

Abstract
The prevalence of neutral mutations implies that biological systems typically have many more genotypes than phenotypes. But, can the way that genotypes are distributed over phenotypes determine evolutionary outcomes? Answering such questions is difficult, in part because the number of genotypes can be hyper-astronomically large. By solving the genotype–phenotype (GP) map for RNA secondary structure (SS) for systems up to length L = 126 nucleotides (where the set of all possible RNA strands would weigh more than the mass of the visible universe), we show that the GP map strongly constrains the evolution of non-coding RNA (ncRNA). Simple random sampling over genotypes predicts the distribution of properties such as the mutational robustness or the number of stems per SS found in naturally occurring ncRNA with surprising accuracy. Because we ignore natural selection, this strikingly close correspondence with the mapping suggests that structures allowing for functionality are easily discovered, despite the enormous size of the genetic spaces. The mapping is extremely biased: the majority of genotypes map to an exponentially small portion of the morphospace of all biophysically possible structures. Such strong constraints provide a non-adaptive explanation for the convergent evolution of structures such as the hammerhead ribozyme. These results present a particularly clear example of bias in the arrival of variation strongly shaping evolutionary outcomes and may be relevant to Mayr's distinction between proximate and ultimate causes in evolutionary biology.
Keywords: genotype–phenotype map, arrival of variation, robustness, bias in development
1. Introduction
Many questions about the limits of evolution hinge not only on what has happened in natural history, but also on counterfactuals: what did not happen, but perhaps could have. Re-run the tape of life [1,2] and what parts of phenotype space—the set of all possible phenotypes [3]—would be occupied? Typically, only a miniscule fraction of the phenotype space has been explored throughout natural history. The reasons given for this phenomenon usually combine adaptive arguments: some parts of phenotype space yield higher fitness than others, with arguments based on contingency: nature has not had time to explore all of phenotype space. However, an evolutionary search does not occur by uniform sampling over phenotypes, but rather by random mutations in the space of genotypes. Does this basic fact alter the way that phenotype space is explored and occupied? To answer such whole genotype–phenotype (GP) map questions is difficult, in part because the number of possible genotypes typically grows exponentially with length, and so rapidly becomes unimaginably vast [4–6], and in part because biological systems with sufficiently tractable GP maps are rare.
One system where progress can be made is the mapping from sequences to the structure of RNA. Although simpler than many biological phenotypes, RNA is interesting and important, because it can fulfil multiple roles both as an information carrier (e.g. messenger RNA and viral RNA) and as functional non-coding RNA (ncRNA) [7], including chemically active catalysts (e.g. ribozymes) and key structural components of larger self-assembled structures (e.g. ribosomal RNA). One reason RNA is so versatile is that it can fold into complex three-dimensional structures. The bonding pattern of these structures is called the secondary structure (SS), which is an important determinant of the three-dimensional structure and biological activity of ncRNA molecules, and so can be treated as a (simplified) phenotype in its own right [8,9] (figure 1).
Figure 1.
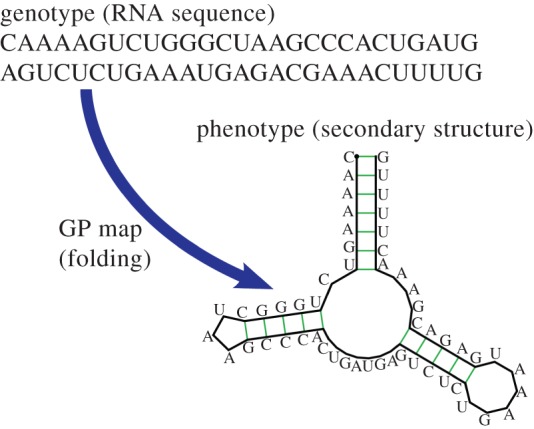
Schematic of the mapping from an RNA sequence to an SS. Here, for an L = 55 type III hammerhead ribozyme with three stems. Any sequence that folds to the same SS topology is considered to map to the same SS phenotype. (Online version in colour.)
Fast algorithms exist to predict the free energy minimum SS for a given sequence, and these are thought to be fairly accurate, especially for shorter strands [10,11]. Moreover, extensive databases exist for functional ncRNA [7]. For these reasons, this computationally tractable yet biologically relevant sequence to structure mapping is uniquely suited for investigating ‘whole genotype–phenotype map’ properties that may point towards general principles relevant for a wider class of systems, and has been extensively studied over the past few decades [6,12–20].
Nevertheless, the RNA sequence to SS GP map also suffers from the exponential growth of the size of genetic space, which has limited prior comprehensive GP studies to fairly small lengths, making direct comparison with evolutionary outcomes difficult. Here, we show that many detailed properties of this GP map can, in fact, be calculated, even for lengths as long as L = 126, where the set of all possible strands would weigh more than the mass of the visible universe (see Methods). One reason this is possible is because the map is highly biased towards a small fraction of phenotypes that take up the majority of genotypes. This bias means that sampling over genotypes (which we will call G-sampling) generates significantly different outcomes from sampling over the morphospace [3] of all shapes (phenotypes; which we will call P-sampling). The existence of such highly peaked distributions in the mapping from genotypes to phenotypes also explains, in a way that is reminiscent of statistical mechanics, why sampling a relatively small number of genotypes is enough to determine certain key properties of RNA SS.
