


Abstract
Small silencing RNAs, including microRNAs, endogenous small interfering RNAs (endo-siRNAs) and Piwi-interacting RNAs (piRNAs), have been shown to play important roles in fine-tuning gene expression, defending virus and controlling transposons. Loss of small silencing RNAs or components in their pathways often leads to severe developmental defects, including lethality and sterility. Recently, non-templated addition of nucleotides to the 3′ end, namely tailing, was found to associate with the processing and stability of small silencing RNAs. Next Generation Sequencing has made it possible to detect such modifications at nucleotide resolution in an unprecedented throughput. Unfortunately, detecting such events from millions of short reads confounded by sequencing errors and RNA editing is still a tricky problem. Here, we developed a computational framework, Tailor, driven by an efficient and accurate aligner specifically designed for capturing the tailing events directly from the alignments without extensive post-processing. The performance of Tailor was fully tested and compared favorably with other general-purpose aligners using both simulated and real datasets for tailing analysis. Moreover, to show the broad utility of Tailor, we used Tailor to reanalyze published datasets and revealed novel findings worth further experimental validation. The source code and the executable binaries are freely available at https://github.com/jhhung/Tailor.
INTRODUCTION
Over the past decade, small silencing RNAs, including microRNAs (miRNAs), endogenous small silencing RNAs (endo-siRNAs) and Piwi-interacting RNAs (piRNAs) have been shown to play indispensable roles in regulating gene expression, protecting against viral infection and preventing mobilization of transposable elements (1–4). Small silencing RNAs exert their silencing function by associating with Argonaute proteins to form RNA-induced silencing complex (RISC), which uses the small RNA guide to find its regulatory targets and reduce gene expression. Although the studies on the biogenesis of small silencing RNAs have made enormous progress in the past decade, the factors controlling their stability and degradation remain elusive.
Recent studies have suggested that non-templated addition to the 3′ end of small silencing RNAs, namely tailing, could play essential roles in this regard. Non-templated 3′ mono- and oligo-uridylation of the pre-microRNAs (pre-miRNAs) regulates miRNA processing by either preventing or promoting Dicer cleavage in flies (5–7). The 3′ mono-uridylation on small interfering RNAs in Caenorhabditis elegans is associated with negative regulation (8). Ameres et al. have demonstrated that highly complementary targets trigger the tailing of miRNAs and eventually lead to their degradation in flies and mammals (9,10); a similar mechanism has been found on some endo-siRNAs as well (11). Identification of tailing events not only suggests the co-evolution of small silencing RNAs and their targets, but also sheds light on the mechanism of their maturation and degradation.
Despite the fact that Next Generation Sequencing (NGS) has greatly facilitated the understanding of RNA tailing, computational detection of non-templated nucleotides from millions of sequencing reads is challenging. The Ketting group used MegaBLAST to align piRNA sequences to the genome and relied on post-processing the reported mismatches to gain insights into tailing (8). However, as a heuristic algorithm, BLAST is not guaranteed to find all the tailing events (12,13) and it is significantly slower than the NGS aligners, like MAQ (14), BWA (15), Bowtie (16) and SOAP (17). The Chen group used an accurate method that iterates between Bowtie alignment and 3′ clipping of unmatched reads (18) to find all the perfect alignments of trimmed reads. A similar approach has been used for removing erroneous bases at 3′ end to increase the sensitivity of detecting miRNAs (19). Let alone that this method inevitably multiples the running time by the maximal length of tails, extra computational works are still needed to retrieve the identity of each trimmed tail. The study by Ameres et al. used a specialized suffix tree data structure to efficiently find all the tails without sacrificing the accuracy. However, due to the high memory footprint of the suffix tree data structure, which is about 16 to 20× of the genome size, the read mapping has to be performed for each chromosome separately (9,20). Extra processing is still required to finalize the alignments from all chromosomes.
Moreover, the task becomes even trickier when technical and biological confounding factors are taken into account for better capturing the true tailing events. For example, it is known that reads from Illumina HiSeq and Genome analyzer platforms have preferential A–C conversions (21,22) and a high error rate at the 3′ end of reads, which frequently leads to uncalled bases, i.e. B-tails (23,24). In addition to these technical artifacts endued by the sequencers, RNA editing is another common post-transcriptional modification in small silencing RNA biology that could perplex the tools with erroneous alignment. There are two major types of RNA editing in mammals, adenosine to inosine (A-to-I) and cytidine to uridine (C-to-U) editing. The major enzymes that catalyze adenosine to inosine are the adenosine deaminases acting on RNA (ADARs), whose main substrates are RNAs with double-stranded structures (25–27). Since many small silencing RNAs are originated from structural RNAs, they are all likely targets of A-to-I editing (28–30). Recent studies have shown that A-to-I editing can occur on the seed region of the miRNAs with fairly high occurrence rate (up to 80% in some cases) and have a direct impact on the selection of their regulatory targets (31,32). Those unmatched bases degenerate the sensitivity and accuracy of short read alignment and have a negative effect on the detection of tailing.
