and of S. aureus (from 2002) [67] is provided in figure 5 to emphasize the overall similarity in the amide-to-amine ratios for cells labelled with l-[ε-15N]Lys in the two Gram-positive organisms where lysine sidechains are amidated in the cell wall (and in lipid II). The amide-to-amine ratio can vary as a function of external stimuli as described in §8.
8. Whole-cell lysine and accumulation of Park's nucleotide
Vancomycin prevents cell-wall synthesis by binding to and sequestering lipid II at the cell surface. During cell-wall synthesis, while still inside the cell, lipid II is anchored to the cell membrane by a lipid tether (an undecaprenyl phosphate) [5]. The lipid carrier is present in limiting quantities and is typically recycled for multiple turnovers of lipid II to the cell surface [73]. Thus, when vancomycin is introduced, lipid II is prevented from being incorporated into the growing glycan strand (preventing the process of transglycosylation) and the C55 lipid tether is not available for newly synthesized lipid II inside the cell. Thus, Park's nucleotide accumulates, akin to a build-up of water resulting from a blocked pipe [74].
Upon treatment with vancomycin, the amine signal from l-[ε-15N]Lys-labelled whole cells increases relative to that of the amide signal (figure 5c), consistent with expectations, and the amide decreases as vancomycin decreases the total cell-wall content during the time of antibiotic treatment. This analysis is simple to execute and requires only lysine-labelled whole cells. From this one observation and in the absence of other information, though, it is possible that an increase in the lysyl amine peak could have arisen simply from overall increased protein content or, as described in §9, from imperfect cell-wall units containing lysine without a glycine bridge that are transported to the cell surface. Alternative strategies, including one that identifies d-Ala–d-Ala pairs as described in §11 have been used to resolve this ambiguity and have shown that d-Ala–d-Ala-containing stems, as in Park's nucleotide, are increased upon treatment with vancomycin [4,47].
9. Cell-wall lysines and detection of imperfect cell-wall units
An unanticipated discovery was made using selective lysine labelling from what were initially thought to be control experiments. The cell-wall morphology and the peptidoglycan structure were determined to be functions of growth stage and glycine availability when cells were grown in SASM [52]. Specifically, S. aureus cells at stationary phase had thicker cell walls compared with cells in exponential phase (figure 6a, top). This had been observed by electron microscopy before this study, but without characterization of the cell-wall composition [75,76]. Using detection by solid-state NMR, cell walls labelled with l-[ɛ-15N]Lys isolated from S. aureus at OD 0.7, OD 2.0, OD 3.6 and OD 4.0 exhibited an increasingly intense amine signal, indicating the emergence of a second population of lysine residues lacking a glycine bridge in the insoluble cell wall (figure 6a, bottom). By OD 4.0, 12% of all the lysine in peptidoglycan lacked a glycine bridge as quantified by NMR. LC–MS could identify the unique molecular mass attributed to this motif in fully digested cell walls (figure 6b), but owing to the insoluble material that typically remains even after extensive hydrolysis, relevant quantification was not possible. The presence of peptidoglycan units without a bridge is a phenomenon that has been observed previously in normal growth conditions for the COL strain of S. aureus, as detected by LC–MS, although it was not studied in detail to understand the origin or manifestation of the modification [19].
Figure 6.
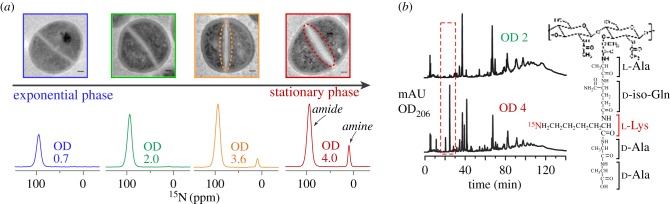
Cell-wall plasticity and glycine-dependent structural alterations in peptidoglycan. (a) Cell walls are thicker in the stationary phase as glycine is depleted from the medium and NMR identified the emergence of PG stems without pentaglycine bridges. (b) HPLC and mass spectrometry analysis identified the alteration in cell-wall digests, which is convenient for rapid identification, but lacks the quantitative power of the NMR analysis. Reproduced with permission from Zhou & Cegelski [52]. Copyright © 2012 American Chemical Society. (Online version in colour.)
Mechanistically, the production of the structurally altered peptidoglycan in the NMR study occurred concomitantly with the depletion of glycine from the medium as monitored by solution NMR of the medium supernatant when grown with [2-13C]Gly. Furthermore, this phenomenon was prevented by the addition of excess glycine to the nutrient medium. Pulse-chase labelling experiments, using l-[ε-13C,15N]lysine added in the stationary phase to cells already growing in the presence of singly labelled l-[ε-15N]Lys, also demonstrated that the structural changes arose within newly synthesized peptidoglycan rather than through the modification of previously synthesized peptidoglycan [52].
FemX is responsible for attaching the glycine bridge to lysine in S. aureus, and a femX knock-out mutation is lethal [77], so it was surprising that S. aureus would continue cell wall assembly without sufficient glycine. Bacteria could have slowed down peptidoglycan synthesis until endogenous glycine synthesis could catch up with the demands of cell-wall assembly in glycine-depleted medium. Yet, they instead transported imperfect peptidoglycan units. Collectively, these observations emphasize the plasticity in bacterial cell-wall assembly and the ability to manipulate peptidoglycan structure with external stimuli. Ultimately, this finding also encourages the design of a strategy to mimic that glycine deprivation in a molecular sense to encourage the transport of stems without bridges to weaken the cell wall.
10. d-Alanine cross-links
The presence of d-amino acids, particularly d-Ala in the S. aureus cell wall results in a d-Ala–Gly pair at the cross-link site, with a unique C–N bond resulting from d-[1-13C]Ala and [15N]Gly labelling. This bond appears only in the cell wall, and thus can be detected in whole-cell spectra even in the context of all the cellular contents and intracellular peptidoglycan precursors (figure 7). Typically, this is performed by taking advantage of the one bond C–N dipolar coupling between the 13C carbonyl of d-Ala and the 15N of glycine. Although not used in the earliest solid-state NMR cell-wall studies [50,67,79], most recent reports (since 2003) using l-Ala or d-Ala isotopic labelling employ the use of a racemase inhibitor (typically alaphosphin) to prevent scrambling of d-Ala labels into l-Ala and vice versa [46–48,78,80,81]. The REDOR difference spectrum, ΔS, represents only nitrogens that are dephased by d-[13C]Ala, i.e. cross-link sites in the cell wall. Penicillin is known to prevent cross-link formation [10,16,82] and, as anticipated, a clear decrease in 13C–15N bond density was observed for cells after treatment with penicillin (figure 7) [78]. A reduction in cross-linking was also observed for the antibiotic oritavancin, whereas vancomycin had no significant effect on the cross-link density.