Perhaps most strikingly, we find that the distributions of several properties of natural ncRNA taken from the function RNA database (fRNAdb) [21], including the number of stems, the mutational robustness and the number of genotypes per SS phenotype, are very similar to what we obtain from random sampling over genotypes, and significantly different from uniform sampling over phenotypes. This result does not mean that natural selection can be ignored, but rather, as we will argue below, that the mapping strongly prescribes which parts of morphospace are presented to natural selection as potential variation. Variation can be selected only if it arrives [22,23].
2. Results
2.1. P-sampling over phenotypes differs significantly from G-sampling over genotypes
We first analyse an exhaustive enumeration of all sequences for L = 20 RNA, the largest system for which this has so far been accomplished. The 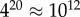 sequences were folded with the Vienna Package [10], and map to
sequences were folded with the Vienna Package [10], and map to 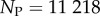 unique bonded SSs and one trivial structure with no bonds, as their free-energy minima [20]. The set of all sequences that map onto a given SS is called the neutral set (NS), and we use Ω to denote the NS size (the number of sequences in the NS). This mapping exhibits strong phenotype bias [6,12,13,17–20]. For example, Ω varies by over 10 orders of magnitude for L = 20 (figure 2).
unique bonded SSs and one trivial structure with no bonds, as their free-energy minima [20]. The set of all sequences that map onto a given SS is called the neutral set (NS), and we use Ω to denote the NS size (the number of sequences in the NS). This mapping exhibits strong phenotype bias [6,12,13,17–20]. For example, Ω varies by over 10 orders of magnitude for L = 20 (figure 2).
Figure 2.
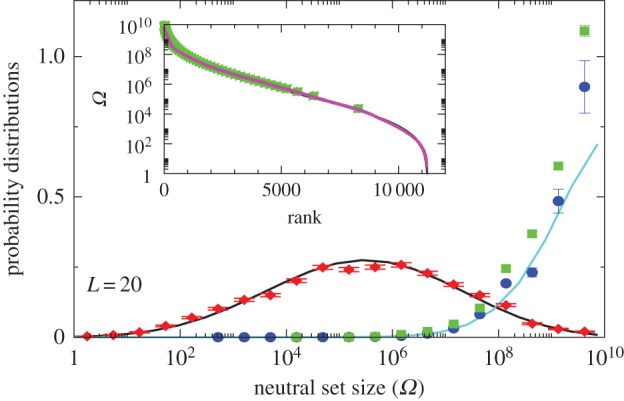
Comparison of P-sampled and G-sampled distributions to natural data for L = 20 RNA. The P-sampled PP(Ω) (red diamonds) measures the probability distribution for a phenotype to have a given NS size Ω. It differs markedly from G-sampled PG(Ω) (blue circles), generated by random sampling over genotypes. Error bars arise from binning data. The black and cyan lines are theoretical approximations to PP(Ω) and PG(Ω), respectively (see Methods). The probability distribution of Ω for the SSs all 7327 (non-trivial) L = 20 sequences for Drosophila melanogaster from the fRNAdb database [21] (green squares) is much closer to the G-sampled PG(Ω) than to the P-sampled PP(Ω). Inset: all 11 218 SS phenotypes (purple triangles) ranked by NS size Ω. There is strong bias, just 5% of phenotypes take up 58% of all genotypes. The 7327 natural data points (green squares) are clustered at lower rank (larger Ω). (Online version in colour.)
Next, we introduce the concept of P-sampling, that is, uniformly sampling over the set of possible phenotypes (the morphospace), and define PP(Ω) as the probability distribution that a randomly chosen phenotype has NS size Ω. We calculate distributions for fixed L and bin data uniformly in 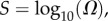 but write PP(Ω) and PG(Ω) for simplicity. PP(Ω) has a maximum when Ω is about half of the maximum value of the exponent
but write PP(Ω) and PG(Ω) for simplicity. PP(Ω) has a maximum when Ω is about half of the maximum value of the exponent 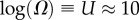 (figure 2). We ignore the trivial structure with no bonds for which
(figure 2). We ignore the trivial structure with no bonds for which 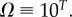 For L = 20,
For L = 20, 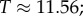 for larger L,
for larger L, 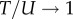 and the probability of finding the trivial structure tends to zero (see Methods).
and the probability of finding the trivial structure tends to zero (see Methods).
Instead of P-sampling, one could also sample uniformly over sequences (genotypes), which we refer to as G-sampling, giving 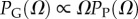 , which highlights structures from the large Ω tail of PP(Ω), as can be seen in figure 2.
, which highlights structures from the large Ω tail of PP(Ω), as can be seen in figure 2.
Novel variation does not arise by uniform random sampling in the morphospace of all physically permissible phenotypes (P-sampling), but instead by processes such as mutation that change genotypes. While evolutionary dynamics do not proceed by simple uniform G-sampling either, recent detailed population genetics calculations for RNA [20] have shown that the rate at which novel variation (a particular SS) arises in an evolving population is, to first order, directly proportional to the NS size of the SS phenotype, and so also to PG(Ω). This proportionality holds for a wide range of mutation rates and population sizes. It was also demonstrated explicitly that strong phenotype bias can overcome fitness differences in an RNA system [20]. Bias should therefore affect outcomes for multiple evolutionary scenarios. Indeed, important prior studies have suggested that natural RNA could have larger than average Ω [17,18]. We took every L = 20 sequence in the fRNAdb database [21] for Drosophila melanogaster (see Methods and electronic supplementary material) and calculated the NS size Ω of its associated SS using the neutral set size estimator (NSSE) from [18]. Not only are SSs with larger Ω overrepresented, but the entire natural distribution is remarkably close to the genotype sampled PG(Ω) (figure 2).
