

Abstract
Purpose
A first-in-human pilot safety and feasibility trial evaluating chimeric antigen receptor (CAR) engineered, autologous primary human CD8+ cytolytic T lymphocytes (CTLs) targeting IL13Rα2 for the treatment of recurrent glioblastoma (GBM).
Experimental Design
Three patients with recurrent GBM were treated with IL13(E13Y)-zetakine CD8+ CTL targeting IL13Rα2. Patients received up to twelve local infusions at a maximum dose of 108 CAR-engineered T cells via a catheter/reservoir system.
Results
We demonstrate the feasibility of manufacturing sufficient numbers of autologous CTL clones expressing an IL13(E13Y)-zetakine CAR for redirected HLA-independent IL13Rα2-specific effector function for a cohort of patients diagnosed with GBM. Intracranial delivery of the IL13-zetakine+ CTL clones into the resection cavity of three patients with recurrent disease was well-tolerated, with manageable temporary CNS inflammation. Following infusion of IL13-zetakine+ CTLs, evidence for transient anti-glioma responses was observed in two of the patients. Analysis of tumor tissue from one patient before and after T cell therapy suggested reduced overall IL13Rα2 expression within the tumor following treatment. MRI analysis of another patient indicated an increase in tumor necrotic volume at the site of IL13-zetakine+ T cell administration.
Conclusion
These findings provide promising first-in-human clinical experience for intracranial administration of IL13Rα2-specific CAR T cells for the treatment of GBM, establishing a foundation on which future refinements of adoptive CAR T cell therapies can be applied.
Keywords: Glioblastoma, Immunotherapy, Chimeric Antigen Receptor (CAR), T cell, IL13Rα2
Introduction
Despite aggressive standard-of-care therapies including surgery, radiation and chemotherapy, glioblastoma (GBM) remains one of the most universally lethal human cancers, with a 5-year survival rate of less than 10% (1). Inevitable treatment failure for most patients is largely attributable to therapy-resistant invasive malignant cells that are responsible for tumor recurrence. Adoptive cell therapy (ACT) with T cells genetically modified to express chimeric antigen receptors (CARs) is a promising therapeutic approach that may be effectively and safely applied to GBM to reduce recurrence rates (2). CAR T cells can be rapidly generated to specifically recognize antigenically-distinct tumor populations independently of pre-existing anti-tumor immunity (3-6), and T cells can migrate through the brain parenchyma to target and kill infiltrative malignant cells (7-9). Recent clinical successes with CAR-engineered T cells mediating durable and complete responses in patients diagnosed with CD19+ malignancies, even under conditions of bulky disease, provide support for further development of this therapeutic approach (10,11). However, broader clinical application to solid tumors, including brain tumors, has proven to be complex and is presently under intense investigation.
Our group has focused on a T cell immunotherapy for GBM targeting IL-13 receptor α2 (IL13Rα2), a monomeric high-affinity IL-13 receptor which is over-expressed by more than 50% of GBM and is a prognostic indicator of poor patient survival (12-15). In the context of molecular subtypes of high-grade gliomas, IL13Rα2 expression is most closely associated with expression of mesenchymal signature genes, which may reflect its association with the proinflammatory characteristics of mesenchymal tumors (12,16). IL13Rα2 is expressed by both stem-like and more differentiated malignant cells (5), as well as by tumor-infiltrating macrophages/myeloid derived suppressor cells (17). Importantly, IL13Rα2 is not expressed at significant levels on normal brain tissue (15,18-20), and early phase clinical trials support the safety and tolerability of targeting IL13Rα2 for the treatment of GBM with vaccine therapy (21) and IL-13-immunotoxins (22), thereby qualifying IL13Rα2 as an attractive immunological therapeutic target.
We have developed an IL13Rα2-specific, MHC-independent CAR, termed IL13-zetakine. This CAR recognizes IL13Rα2 via a membrane-tethered IL13 ligand mutated at a single site (E13Y) to reduce potential binding to the more widely expressed IL13Rα1/R4α complex (23,24), and initiates cytolytic killing via an intracellular CD3ζ T cell-activating domain. IL13-zetakine+ cytotoxic T lymphocytes (CTL) possess MHC-independent, IL13Rα2-specific anti-glioma cytolytic activity, secrete Tc1 cytokines and proliferate in vitro upon engagement of IL13Rα2-expressing targets, and mediate regression of established human GBM xenografts in vivo (5,23). IL13-zetakine+ CTL also target IL13Rα2+ glioma stem-like cancer initiating cells in vitro and eliminate glioma-initating activity in an orthotopic mouse tumor model (5).
These preclinical studies have culminated in the completion of this first-in-human pilot safety and feasibility study evaluating intracranial adoptive transfer of autologous IL13-zetakine+ CD8+ T cells in patients with recurrent glioblastoma. Here we report our clinical experience treating three patients using repetitive intracavitary administration of IL13Rα2-specific CD8+ CAR T cell clones following tumor resection.
Materials and Methods
Study Design and Research Participants
This single-institution first-in-human pilot safety and feasibility study was conducted from 2008-2011. All participating patients gave written informed consent. The clinical protocol was approved by the City of Hope Institutional Review Board, conducted under an Investigational New Drug Application (IND 10109), and registered at ClinicalTrials.gov (NCT00730613).
Eligible patients were adults (18-70 yrs) with recurrent or refractory unifocal supratentorial grade III or IV glioma whose tumors did not show communication with ventricles/CSF pathways and were amenable to resection. Patients were required to have a survival expectation of greater than 3 months, a Karnofsky performance status (KPS) equal to or greater than 70, to be steroid independent, and to have completed primary therapy (≥ 2 weeks) recovering from all acute side effects prior to enrollment. Participation in this trial was independent of IL13Rα2 tumor antigen status.
Patients were enrolled following initial diagnosis of high-grade glioma (WHO grade III or IV), at which time they underwent leukapheresis for collection of peripheral blood mononuclear cells (PBMC). These cells were used to engineer CD8+ CTLs to express the IL13-zetakine CAR and the ancillary HyTK selection/suicide fusion protein (23). Subsequently, the release tested therapeutic IL13-zetakine/HyTK T cells were cryopreserved and stored for later use. At the time of first recurrence of the tumor, the research participant underwent resection of tumor along with placement of a Rickham reservoir/catheter. Concurrently, the therapeutic clone was thawed, re-expanded in vitro using rapid expansion method (REM) stimulation. Following recovery from surgery and post baseline MR imaging, the IL13-zetakine+ CD8+ CTLs were administered directly into the resection cavity via the indwelling catheter (Supplementary Fig. S1 and Supplementary Methods). Cells were manually injected into the Rickham reservoir using a 21 gauge butterfly needle to deliver a 2 mL volume over 5-10 minutes, followed by 2 mL flush with preservative free normal saline over 5 minutes.
The protocol treatment plan specified an intra-patient dose escalation schedule with a target of 12 CAR T cell doses administered intracranially over a 5 week period comprised of weekly treatment cycles (Fig. 1A). During cycles 1, 2, 4 and 5, T cell infusions were performed on days 1, 3 and 5 of the cycle week, and week 3 was a rest cycle. For safety, in cycle 1 we utilized an intrapatient dose escalation strategy, with CAR T cell doses of 107, 5 × 107 and 108 cells per infusion administered on days 1, 3 and 5 respectively, and this was followed by 9 additional CAR T cell infusions of 108 cells over 4 weeks. Imaging to assess response was performed during the week 3 rest cycle and after week 5. The guidelines provided in the NCI Common Toxicity Criteria version 2.0 (https://ctep.ifo.nih.gov/l) were followed for the monitoring of toxicity and adverse event reporting.