Most of the current methods simply ignore those confounding factors and rely on adapting existing, less specialized tools with extensive post-processing and as a consequence the performance, usefulness and application of tailing analysis is seriously compromised. A fast, accurate and straightforward approach to study tailing is still in need. To ease the cost of performing tailing analysis with dramatically increasing sequencing throughput, we here introduce Tailor—a framework that preprocesses and maps sequences to a reference, distinguishes tails from mismatches or bad alignments with a novel algorithm and reports both perfect and tailed alignment simultaneously without loss of information. Tailor is capable of analyzing the non-templated tailing for miRNA and other types of small RNAs and produce publication-quality summary figures. In addition, to better demonstrate the utility of Tailor, we reanalyzed published datasets with Tailor and unearthed several interesting observations (see Applications—case studies in Results). Although the findings still require thorough experimental validation, it is clear that Tailor would help expand the scope of the study of small silencing RNAs.
MATERIALS AND METHODS
Datasets
Illumina sequencing data of small RNAs from Drosophila melanogaster hen1 (SRR029608, SRR029633), Danio rerio hen1 (SRR363984–5), Arabidopsis hen1 and heso1 (SRP010683) and Ago2 associated small RNAs in cytoplasmic (SRR529097) and nuclear fraction (SRR529100) of HeLa were obtained from NCBI Sequence Read Archive. The length distribution of the simulated confounded reads was from the D. melanogaster Ago3 associated small RNAs extracted from ovaries (SRR916073). In-house program was used to trim the 3′ adaptors and filter the reads with low quality. Randomly distributed reads from fruitfly genome was generated by ArtificialFastqGenerator (33). Ten millions reads were randomly chosen using seqtk (github.com/lh3/seqtk.git) with options ‘sample -s100 -10000000’. To remove multiple mapping reads in some simulation datasets, we used Bowtie iteratively before and after the tail appending and seed mutation to assure each read has only one occurrence in the reference.
Rationale
The principle of detecting non-templated bases at the 3′ end of reads is basically to find the longest common prefix (LCP) between the read and each of the suffixes of the reference and then report the remainder on the read as a tail. Given a read R (M base pairs [bp] long) and all the suffixes (Si) of a reference sequence G (N bp long), one can find the LCP between R and Si by finding the longest consecutive matches from the first base to the last. Since there are totally N suffixes of G, a trivial solution needs at worse M*N times of comparison to find the LCP of R and G; however the performance is unacceptably slow when G is as large as a human genome. Using index structures, such as the suffix tree or suffix array, finding LCPs between the NGS reads and the reference can be solved much more efficiently (9,34).
Recently, the Full-text index in Minute space (FM-index) derived from the Burrows-Wheeler transform (BWT) (35–37) is widely used in many NGS applications (15–17). The FM-index is both time and space efficient and can be built from a suffix array and requires only 3 to 4 bits per base to store the index. A more detailed introduction of building the FM-index of long biological sequences is given in the Supplementary Materials. However, since the FM-index is originally designed for matching all bases of a read to a substring of the reference, it cannot be used directly for finding tails. One straightforward solution is to align reads without those non-templated bases by repeatedly removed one last base in each round of the alignment process until at least one perfect hit is found (18), but the approach scarifies the speed greatly and requires extensive post-processing. To benefit from the space and time efficiency of the FM-index, we further modified its matching procedure and adapted the error tolerant strategy proposed by Langmead et al. (16) to devise an FM-index based tail detection algorithm, Tailor, which is specialized in capturing the non-templated bases at the 3′ end of reads with confounding factors, such as sequencing errors and RNA editing.
Read mapping algorithm of Tailor
The system flow of the Tailor algorithm is outlined in Figure 1. Since searching within the FM-index initiates from the 3′ end of the query string (i.e. the read) (36), where the non-templated nucleotides append, Tailor first makes the reverse-complement of the query sequence so that searching starts from the original 5′ end to avoid excessive exhaustive search at the early stage. To do so, the reference should be reversed complemented as well, and the coordinate of each alignment should be calculated accordingly. To allow searching against both strands simultaneously and improves the speed, Tailor concatenates the plus and minus strands of the reference and constructs one index instead of two (Figure 1A and Supplementary Materials). Tailor also stores a part of the suffix array similar to other FM-index based aligners (16,38–40) to achieve fast calculation of the text shift for getting the coordinate of each occurrence. Any alignment whose prefix matching portion exceeds the boundary of the mapped chromosome is filtered. The searching continues until either it matches all the characters of the query to the reference (i.e. the perfect matching) or no more bases can be matched (i.e. the prefix matching). In the latter case, Tailor backtracks to the previous matched position and exhaustively enumerates all the possible prefix matches. The unmatched part remained in the query is reported as a tail (Figure 1B).
Figure 1.

BWT-based tailing detection algorithm. (A) Procedure of constructing the FM-index from a reference sequence. (B) Procedure of query searching using the FM-index. Searching starts from the 3′ end of a reverse-complemented query. Green letters indicate the non-templated tail. Red letters indicate the positions being matched against the index. When a non-templated letter is spotted as in step 4, the algorithm backtracks to previous step and reports all the hits and marks the unmatched string as ‘tail’.
Clearly, this strategy is vulnerable to confounding factors, since the first mismatch encountered directly defines the remainder as the tail, which can be very misleading. To accommodate possible sequencing errors or RNA editing events in a read, we devised specialized selection rules as depicted in Figure 2. For each read, the first S (S = 18 by default) bases at its 5′ portion is defined as the seed (Figure 2A). Given the fact that sequencing errors tend to occur at the 3′ end (23,24) and RNA editing events in miRNAs are enriched at the other end (i.e. the seed region) (30–32), the selection rules behave according to whether or not the first mismatch appears in the seed (Figure 2B).
Figure 2.