2.2. Sampling for lengths up to L = 126
While these results for L = 20 are suggestive, distributions for larger length RNA are needed to ensure that we are not just observing database biases or artefacts of the short length. Because the number of sequences grows exponentially with increasing L, exhaustive enumeration is not an option for lengths much larger than L = 20. Instead, we estimate the NS size distributions by randomly sampling genotypes, folding them into an SS, and then measuring their NS size with the NSSE (Methods). This process naturally generates PG(Ω); PP(Ω) can be backed out by dividing by Ω and normalizing. However, it is hard to sample SSs with small Ω, so PP(Ω) is only partially determined. To make progress, we use a simple analytical ansatz based on a log-binomial approximation to the distributions (see Methods), which works well for both PG(Ω) and PP(Ω) for L = 20 (figure 2). We compare this approximation with sampled data for L = 20 up to L = 126 (figure 3 and electronic supplementary material, figures S1 and S2). In each case, the analytic fit to PG(Ω) is excellent, and the fit to PP(Ω) works well, giving confidence that this form provides a reasonable approximation to the full PP(Ω).
Figure 3.

Neutral set size distributions illustrate how bias constrains natural RNA secondary structures. Sampled distributions PP(Ω) (red diamonds) and PG(Ω) (blue circles) and analytic approximations to PP(Ω) (black lines) and PG(Ω) (cyan lines) are compared with ncRNA from the fRNAdb database [21] (green squares). For each length (denoted in the top corners), the data are shown both on a lin–log and log–log graph to highlight different parts of the distributions. The natural data are remarkably close to the G-sampled PG(Ω), but quite far from the P-sampled PP(Ω). The number of natural structures plotted are 7327 for L = 20 (which is repeated from figure 2 for clarity of comparison); 658 for L = 40; 472 for L = 50; 350 for L = 60; 2263 for L = 70; 553 for L = 80; 731 for L = 90; 891 for L = 100; and 291 for L = 126. In each case, all structures in the fRNAdb [21] database are used, except for L = 20, where only structures for Drosophila melanogaster are used, and for L = 55 and L = 126, where we also plot a smaller curated dataset of 213 and 172 structures, respectively (magenta triangles), where the SS is known to be important (see electronic supplementary material). Smaller numbers of data lead to larger binning error bars, but the curated sets are clearly very similar to the full set of fRNAdb structures. (Online version in colour.)
2.3. Entropy and the bias ratio
The PG(Ω) distributions can be further quantified by the Shannon entropy 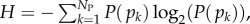 where P(pk) is the probability of choosing one of the NP possible phenotypes pk by G-sampling. The exponential of the entropy, 2H, is used in statistical physics and information theory [24] as a measure of the effective number of states. Because
where P(pk) is the probability of choosing one of the NP possible phenotypes pk by G-sampling. The exponential of the entropy, 2H, is used in statistical physics and information theory [24] as a measure of the effective number of states. Because 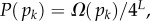 it follows straightforwardly that
it follows straightforwardly that
| 2.1 |
where 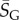 is a G-sampled average of S = log(Ω). Thus to measure the ‘effective number’ of phenotypes that take up the majority of genotypes, one only needs to determine
is a G-sampled average of S = log(Ω). Thus to measure the ‘effective number’ of phenotypes that take up the majority of genotypes, one only needs to determine 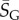 . This can be done rapidly, because the G-sampled distributions are sharply peaked in a manner reminiscent of statistical mechanics [24].
. This can be done rapidly, because the G-sampled distributions are sharply peaked in a manner reminiscent of statistical mechanics [24].
As an example of how the highly peaked nature of these distributions facilitates the calculation of averages, consider the case of L = 126. We find that 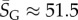 with standard deviation (s.d.)
with standard deviation (s.d.) 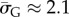 , so that 95% of SS structures found by G-sampling have S in range 51.5 ± 4.2, which is very narrow compared with the full range [0, 64.5] and comparatively far (about 6 s.d.) from the phenotype averaged
, so that 95% of SS structures found by G-sampling have S in range 51.5 ± 4.2, which is very narrow compared with the full range [0, 64.5] and comparatively far (about 6 s.d.) from the phenotype averaged 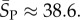 Rather strikingly, this implies that, even though the set of all possible L = 126 nucleotide sequences would weigh more than the mass of the observable universe (see Methods), sampling the NS size for just 10 randomly chosen L = 126 structures is enough to fix the exponent
Rather strikingly, this implies that, even though the set of all possible L = 126 nucleotide sequences would weigh more than the mass of the observable universe (see Methods), sampling the NS size for just 10 randomly chosen L = 126 structures is enough to fix the exponent 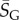 to within about 1.3% relative accuracy, which allows us to determine, for example, the number of relevant states 2H via equation (2.1).
to within about 1.3% relative accuracy, which allows us to determine, for example, the number of relevant states 2H via equation (2.1).
To further quantify the bias, we introduce the bias ratio
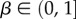 as
as
| 2.2 |
which can be interpreted as the ratio of the effective number of phenotypes that take up most of the sequences to the total number of phenotypes NP. For example, if all Ω are equal, then 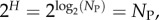 β = 1, and bias is weak. On the other hand, if just one phenotype accounts for nearly all the genotypes, then 2H ≈ 20 = 1,
β = 1, and bias is weak. On the other hand, if just one phenotype accounts for nearly all the genotypes, then 2H ≈ 20 = 1, 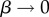 and bias is very strong.
and bias is very strong.