Figure 7.
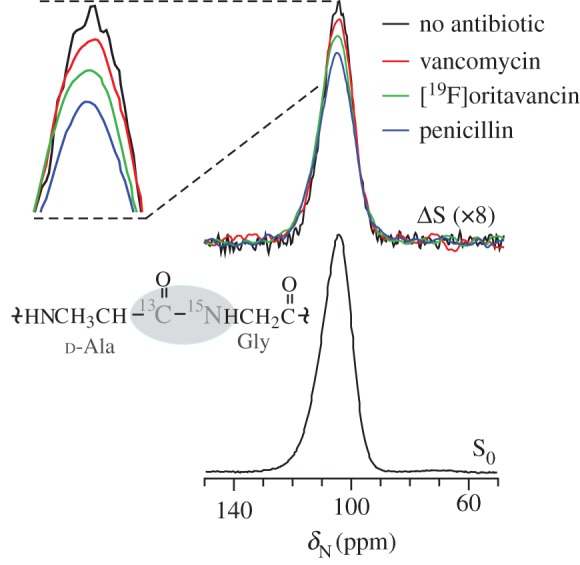
The influence of antibiotics on cross-links detected by whole-cell NMR. REDOR can quantify the density of Gly-d-Ala cross-links in whole cells using [15N]Gly and d-[1-13C]Ala labelling. Of all the glycines in the cell, a d-Ala–Gly pair appears only at the cross-link site in peptidoglycan, resulting in a uniquely labelled 13C–15N bond. The REDOR difference spectrum (ΔS) reports on the relative number of cross-links. Penicillin has the largest effect and inhibits cross-link formation. Vancomycin has no significant effect and oritavancin exhibits intermediate inhibition of cross-linking. Adapted fom Kim et al. [78] with permission from Elsevier. (Online version in colour.)
Bernhardt and co-workers [83] recently described an alternative model to understanding the killing action of beta-lactam antibiotics, specifically for mecillinam in E. coli, using pulse-chase labelling experiments with radioactive [3H]-labelled meso-diaminopimelic acid, which in E. coli takes the place of lysine in the peptidoglycan and directly cross-links to the d-Ala in a neighbouring stem. Although penicillin does inhibit transpeptidation and cross-linking, the authors pointed to prior studies in which cells are killed but without cell-wall thinning and lysis, and used their new results to suggest that aberrant functioning of the balance between cell-wall assembly and degradation may serve as the major killing mechanism. Specifically, an active and futile cycle of cell-wall synthesis coupled with degradation and turnover induces the depletion of cellular peptidoglycan precursors, taxing cellular resources and inducing toxicity. This work focused on mecillinam-treated E. coli and identified key genetic and molecular determinants for these phenomena [83].
Interestingly, solid-state NMR measurements of penicillin-treated A. viridans cell walls and whole cells in 1985 by Wilson et al. [72] also resulted in unanticipated results wherein mature peptidoglycan production increased in the presence of 0.2 μg ml−1 penicillin, even though the cross-link density was reduced. Furthermore, they found that the diminution in cross-linking levelled off before the total inhibition of growth and they similarly invoked that another mechanism must be responsible for the cell-killing action [72]. No major changes in cellular peptidoglycan precursors (measured by lysine labelling) were observed. This 1985 work concluded that the data could be explained by the presence of a ‘feedback mechanism which normally limits peptidoglycan production’ and that penicillin could be resulting in ‘metabolic disruption, which redirects a major portion of the cell's resources and metabolites to the cell wall at higher penicillin concentrations, [contributing] to the drug's lethal action on this bacterium’ [72]. One cannot extrapolate these exact results directly to S. aureus, but future work can evaluate such phenomena by solid-state NMR in S. aureus. The quantification possible with the solid-state NMR methodology and the non-perturbative method of analysis positions it as a powerful analysis tool for these types of investigations of cell-wall assembly and antibiotic activity.
11. Spectroscopic identification of distinct d-Ala contributions in whole cells
An approach to dissect and define the different d-Ala contributions in whole cells was introduced for S. aureus [46,78] and later extended to Enterococcus faecium [71], as highlighted in figure 8. In whole cells, there are four types of d-Ala as shown in figure 8: (i) the peptide carbonyl in cross-linked stems ending in d-Ala (blue); (ii) the peptide carbonyl of d-Ala-4 of d-Ala–d-Ala stems (red); (iii) the d-Ala-5 terminal carboxyl of those stems (green) and (iv) d-Ala in lipoteichoic acid and wall teichoic acid (grey). The full-echo spectrum is shown in black and contains all d-Ala contributions. The d-Ala contributions from peptide stems can be analytically determined by spectral selection. For example, d-Ala that is cross-linked to Gly was selected by 13C{15N}REDOR in cells labelled with d-Ala and Gly (figure 8, blue). By difference, the remaining d-Ala contributions are attributed to teichoic acid d-Ala. For this review, we include a spectral comparison of the 13C NMR spectrum of d-[1-13C]Ala-labelled whole cells with that of isolated cell walls in which esterified d-Ala is lost from teichoic acid, further confirming the identification of the right-side shoulder in whole cells as arising from teichoic acid d-Ala. This analysis can be used to compare the overall d-Ala contributions in other whole cell samples and the extent of d-Ala present in teichoic acids without cellular perturbations.
Figure 8.