Fig. 1. Treatment schema and IL13-zetakine+ CTL manufacturing.
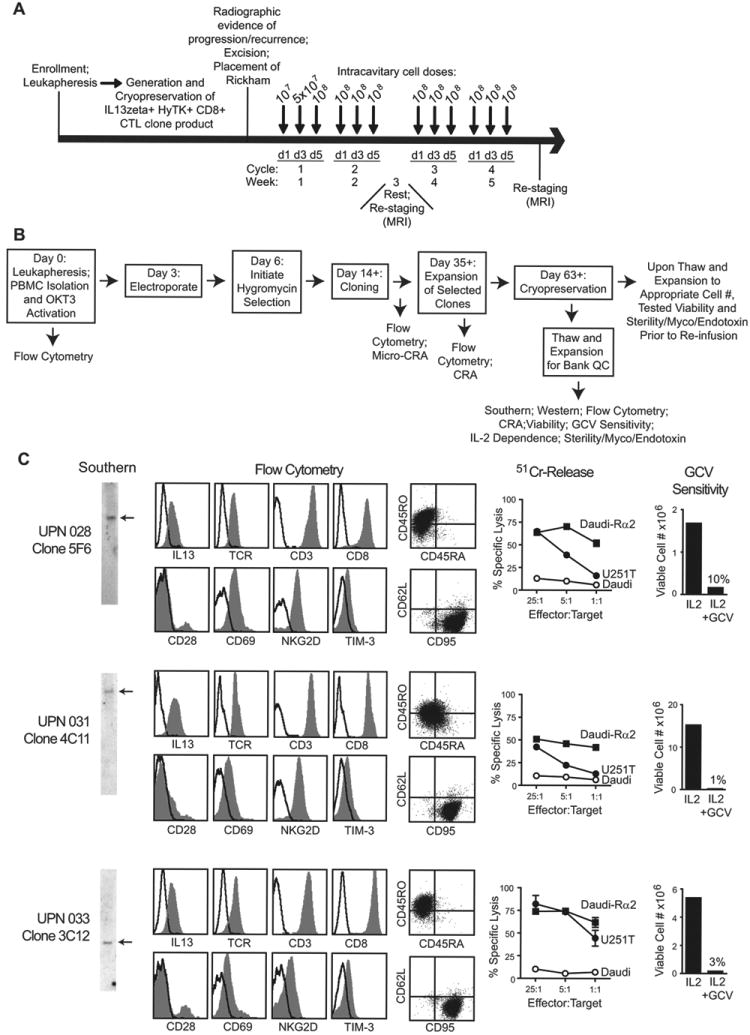
A, Four weekly cycles of intracavitary cell doses were administered after enrolled patients experienced recurrence and underwent tumor excision with placement of a Rickham catheter. Patients had a week of rest for brain imaging between cycles 2 and 3. B, Schematic of the manufacturing process, with day of each step(s) and in-process analyses indicated. CRA, chromium release assay; GCV, ganciclovir; Myco, mycoplasma; OKT3, a CD3 agonistic antibody used to activate T cells; PBMC, peripheral blood mononuclear cell. C, Characterization of the three cell products administered to patients with recurrent glioblastoma. Depicted from left to right: Southern blots of T cell genomic DNA using a hygomycin-specific probe (detecting HyTK selection/suicide fusion gene) showing existence of single bands as indicated by arrows; flow cytometric analysis of surface CAR expression using anti-IL13 antibody, or the T cell markers TCR-αβ, CD3, CD8, CD28, CD45RO, CD45RA, CD62L, CD69, CD95, NKG2D, and TIM-3 where isotype control staining is indicated with the open histograms or quadrant placement; ability of CTL clones to lyse IL13Rα2+ targets U251T and Daudi-13Rα2, but not non-engineered Daudi cells, determined in a 4-hour 51Cr-release assay; and ganciclovir (GCV) sensitivity using a flow-cytometry based assay for viable cell numbers after 14 days of culture with either rHuIL-2 or rHuIL-2 + GCV.
Clinical IL13-zetakine CAR vector and T cell manufacturing
A schematic of the T cell manufacturing process is provided in Figure 1B. The plasmid vector, encoding the IL13-zetakine CAR and the selection/suicide HyTK transgenes, has been described previously (23). Briefly, the IL13-zetakine CAR is composed of the human GM-CSF receptor alpha chain leader peptide, human IL-13(E13Y) mutein, human IgG4 Fc linker, human CD4 transmembrane domain, and the cytoplasmic domain of human CD3 zeta. For CAR T cell manufacturing, PBMC were stimulated with OKT3 (Janssen Biotech, Inc., Horsham, PA), electroporated with linearized IL13-zetakine/HyTk DNA, selected in hygromycin (InvivoGen, San Diego, CA), and plated at limiting dilution to isolate single CD8+ CAR+ T cell clones. Subsequently, CAR T cell clones were expanded using a 14-day cycle of REM consisting of OKT3, rhIL-2 (Novartis Oncology, East Hanover, NJ), and irradiated feeders as previously described (25), the only exception being the use of 100 mg/mL hygromycin in place of G418. Irradiated feeders were authenticated for viability, growth, sterility, and lack of mycoplasma prior to cryopreservation as a GMP bank, and thawed cells were cultured for less than 6 months prior to use in a REM cycle. A summary of the quality control tests performed and the requisite test results for T cell product release are summarized in Table S1 and Supplementary Methods.
Clinical Imaging
MRI scans of the brain, including fluid attenuated inversion recovery (FLAIR) and post-gadolinium T1-weighted sequences were acquired on a GE Signa Excite scanner (1.5 Tesla). Regions of contrast-enhancing or necrotic tumor were outlined by a radiologist, and the corresponding volumes were automatically determined using a GE AW workstation running AW version 4.3 software. Imaging with 18F-fluorodeoxyglucose (18F-FDG) was performed using a GE Advance NXi PET scanner (15 cm axial field of view, slice separation 4.3 mm) or a GE Discovery STe 16 PET-CT scanner (15 cm axial field of view, slice separation 3.3 mm).
Protein and mRNA analyses
IL13Rα2 immunohistochemistry (IHC) was performed on 5 µm-sections of formalin-fixed paraffin-embedded specimens as previously described (5). The tumor regions (tumor cells >60%) were determined by a clinical neuropathologist. For Figure 2A and B, IL13Rα2 expression was calculated for defined tumor areas as integrated OD (rIOD)/Lumen.pixel2) using the DAB plug-in for ImagePro Premier v9.1 (Media Cybernetics). For Figure 4C, IL13Rα2 expression levels were measured in individual pixels using the color deconvolution function in Fiji (http://fiji.sc/Colour_Deconvolution) to extract the DAB channel, and by measuring staining density on a per pixel basis in neuropathologist-defined tumor areas using ImagePro Premier v9.1.
Fig. 2. Tumor IL13Rα2 expression for each patient.