The number of phenotypes NP can be estimated from PP(Ω); 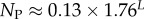 fits the data well (see Methods). From this, we find β ≈ 0.25 × 0.91L, which shrinks with increasing L. Typically, we find that about 75% of genotypes map to the ‘effective number’
fits the data well (see Methods). From this, we find β ≈ 0.25 × 0.91L, which shrinks with increasing L. Typically, we find that about 75% of genotypes map to the ‘effective number’ 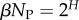 of structures. Nevertheless,
of structures. Nevertheless, 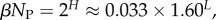 and continues to grow exponentially. For L = 55,
and continues to grow exponentially. For L = 55, 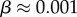 and
and 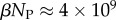 of the
of the 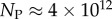 phenotypes take up about 75% of the genotypes. For L = 126,
phenotypes take up about 75% of the genotypes. For L = 126, 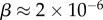 and
and 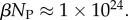 Even though bias greatly reduces the effective number of phenotypes accessible to mutations, their number continues to grow exponentially, and so can still be extremely large for this system.
Even though bias greatly reduces the effective number of phenotypes accessible to mutations, their number continues to grow exponentially, and so can still be extremely large for this system.
2.4. Comparison with fRNAdb database of functional non-coding RNA
To explore how this bias plays out in nature, we took, for lengths ranging from L = 20 to 126, every sequence in the fRNAdb database [21] and calculated the NS size Ω of its associated SS. The correspondence between PG(Ω) from random sampling and the distribution of Ω from the database is striking (figure 3 and electronic supplementary martial, figures S1 and S2). Not only are these natural RNAs mainly drawn from the minuscule fraction β of SSs with large Ω, but their distribution is remarkably close to what one would obtain from randomly sampling genotypes. Rank plots for L = 55 (figure 4) further illustrate how natural ncRNAs are constrained mainly to the set of 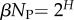 ‘relevant structures’.
‘relevant structures’.
Figure 4.

Ω versus rank plot for all 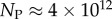 possible SS structures for L = 55 shown as (a) log–lin and (b) log–log plots. The black line is from the analytical approximation and the green squares denote the natural data. The horizontal red dotted-dashed line is
possible SS structures for L = 55 shown as (a) log–lin and (b) log–log plots. The black line is from the analytical approximation and the green squares denote the natural data. The horizontal red dotted-dashed line is 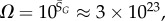 near the peak of the PG(Ω) distribution for L = 55 in figure 3. The 13 L = 55 hammerhead ribozyme structures from the fRNA database (red squares) are clustered near this peak. The vertical blue dotted line in (b) denotes
near the peak of the PG(Ω) distribution for L = 55 in figure 3. The 13 L = 55 hammerhead ribozyme structures from the fRNA database (red squares) are clustered near this peak. The vertical blue dotted line in (b) denotes 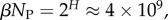 the ‘effective’ number of SSs. This set of
the ‘effective’ number of SSs. This set of 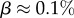 of all structures captures the majority (≈75%) of natural structures. (Online version in colour.)
of all structures captures the majority (≈75%) of natural structures. (Online version in colour.)
These results are, at first glance, surprising, because many ncRNAs have well-defined functional roles and so will have been subject to natural selection for which there exists extensive evidence [7]. Some insight may be gleaned from an exchange that occurred not long after the start of the molecular revolution in biology. Frank Salisbury [4] expressed doubt that evolutionary search could work, because genetic spaces are typically exponentially large, and, he claimed, are probably sparsely populated with functional phenotypes. In a famous response [5], John Maynard Smith pointed out that evolution is aided by neutral mutations, because these imply many fewer phenotypes than genotypes and also connect genotypes into neutral networks that can be explored by evolving populations. Phenotype bias may further facilitate evolutionary search by limiting potential outcomes. But, taken together, this still does not counter the heart of Salisbury's objection, namely that functional phenotypes are extremely rare and so hard to find.
Here, we instead suggest that the reason the ‘null model’ PG(Ω) predicts what is found in nature so accurately is that ‘good enough’ SSs are relatively easy to find. Of course, the full functional ncRNA phenotype typically has a smaller NS size than the SS does, because it requires further constraints on the sequence to achieve functionality [8]. We postulate that while natural selection creates function by acting on such sequence constraints, it also automatically draws from a palette of easily accessible SS variation that is strongly pre-sculpted by the mapping (see [14] for a similar suggestion). Another possibility might be that the correspondence with PG(Ω) simply means that the SS has no adaptive value. However, this scenario is unlikely, because it is well established that the SS is an important determinant of the three-dimensional structure [6,8,9], which, in turn, helps determine function. Moreover, the correlation with PG(Ω) remains strong when we curate the database for structures where SSs is known to be important. Altogether, this suggests that RNA SSs that facilitate biological function are—contra Salisbury—not rare, at least not within the set of ‘relevant structures’ most easily accessible to random mutations.
Salisbury's arguments are also undermined by in vitro evolution experiments that selected random RNA strands for self-cleaving catalytic activity and found that the hammerhead ribozyme repeatedly emerged, suggesting convergent evolution [25]. The hammerhead ribozyme appears so frequently in all three kingdoms of life that it has recently been termed ubiquitous [26]. We examined the 13 hammerhead ribozymes from the natural dataset [21] for L = 55, finding that 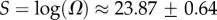 4, very close to the peak of the G-sampled distribution at
4, very close to the peak of the G-sampled distribution at 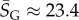 (see L = 55 panel in figures 3 and 4). We postulate that evolutionary convergence is observed in these experiments and in nature, not so much because the hammerhead ribozyme is fitter than other possible self-cleaving enzymes but, rather, because it is particularly easy to find. There may even be many other self-cleaving ribozymes among the 99.9% of L = 55 structures that evolution is unlikely to search through [25]. It would in fact be interesting to devise artificial methods to search for such undiscovered ribozymes [27,28].