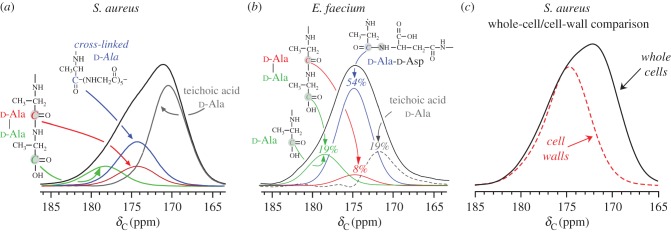
d-Ala deconvolution and contributions in S. aureus and E. faecium. (a,b) Complete accounting of whole-cell d-alanine is possible by experimental deconvolution using [15N]Gly/d-[1-13C]Ala-labelled whole cells. d-Ala contributions from peptide stems are analytically determined by spectral selection. By difference, the remaining d-Ala contributions are attributed to teichoic acid d-Ala. (c) Direct comparison of the 13C NMR spectrum of whole cells with isolated cell walls in which esterified d-Ala is lost from teichoic acid confirms the identification of teichoic acid d-Ala. Adapted from Kim et al. [78] (with permission from Elsevier) and [71] (Copyright © 2008 American Chemical Society). (Online version in colour.)
12. Lysine–glycine contacts reveal compact glycine bridges in the cell wall
A combination of l-[ɛ-15N]lysine–[1-13C]glycine labelling resulted in the discovery that the pentaglycine bridge in S. aureus cell walls is compact [50]. Using a REDOR evolution time to detect one-bond couplings, one observes that 20% of the glycine 13C signal is dephased by lysine. This is expected as only one of five glycines in the bridge is one bond away from the lysyl nitrogen. Extending the REDOR evolution time in subsequent measurements, 60% dephasing was realized by 15 ms, consistent with most of the glycyl carbonyls being within 5 Å, and established that in intact cell walls, the bridge is compact, with the lysyl amine approximately 5 Å from the farthest glycyl carbonyl, similar to the distance for an α-helical arrangement. The chemical shift of the glycyl carbonyl is additionally consistent with an α-helical conformation for the bridge [79,84]. Such a measurement is important to make in intact cell walls as a glycine bridge in a soluble cell-wall surrogate is not cross-linked and spatially confined as it is in the cross-linked network of the native cell wall [85]. This result begins to address elements of the three-dimensional architecture of the cell wall, but resulted from the same simple control experiments used to confirm labelling enrichment and glycine bridge lengths. In addition, Sharif et al. [86–88] found that a vancomycin-intermediate-resistant strain (S. aureus Mu50) had a more compact peptidoglycan than in a methicillin-resistant strain (S. aureus BB225). The conformational differences in these cell walls could have implications for the modes of action of antibiotics that interact with the glycine bridge.
13. Antibiotic modes of action and antibiotic-cell-wall complexes
An important application of solid-state NMR to study the bacterial cell wall has been its use in elucidating the modes of action of cell-wall synthesis inhibitors and in mapping the binding of antibiotics to components of the Gram-positive cell wall. These studies have been particularly useful in understanding vancomycin-like glycopeptides developed to treat vancomycin-intermediate or vancomycin-resistant S. aureus (VISA and VRSA, respectively). Vancomycin, oritavancin and plusbacin-A3 are some of the specific glycopeptide antibiotics that have been studied [46,48,67,79]. Although an entire review could be dedicated to these studies (two recent reviews did cover some of this work [28,39]), we briefly summarize some of the key discoveries and strategies here to highlight the extensive work and discoveries in this area. Two major strategies have been employed in efforts to dissect inhibitor modes of action.
To elucidate modes of action, the composition of whole cells or cell walls can be examined, as illustrated already by examples in §§4, 7 and 10. For example, although early biochemical studies of vancomycin's mode of action suggested it may work by blocking transpeptidase activity in cell wall assembly, NMR studies of S. aureus whole cells labelled with l-Lys, d-Ala and Gly revealed that cross-linking did not diminish appreciably when cells were treated with vancomycin (figure 7). Rather, the full panel of solid-state NMR data supported other existing biochemical evidence that vancomycin blocked transglycosylation by sequestration of lipid II in S. aureus [67].
14. The influence of oritavancin and structural analogues
(a). Oritavancin
Oritavancin (now called Orbactiv™) received FDA approval in August 2014 for the treatment of acute bacterial skin infections. Oritavancin differs from vancomycin by two substituents peripheral to the primary d-Ala–d-Ala binding site, yet oritavancin was known to be effective at killing vancomycin-resistant enterococci with altered stem termini (d-Ala–d-Lac) [89]. Beginning in 2002, solid-state NMR experiments were implemented to examine the mode of action of oritavancin on cell-wall composition and to develop a structural understanding of its binding and mechanism in preventing cell-wall assembly [46,79]. Tracking the fate of cell-wall NMR signatures indicated a dual mode of action in which oritavancin prevents both transpeptidation and transglycosylation, with an increase in Park's nucleotide (as in figure 6) and a decrease in cross-linking (figure 7) [78]. A fluorinated drug, [19F]oritavancin (also referred to as LY333328) (figure 9), bound to both isolated cell walls and whole cells with high affinity, and the substitution of Cl in the native compound with 19F did not perturb antibiotic activity. Distance measurements between an incorporated 19F on the biphenyl substituent and 13C and 15N labels in the peptidoglycan revealed that the biphenyl substituent bound along the pentaglycine bridge with the fluorine closest to the d-Ala–Gly cross-link site of a neighbouring strand to which the drug was bound (figure 9). Thus, the biphenyl group provided a secondary binding site, and this was demonstrated to be sufficient for activity when the primary d-Ala–d-Ala binding site was abrogated [68]. Results from several studies on isolated cell-wall complexes as well as complexes in whole cells provided complementary distance and orientation parameters to build a three-dimensional oritavancin–peptidoglycan model (figure 9).
Figure 9.

Structural model of oritavancin bound to peptidoglycan. The structural model of peptidoglycan-bound oritavancin was generated from molecular dynamics coupled with key NMR distances measured in antibiotic complexes with cell walls and whole cells. Adapted from Toke et al. [33]. (Online version in colour.)
Experiments were designed to detect possible positioning of the diphenyl moiety as a type of anchor in the cell membrane, a mechanism that had been proposed for oritavancin. Solid-state NMR measurements in whole cells did not reveal contact between the 19F and 31P of lipids, which would be anticipated in a membrane-anchor arrangement. In experiments with protoplasts, with the outer layers of cell wall stripped away, 19F–31P contact could be observed as all the oritavancin must bind near to the membrane surface and some anchoring could also be possible in that context [57]. Yet, with the cell wall intact, oritavancin appears to bind to the cell wall without appreciable membrane localization. Additionally, oritavancin dimers were not detected as had been observed in in vitro experiments with small soluble cell-wall units [90,91]. The determination that oritavancin harnessed a secondary binding site in the cell wall, along the pentaglycine bridge, helped in understanding the structure-based mode of action of this important and now clinically available antibiotic.