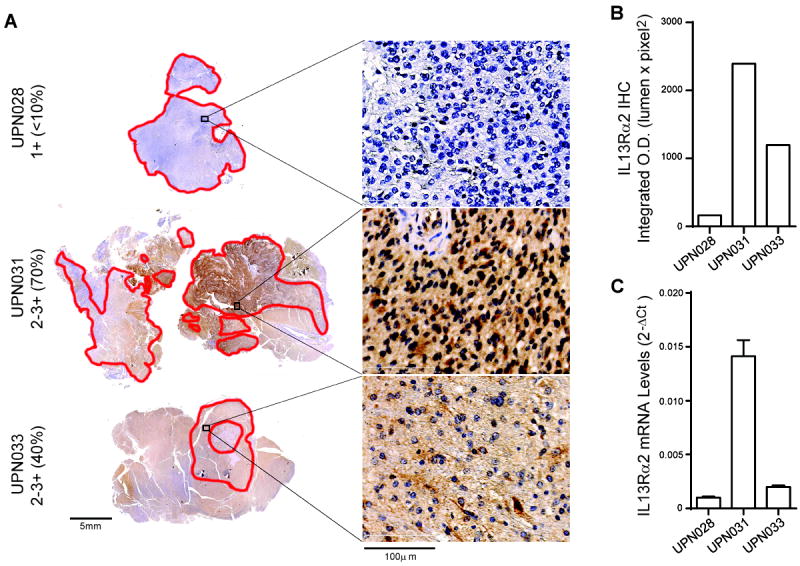
A, Immunohistochemical staining of IL13Rα2 on paraffin-embedded primary patient derived brain tissues. Sections were scored blindly by a neuropathologist for staining intensity (0, not detected; 1+, low; 2+, moderate; 3+, high), and percentages of positive cells are indicated. B, Quantification of DAB intensity (lumen × pixel2) for dense tumor regions (>60% tumor, red outline). C, Gene expression of IL13Rα2 mRNA levels evaluated by qPCR using Taqman gene expression assay.
Fig. 4. Effect of CAR T cell therapy on IL13Rα2 expression by the tumor of UPN031.
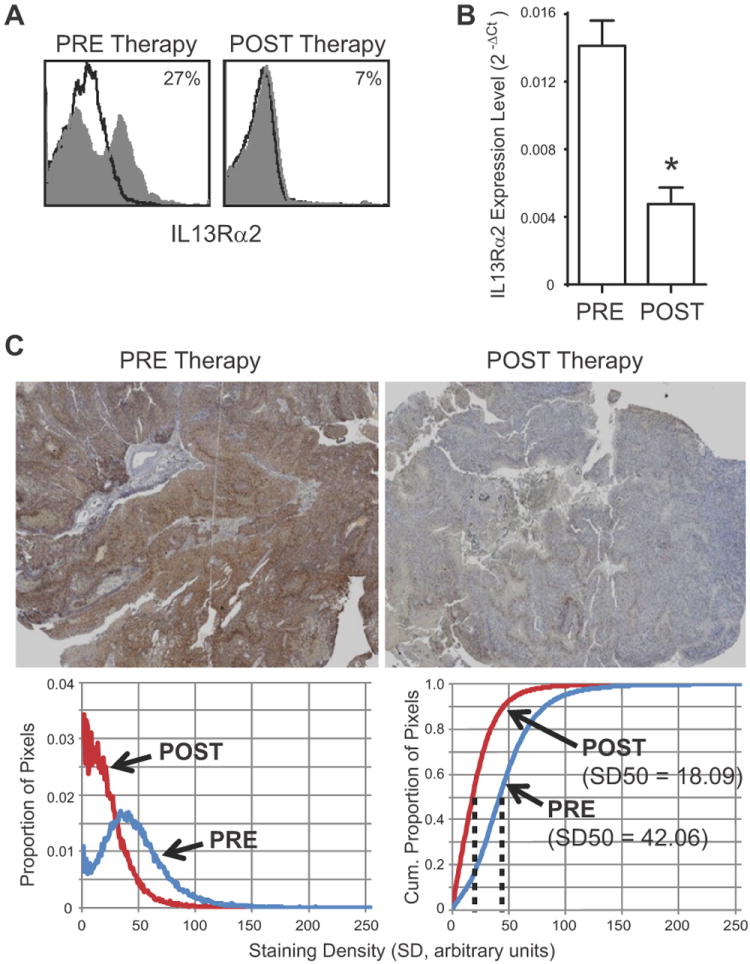
Primary patient derived brain tumor tissue from UPN031 was excised at day 8 (PRE Therapy) and day 184 (POST Therapy) according to the timeline depicted in Figure S2B. A, Flow cytometric analysis of IL13Rα2 surface expression by freshly-dissociated tumor cells using fluorochrome-conjugated anti-IL13Rα2 antibody. Percentage of immunoreactive cells (grey) above isotype control (black line) is indicated in each histogram. B, Gene expression of IL13Rα2 mRNA levels evaluated by qPCR using Taqman gene expression assay. C, Immunochemical staining (IL13Rα2-specific DAB with hematoxylin counterstain; top) and histograms of IL13Rα2-specific DAB staining density (below). Left, proportion of pixels with IL13Rα2-specific DAB staining between 0 (clear) and 255 (opaque). Right, cumulative proportions of pixels with IL13Rα2-specific DAB staining, with median values (SD50) indicated.
To quantify IL13Rα2 mRNA levels, RNA samples were isolated from frozen tumor samples using the Mini RNeasy Kit (Qiagen), and cDNA was synthesized using the Sensiscript RT Kit (Qiagen). Real-time PCR analysis was performed in triplicate using Taqman gene expression assay (Life Technologies) with probe sets for IL13Rα2 (ID Hs00152924) and GAPDH (ID Hs02758991).
Cell surface expression of IL13Rα2 was detected by flow cytometry using goat polyclonal anti-IL13Rα2 (AF146, R&D Systems), followed by FITC-conjugated mouse anti-goat Ig (Jackson ImmunoResearch). Samples were run on a FACS Caliber (BD Biosciences), and percentages of immunoreactive cells were calculated using the subtraction method via FCS Express version 3 software (De Novo Software). For other surface phenotyping of CAR T cells, see Supplementary Methods.
Results
Characterization of Autologous IL13-zetakine+ CTL clones
Characterization of the three manufactured IL13-zetakine/HyTK CAR CD8+ T cell clones used for therapy are shown in Figure 1C. Manufactured CAR T cell lines were evaluated for clonality and single-site integration by Southern blot analysis, and for uniform cell surface expression of the IL13-zetakine CAR by flow cytometry. Flow cytometry demonstrated that the CAR T cell clones uniformly expressed the T cell receptor-αβ, CD3 and CD8, and displayed an activated effector T cell phenotype, as they expressed CD45RO, CD69 and CD95, but were negative for CD45RA, and central memory markers CD62L, CCR7 and CD127 (Fig. 1C and data not shown). The CAR T cell clones all expressed the NKG2D co-stimulatory receptor, with a portion of each population also expressing CD28 (Fig. 1C). Tim-3, a marker of T cell exhaustion and senescence, was also expressed at low levels by all three lines (Fig. 1C), but the CAR T cells were negative for other markers of T cell exhaustion including KLRG-1 and PD-1 (data not shown).