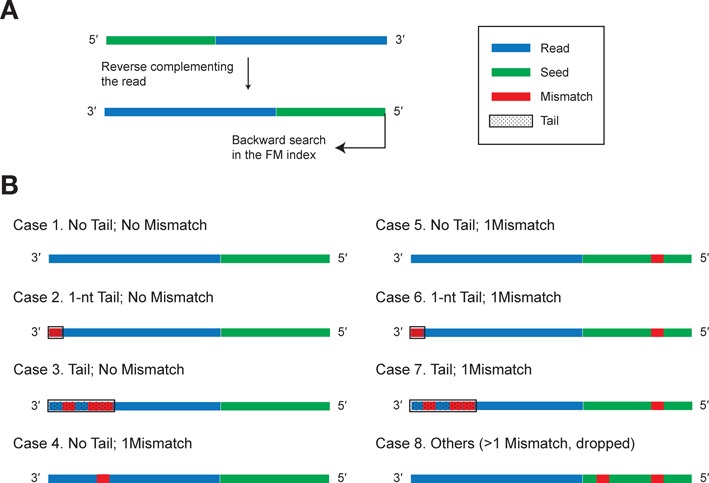
Error tolerance filtering rules. (A) Reads would have to be reverse-complemented before searching. The corresponding seed region is highlighted in green. (B) Eight rules for determining tails. See the main text for more details.
If the first mismatch is not in the seed region, it is regarded as either the first base of the tail or a sequencing error. In the case that the mismatch is at the last base, it is directly deemed as a valid tail (Case 2 in Figure 2B). If the tail is longer than 1 nucleotide (nt), it will be further scanned to make sure that the sequence of the tail consists of multiple non-templated nucleotides (Case 3). If the tail is only one nucleotide different from the reference, no tail but a mismatch will be reported (Case 4). Note that in order to differentiate tails from sequencing error, a filtering step based on the quality is necessary to avoid type I error and has been included in Tailor's pipeline (see below; Analysis pipeline). Our current algorithm cannot differentiate the circumstance that the tailed sequence is identical to the genome sequence. This problem is unlikely to be solved computationally and experimental solutions are expected to be more effective (e.g. using mutant with a defective tailing pathway).
On the other hand, if the first mismatch is in the seed, where RNA editing events occur frequently, the backtracking search will be reinitiated and looks for an LCP started from the succeeding base after the mismatch. If no mismatch is found in the reinitiated search, no tail but a mismatch is reported (Case 5). If a mismatch is occurred outside the seed, the reminder is reported as a tail (Case 6 and 7); otherwise, the read is dropped (Case 8). Note that the scenario that Case 4 with another mismatch in the seed is not allowed (i.e. two mismatches as in Case 8), since in principle we want to endow Tailor an error tolerance strategy consistent to that of conventional approaches under the one mismatch setting (e.g. −v 1 in Bowtie).
Implementation
We implemented the core of the Tailor aligner using C++ with built-in support for multithreading. Since Tailor concatenates both strands of the chromosomes into one long reference, whose length could exceed the maximum number represented by 32 bits, we have to use 64 bits to store the indexes in all the relevant data structures, which require about 2X memory footprint than that of other FM-index based aligners. To backward compatible with the algorithm introduced in Ameres et al. (9), which allow only case 1, 2 and 3 in Figure 2, an option (−v) is needed to turn on the detection of other cases. Tailor has a similar command line interface like other NGS aligners and reports alignment in the SAM (41) format. A tail is described as ‘soft-clipping’ in CIGAR and the sequences are reported under ‘TL:Z:’ in the optional fields. Mismatches, if allowed (−v), will be reported in the ‘MD’ tag (see Supplementary Materials for more details). Tailor is freely available on GitHub (http://jhhung.github.io/Tailor/) under GNU General Public License 2. All the scripts used in preparing this manuscript have also been included in the same GitHub repository. The tailing pipelines were implemented in shell scripting language and R.
Test environment and software
All software tests were performed in the x86_64 Centos environment with 24 cores and 48G of memory. The Bowtie software used in this study is version 1.0.0, 64-bit. The version of BWA used is 0.7.5a-r405. The version of Tailor used is 1.0.0. All commands for all the tests are listed in the Supplementary Materials.
RESULTS
Performance without confounding factors
To begin with, we ignored confounding factors in the following tests to compare with conventional approaches first. To assess the aligning speed directly, we indiscriminately generated 10 millions of perfectly genome-matching reads from the D. melanogaster genome (simulated tail-free dataset) (33) and randomly appended 1–4 genome-unmatched nucleotides to the 3′ ends (simulated tailed dataset). We compared Tailor with two most popular BWT aligners Bowtie and BWA by applying them on simulated small RNA datasets (Figure 3A). For the simulated tail-free dataset, Tailor outperformed Bowtie and BWA in five thread settings (using 2, 4, 8, 12 and 24 threads; Figure 3A, top. All the running time plotted was the average of the actual running time of five repeated experiments). But for the simulated tailed dataset, Bowtie ran slightly faster than Tailor possibly due to the fact the it reported no alignment and did not perform any disk writing (Figure 3A, bottom). We also performed the speed test with real small RNA sequencing data from hen1+/− and hen1−/− fruitfly and zebrafish (see Datasets in ‘Materialsand Methods’ section) (Figure 3B). hen1 encodes for a methyl-transferase that adds a methyl group to the 3′ end of siRNA and piRNA at the 2′-O position and prevents tailing (9,42). For both hen1+/− and hen1−/− libraries, Tailor outperformed Bowtie and BWA and reproduced the published result that siRNAs, but not miRNAs, were subjected to tailing in the absence of hen1 (Supplementary Figure S1). Please note that Bowtie and BWA in the speed test setting here were not capable of detecting non-templated tails. These tests were just used to compare their execution speed but not functionality.