(b). Altered positioning for oritavancin-like analogues
Similar metabolic and structural analyses as those performed with oritavancin were also performed for a panel of other important structurally related fluorinated vancomycin derivatives, including fluorophenylbenzyl-vancomycin (FPBV) and derivatives with altered hydrophobic chains (LCTA-1110 and LCTA-1049, and LY309687) [46,92]. These antibiotics differ by the placement and length of hydrophobic aromatic moieties, and/or the presence of an additional vancosamine sugar. All of the antibiotics bound peptidoglycan in a relatively similar manner, but the orientation and placement of the hydrophobic substituent and its fluorine relative to the glycine bridge varied slightly. The fluorine of FPBV, for example, was 6 Å from the Gly–Lys bridgelink and 11 Å from the glycine in the d-Ala–Gly cross-link, whereas LY309687's fluorine label had corresponding values of 8 and 10 Å, implying a different conformation where the 19F is not looking down the bridge collinearly, but is positioned out along the centre of the bridge such that it has a more similar distance to each end of the pentaglycine bridge.
For glycopeptides where the hydrophobic chain was alkyl-based rather than an aryl-based substituent (e.g. FNCE or plusbacin-A3), the hydrophobic chain still positioned itself along the glycine bridge [48,93]. Furthermore, the effective length of the chain was important for binding: chains that were either too long or too short had lower binding affinity for whole cells and higher MICs [48,92]. REDOR showed that the fluorine labels of drugs that were too short or too long had poor contact to the glycine bridges and required distance distributions to fit the structural data, indicative of heterogeneous placement. By contrast, oritavancin and FPBV, with appropriate medium-length hydrophobic substituents, had homogeneous binding, with the fluorine label detected in better-defined positions, correlated with more potent activity.
These studies of oritavancin and related antibiotics also helped resolve a puzzle regarding the relatively poor binding to l-Lys–d-Ala–d-Ala substrates in solution when compared with that of vancomycin [94,95]. They may not bind the l-Lys–d-Ala–d-Ala substrates in solution as strongly as vancomycin, but they can bind to the peptidoglycan with higher affinity because their hydrophobic sidechains interact with the glycine bridge of S. aureus. The additional steric bulk of the antibiotic is presumably responsible for its ability to prevent transpeptidation (a reduction in cross-linking is observed by solid-state NMR), leading to enhanced antibiotic activity, even in vancomycin-resistant bacteria.
Finally, while these studies all focused on S. aureus and vancomycin derivatives, the solid-state NMR approach is general, and can be applied to other antibiotics or bacteria. For example, oritavancin was found to have a single mode of action in E. faecium, where it bound the bridges of the peptidoglycan, particularly nascent peptidoglycan, without blocking transglycosylation or needing to bind d-Ala–d-Ala stems [96]. A solid-state NMR study of plusbacin-A3, a glycopeptide unrelated to vancomycin, found that it, too, had a dual mode of action [48]. It also appeared to inhibit teichoic acid synthesis, reducing the d-alanine associated with teichoic acid (using the measurements in figure 8). Thus, solid-state NMR has considerable power and potential for studying antibiotic modes of action in intact whole-cell systems.
15. Outlook
Solid-state NMR has been applied most extensively to S. aureus and Gram-positive organisms, yet it is also poised to address questions of cell-wall composition and architecture in other organisms. Naturally, it should complement other measurements including microscopy and biochemical analyses, but is unique in being able to obtain quantitative parameters of architecture and metabolism in intact cells and cell walls without perturbation. Finally, we see from results with penicillin, vancomycin, oritavancin and others, that there continues to be much to learn from and about existing antibiotics. In a chemical genetics spirit, these lessons ultimately help us to understand function and may drive the identification and development of new antibiotics.
Acknowledgements
L.C. acknowledges support from the National Institutes of Health through the NIH Director's New Innovator Award Programme (1-DP2-OD007488).
Competing interests
We declare we have no competing interests.
Funding
We received no funding for this study.
References
- 1.Vollmer W, Blanot D, de Pedro MA. 2008. Peptidoglycan structure and architecture. FEMS Microbiol. Rev. 32, 149–167. ( 10.1111/j.1574-6976.2007.00094.x) [DOI] [PubMed] [Google Scholar]
- 2.Lowy FD. 1998. Staphylococcus aureus infections. N. Engl. J. Med. 339, 520–532. ( 10.1056/NEJM199808203390806) [DOI] [PubMed] [Google Scholar]
- 3.Klevens RM, et al. 2007. Invasive methicillin-resistant Staphylococcus aureus infections in the United States. JAMA 298, 1763–1771. ( 10.1001/jama.298.15.1763) [DOI] [PubMed] [Google Scholar]
- 4.Boucher HW, Talbot GH, Bradley JS, Edwards JE, Gilbert D, Rice LB, Scheld M, Spellberg B, Bartlett J. 2009. Bad bugs, no drugs: no ESKAPE! An update from the Infectious Diseases Society of America. Clin. Infect. Dis. 48, 1–12. ( 10.1086/595011) [DOI] [PubMed] [Google Scholar]
- 5.Wright GD. 2012. Antibiotics: a new hope. Chem. Biol. 19, 3–10. ( 10.1016/j.chembiol.2011.10.019) [DOI] [PubMed] [Google Scholar]