The manufactured CAR T cell clones exhibited redirected killing against the IL13Rα2-positive U251T glioma cell line, as well as Daudi cells genetically modified to express IL13Rα2, but not control IL13Rα2-negative Daudi cells. Interestingly, there was no direct correlation between cell surface expression of the CAR and percent IL13Rα2-specific cytotoxicity (e.g., compare low-expressing clone 3C12 of UPN033 with higher-expressing clone 4C11 of UPN031; Fig. 1C). Furthermore, all three of the lines were sensitive to ganciclovir-mediated ablation based on the co-expression of herpes simplex virus (HSV) thymidine kinase contained within the CAR/HyTk plasmid vector used for gene-modification.
Treatment Overview
The primary objective of this study was to assess the feasibility, safety and toxicity of intracranial administration of autologous IL13Rα2-specific CAR CD8+ T cell clones for the treatment of recurrent high-grade glioma (WHO grade III and IV) following tumor resection. Cell products meeting all quality control tests (Supplementary Table S1) were generated for 10 of the 13 patients enrolled on this study; three T cell products (UPN024, UPN029, UPN032) did not complete product release due to patient disease progression prior to completion of CAR T cell manufacturing (Supplementary Table S2). Of the 10 remaining enrolled patients, three (UPN025, UPN027, UPN030) did not meet T cell infusion eligibility criteria due to multifocal disease, death, and steroid dependence, respectively, and therefore did not receive their IL13-zetakine+ CTL product. An additional three patients (UPN026, UPN034, UPN038) did not develop disease progression before the protocol was closed to treatment (Supplementary Table S2), and one patient (UPN036) voluntarily withdrew from the study at the time of progression. The three remaining patients (UPN028, UPN031, UPN033) were treated on this study. An overview of manufactured products and patient treatment/non-treatment rationale is provided in Supplementary Table S2 and Supplementary Fig. S2. For the three patients that were treated on this trial, patient characteristics, including diagnosis and prior therapy, are summarized in Supplementary Table S3. For these three patients, analyses of excised tumors collected at the time of Rickham catheter placement revealed a range of IL13Rα2 antigen expression levels as determined by both IHC (Fig. 2A and B) and qPCR (Fig. 2C) with UPN028 exhibiting the lowest, UPN033 intermediate, and UPN031 the highest level of antigen expression. Of interest, our previous studies showed that low-passage stem-like or differentiated glioma cell lines derived from IL13Rα2-positive (by IHC) UPN033 maintained IL13Rα2 expression, and were recognized and killed by this patient’s autologous IL13-zetakine+ CTLs, thus suggesting that the differentiation status of malignant cells in the tumor does not impact IL13Rα2 antigen expression or targetability (5).
Safety of IL13-zetakine+ CAR T cell Administration
The intrapatient dose escalation schedule depicted in Figure 1A was executed as planned for two of three patients (UPN031, UPN033), who received a full course of 12 escalating intracavitary doses of 107 to 108 IL13-zetakine+ CTL. The other patient (UPN028) received all but one 108 cell dose at day 1 of cycle 3 due to transient worsening of a headache (for individual patient treatment regimens, see Supplementary Fig. S3). There were no Grade 3 or higher adverse events with possible correlation to administration of either 107 or 5×107 T cells. However, at the 108 T cell dose, two cases of Grade 3 headache occurred in one subject (UPN028), which were possibly attributed to T cell administration (Table 1). There was also one Grade 3 neurologic event (UPN031), which included shuffling gait and tongue deviation, possibly attributed to T cell administration (Table 1). This neurological event occurred the day after the twelfth and final CAR T cell infusion, required hospital admission, and was treated with a single infusion of 10 mg intravenous dexamethasone. At follow-up four days later, the patient remained stable and did not require additional dexamethasone for this adverse event. While transient neurologic worsening of symptoms was anticipated in this protocol, and transient MRI gadolinium (Gd)-enhancement and increased FLAIR signal had been seen in the first two patients following T cell therapy, this subject (UPN031) had the highest level of IL13Rα2 expression, which may have elicited in a stronger inflammatory response. Alternatively, tumor recurrence in UPN031, confirmed with a third surgery 14 weeks after the T cell treatment, may also have contributed to the development of these neurologic adverse events. UPN033, whose GBM expression of IL13Rα2 was documented by IHC and qPCR (Fig. 2), did not exhibit any adverse events of Grade 3 or higher attributable to T cell administration (Table 1). In summary, we conclude that repetitive dosing of up to 12 infusions of IL13Rα2-specific CAR CD8+ T cell clones, with cumulative doses of 10.6 × 108 T cells administered into the resected tumor cavity, were well-tolerated and exhibited an acceptable safety profile with limited transient adverse events.
Table 1.
Safety and Tolerability
| Patient | T-cell Doses | Maximum Tolerated T-cell Dose | Cumulative T-cell Dose | Adverse Eventa | Survival Post Relapse (months) | |
|---|---|---|---|---|---|---|
| Intracavitary Delivery | UPN028 | 11 | 108 | 9.6×108 | Headaches | 10.3 |
| UPN031 | 12 | 108 | 10.6×108 | Neurologic – Other b | 8.6 | |
| UPN033 | 12 | 108 | 10.6 ×108 | None | 13.9 | |
|
| ||||||
| Intratumoral Delivery | UPN033 (T cell tx at distant sits of recurrence) | 5 | 108 | 3.75×108 | Low WBC Headache Fatigue | 13.9c |
Only events of Grade 3 or higher, according to the NCI Common Toxicity Criteria, with possible or higher attribution to the T cell administration are reported.
Shuffling gait and tongue deviation to the left.
Survival after second biopsy/relapse detected on day 64 as related to timeline in Figure S1D was 11.8 months.
For all patients, adoptive transfer of autologous IL13-zetakine+ CTL clones resulted in brain inflammation at the site of T cell infusion, as detected by increased MRI Gd-enhancement and increased signal on FLAIR images. This increased signal change is believed to be a consequence of the therapeutic cells and not indicative of disease progression, since the MRI signal change was observed immediately followed the first two cycles of six total infusions for all three patients, and was transient, subsiding within a few months of T cell infusion for UPN028 and UPN033 who did not develop tumor recurrence at the treatment site (Fig. 3A and B). For UPN031, whose tumor had the highest IL13Rα2 tumor expression, the inflammatory reaction to T cell therapy was more pronounced than in the other two subjects (Fig. 3C). Again, these early MRI enhancements were most consistent with T cell related inflammation and not tumor recurrence, since regions of enhancement showed low PET activity (Supplementary Fig. S4A). However, in contrast to the other patients, the CNS inflammatory reaction for UPN031 did not substantially subside, and disease recurrence was confirmed following a third craniotomy. Within these three patients, the degree of brain inflammation appeared to correlate with IL13Rα2 antigen expression, since inflammation was most pronounced in the two patients with the highest expression of IL13Rα2 (UPN031, UPN033). While this inflammation may have resulted from other factors, such as the infusion vehicle or non-specific T cell activity, these factors were similar in all three patients.
Fig. 3. Brain MRIs detected inflammation following IL13-zetakine+ CTL administration.