Figure 3.
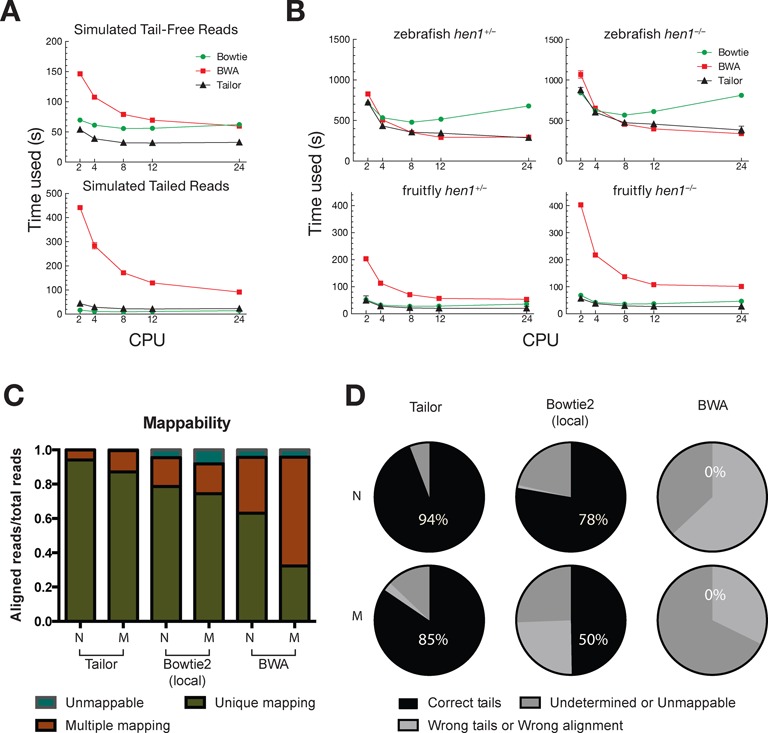
Speed comparison between Tailor and others software. (A) Speed comparison between Tailor, BWA and Bowtie using simulated 18–23 nt small RNA with (top) or without (bottom) non-templated tails. Tailor ran with the default setting, which allows no mismatch in the middle of the query. Tailed alignments were reported if perfect match could not be found. Bowtie ran with ‘−a –best –strata −v 0’ setting to allow no mismatch while report all best alignments. BWA ran with the default setting. Five different CPU settings were used and the running time was plotted. Three replicates were performed. (B) Speed comparison between Tailor, BWA and Bowtie (commands can be found in Supplementary Materials) using published small RNA Illumina NGS libraries from hen1+/− and hen1−/− mutants in fruitfly and zebrafish. Same settings were used as in (A). (C) The mappability of the normal (N) and mutated (M) datasets aligned by Tailor, Bowtie2 (with local alignment) and BWA. Multiple mapping was deemed as misalignment since each read was guaranteed to have only one occurrence in the reference. (D) The unique mapping reads shown in (C) were further examined to make sure they were aligned correctly and with proper tails reported (correct tails); unique mapping reads that didn't have correct alignment or tails were categorized another group (wrong tails/wrong alignment). The unmappable and multiple mapping reads were grouped together (undetermined or unmappable).
To prove the accuracy of Tailor when confounding factors were not considered, we then used either Tailor or the Chen method to identify the non-templated tailing events (18). To achieve maximal speed of the Chen method to our best knowledge, we used the ‘-3 k’ option of Bowtie to clip k bases off from the 3′ end of each read. This strategy avoided calling secondary programs and ensured that minimal computational work was done other than Bowtie mapping. We started the alignment by setting k to 0. After the initial mapping, the unaligned reads were realigned with an incremented k (k = 1). This process was repeated four times. In the last iteration, four nucleotides were trimmed off from the 3′ end (k = 4) and all the tailed reads should have been mapped at this point. In the simulation test, this method finished in 67 ± 1 s with Bowtie been called five times (k = 0–4). Not surprisingly, directly mapping by Tailor finished in 22 ± 1 seconds in the same computational environment. Both methods reported the same coordinates. However, in such setting, Chen method was not able to identify the tails, which requires considerable computational work and time to retrieve from the raw reads. In contrast, Tailor revealed the length and the identity of the tails in the alignment output directly (see Supplementary Materials).
Performance with error tolerance
It is arguable that some NGS aligners that support local alignment, such as Bowtie2 (38) and BWA, can recover those tails with error tolerance. We simulated two datasets (one normal, one mutated, see below) whose distribution of read length follows that of the real small RNA sequencing dataset (43) (see Datasets in ‘Materials and Methods’ section; and also Supplementary Figure S2). For the normal dataset, two million reads were randomly sampled from the reference genome. We intentionally kept reads having just one unique occurrence in the genome and then appended a 1–4 nt non-templated tail on each read. For the mutated dataset, a similar procedure was used to generate another two million reads, but one additional step was added: we introduced one substitution in the nucleotides 2–8 of each read to simulate an RNA editing event as suggested by Vesely et al. (32). Again, this substitution was picked carefully to have only one occurrence in the genome with exactly one mismatch. The simulation guaranteed that there existed only one best alignment to the reference for each read in both datasets (see Datasets in ‘Materials and Methods’ section).