(see L = 55 panel in figures 3 and 4). We postulate that evolutionary convergence is observed in these experiments and in nature, not so much because the hammerhead ribozyme is fitter than other possible self-cleaving enzymes but, rather, because it is particularly easy to find. There may even be many other self-cleaving ribozymes among the 99.9% of L = 55 structures that evolution is unlikely to search through [25]. It would in fact be interesting to devise artificial methods to search for such undiscovered ribozymes [27,28].
2.5. Distribution of stacks
Are the 2H relevant structures different from the whole set of NP possible structures? Our arguments above suggest that this should be the case for properties that correlate with NS size. Previous studies (typically on much smaller datasets) have found that structural features (e.g. distributions of stack and loop sizes) of the natural and random SSs are quite similar [13], and that natural and random rRNA share strong similarities in the sequence nucleotide composition of SS motifs such as stems, loops and bulges [16], although natural RNAs are more stable than random RNAs [14]. Indeed, we find that the natural RNAs have slightly more bonds than in G-sampled structures (see electronic supplementary material, figure S3c,d).
We find that Ω correlates negatively with the number of stacks (i.e. sets of contiguous base-pairs) K (see electronic supplementary material, figure S6). The natural distribution of stacks closely follows the G-sampled distribution, but differs markedly from the P-sampled distribution (figure 4a). For example, the hammerhead ribozyme has three stems, close to the most likely number by random G-sampling for L = 55, but much less than the P-sampled average of ≈10. Bias means that it will be difficult for evolution to find L = 55 structures with a large number of stacks, again raising the question of what kind of functionality is possible in principle that cannot be reached by evolution because of such phenotype bias constraints.
As an independent check on our stack predictions, we also obtained 214 natural experimentally determined SSs from the STRAND RNA database [29], and plot the experimentally determined number of stacks versus length L in figure 5b. The majority of experimentally determined structures have numbers of stacks that are within 1 s.d. of the mean calculated from G-sampling, as one would expect if our theoretical predictions were accurate. By contrast, the experimentally determined number of stacks differs significantly from P-sampling estimates.
Figure 5.

Natural stack distributions for ncRNA correlate with G-sampling but not with P-sampling. (a) Distribution of the number of stacks for L = 55 via G-sampling (blue circles) and P-sampling (black diamonds). Natural data (green squares) are remarkably close to the G-sampled distribution, and are drawn from a tiny fraction of the full morphospace of structures (the P-sampled distribution) with small numbers of stacks. The red diamonds are from a combinatorial estimate of stack number [30] that helps corroborate our P-sampling result. (b) Comparison with experimentally measured structures: the brown squares denote the number of stacks experimentally determined for 214 non-pseudo-knotted structures with L > 20 which were taken from the RNA STRAND database [29]. The cyan diamonds show 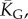 the mean number of stacks calculated by G-sampling with the Vienna package [10], which can be accurately fitted with
the mean number of stacks calculated by G-sampling with the Vienna package [10], which can be accurately fitted with 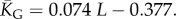 The blue diamonds show the G-sampled number of stacks 1 s.d. above or below the mean. The natural data (brown squares) from the RNA STRAND database are consistent with the G-sampled theoretical data. By contrast these natural data are far away from the expected number of stacks from P-sampling, shown here for an estimate that uses the linear relationship between the P-sampled distribution of log(Ω) and K (electronic supplementary material, figure S6) to infer
The blue diamonds show the G-sampled number of stacks 1 s.d. above or below the mean. The natural data (brown squares) from the RNA STRAND database are consistent with the G-sampled theoretical data. By contrast these natural data are far away from the expected number of stacks from P-sampling, shown here for an estimate that uses the linear relationship between the P-sampled distribution of log(Ω) and K (electronic supplementary material, figure S6) to infer 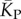 (red diamonds), which are well described by a linear fit
(red diamonds), which are well described by a linear fit 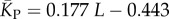 (solid red line). Independent estimates of
(solid red line). Independent estimates of 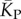 come from [30], and include an asymptotic measure
come from [30], and include an asymptotic measure 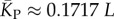 (dashed-dotted line) (see electronic supplementary material). The close agreement between the two independent methods for estimating the mean number of stacks from P-sampling gives us further confidence in our fits to the full P-sampled distribution. (Online version in colour.)
(dashed-dotted line) (see electronic supplementary material). The close agreement between the two independent methods for estimating the mean number of stacks from P-sampling gives us further confidence in our fits to the full P-sampled distribution. (Online version in colour.)
2.6. Distribution of mutational robustness
Interestingly, the bias towards larger Ω also leads to structures with larger mutational robustness [6,12], and again natural data closely follow G-sampled distributions (figure 6). Larger robustness is considered to be advantageous [6], so that, in this important way, phenotype bias facilitates evolution. It is also interesting to note just how large the mutational robustness is for these data. For L = 55, there are of the order of 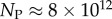 phenotypes, so that the mean probability of finding a phenotype by randomly picking a genotype is of the order of 1 × 10−13. Instead, for both the G-sampled and the P-sampled structures, the probability of a nearest neighbour generating the same phenotypes is of the order of 1012 times higher than random chance. As recently emphasized in [31], this large difference arises from genetic correlations, and typically lifts the robustness well over the minimal threshold
phenotypes, so that the mean probability of finding a phenotype by randomly picking a genotype is of the order of 1 × 10−13. Instead, for both the G-sampled and the P-sampled structures, the probability of a nearest neighbour generating the same phenotypes is of the order of 1012 times higher than random chance. As recently emphasized in [31], this large difference arises from genetic correlations, and typically lifts the robustness well over the minimal threshold 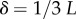 [5] (
[5] (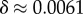 for L = 55) needed to generate large connected neutral networks.
for L = 55) needed to generate large connected neutral networks.