- 6.Bush K, et al. 2011. Tackling antibiotic resistance. Nat. Rev. Microbiol. 9, 894–896. ( 10.1038/nrmicro2693) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Walsh CT, Wencewicz TA. 2014. Prospects for new antibiotics: a molecule-centered perspective. J. Antibiot. (Tokyo) 67, 7–22. ( 10.1038/ja.2013.49) [DOI] [PubMed] [Google Scholar]
- 8.Lovering AL, Safadi SS, Strynadka NC. 2012. Structural perspective of peptidoglycan biosynthesis and assembly. Annu. Rev. Biochem. 81, 451–478. ( 10.1146/annurev-biochem-061809-112742) [DOI] [PubMed] [Google Scholar]
- 9.Kahan FM, Kahan JS, Cassidy PJ, Kropp H. 1974. Mechanism of action of fosfomycin (phosphonomycin). Ann. NY Acad. Sci. 235, 364–386. ( 10.1111/j.1749-6632.1974.tb43277.x) [DOI] [PubMed] [Google Scholar]
- 10.Tipper DJ, Strominger JL. 1965. Mechanism of action of penicillins: a proposal based on their structural similarity to acyl-d-alanyl-d-alanine. Proc. Natl Acad. Sci. USA 54, 1133–1141. ( 10.1073/pnas.54.4.1133) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yocum RR, Rasmussen JR, Strominger JL. 1980. The mechanism of action of penicillin: penicillin acylates the active-site of Bacillus stearothermophilus d-alanine carboxypeptidase. J. Biol. Chem. 255, 3977–3986. [PubMed] [Google Scholar]
- 12.Brown S, Santa Maria JP, Walker S. 2013. Wall teichoic acids of Gram-positive bacteria. Annu. Rev. Microbiol. 67, 313–336. ( 10.1146/annurev-micro-092412-155620) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Navarre WW, Schneewind O. 1999. Surface proteins of Gram-positive bacteria and mechanisms of their targeting to the cell wall envelope. Microbiol. Mol. Biol. Rev. 63, 174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bumsted RM, et al. 1968. Biosynthesis of the peptidoglycan of bacterial cell walls. X. Further study of the glycyl transfer ribonucleic acids active in peptidoglycan synthesis in Staphylococcus aureus. J. Biol. Chem. 243, 779–782. [PubMed] [Google Scholar]
- 15.Siewert G, Strominger JL. 1968. Biosynthesis of the peptidoglycan of bacterial cell walls. XI. Formation of the isoglutamine amide group in the cell walls of Staphylococcus aureus. J. Biol. Chem. 243, 783–790. [PubMed] [Google Scholar]
- 16.Tipper DJ, Strominger JL. 1968. Biosynthesis of the peptidoglycan of bacterial cell walls. XII. Inhibition of cross-linking by penicillins and cephalosporins: studies in Staphylococcus aureus in vivo. J. Biol. Chem. 243, 3169–3179. [PubMed] [Google Scholar]
- 17.Glauner B. 1988. Separation and quantification of muropeptides with high-performance liquid chromatography. Anal. Biochem. 172, 451–464. ( 10.1016/0003-2697(88)90468-X) [DOI] [PubMed] [Google Scholar]
- 18.Patti GJ, Chen J, Gross ML. 2009. Method revealing bacterial cell-wall architecture by time-dependent isotope labeling and quantitative liquid chromatography/mass spectrometry. Anal. Chem. 81, 2437–2445. ( 10.1021/ac802587r) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.de Jonge BL, et al. 1992. Peptidoglycan composition of a highly methicillin-resistant Staphylococcus aureus strain. The role of penicillin binding protein 2A. J. Biol. Chem. 267, 11 248–11 254. [PubMed] [Google Scholar]
- 20.Boneca IG, et al. 2000. Characterization of Staphylococcus aureus cell wall glycan strands, evidence for a new beta-N-acetylglucosaminidase activity. J. Biol. Chem. 275, 9910–9918. ( 10.1074/jbc.275.14.9910) [DOI] [PubMed] [Google Scholar]
- 21.Laws DD, Bitter HM, Jerschow A. 2002. Solid-state NMR spectroscopic methods in chemistry. Angew. Chem. Int. Ed. Engl. 41, 3096–3129. () [DOI] [PubMed] [Google Scholar]
- 22.Reichhardt C, Cegelski L. 2014. Solid-state NMR for bacterial biofilms. Mol. Phys. 112, 887–894. ( 10.1080/00268976.2013.837983) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McDermott A. 2009. Structure and dynamics of membrane proteins by magic angle spinning solid-state NMR. Annu. Rev. Biophys. 38, 385–403. ( 10.1146/annurev.biophys.050708.133719) [DOI] [PubMed] [Google Scholar]
- 24.Renault M, Tommassen-van Boxtel R, Bos MP, Post JA, Tommassen J, Baldus M. 2012. Cellular solid-state nuclear magnetic resonance spectroscopy. Proc. Natl Acad. Sci. USA 109, 4863–4868. ( 10.1073/pnas.1116478109) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Durr UHN, Gildenberg M, Ramamoorthy A. 2012. The magic of bicelles lights up membrane protein structure. Chem. Rev. 112, 6054–6074. ( 10.1021/cr300061w) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yamamoto K, Caporini MA, Im S-C, Waskell L, Ramamoorthy A. 2015. Cellular solid-state NMR investigation of a membrane protein using dynamic nuclear polarization. Biochim. Et Biophys. Acta-Biomembr. 1848, 342–349. ( 10.1016/j.bbamem.2014.07.008) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vogel EP, Weliky DP. 2013. Quantitation of recombinant protein in whole cells and cell extracts via solid-state NMR spectroscopy. Biochemistry 52, 4285–4287. ( 10.1021/bi4007034) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kim SJ, Chang J, Singh M. 2015. Peptidoglycan architecture of Gram-positive bacteria by solid-state NMR. Biochim. Biophys. Acta 1848, 350–362. ( 10.1016/j.bbamem.2014.05.031) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kern T, et al. 2010. Dynamics characterization of fully hydrated bacterial cell walls by solid-state NMR: evidence for cooperative binding of metal ions. J. Am. Chem. Soc. 132, 10 911–10 919. ( 10.1021/ja104533w) [DOI] [PubMed] [Google Scholar]