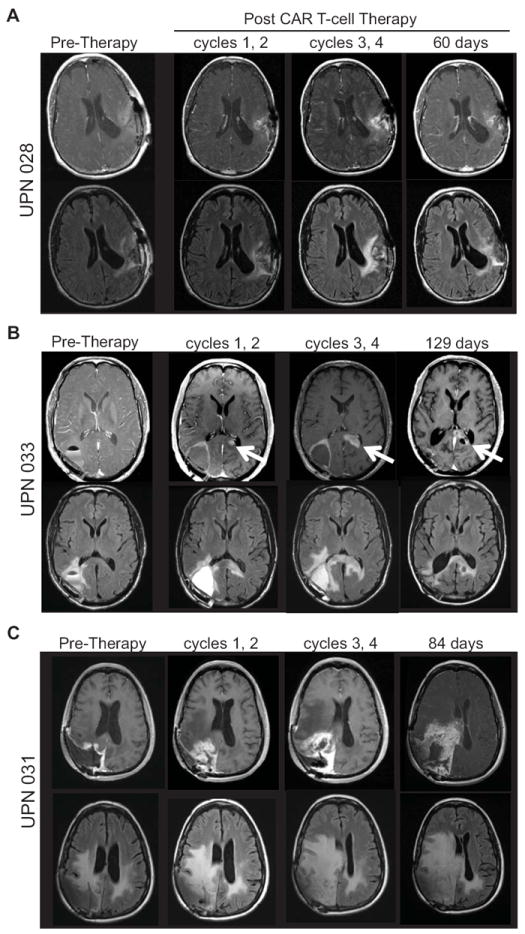
MRI for (A) UPN028 (B) UPN033, and (C) UPN031 before and after intracranial administration of IL13-zetakine+IL13-zetakine+ CD8+ CTL. MRI images are shown pre-therapy, i.e., post surgical resection and prior to T cell administration (within 12 days), as well as following cycles 1 and 2 (within 10 days), following cycles 3 and 4 (within 5 days), and 60 days or more after the last T cell treatment. Top rows, post Gd T1-weighted images demonstrating changes in contrast uptake at the site of T cell injections over treatment duration. Bottom rows, FLAIR images without Gd highlighting progression of cerebral edema following T cell therapy. White arrow indicates site of tumor recurrence for UPN033.
Treatment of UPN033 has been previously published in a manuscript describing the use of 18F-FHBG PET to detect therapeutic CTLs via the HSV tk gene included as part of the IL13-zetakine/HyTK expression construct (9). As we previously noted for this patient, following CAR T cell administration into the resection cavity a 18F-FDG PET-avid, MR enhancing lesion was detected contralaterally across the splenium of the corpus collosum rather than adjacent to the CAR-treated tumor cavity (Fig. 3B and 5A, and Supplementary Fig. S4B). Although the inflammatory reaction surrounding the resection cavity subsided over time, the lesion in the corpus collosum continued to progress. UPN033 was, therefore, taken off protocol 20 days after the last CAR T cell infusion, and received additional radiation, bevacizumab and BCNU over a five month period (Supplementary Fig. S3D). Following further tumor progression, this patient was enrolled on a separate single subject protocol in which the enhancing lesion was biopsied to confirm tumor recurrence, and IL13-zetakine+ CTL clones were administered directly into the tumor. In this second protocol, UPN033 received a series of five daily infusions of autologous IL13-zetakine+ CTL with escalating intratumoral doses ranging from 2.5 × 107 to 1 × 108 cells delivered directly into the recurrent tumor site via an externalized catheter (2 mL of cells were infused over 4 hours) (Supplementary Fig. S3D). Grade 3 or higher adverse events that were possibly associated with T cell administration included transient leukopenia, headaches during the infusion period, and fatigue (Table 1), again indicating an acceptable safety profile.
Fig. 5. MR Imaging of UPN033 shows increased necrotic tumor volume following administration of IL13-zetakine+ CD8+ T cell clones.
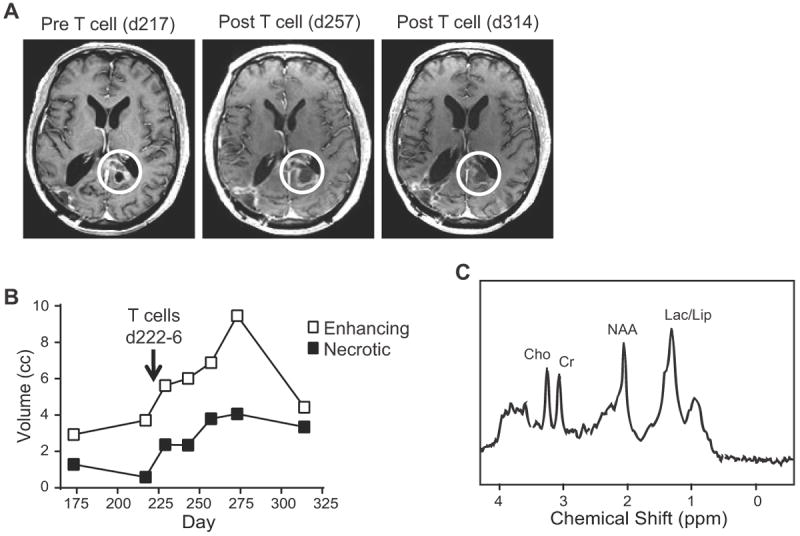
A, T1-weighted post-gadolinium MRI before and after the 5 daily infusions of autologous IL13 (E13Y)-zetakine+/HyTK+ CD8+ CTL. B, MRS analysis of enhancing volume (indicative of neuroinflammation) and necrotic volume at the recurrent site following re-treatment with T cells. Lack of tumor recurrence at the original treatment site (right occipital) and increase in necrosis at the recurrent site (left corpus callosum, white circles in A) are highly suggestive of therapeutic activity. C, Single voxel MR spectroscopy with a pane over the lesion medial to the atrium of the left lateral ventricle on day 314. Choline (Cho), creatine (Cr), N-acetyl-l-aspartate (NAA), and lacate/lipid (Lac/Lip) peaks are indicated.
Evidence for anti-tumor bioactivity following adoptive transfer of IL13-zetakine+ CTLs
Decreased tumor IL13Rα2 expression following therapy
Of the three patients treated on NCT00730613, the tumor excised from UPN031 prior to therapy had the highest IL13Rα2 expression (Fig. 2). As mentioned above, this patient required another craniotomy for tumor resection approximately 14 weeks after CAR T cell therapy. In recurrent tumor tissue adjacent to the T cell injection site, IL13Rα2 expression was significantly decreased from pre-T cell therapy levels. This was initially observed by flow cytometry of freshly excised tumor (Fig. 4A), and confirmed by both qPCR analysis of tumor samples (Fig. 4B), and IHC staining of paraffin embedded histological sections (Fig. 4C). While in this single patient we cannot rule out confounding contributions of local sampling within a heterogeneous tumor, we suggest that autologous IL13-zetakine+ CTL successfully targeted IL13Rα2-expressing tumor cells and reduced overall IL13Rα2 expression. These data thus provide corroborating evidence for an IL13-zetakine+ CTL-mediated anti-tumor response, with treatment failure in this patient due to recurrence/progression of IL13Rα2-negative or low tumor cells. Tumor recurrence could also be impacted by lack of significant expansion and/or survival of therapeutic CAR T cells, as indicated by our ability to specifically detect only low levels of the adoptively transferred T cells in the recurrent tumor tissue analyzed 14-weeks after CAR T cell therapy and post dexamethosome treatment (Supplementary Fig. S5).