Then we examined the mappability of these datasets by Tailor (with −v option), Bowtie2 and BWA (See Figure 3C). Tailor clearly reported more unique mapping reads than others especially in the mutated datasets. When we looked closer to those reads that were mapped to multiple positions, we found Bowtie2 and BWA were more likely to align the tails to the reference than Tailor and create many alternative alignments. Note that the seed region setting was used to aid all three tools for the alignment (S = 20 and −v in Tailor and the equivalences in Bowtie2 and BWA; mismatches in the seed region were allowed) and all tools should try to align the first 20 nt of each read to the genome, but Bowtie2 and BWA still generated suboptimal alignments. The execution time of three aligners with the error tolerant setting is depicted in Supplementary Figure S3. The complete commands for running all the tests are listed in Supplementary Materials.
We further checked whether the alignments and the tails were correctly reported. As shown in Figure 3D, Tailor was the only tool that gave satisfactory results reporting correct alignments and tails in the mutated dataset. There was no information in the output of BWA to recover the tails, and since most of the reads were aligned to multiple locations, it was expected that extensive post-processing would be needed for extracting the tails. The simulation clearly shows that Tailor is the only practical solution for doing tailing analysis with confounding factors.
Analysis pipeline
In order to provide a thorough and straightforward tailing analysis of deep sequencing libraries to the scientific community, we developed the interface of Tailor to take FastQ files as input and produce publication-ready figures. The flowchart of the pipeline is summarized in Supplementary Figure S4A. In brief, the input reads, with barcodes and adaptors removed, are subject to a quality-filtering step based on a PHRED score threshold provided by the user (e.g. to get rid of B-tails). The pipeline then applies Tailor to align the high-quality reads to the reference. The information on the length and identity of tails are then retrieved from the SAM formatted output and summarized to a tabular text file. Additionally, the alignments are assigned to different genomic features (miRNAs, exons, introns, etc.) using BEDTools (44). Tails from different categories are summarized. Publication quality figures depicting the length distribution are drawn using R package ggplot2 (23) (Supplementary Figure S4B). The pipeline also offers microRNA specific analysis. Balloon plots describing the 5′ and 3′ relative positions and the tails length are provided for a comprehensive overview (Supplementary Figure S4C).
Applications—case studies
To prove the utility of Tailor, we applied Tailor to reanalyze several publicly available small RNA sequencing datasets and revealed new facts about the data that has not been reported yet. In plants, HUA ENHANCER 1 (HEN1) methylates both miRNA and siRNA at their 3′ ends to protect them from non-templated uridylation catalyzed by HEN1 SUPPRESSOR1 (HESO1), a terminal nucleotidyl transferase that favors uridine as substrate (18,45). We applied Tailor on small RNA sequencing libraries from WT, hen1−/− and hen1−/−;heso1−/− cells of Arabidopsis and the results showed that siRNAs were subjected to both non-templated uridylation and cytosylation without HEN1 while miRNAs were mainly subjected to uridylation. Furthermore, the loss of HESO1 only reduced the uridylation but not cytosylation of siRNAs, suggesting the existence of additional nucleotidyl transferase that prefers cytosine as substrates (Figure 4A).
Figure 4.
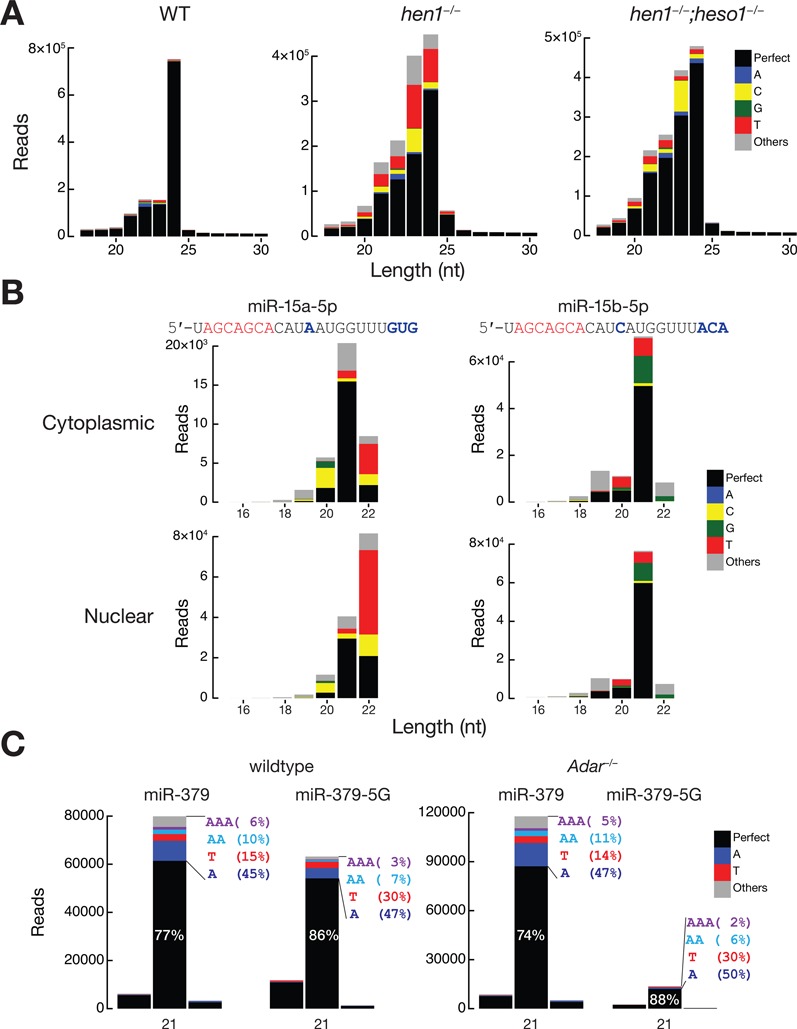
Applications of Tailor and the accompanying shell pipeline. (A) Length distribution of mRNA-derived small RNA reads with tailing information from wild-type, hen1 mutant and hen1, heso1 double mutant tissues from Arabidopsis. Raw read counts are shown without normalization. Perfect match and tailed reads are indicated in different colors. (B) Length distribution of Ago2 associated Hsa-miR-15a (left) and Hsa-miR-15b (right) in cytoplasm (top) and nucleus (bottom) fraction of HeLa cell. Raw read count are shown without normalization. Note that since the authors of these libraries used poly-adenylation instead of 3′ ligation in their cloning strategy, it was impractical to identify A tailing. (C) Tail composition for miR-379 and the edited form (miR-379–5G) in wild-type and Adar−/− libraries.