- 30.Ramamoorthy A. 2009. Beyond NMR spectra of antimicrobial peptides: dynamical images at atomic resolution and functional insights. Solid State Nucl. Magn. Reson. 35, 201–207. ( 10.1016/j.ssnmr.2009.03.003) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bechinger B, Salnikov ES. 2012. The membrane interactions of antimicrobial peptides revealed by solid-state NMR spectroscopy. Chem. Phys. Lipids 165, 282–301. ( 10.1016/j.chemphyslip.2012.01.009) [DOI] [PubMed] [Google Scholar]
- 32.Hong M, Su YC. 2011. Structure and dynamics of cationic membrane peptides and proteins: insights from solid-state NMR. Protein Sci. 20, 641–655. ( 10.1002/pro.600) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Toke O, Cegelski L, Schaefer J. 2006. Peptide antibiotics in action: investigation of polypeptide chains in insoluble environments by rotational-echo double resonance. Biochim. Biophys. Acta-Biomembr. 1758, 1314–1329. ( 10.1016/j.bbamem.2006.02.031) [DOI] [PubMed] [Google Scholar]
- 34.Schaefer J, Stejskal EO. 1976. Carbon-13 nuclear magnetic resonance of polymers spinning at the magic angle. J. Am. Chem. Soc. 98, 1031–1032. ( 10.1021/ja00420a036) [DOI] [Google Scholar]
- 35.Yannoni CS. 1982. High-resolution NMR in solids: the CPMAS experiment. Acc. Chem. Res. 15, 201–208. ( 10.1021/ar00079a003) [DOI] [Google Scholar]
- 36.McCrate OA, Zhou X, Reichhardt C, Cegelski L. 2013. Sum of the parts: composition and architecture of the bacterial extracellular matrix. J. Mol. Biol. 425, 4286–4294. ( 10.1016/j.jmb.2013.06.022) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Reichhardt C, Fong JCN, Yildiz F, Cegelski L. 2015. Characterization of the Vibrio cholerae extracellular matrix: a top-down solid-state NMR approach. Biochim. Biophys. Acta 1848, 378–383. ( 10.1016/j.bbamem.2014.05.030) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.De Paëpe G. 2012. Dipolar recoupling in magic angle spinning solid-state nuclear magnetic resonance. Annu. Rev. Phys. Chem. 63, 661–684. ( 10.1146/annurev-physchem-032511-143726) [DOI] [PubMed] [Google Scholar]
- 39.Cegelski L. 2013. REDOR NMR for drug discovery. Bioorg. Med. Chem. Lett. 23, 5767–5775. ( 10.1016/j.bmcl.2013.08.064) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cegelski L. 2015. Bottom-up and top-down solid-state NMR approaches for bacterial biofilm matrix composition. J. Magn Reson. 253, 91–97. ( 10.1016/j.jmr.2015.01.014) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schanda P, Triboulet S, Laguri C, Bougault CM, Ayala I, Callon M, Arthur M, Simorre J-P. 2014. Atomic model of a cell-wall cross-linking enzyme in complex with an intact bacterial peptidoglycan. J. Am. Chem. Soc. 136, 17 852–17 860. ( 10.1021/ja5105987) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yu TY, et al. 2008. 15N{31P} REDOR NMR studies of the binding of phosphonate reaction intermediate analogues to Saccharomyces cerevisiae lumazine synthase. Biochemistry 47, 13 942–13 951. ( 10.1021/bi8015789) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McDowell LM, Studelska DR, Poliks B, O'Connor RD, Schaefer J. 2004. Characterization of the complex of a trifluoromethyl-substituted shikimate-based bisubstrate inhibitor and 5-enolpyruvylshikimate-3-phosphate synthase by REDOR NMR. Biochemistry 43, 6606–6611. ( 10.1021/bi049685w) [DOI] [PubMed] [Google Scholar]
- 44.McDowell LM, et al. 2003. Human factor Xa bound amidine inhibitor conformation by double rotational-echo double resonance nuclear magnetic resonance and molecular dynamics simulations. J. Med. Chem. 46, 359–363. ( 10.1021/jm0202324) [DOI] [PubMed] [Google Scholar]
- 45.Jiang YL, et al. 2004. Recognition of an unnatural difluorophenyl nucleotide by uracil DNA glycosylase. Biochemistry 43, 15 429–15 438. ( 10.1021/bi0483864) [DOI] [PubMed] [Google Scholar]
- 46.Kim SJ, Cegelski L, Preobrazhenskaya M, Schaefer J. 2006. Structures of Staphylococcus aureus cell-wall complexes with vancomycin, eremomycin, and chloroeremomycin derivatives by 13C{19F} and 15N{19F} rotational-echo double resonance. Biochemistry 45, 5235–5250. ( 10.1021/bi052660s) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cegelski L, Steuber D, Mehta AK, Kulp DW, Axelsen PH, Schaefer J. 2006. Conformational and quantitative characterization of oritavancin-peptidoglycan complexes in whole cells of Staphylococcus aureus by in vivo 13C and 15N labeling. J. Mol. Biol. 357, 1253–1262. ( 10.1016/j.jmb.2006.01.040) [DOI] [PubMed] [Google Scholar]
- 48.Kim SJ, et al. 2013. The isotridecanyl side chain of plusbacin-A3 is essential for the transglycosylase inhibition of peptidoglycan biosynthesis. Biochemistry 52, 1973–1979. ( 10.1021/bi4000222) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Garbow JR, Jacob GS, Stejskal EO, Schaefer J. 1989. Protein dynamics from chemical shift and dipolar rotational spin-echo nitrogen-NMR. Biochemistry 28, 1362–1367. ( 10.1021/bi00429a063) [DOI] [PubMed] [Google Scholar]
- 50.Tong G, Pan Y, Dong H, Pryor R, Wilson GE, Schaefer J. 1997. Structure and dynamics of pentaglycyl bridges in the cell walls of Staphylococcus aureus by 13C–15N REDOR NMR. Biochemistry 36, 9859–9866. ( 10.1021/bi970495d) [DOI] [PubMed] [Google Scholar]
- 51.Kern T, et al. 2008. Toward the characterization of peptidoglycan structure and protein-peptidoglycan interactions by solid-state NMR spectroscopy. J. Am. Chem. Soc. 130, 5618–5619. ( 10.1021/ja7108135) [DOI] [PubMed] [Google Scholar]