Evidence of tumor responses by MRI
All patients treated in this trial had undergone gross total tumor resection prior to T cell infusions and therefore direct evaluation of anti-tumor responses was not feasible. However, as tumor recurrence at the margin of the resection cavity is observed in most patients with GBM, we monitored the patients with frequent serial images post therapy. In 2 of the 3 subjects (UPN028, UPN033), tumor did not recur at the border of the resected cavity near the T cell infusion sites as expected (Fig. 3, and data not shown), suggesting anti-tumor activity of CAR T cells. Further, as discussed above, the recurrent tumor adjacent to the treatment cavity for UPN031 showed significantly lower expression levels of IL13Rα2, implying selective targeting of IL13Rα2-expressing tumor cells.
Re-treatment of UPN033, comprising five daily infusions of IL13-zetakine+ CD8+ CTLs directly into the recurrent tumor mass within the splenium of the left corpus callosum, provided an opportunity to evaluate the therapeutic response of CAR T cells within an intact tumor. This recurrent lesion had been treated with radiation, chemotherapy (BCNU) and anti-angiogenic therapy (bevacizumab) 1.5 to 3 months prior to T cell infusion (-89 days for radiation, and -44 days for bevicizumab/BCNU). Immediately following T cell administration, there was a transient inflammatory response on MRI, as well as an increase in necrotic volume which persisted for several months (Fig. 5A and B). Although necrosis is a feature of GBM, the rapid onset of the inflammatory response, along with the persistent necrosis observed shortly after T cell infusion, suggest that these responses were T-cell mediated rather than due to tumor progression or previous treatments. Further, single voxel MR spectroscopy over the lesion 88 days after the final T cell infusion (day 314) showed a significant elevation of the lactate and lipid peaks, confirming necrosis, together with very little elevation of the choline peak relative to the creatine peak, suggesting low to negligible tumor burden (Fig. 5C). Collectively, these observations are highly suggestive of anti-tumor activity of IL13Rα2-specific CAR T cell therapy.
Discussion
In this study we present the first-in-human evaluation of local administration of CAR T cells to treat high-grade glioma. CAR T cell therapies have demonstrated remarkable clinical success in the treatment of CD19+ hematological malignancies, with complete response rates reported for the majority of patients with acute lymphoblastic leukemias (10,26). A major goal for this field of research is the successful application of CAR T cell therapy to a range of solid tumors. Achieving this goal will require overcoming critical barriers to therapeutic success including, but not limited to, tumor antigen selection, tumor heterogeneity, and the immunosuppressive tumor microenvironment. Our group is focusing on the development of CAR T cell therapy for the treatment of high-grade glioma, a tumor class that is essentially incurable with current standard-of-care therapies. While preclinical studies had demonstrated that CAR T cells can have potent anti-glioma responses in orthotopic mouse models (5,23), this approach had yet to be validated in a patient context. Here we summarize our experience treating three patients with IL13Rα2-specific CAR T cells for recurrent glioblastoma. We demonstrate feasibility and safety of repetitive intracranial delivery of autologous IL13-zetakine+ CD8+ CTL clones, and further report findings of transient anti-tumor activity for some patients in the absence of serious adverse events.
CAR T cell immunotherapy has the potential to improve treatment outcomes for patients with high-grade glioma. One of the major design challenges for glioma immunotherapy is the identification of tumor-specific target antigens. The ideal antigen would be expressed by the majority of tumor cells, but absent from normal brain tissue, thus reducing the potential for off-tumor CNS toxicities. Promising cell surface markers that are being pursued for development of CAR based T cell therapy to treat high-grade glioma include: IL13Rα2, HER2, EphA2 and EGFRvIII (3,4,6,23). In this study, we focused on IL13Rα2, as previous studies have shown that IL13Rα2 is over-expressed by greater than 50% of glioblastomas, its expression increases with malignancy grade, and it is associated with decreased long-term survival (12,15). Further, IL13Rα2 is expressed independently of tumor differentiation status, having been identified on stem-like malignant cells and more differentiated counterparts (5). Importantly, IL13Rα2 is not expressed at significant levels by normal brain tissue (15,19,20). The clinical experience treating three patients reported here further validates IL13Rα2 as a suitable target molecule for CAR T cell immunotherapy by establishing patient tolerance and acceptable safety profiles for repetitive intracranial infusions of IL13Rα2-specific CAR T cells, with adverse events being transient and manageable. Further, we provide proof-of-principle that CAR T cell targeting of a documented IL13Rα2-positive tumor can elicit tumor regression.
For this trial, autologous IL13-zetakine+ CD8+ CTL clones were manufactured for each patient using a process that included DNA electroporation, followed by drug selection and ex vivo expansion using OKT3 and cell feeders. This process was feasible, with IL13-zetakine+ CD8+ T cell clones successfully generated for all patients (10 of 13) who did not progress within the manufacturing time-frame, however these methodologies were cumbersome and took over 3-4 months to generate the final therapeutic product for each patient. To accommodate this delay, patients on this trial were enrolled following their primary diagnosis to give sufficient time for T cell manufacturing prior to disease progression/recurrence. The overall time needed for clinical manufacture becomes an important issue for patients with recurrent glioblastoma, who have a very limited therapeutic timeline. One strategy that could expedite CAR T cell therapy is to develop an “off-the-shelf” allogeneic CAR T cell product that would be available for administration shortly after tumor recurrence (27). Our group, in collaboration with Sangamo Biosciences, has recently shown the feasibility of infusing allogeneic IL13-zetakine+ CAR T cells while maintaining patients on steroids to reduce the possibility of CAR T cell rejection (manuscript in preparation). Alternatively, newer manufacturing strategies using CD3/CD28 beads that have reduced manufacturing times to less than 2-weeks, suggesting that with these further improvements enrollment at time of recurrence would be practical for autologous CAR T cell therapy.
Route of delivery is an additional unresolved issue related to the application of CAR T cell therapy for brain tumors. In this pilot safety and feasibility study we delivered the CAR T cells intracranially via an implanted reservoir/catheter system. This enabled repetitive dosing of the CAR T cells directly into the resected tumor cavity, and, for the second treatment of UPN033, the site of recurrent tumor. Repetitive delivery of up to 1.6 × 109 total CAR T cells was well-tolerated by all three patients, as was repetitive local delivery into the site of active disease for UPN033. Further, there were no immediate or acute device-related adverse events, such as occlusion, malfunction, or infection.
For the optimal therapeutic response, it is imperative that the CAR T cells are able to migrate to distant sites of infiltrative and/or multi-focal disease. The delivery strategy best able to target infiltrative and/or multifocal disease is not yet evident. Although the standard imaging modalities reported here could not evaluate T-cell trafficking using the reservoir/catheter system, our collaborators on this clinical study have previously provided evidence, using 18F-FHBG PET, that CAR/HyTk T cells were detected at the site of injection, as well as at a recurrent secondary site near the corpus callosum. This observation suggests the potential trafficking of CAR T cells to distant sites within the brain after intratumoral injection (9). In an alternative approach, both the NCI and Baylor groups, in ongoing phase I clinical trials evaluating EGFRvIII and HER2-specific CAR T cells (NCT01454596; NCT0110905), are delivering the CAR T cells intravenously (i.v.) for the treatment of glioblastoma. Indeed, i.v. administered melanoma-specific tumor infiltrating lymphocyte-derived T cells provide proof-of-principle for the capacity of adoptively transferred T cells to traffic to the brain and mediate regression of brain metastases (8). Thus, as future studies evaluate the treatment of brain tumors with CAR T cell therapies it will be important to compare differences in T cell homing, persistence and anti-tumor responses with different T cell delivery methodologies.