We then applied Tailor to two NGS libraries that cloned Ago2 associated small RNA from nuclear and cytoplasmic fraction of HeLa cells respectively (46). Since RNAs were cloned using poly-A polymerase instead of 3′ adaptor ligation in the library preparation, A-tails were unable to be recovered computationally. Although most miRNAs showed very similar length distribution and tailing frequency between these two samples, one miRNA, miR-15a, exhibited a distinct pattern. In cytoplasm, miR-15a was mostly 21 nt long and had modest U tailing for its 22-mer isoform. Surprisingly, in the nuclear fraction, miR-15a peaked at 22 nt and showed strong U tailing (Figure 4B). In addition, miR-15b, which shares its seed sequence with miR-15a and only has one nucleotide different from miR-15a in the first 19 nt of its mature sequence, did not exhibit obvious variation between the two samples. This suggests that, either 9–12 nt, also known as the ‘central site’ or the 3′ end of guide miRNA play an important role in tailing regulation.
Finally, we applied Tailor to study the possible relationship between RNA editing and tailing in microRNAs. The miRNA libraries were constructed from the whole brain tissue cells dissected from Adar2−/− and wild-type mice (32). Adar2 is known for its strongest effects on miRNA abundance and editing among the three isoforms of ADARs (47). One of the highly expressed ADAR substrates, miR-379, was shown to be directly edited at the nucleotide five within the seed region and about half of the mature miR-379 were edited by ADAR2 (32). As expected, the edited form of miR-379 (i.e. miR-379–5G) was greatly reduced in Adar−/− mice. Surprisingly, we found that the normal miR-379 has much more tailing than miR-379–5G (see Figure 4C). Mono-A and poly-A tails (the bluish portion) were depleted in miR-379–5G, which raises the probability that ADARs and the A-to-I editing could affect the affinity between the miRNAs and the unknown enzymes responsible for adenylylating the 3′ end. Since the proportion of different types of tails was unchanged upon Adar2 knockout, the tailing machinery is less likely modulated by ADAR2 directly but by the subsequent factors after editing in the seed, such as differential targeting, RNA stability change or miRNA-Argonaute sorting (1,48).
DISCUSSION
Tailing is a molecular phenomenon that associates with the function, processing and stability of many small RNAs. Computational identification of the tailed sequences from the millions of NGS reads has been proven to be challenging and time-consuming. We herein present a tailing analysis framework, Tailor, which aligns reads to the reference genome, reports tailing events simultaneously and visualizes analysis results. We assessed the accuracy of Tailor by comparing it with the Chen method with simulated reads and found they generated exactly the same results while Tailor only used a third of the time to align and provided more information comparing to the alternative.
When confounding factor was ignored, Tailor was not slower than other well-known fast general-purpose mappers in our tests. We demonstrated that Tailor executed in a speed that was very competitive to, if not better than, Bowtie and BWA, while providing more functionalities for detecting tailing events. When confounding factors was presented in the reads, it was arguable that advanced NGS aligners that support the local alignment mode (e.g. Bowtie2) could be competent in finding tails, but we tested them with simulated reads and showed that Tailor performed significantly better in both accuracy and efficiency.
Tailor's shell-based framework takes raw reads as input and produces comprehensive tailing analysis results and publication quality figures. We reproduced known conclusions drawn from the published tailing study by the pipeline with little extra scripting and post-processing. We also applied the pipeline to other datasets and shed light on other possibilities of the functional roles of tailing, such as involving in RNA processing, transport, decay and storage by interacting with other RNA binding proteins (49).
Our aims to design Tailor are to reduce the cost of doing tailing analysis and reinforce or even replace the conventional computational procedure in analyzing all short non-coding RNAs. We expect that Tailor could be applied to a broader scope and subsequently facilitate the understanding of biological processes related to tailing.
AVAILABILITY
Source code as an Open Source project: http://jhhung.github.io/Tailor.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
We thank the members of the Hung, Weng and Zamore laboratories for helpful discussion and critical testing.
FUNDING
National Institutes of Health [GM62862, GM65236 to P.D.Z., in part and P01HD078253 to Z.W.]; National Science Council [103-2221-E-009 -128 -MY2 to J.H.H.]. Funding for open access charge: National Science Council [103-2221-E-009 -128 -MY2 to J.H.H.].