Figure 6.
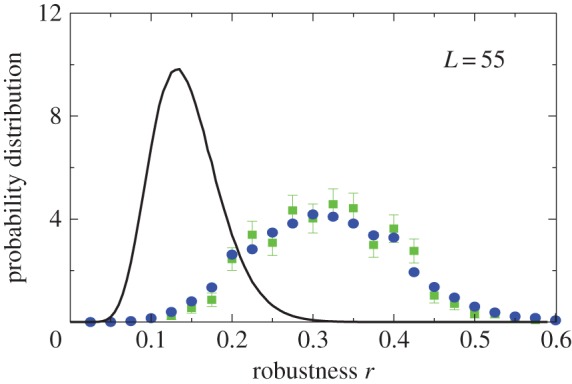
Natural robustness distributions for ncRNA correlate with G-sampling but not with P-sampling. The distribution of robustness, defined as the fraction of mutations r that retain the same SS phenotype, is given for L = 55 via P-sampling (black line) and G-sampling (blue circles). Natural data (green squares) are remarkably close to the G-sampled distribution, and are considerably more robust than the average of structures in the full morphospace. We also note that most phenotypes have a robustness that is above the threshold (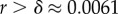 ) needed for the formation of connected neutral networks [31]. (Online version in colour.)
) needed for the formation of connected neutral networks [31]. (Online version in colour.)
3. Discussion
By solving properties of the GP map from sequences to RNA SSs for strands up to L = 126 nucleotides in length, we show explicitly that the vast majority of sequences map to an exponentially small fraction of all possible phenotypes (a summary of scaling forms for key properties of RNA can be found in table 1). One consequence of this strong bias is that only an exponentially small proportion of the morphospace of possible structures can ever be presented to natural selection. Even if one could re-run the tape of life over again multiple times, many structures that are physically feasible and potentially biochemically functional are extremely unlikely to appear simply because they are inaccessible to evolutionary search.
Table 1.
Large L scaling of some key quantities for RNA SSs. For the number of sequences Ω for the NS of the largest non-trivial structure, defined as Ω = 10U, we find 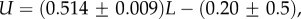 whereas for the trivial structure, with Ω = 10T, we find
whereas for the trivial structure, with Ω = 10T, we find 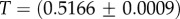
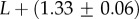 so that U becomes relatively more close to T as L increases. The G-sampled mean of
so that U becomes relatively more close to T as L increases. The G-sampled mean of 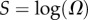 scales as
scales as 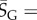
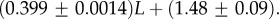 For large L,
For large L, 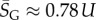 , whereas for P-sampling
, whereas for P-sampling 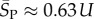 so that
so that 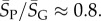 Similarly, the standard deviations of log(Ω) can be directly calculated, and in the large L limit tend to
Similarly, the standard deviations of log(Ω) can be directly calculated, and in the large L limit tend to 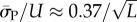 and
and 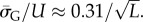 This explains analytically what can be observed qualitatively in figure 3 and the electronic supplementary material, figures S1 and S2: the PG(Ω) distribution is slightly narrower than the PP(Ω) distribution. As L increases both distributions become more sharply peaked relative to the total range [0, U] and PG(Ω) highlights SS phenotypes that are deeper into the tails of the PP(Ω) distribution (and vice versa).
This explains analytically what can be observed qualitatively in figure 3 and the electronic supplementary material, figures S1 and S2: the PG(Ω) distribution is slightly narrower than the PP(Ω) distribution. As L increases both distributions become more sharply peaked relative to the total range [0, U] and PG(Ω) highlights SS phenotypes that are deeper into the tails of the PP(Ω) distribution (and vice versa).
| quantity | large L scaling form |
|---|---|
| total number of genotypes | NG = 4L |
| total number of SS phenotypes | 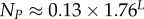 |
| mean Ω | 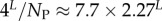 |
| largest non-trivial Ω | 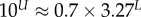 |
| Ω for the trivial structure | 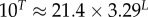 |
| probability to sample trivial structure | 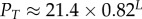 |
| Ω near peak for phenotype sampling | 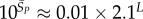 |
| Ω near peak for genotype sampling | 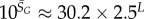 |
| Shannon entropy of distribution | 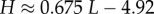 |
| ‘effective number’ of SS phenotypes | 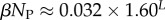 |
| bias parameter | 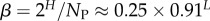 |
| G-sampled mean number of stacks | 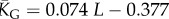 |
| P-sampled mean number of stacks | 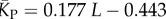 |
While the existence of bias in the RNA GP map has been known for quite some time [6,12–20], and studies using smaller amounts of natural data have suggested that aspects of the mapping are reflected in nature [13,14,16–18], our ability to calculate GP map properties for much larger L allows us to make a comprehensive comparison with the fRNAdb database for functional ncRNA [21]. While one might expect that, owing to bias, large Ω structures are relatively more plentiful in nature [17,18], perhaps the most surprising result of this study is just how closely the natural data follow the prediction of uniform G-sampling over genotypes. We find that the distribution of NS size Ω, the number of stacks S and the mutational robustness r of naturally occurring ncRNA all closely follow the G-sampled distributions, and deviate significantly from the P-sampled distributions. The distribution of bonds also deviates strongly from the P-sampled distribution, but, in contrast to the other distributions above, it also exhibits a small deviation from the G-sampled distribution, with natural RNA having slightly more bonds [14] (see electronic supplementary material, figure S3c,d).