- 52.Zhou X, Cegelski L. 2012. Nutrient-dependent structural changes in S. aureus peptidoglycan revealed by solid-state NMR spectroscopy. Biochemistry 51, 8143–8153. ( 10.1021/bi3012115) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nygaard R, Romaniuk JAH, Rice DM, Cegelski L. 2015. Spectral snapshots of bacterial cell-wall composition and the influence of antibiotics by whole-cell NMR. Biophys. J. 108, 1380–1389. ( 10.1016/j.bpj.2015.01.037) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Meredith TC, Swoboda JG, Walker S. 2008. Late-stage polyribitol phosphate wall teichoic acid biosynthesis in Staphylococcus aureus. J. Bacteriol. 190, 3046–3056. ( 10.1128/JB.01880-07) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mirelman D, Beck BD, Shaw DR. 1970. The location of the d-alanyl ester in the ribitol teichoic acid of Staphylococcus aureus. Biochem. Biophys. Res. Commun. 39, 712–717. ( 10.1016/0006-291X(70)90263-9) [DOI] [PubMed] [Google Scholar]
- 56.Coles NW, Gross R. 1973. Preparation of metabolically active Staphylococcus aureus protoplasts by use of the Aeromonas hydrophila lytic enzyme. J. Bacteriol. 115, 746–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kim SJ, Singh M, Schaefer J. 2009. Oritavancin binds to isolated protoplast membranes but not intact protoplasts of Staphylococcus aureus. J. Mol. Biol. 391, 414–425. ( 10.1016/j.jmb.2009.06.033) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Morath S, von Aulock S, Hartung T. 2005. Structure/function relationships of lipoteichoic acids. J. Endotoxin Res 11, 348–356. ( 10.1177/09680519050110061001) [DOI] [PubMed] [Google Scholar]
- 59.Grif K, et al. 2001. In vitro activity of fosfomycin in combination with various antistaphylococcal substances. J. Antimicrob. Chemother. 48, 209–217. ( 10.1093/jac/48.2.209) [DOI] [PubMed] [Google Scholar]
- 60.Gisin J, Schneider A, Nägele B, Borisova M, Mayer C. 2013. A cell wall recycling shortcut that bypasses peptidoglycan de novo biosynthesis. Nat. Chem. Biol. 9, 491–493. ( 10.1038/nchembio.1289) [DOI] [PubMed] [Google Scholar]
- 61.Contreras A, Barbacid M, Vazquez D. 1974. Binding to ribosomes and mode of action of chloramphenicol analogues. Biochim. Biophys. Acta 349, 376–388. ( 10.1016/0005-2787(74)90124-5) [DOI] [PubMed] [Google Scholar]
- 62.Takahashi H, Ayala I, Bardet M, De Paëpe G, Simorre J-P, Hediger S. 2013. Solid-state NMR on bacterial cells: selective cell wall signal enhancement and resolution improvement using dynamic nuclear polarization. J. Am. Chem. Soc. 135, 5105–5110. ( 10.1021/ja312501d) [DOI] [PubMed] [Google Scholar]
- 63.Wang T, Park YB, Caporini MA, Rosay M, Zhong L, Cosgrove DJ, Hong M. 2013. Sensitivity-enhanced solid-state NMR detection of expansin's target in plant cell walls. Proc. Natl Acad. Sci. USA 110, 16 444–16 449. ( 10.1073/pnas.1316290110) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ni QZ, et al. 2013. High frequency dynamic nuclear polarization. Acc. Chem. Res. 46, 1933–1941. ( 10.1021/ar300348n) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bui NK, Eberhardt A, Vollmer D, Kern T, Bougault C, Tomasz A, Simorre J-P, Vollmer W. 2012. Isolation and analysis of cell wall components from Streptococcus pneumoniae. Anal. Biochem. 421, 657–666. ( 10.1016/j.ab.2011.11.026) [DOI] [PubMed] [Google Scholar]
- 66.Breukink E, de Kruijff B. 2006. Lipid II as a target for antibiotics. Nat. Rev. Drug Discov. 5, 321–332. ( 10.1038/nrd2004) [DOI] [PubMed] [Google Scholar]
- 67.Cegelski L, Kim SJ, Hing AW, Studelska DR, O'Connor RD, Mehta AK, Schaefer J. 2002. Rotational-echo double resonance characterization of the effects of vancomycin on cell wall synthesis in Staphylococcus aureus. Biochemistry 41, 13 053–13 058. ( 10.1021/bi0202326) [DOI] [PubMed] [Google Scholar]
- 68.Pan Y, et al. 1993. Cross-links in cell walls of Bacillus subtilis by rotational-echo double-resonance 15N NMR. J. Biol. Chem. 268, 18 692–18 695. [PubMed] [Google Scholar]
- 69.Jacob GS, Schaefer J, Wilson GE Jr. 1983. Direct measurement of peptidoglycan cross-linking in bacteria by 15N nuclear magnetic resonance. J. Biol. Chem. 258, 10 824–10 826. [PubMed] [Google Scholar]
- 70.Hing A, Vega S, Schaefer J. 1993. Measurement of heteronuclear dipolar coupling by transferred-echo double-resonance NMR. J. Magn. Reson. 103, 151–162. ( 10.1006/jmra.1993.1146) [DOI] [Google Scholar]
- 71.Patti GJ, Kim SJ, Schaefer J. 2008. Characterization of the peptidoglycan of vancomycin-susceptible Enterococcus faecium. Biochemistry 47, 8378–8385. ( 10.1021/bi8008032) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wilson GE Jr, Jacob GS, Schaefer J. 1985. Solid-state 15N NMR studies of the effects of penicillin on cell-wall metabolism of Aerococcus viridans (Gaffkya homari). Biochem. Biophys. Res. Commun. 126, 1006–1012. ( 10.1016/0006-291X(85)90285-2) [DOI] [PubMed] [Google Scholar]
- 73.Siewert G, Strominger JL. 1967. Bacitracin: an inhibitor of the dephosphorylation of lipid pyrophosphate, an intermediate in the biosynthesis of the peptidoglycan of bacterial cell walls. Proc. Natl Acad. Sci. USA 57, 767–773. ( 10.1073/pnas.57.3.767) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Strominger JL. 1957. Microbial uridine-5′-pyrophosphate N-acetylamino sugar compounds. I. Biology of the penicillin-induced accumulation. J. Biol. Chem. 224, 509–523. [PubMed] [Google Scholar]
- 75.Turner RD, Ratcliffe EC, Wheeler R, Golestanian R, Hobbs JK, Foster SJ. 2010. Peptidoglycan architecture can specify division planes in Staphylococcus aureus. Nat. Commun. 1, 26 ( 10.1038/ncomms1025) [DOI] [PubMed] [Google Scholar]