Conflict of interest statement. None declared.
REFERENCES
- 1.Ameres S.L., Zamore P.D. Diversifying microRNA sequence and function. Nat. Rev. Mol. Cell Biol. 2013;14:475–488. doi: 10.1038/nrm3611. [DOI] [PubMed] [Google Scholar]
- 2.Castel S.E., Martienssen R.A. RNA interference in the nucleus: roles for small RNAs in transcription, epigenetics and beyond. Nat. Rev. Genet. 2013;14:100–112. doi: 10.1038/nrg3355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Luteijn M.J., Ketting R.F. PIWI-interacting RNAs: from generation to transgenerational epigenetics. Nat. Rev. Genet. 2013;14:523–534. doi: 10.1038/nrg3495. [DOI] [PubMed] [Google Scholar]
- 4.Ghildiyal M., Zamore P.D. Small silencing RNAs: an expanding universe. Nat. Rev. Genet. 2009;10:94–108. doi: 10.1038/nrg2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Heo I., Ha M., Lim J., Yoon M.J., Park J.E., Kwon S.C., Chang H., Kim V.N. Mono-uridylation of pre-microRNA as a key step in the biogenesis of group II let-7 microRNAs. Cell. 2012;151:521–532. doi: 10.1016/j.cell.2012.09.022. [DOI] [PubMed] [Google Scholar]
- 6.Heo I., Joo C., Cho J., Ha M., Han J., Kim V.N. Lin28 mediates the terminal uridylation of let-7 precursor MicroRNA. Mol. Cell. 2008;32:276–284. doi: 10.1016/j.molcel.2008.09.014. [DOI] [PubMed] [Google Scholar]
- 7.Heo I., Joo C., Kim Y.K., Ha M., Yoon M.J., Cho J., Yeom K.H., Han J., Kim V.N. TUT4 in concert with Lin28 suppresses microRNA biogenesis through pre-microRNA uridylation. Cell. 2009;138:696–708. doi: 10.1016/j.cell.2009.08.002. [DOI] [PubMed] [Google Scholar]
- 8.van Wolfswinkel J.C., Claycomb J.M., Batista P.J., Mello C.C., Berezikov E., Ketting R.F. CDE-1 affects chromosome segregation through uridylation of CSR-1-bound siRNAs. Cell. 2009;139:135–148. doi: 10.1016/j.cell.2009.09.012. [DOI] [PubMed] [Google Scholar]
- 9.Ameres S.L., Horwich M.D., Hung J.H., Xu J., Ghildiyal M., Weng Z., Zamore P.D. Target RNA-directed trimming and tailing of small silencing RNAs. Science. 2010;328:1534–1539. doi: 10.1126/science.1187058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xie J., Ameres S.L., Friedline R., Hung J.H., Zhang Y., Xie Q., Zhong L., Su Q., He R., Li M., et al. Long-term, efficient inhibition of microRNA function in mice using rAAV vectors. Nat. Methods. 2012;9:403–409. doi: 10.1038/nmeth.1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ameres S.L., Hung J.H., Xu J., Weng Z., Zamore P.D. Target RNA-directed tailing and trimming purifies the sorting of endo-siRNAs between the two Drosophila Argonaute proteins. RNA. 2011;17:54–63. doi: 10.1261/rna.2498411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Altschul S.F., Gish W., Miller W., Myers E.W., Lipman D.J. Basic local alignment search tool. J. Mol. Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 13.Zhang Z., Schwartz S., Wagner L., Miller W. A greedy algorithm for aligning DNA sequences. J. Comput. Biol. 2000;7:203–214. doi: 10.1089/10665270050081478. [DOI] [PubMed] [Google Scholar]
- 14.Li H., Ruan J., Durbin R. Mapping short DNA sequencing reads and calling variants using mapping quality scores. Genome Res. 2008;18:1851–1858. doi: 10.1101/gr.078212.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Langmead B., Trapnell C., Pop M., Salzberg S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li R., Li Y., Kristiansen K., Wang J. SOAP: short oligonucleotide alignment program. Bioinformatics. 2008;24:713–714. doi: 10.1093/bioinformatics/btn025. [DOI] [PubMed] [Google Scholar]
- 18.Zhao Y., Yu Y., Zhai J., Ramachandran V., Dinh T.T., Meyers B.C., Mo B., Chen X. The Arabidopsis nucleotidyl transferase HESO1 uridylates unmethylated small RNAs to trigger their degradation. Curr. Biol. 2012;22:689–694. doi: 10.1016/j.cub.2012.02.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Marco A., Griffiths-Jones S. Detection of microRNAs in color space. Bioinformatics. 2012;28:318–323. doi: 10.1093/bioinformatics/btr686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ukkonen E. Online Construction of Suffix Trees. Algorithmica. 1995;14:249–260. [Google Scholar]
- 21.Dohm J.C., Lottaz C., Borodina T., Himmelbauer H. Substantial biases in ultra-short read data sets from high-throughput DNA sequencing. Nucleic Acids Res. 2008;36:e105. doi: 10.1093/nar/gkn425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Qu W., Hashimoto S., Morishita S. Efficient frequency-based de novo short-read clustering for error trimming in next-generation sequencing. Genome Res. 2009;19:1309–1315. doi: 10.1101/gr.089151.