Why does it appear as if we can virtually ignore natural selection with G-sampling? We postulate that, even though the number of relevant structures remains extremely large, SSs that are good enough for function are nonetheless abundant, most likely because small differences between them do not matter that much. Instead, some broader coarse-grained structural features are likely to be sufficient for many functional roles. We suggest that natural selection works on this pre-sculpted variation mainly by further refining parts of the sequence. Some evidence for this picture can be gleaned by the fact that natural structures have slightly more bonds than G-sampled RNA structures do, suggesting selection for greater thermal stability [14].
Interestingly, the fact that many properties of ncRNA SSs so closely follow the G-sampled distribution is direct evidence against claims that the hyper-astronomically large size of genotype space makes functional structures virtually impossible to find. Such assertions were perhaps most famously made by Salisbury [4] over 45 years ago. Despite an illuminating response by Maynard Smith [5], and much experimental evidence to the contrary, such ‘arguments from large numbers’ remain a popular trope in anti-evolutionary polemics today.
The ease with which we can calculate distributions of RNA properties suggests an analogy to the concept of ergodicity in statistical physics, which means that ensemble averages are equivalent to time-averages. When ergodicity (approximately) holds, then it is often true in practice that sampling (via computer simulation for example) a relatively small number of ‘typical’ states (small compared with the total number of possible microstates) is sufficient to accurately calculate ensemble averages of key properties. Something akin to ergodicity may be operating in our case, because statistical averaging via G-sampling is close to the snapshot of many trajectories over time that we observe in the database, the latter being analogous to a time-average. On the other hand, evolution is not an equilibrium process. Indeed, the exponential bias in the GP map suggests that evolutionary waiting times for more rare phenotypes to appear also grows exponentially [20]. Thus, the number of novel phenotypic possibilities will continue to (slowly) increase with time. With large waiting times, contingency is more likely to play an important role. So if a certain evolutionary change is predicated on one of these rare phenotype fixing, then the process of waiting for these innovations may be non-ergodic.
The suggestion that biases in development or other internal processes could strongly affect evolutionary outcomes has traditionally been highly contentious (see [32,33] for an overview). For example, in a recent exchange, entitled: ‘Does evolutionary theory need a rethink?’, Laland et al. [34] argue in favour of the thesis by, among other things, advocating for the importance of developmental bias. In their rejoinder, Wray et al. [35] write: ‘Lack of evidence also makes it difficult to evaluate the role that developmental bias may have in the evolution (or lack of evolution) of adaptive traits’, and call for new evidence: ‘The best way to elevate the prominence of genuinely interesting phenomena such as … developmental bias … is to strengthen the evidence for their importance’. While the RNA system we study here is much simpler than a typical developmental system, that vice is also a virtue, because it allows us to make detailed calculations of the whole morphospace that can then be closely compared with natural data. Thus, RNA SS provides perhaps the clearest and most unambiguous evidence for the importance of bias in shaping evolutionary outcomes.
Bias in the GP map constrains outcomes and so naturally suggests one mechanism for homoplasy (where similar biological forms evolve independently) [36]. The causes of homoplasy are sometimes elaborated in the context of the difference between parallel evolution, where homoplasy is thought to occur because two organisms share a common genetic heritage, and convergence proper, where the same solution is found by different genetic means, and where the primary causal force is usually attributed to selection. While this binary distinction may be too simplistic (see [36–39] for some recent discussion), very roughly, parallel evolution is thought to be considerably weaker evidence than true convergent evolution is for the idea that re-running the tape of life would generate similar outcomes. Interestingly, the bias in the GP map discussed here does not fit into this twofold demarcation at all. Re-run the tape of life, and as long as RNA graces the replay, so will a very similar suite of molecular shapes. But the reason for this repetition is not a contingent common genetic history, nor the Allmacht of selection [40], but rather a different kind of ‘deep structure in biology’ [41].
Our ability to make detailed predictions about evolutionary outcomes as well as counterfactuals for RNA may also shed light on Mayr's famous distinction between proximate and ultimate causes in biology [42], which has been the subject of much recent debate in the literature [43]. This distinction has historically been used to argue against the role of developmental bias in determining evolutionary outcomes [33,44]. While, as also mentioned above, the RNA GP map is much simpler than a typical developmental system, it is instructive to consider how the bias described in this paper plays into Mayr's ultimate–proximate distinction. For example, what is the cause of convergent evolution of the hammerhead ribozyme [25]? The ultimate cause for a self-cleaving ribozyme emerging in populations or in in vitro experiments is surely natural selection for self-cleaving catalytic activity. But why is a three-stack structure repeatedly found, and not say a 10-stack structure, even though the latter are much more common in the morphospace? The cause here is not natural selection per se because it is unlikely that an efficient 10-stack ribozyme is biophysically or biochemically impossible to make. Instead, the explanatory force [44,45] for the ‘why’ question is mainly carried by a proximate GP mapping constraint, namely that the frequency with which 10-stack structures arise as potential variation is many orders of magnitude lower than the frequency with which three-stack structures do. Because the fittest can only be selected and survive if they arrive in the first place [22,23], the evolutionary mechanism that leads to convergence here might be better termed the arrival of the frequent [20].