- 76.Williams I, et al. 1999. Flow cytometry and other techniques show that Staphylococcus aureus undergoes significant physiological changes in the early stages of surface-attached culture. Microbiology 145, 1325–1333. ( 10.1099/13500872-145-6-1325) [DOI] [PubMed] [Google Scholar]
- 77.Rohrer S, Ehlert K, Tschierske M, Labischinski H, Berger-Bachi B. 1999. The essential Staphylococcus aureus gene fmhB is involved in the first step of peptidoglycan pentaglycine interpeptide formation. Proc. Natl Acad. Sci. USA 96, 9351–9356. ( 10.1073/pnas.96.16.9351) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kim SJ, et al. 2008. Oritavancin exhibits dual mode of action to inhibit cell-wall biosynthesis in Staphylococcus aureus. J. Mol. Biol. 377, 281–293. ( 10.1016/j.jmb.2008.01.031) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kim SJ, Cegelski L, Studelska DR, O'Connor RD, Mehta AK, Schaefer J. 2002. Rotational-echo double resonance characterization of vancomycin binding sites in Staphylococcus aureus. Biochemistry 41, 6967–6977. ( 10.1021/bi0121407) [DOI] [PubMed] [Google Scholar]
- 80.Kim SJ, Singh M, Sharif S, Schaefer J. 2014. Cross-link formation and peptidoglycan lattice assembly in the FemA mutant of Staphylococcus aureus. Biochemistry 53, 1420–1427. ( 10.1021/bi4016742) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Singh M, Kim SJ, Sharif S, Preobrazhenskaya M, Schaefer J. 2015. REDOR constraints on the peptidoglycan lattice architecture of Staphylococcus aureus and its FemA mutant. Biochim. Biophys. Acta 1848, 363–368. ( 10.1016/j.bbamem.2014.05.025) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Strominger JL, Willoughby E, Kamiryo T, Blumberg PM, Yocum RR. 1974. Penicillin-sensitive enzymes and penicillin-binding components in bacterial cells. Ann. NY Acad. Sci. 235, 210–224. ( 10.1111/j.1749-6632.1974.tb43267.x) [DOI] [PubMed] [Google Scholar]
- 83.Cho H, Uehara T, Bernhardt TG. 2014. Beta-lactam antibiotics induce a lethal malfunctioning of the bacterial cell wall synthesis machinery. Cell 159, 1300–1311. ( 10.1016/j.cell.2014.11.017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Saito H. 1986. Conformation-dependent 13C chemical shifts: a new means of conformational characterization as obtained by high-resolution solid-state 13C NMR. Magn. Reson. Chem. 24, 835–852. ( 10.1002/mrc.1260241002) [DOI] [Google Scholar]
- 85.Meroueh SO, Bencze KZ, Hesek D, Lee M, Fisher JF, Stemmler TL, Mobashery S. 2006. Three-dimensional structure of the bacterial cell wall peptidoglycan. Proc. Natl Acad. Sci. USA 103, 4404–4409. ( 10.1073/pnas.0510182103) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Sharif S, Kim SJ, Labischinski H, Schaefer J. 2009. Characterization of peptidoglycan in fem-deletion mutants of methicillin-resistant Staphylococcus aureus by solid-state NMR. Biochemistry 48, 3100–3108. ( 10.1021/bi801750u) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Sharif S, Kim SJ, Labischinski H, Chen J, Schaefer J. 2013. Uniformity of glycyl bridge lengths in the mature cell walls of FEM mutants of methicillin-resistant Staphylococcus aureus. J. Bacteriol. 195, 1421–1427. ( 10.1128/JB.01471-12) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Sharif S, Singh M, Kim SJ, Schaefer J. 2009. Staphylococcus aureus peptidoglycan tertiary structure from carbon-13 spin diffusion. J. Am. Chem. Soc. 131, 7023–7030. ( 10.1021/ja808971c) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Allen NE, LeTourneau DL, Hobbs JN Jr. 1997. Molecular interactions of a semisynthetic glycopeptide antibiotic with d-alanyl-d-alanine and d-alanyl-d-lactate residues. Antimicrob. Agents Chemother. 41, 66–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Williams DH, et al. 1998. An analysis of the origins of a cooperative binding energy of dimerization. Science 280, 711–714. ( 10.1126/science.280.5364.711) [DOI] [PubMed] [Google Scholar]
- 91.Sharman GJ, et al. 1997. The roles of dimerization and membrane anchoring in activity of glycopeptide antibiotics against vancomycin-resistant bacteria. J. Am. Chem. Soc. 119, 12 041–12 047. ( 10.1021/ja964477f) [DOI] [Google Scholar]
- 92.Kim SJ, Schaefer J. 2008. Hydrophobic side-chain length determines activity and conformational heterogeneity of a vancomycin derivative bound to the cell wall of Staphylococcus aureus. Biochemistry 47, 10 155–10 161. ( 10.1021/bi800838c) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kim SJ, Tanaka KSE, Dietrich E, Rafai Far A, Schaefer J. 2013. Locations of the hydrophobic side chains of lipoglycopeptides bound to the peptidoglycan of Staphylococcus aureus. Biochemistry 52, 3405–3414. ( 10.1021/bi400054p) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Good VM, Gwynn MN, Knowles DJ. 1990. MM 45289, a potent glycopeptide antibiotic which interacts weakly with diacetyl-l-lysyl-d-alanyl-d-alanine. J. Antibiot. (Tokyo) 43, 550–555. ( 10.7164/antibiotics.43.550) [DOI] [PubMed] [Google Scholar]
- 95.Allen NEL, Hobbs DL. 1997. The role of hydrophobic side chains as determinants of antibacterial activity of semisynthetic glycopeptide antibiotics. J. Antibiot. (Tokyo) 50, 677–684. ( 10.7164/antibiotics.50.677) [DOI] [PubMed] [Google Scholar]
- 96.Patti GJ, Kim SJ, Yu T-Y, Dietrich E, Tanaka KSE, Parr TR, Far AR, Schaefer J.. 2009. Vancomycin and oritavancin have different modes of action in Enterococcus faecium. J. Mol. Biol. 392, 1178–1191. ( 10.1016/j.jmb.2009.06.064) [DOI] [PMC free article] [PubMed] [Google Scholar]