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Minoche A.E., Dohm J.C., Himmelbauer H. Evaluation of genomic high-throughput sequencing data generated on Illumina HiSeq and genome analyzer systems. Genome Biol. 2011;12:R112. doi: 10.1186/gb-2011-12-11-r112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Le H.S., Schulz M.H., McCauley B.M., Hinman V.F., Bar-Joseph Z. Probabilistic error correction for RNA sequencing. Nucleic Acids Res. 2013;41:e109. doi: 10.1093/nar/gkt215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Blow M., Futreal P.A., Wooster R., Stratton M.R. A survey of RNA editing in human brain. Genome Res. 2004;14:2379–2387. doi: 10.1101/gr.2951204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim D.D., Kim T.T., Walsh T., Kobayashi Y., Matise T.C., Buyske S., Gabriel A. Widespread RNA editing of embedded alu elements in the human transcriptome. Genome Res. 2004;14:1719–1725. doi: 10.1101/gr.2855504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Morse D.P., Aruscavage P.J., Bass B.L. RNA hairpins in noncoding regions of human brain and Caenorhabditis elegans mRNA are edited by adenosine deaminases that act on RNA. Proc. Natl. Acad. Sci. U.S.A. 2002;99:7906–7911. doi: 10.1073/pnas.112704299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Blow M.J., Grocock R.J., van Dongen S., Enright A.J., Dicks E., Futreal P.A., Wooster R., Stratton M.R. RNA editing of human microRNAs. Genome Biol. 2006;7:R27. doi: 10.1186/gb-2006-7-4-r27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Luciano D.J., Mirsky H., Vendetti N.J., Maas S. RNA editing of a miRNA precursor. RNA. 2004;10:1174–1177. doi: 10.1261/rna.7350304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Warnefors M., Liechti A., Halbert J., Valloton D., Kaessmann H. Conserved microRNA editing in mammalian evolution, development and disease. Genome Biol. 2014;15:R83. doi: 10.1186/gb-2014-15-6-r83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kume H., Hino K., Galipon J., Ui-Tei K. A-to-I editing in the miRNA seed region regulates target mRNA selection and silencing efficiency. Nucleic Acids Res. 2014;42:10050–10060. doi: 10.1093/nar/gku662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vesely C., Tauber S., Sedlazeck F.J., Tajaddod M., von Haeseler A., Jantsch M.F. ADAR2 induces reproducible changes in sequence and abundance of mature microRNAs in the mouse brain. Nucleic Acids Res. 2014;42:12155–12168. doi: 10.1093/nar/gku844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Frampton M., Houlston R. Generation of artificial FASTQ files to evaluate the performance of next-generation sequencing pipelines. PLoS One. 2012;7:e49110. doi: 10.1371/journal.pone.0049110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Burrows M., Wheeler D.J. Technical Report 124. Palo Alto, CA: DEC, Digital Systems Research Center; 1994. A block-sorting lossless data compression algorithm. [Google Scholar]
- 36.Ferragina P., Manzini G. Opportunistic data structures with applications. Ann. IEEE Symp. 2000:390–398. [Google Scholar]
- 37.Karkkainen J. Fast BWT in small space by blockwise suffix sorting. Theor. Comput. Sci. 2007;387:249–257. [Google Scholar]
- 38.Langmead B., Salzberg S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vyverman M., De Baets B., Fack V., Dawyndt P. Prospects and limitations of full-text index structures in genome analysis. Nucleic Acids Res. 2012;40:6993–7015. doi: 10.1093/nar/gks408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li H., Homer N. A survey of sequence alignment algorithms for next-generation sequencing. Brief. Bioinform. 2010;11:473–483. doi: 10.1093/bib/bbq015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., Genome Project Data Processing, S. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li J., Yang Z., Yu B., Liu J., Chen X. Methylation protects miRNAs and siRNAs from a 3′-end uridylation activity in Arabidopsis. Curr. Biol. 2005;15:1501–1507. doi: 10.1016/j.cub.2005.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Huang H., Li Y., Szulwach K.E., Zhang G., Jin P., Chen D. AGO3 Slicer activity regulates mitochondria-nuage localization of Armitage and piRNA amplification. J. Cell Biol. 2014;206:217–230. doi: 10.1083/jcb.201401002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Quinlan A.R., Hall I.M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ren G., Chen X., Yu B. Uridylation of miRNAs by hen1 suppressor1 in Arabidopsis. Curr. Biol. 2012;22:695–700. doi: 10.1016/j.cub.2012.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ameyar-Zazoua M., Rachez C., Souidi M., Robin P., Fritsch L., Young R., Morozova N., Fenouil R., Descostes N., Andrau J.C., et al. Argonaute proteins couple chromatin silencing to alternative splicing. Nat. Struct. Mol. Biol. 2012;19:998–1004. doi: 10.1038/nsmb.2373. [DOI] [PubMed] [Google Scholar]
- 47.Vesely C., Tauber S., Sedlazeck F.J., von Haeseler A., Jantsch M.F. Adenosine deaminases that act on RNA induce reproducible changes in abundance and sequence of embryonic miRNAs. Genome Res. 2012;22:1468–1476. doi: 10.1101/gr.133025.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dueck A., Ziegler C., Eichner A., Berezikov E., Meister G. microRNAs associated with the different human Argonaute proteins. Nucleic Acids Res. 2012;40:9850–9862. doi: 10.1093/nar/gks705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gerstberger S., Hafner M., Tuschl T. A census of human RNA-binding proteins. Nat. Rev. Genet. 2014;15:829–845. doi: 10.1038/nrg3813. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.