While the primary objective of our pilot clinical study was assessment of safety, we observed encouraging evidence of transient anti-glioma activity, including increased necrotic volume by MRI, significant loss of IL13Rα2+ tumor cell expression following CAR T cell administration, and detection of transferred T cells at tumor microfoci removed from the site of injection (9). Although a survival benefit could not be established with such a small cohort of patients, the three patients treated had a mean survival of 11 months after relapse, with best survival of almost 14 months (Table 1).
This study also highlights several barriers to more durable clinical outcomes. First, we find that tumor heterogeneity may contribute to relapse by providing pools of target-deficient tumor cells that can then expand, as exemplified by the IL13Rα2-low tumor that recurred in UPN031. The recurrence of antigen negative glioblastoma is not unique to our study or specific for CAR T cell immunotherapy, since it has also been observed following EGFRvIII-targeted vaccination strategies (28,29). To address this issue, we and others are pursuing CAR T cell approaches that target multiple antigens (30). Second, our data indicate that T cell persistence may be another important factor (5). UPN031, whose recurrent tumor was resected 14-weeks following the last T cell infusion, displayed very low levels of CAR T cell persistence (Supplemental Fig. S5). While administration of dexamethosone may have influenced T cell persistence for this patient, this lack of therapeutic T cell persistence is also consistent with observations from our orthotopic xenograft mouse models which indicate low survival/persistence 14-days after i.c. administration of IL13-zetakine+ CTL clones (5). Further, previous clinical studies administering first-generation CAR T cells manufactured using a similar platform, and administered i.v., also identified limited T cell persistence as a barrier to durable clinical efficacy (31,32). To address this limitation, we have recently optimized the IL13Rα-specific CAR T cells, incorporating several innovations that improve both T cell persistence and anti-tumor efficacy in orthotopic glioma mouse models, including engineering T cells to express a costimulatiory CAR, starting with a population of central memory T cells, and utilizing CD3/CD28 beads in a manufacturing platform that limits ex vivo expansion time (manuscript in preparation). Finally, to improve the delivery methodology, our group is developing devices that allow for large infusions of therapeutic cells directly into tumors (B. Badie, unpublished data).
In summary, our clinical experience treating three research participants with recurrent glioblastoma has demonstrated the feasibility of engineering, expanding and locally delivering autologous CAR+ T cells without the development of serious therapy-related side-effects, and has provided evidence for transient anti-glioma responses. These findings support the potential of IL13Rα2-specific CAR therapy for the treatment of glioblastoma. Future studies will focus on improving T cell persistence, tumor targeting and efficient delivery.
Supplementary Material
Translational Relevance.
Inadequate standard-of-care therapies for glioblastoma (GBM), the most aggressive and common type of glioma, have resulted in an unacceptably poor median overall survival of approximately 15 months following primary diagnosis. Immunotherapeutic targeting of brain tumors offers the opportunity to redirect the potency and specificity of the immune system to improve therapeutic outcomes. However, clinical realization of this potential is still forthcoming. Here we report a first-in-human pilot safety and feasibility study evaluating chimeric antigen receptor (CAR)-engineered IL13Rα2-specific primary human autologous CD8+ cytolytic T lymphocytes (CTLs) for the treatment of recurrent GBM. We provide evidence for feasibility of this treatment approach, and transient anti-glioma activity in the absence of serious adverse events, thus establishing the foundation for further development of this IL13Rα2-specific CAR T cell therapy.
Acknowledgments
The authors would like to thank the COH Anatomic Pathology Core, Sofia Loera and Emerald Wu for technical support; Sandra Thomas for manuscript editing; and Andrew Raubitschek, Brenda Williams and John Zaia for clinical trial support.
Financial Support: This work was supported by NIH grants P30 CA33572, R01 CA103959, R01 CA155769, R21 NS081594 and R21 CA189223; and California Institute for Regenerative Medicine (CIRM) grant TR3-05641.
Footnotes
Conflict of Interest:
MCJ is an inventor of licensed intellectual property pertaining to the results presented in this paper (patent US7514537 B2) and a co-founder/equity of ZetaRx Biosciences, Inc. All other authors have no conflicts of interest to disclose.
References
- 1.Dolecek TA, Propp JM, Stroup NE, Kruchko C. CBTRUS statistical report: primary brain and central nervous system tumors diagnosed in the United States in 2005-2009. Neuro Oncol. 2012;14(Suppl 5):v1–49. doi: 10.1093/neuonc/nos218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bielamowicz K, Khawja S, Ahmed N. Adoptive cell therapies for glioblastoma. Front Oncol. 2013;3:275. doi: 10.3389/fonc.2013.00275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ahmed N, Salsman VS, Kew Y, Shaffer D, Powell S, Zhang YJ, et al. HER2-specific T cells target primary glioblastoma stem cells and induce regression of autologous experimental tumors. Clin Cancer Res. 2010;16:474–85. doi: 10.1158/1078-0432.CCR-09-1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sampson JH, Choi BD, Sanchez-Perez L, Suryadevara CM, Snyder DJ, Flores CT, et al. EGFRvIII mCAR-modified T-cell therapy cures mice with established intracerebral glioma and generates host immunity against tumor-antigen loss. Clin Cancer Res. 2014;20:972–84. doi: 10.1158/1078-0432.CCR-13-0709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brown CE, Starr R, Aguilar B, Shami AF, Martinez C, D’Apuzzo M, et al. Stem-like Tumor-Initiating Cells Isolated from IL13Ralpha2 Expressing Gliomas Are Targeted and Killed by IL13-Zetakine-Redirected T Cells. Clin Cancer Res. 2012;18:2199–209. doi: 10.1158/1078-0432.CCR-11-1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chow KK, Naik S, Kakarla S, Brawley VS, Shaffer DR, Yi Z, et al. T cells redirected to EphA2 for the immunotherapy of glioblastoma. Mol Ther. 2013;21:629–37. doi: 10.1038/mt.2012.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brown CE, Vishwanath RP, Aguilar B, Starr R, Najbauer J, Aboody KS, et al. Tumor-Derived Chemokine MCP-1/CCL2 Is Sufficient for Mediating Tumor Tropism of Adoptively Transferred T Cells. J Immunol. 2007;179:3332–41. doi: 10.4049/jimmunol.179.5.3332. [DOI] [PubMed] [Google Scholar]
- 8.Hong JJ, Rosenberg SA, Dudley ME, Yang JC, White DE, Butman JA, et al. Successful treatment of melanoma brain metastases with adoptive cell therapy. Clin Cancer Res. 2010;16:4892–8. doi: 10.1158/1078-0432.CCR-10-1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yaghoubi SS, Jensen MC, Satyamurthy N, Budhiraja S, Paik D, Czernin J, et al. Noninvasive detection of therapeutic cytolytic T cells with 18F-FHBG PET in a patient with glioma. Nat Clin Pract Oncol. 2009;6:53–8. doi: 10.1038/ncponc1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Maus MV, Grupp SA, Porter DL, June CH. Antibody-modified T cells: CARs take the front seat for hematologic malignancies. Blood. 2014;123:2625–35. doi: 10.1182/blood-2013-11-492231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ramos CA, Savoldo B, Dotti G. CD19-CAR trials. Cancer J. 2014;20:112–8. doi: 10.1097/PPO.0000000000000031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brown CE, Warden CD, Starr R, Deng X, Badie B, Yuan YC, et al. Glioma IL13Ralpha2 is associated with mesenchymal signature gene expression and poor patient prognosis. PLoS One. 2013;8:e77769. doi: 10.1371/journal.pone.0077769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Thaci B, Brown CE, Binello E, Werbaneth K, Sampath P, Sengupta S. Significance of interleukin-13 receptor alpha 2-targeted glioblastoma therapy. Neuro Oncol. 2014 doi: 10.1093/neuonc/nou045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wykosky J, Gibo DM, Stanton C, Debinski W. Interleukin-13 receptor alpha 2, EphA2, and Fos-related antigen 1 as molecular denominators of high-grade astrocytomas and specific targets for combinatorial therapy. Clin Cancer Res. 2008;14:199–208. doi: 10.1158/1078-0432.CCR-07-1990. [DOI] [PubMed] [Google Scholar]
- 15.Debinski W, Gibo DM, Hulet SW, Connor JR, Gillespie GY. Receptor for interleukin 13 is a marker and therapeutic target for human high-grade gliomas. Clin Cancer Res. 1999;5:985–90. [PubMed] [Google Scholar]
- 16.He CH, Lee CG, Dela Cruz CS, Lee CM, Zhou Y, Ahangari F, et al. Chitinase 3-like 1 regulates cellular and tissue responses via IL-13 receptor alpha2. Cell Rep. 2013;4:830–41. doi: 10.1016/j.celrep.2013.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fichtner-Feigl S, Terabe M, Kitani A, Young CA, Fuss I, Geissler EK, et al. Restoration of tumor immunosurveillance via targeting of interleukin-13 receptor-alpha 2. Cancer Res. 2008;68:3467–75. doi: 10.1158/0008-5472.CAN-07-5301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jarboe JS, Johnson KR, Choi Y, Lonser RR, Park JK. Expression of interleukin-13 receptor alpha2 in glioblastoma multiforme: implications for targeted therapies. Cancer Res. 2007;67:7983–6. doi: 10.1158/0008-5472.CAN-07-1493. [DOI] [PubMed] [Google Scholar]
- 19.Joshi BH, Puri RA, Leland P, Varricchio F, Gupta G, Kocak M, et al. Identification of interleukin-13 receptor alpha2 chain overexpression in situ in high-grade diffusely infiltrative pediatric brainstem glioma. Neuro Oncol. 2008;10:265–74. doi: 10.1215/15228517-2007-066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kawakami M, Kawakami K, Takahashi S, Abe M, Puri RK. Analysis of interleukin-13 receptor alpha2 expression in human pediatric brain tumors. Cancer. 2004;101:1036–42. doi: 10.1002/cncr.20470. [DOI] [PubMed] [Google Scholar]
- 21.Iwami K, Shimato S, Ohno M, Okada H, Nakahara N, Sato Y, et al. Peptide-pulsed dendritic cell vaccination targeting interleukin-13 receptor alpha2 chain in recurrent malignant glioma patients with HLA-A*24/A*02 allele. Cytotherapy. 2012;14:733–42. doi: 10.3109/14653249.2012.666633. [DOI] [PubMed] [Google Scholar]
- 22.Kunwar S, Prados MD, Chang SM, Berger MS, Lang FF, Piepmeier JM, et al. Direct intracerebral delivery of cintredekin besudotox (IL13-PE38QQR) in recurrent malignant glioma: a report by the Cintredekin Besudotox Intraparenchymal Study Group. J Clin Oncol. 2007;25:837–44. doi: 10.1200/JCO.2006.08.1117. [DOI] [PubMed] [Google Scholar]
- 23.Kahlon KS, Brown C, Cooper LJ, Raubitschek A, Forman SJ, Jensen MC. Specific recognition and killing of glioblastoma multiforme by interleukin 13-zetakine redirected cytolytic T cells. Cancer Res. 2004;64:9160–6. doi: 10.1158/0008-5472.CAN-04-0454. [DOI] [PubMed] [Google Scholar]
- 24.Debinski W, Thompson JP. Retargeting interleukin 13 for radioimmunodetection and radioimmunotherapy of human high-grade gliomas. Clin Cancer Res. 1999;5:3143s–7s. [PubMed] [Google Scholar]
- 25.Jensen MC, Clarke P, Tan G, Wright C, Chung-Chang W, Clark TN, et al. Human T lymphocyte genetic modification with naked DNA. J Mol Ther. 2000;1:49–55. doi: 10.1006/mthe.1999.0012. [DOI] [PubMed] [Google Scholar]
- 26.Davila ML, Bouhassira DC, Park JH, Curran KJ, Smith EL, Pegram HJ, et al. Chimeric antigen receptors for the adoptive T cell therapy of hematologic malignancies. Int J Hematol. 2014;99:361–71. doi: 10.1007/s12185-013-1479-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Torikai H, Reik A, Liu PQ, Zhou Y, Zhang L, Maiti S, et al. A foundation for universal T-cell based immunotherapy: T cells engineered to express a CD19-specific chimeric-antigen-receptor and eliminate expression of endogenous TCR. Blood. 2012;119:5697–705. doi: 10.1182/blood-2012-01-405365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sampson JH, Aldape KD, Archer GE, Coan A, Desjardins A, Friedman AH, et al. Greater chemotherapy-induced lymphopenia enhances tumor-specific immune responses that eliminate EGFRvIII-expressing tumor cells in patients with glioblastoma. Neuro Oncol. 2011;13:324–33. doi: 10.1093/neuonc/noq157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sampson JH, Heimberger AB, Archer GE, Aldape KD, Friedman AH, Friedman HS, et al. Immunologic escape after prolonged progression-free survival with epidermal growth factor receptor variant III peptide vaccination in patients with newly diagnosed glioblastoma. J Clin Oncol. 2010;28:4722–9. doi: 10.1200/JCO.2010.28.6963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hegde M, Corder A, Chow KK, Mukherjee M, Ashoori A, Kew Y, et al. Combinational targeting offsets antigen escape and enhances effector functions of adoptively transferred T cells in glioblastoma. Mol Ther. 2013;21:2087–101. doi: 10.1038/mt.2013.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jensen MC, Popplewell L, Cooper LJ, DiGiusto D, Kalos M, Ostberg JR, et al. Antitransgene rejection responses contribute to attenuated persistence of adoptively transferred CD20/CD19-specific chimeric antigen receptor redirected T cells in humans. Biol Blood Marrow Transplant. 2010;16:1245–56. doi: 10.1016/j.bbmt.2010.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Park JR, Digiusto DL, Slovak M, Wright C, Naranjo A, Wagner J, et al. Adoptive transfer of chimeric antigen receptor re-directed cytolytic T lymphocyte clones in patients with neuroblastoma. Mol Ther. 2007;15:825–33. doi: 10.1038/sj.mt.6300104. [DOI] [PubMed] [Google Scholar]
- 33.Cooper LJ, Topp MS, Serrano LM, Gonzalez S, Chang WC, Naranjo A, et al. T-cell clones can be rendered specific for CD19: toward the selective augmentation of the graft-versus-B-lineage leukemia effect. Blood. 2003;101:1637–44. doi: 10.1182/blood-2002-07-1989. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.