We also note that the mapping constraint described here differs from classical physical constraints, which would act on the whole morphospace, and from phyletic constraints, which are contingent on evolutionary histories [46]. This mapping constraint has some resemblance to classical morphogenetic constraints which also bias the arrival of variation [47]. But it also differs, because the latter are conceptualized at the level of phenotypes and developmental processes, and may have been shaped by prior selection, whereas the former constraint is a fundamental property of the mapping from genotypes to phenotypes and was not selected for (except perhaps at the origin of life itself).
Finally, strong phenotype bias is also found in model GP maps for protein tertiary [48,49] and quaternary structure [50], gene regulatory networks [51,52] and development [53], suggesting that some of the results discussed in this paper for RNA may hold more widely in biology. While all these GP maps only capture a tiny fraction of the full biological complexity of an organism, if the bias is also (exponentially) strong, then its effects on the rate with which novel variation appears are likely to persist even when further biological details are included. Although more evidence needs to be gathered before making firm pronouncements, it may well be that we need to let go of the commonly held expectation that variation is isotropic in phenotype space or morphospace [54]. Or to put it more provocatively, perhaps our null models should by default assume that variation is highly anisotropic and biased towards certain outcomes over others, unless there is direct evidence to the contrary.
4. Methods
4.1. Folding RNA structures
To map a sequence to an SS, we use the Vienna package [10] with all parameters set to their default values (e.g. the temperature T = 37°C). This software employs dynamic programming techniques to efficiently fold sequences based on thermodynamic rules. Methods of this type are widely used, have been extensively tested and are thought to be relatively accurate. For example, a related method [55] was recently shown to correctly predict 73 ± 9% of canonical base pairs for a database of known RNA structures up to lengths of 700 nucleotides. Generally, these methods are expected to work better for shorter strands [56], and should work well for the lengths we explore. However, they typically cannot correctly predict pseudo-knots. Knowledge-based methods that also take input from known structures and other information may be more accurate for predicting the structures of individual sequences [57], but such methods could introduce biases for GP maps because they take input from natural structures. Thermodynamically based methods such as the Vienna package are therefore probably better suited for working out global properties of the entire GP map, including structures that have not (yet) been found in nature. For these reasons, this package has been most frequently used in studies of full GP maps [6,12–20]. To check our Vienna package results, we compared RNA structure package [55] calculations for the G-distributions of stacks and bonds (electronic supplementary material, figure S3), finding very similar distributions. We also compared the two packages for other motifs like bulges, loops and junctions, finding again very similar predictions (graphs are not shown).
4.2. Exponential growth of the number of strands with strand length L
To illustrate how rapidly the number of sequences grows with length consider the following: there are L4L nucleotides in the set of all possible sequences of length L. The mean mass of a single RNA nucleotide is about 5 × 10−23 kg, so that, for example, the set of all L = 55 strands weighs about 3.8 × 1010 kg, the set of all L = 79 strands weighs about 1.5 × 1025 kg or about 2.6 times the mass of our Earth, whereas the set of all L = 126 RNA strands would have an almost unimaginably large mass of about 5 × 1053 kg, more than the mass of the observable Universe, which is estimated to be about 1053 kg.
4.3. Generating distributions PG(Ω) and PP(Ω) by sampling
We used a standard (Python) random number generator to create sets of random sequences. For each sequence, we used the Vienna package to find the lowest free energy SS. To determine Ω for each SS, we used the NSSE described in [18], which employs sampling techniques together with the inverse fold algorithm from the Vienna package. We used default settings except for the total number of measurements (set with the -m option), which we set to 1 instead of the default 10 for the sake of speed. We checked that this has a negligible effect (typically less than 1%) on the accuracy of our distributions. We also checked the NSSE against the full enumeration for L = 20, finding an agreement of R2 = 0.97 for structures with Ω larger than the average; it performs slightly less well for rare structures. For longer lengths, the number of samples were: 105 for L = 30, 3 × 105 for L = 40, 20 000 for L = 35−80, 5000 for L = 85−100 and 1000 for L = 126. Sequences that generate the trivial structure are discarded. A small fraction of sequences (which increases with increasing length) were also discarded owing to the inverse folding package failing to converge (see electronic supplementary material).
To generate the PG(Ω) distribution, we partition the support of the distribution into bins which are uniform on an 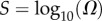 scale. We then determine the probability mass PG(Ω) in each bin. Error bars are simply statistical: there is a trade-off between making smaller bins to give a greater resolution and minimizing statistical errors that increase when there are fewer measurements per bin. Bins with too few sampled points were typically not included in the graphs to avoid large error bars. PP(Ω) is generated from the sampled data by dividing though by Ω (measured at the midpoint of the bin). PP(Ω) is normalized with the NP calculated from the analytic approximation to PP(Ω) (see below).
scale. We then determine the probability mass PG(Ω) in each bin. Error bars are simply statistical: there is a trade-off between making smaller bins to give a greater resolution and minimizing statistical errors that increase when there are fewer measurements per bin. Bins with too few sampled points were typically not included in the graphs to avoid large error bars. PP(Ω) is generated from the sampled data by dividing though by Ω (measured at the midpoint of the bin). PP(Ω) is normalized with the NP calculated from the analytic approximation to PP(Ω) (see below).
4.4. Analytical fit to neutral set size distributions
For analytic fits, we make a simple log-binomial ansatz:
| 4.1 |
where 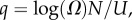 10U is the largest non-trivial Ω, and N and pP are parameters that are fitted to measured distributions. In other words, the probability that a P-sampled SS is found with S = log(Ω) is distributed binomially. By definition
10U is the largest non-trivial Ω, and N and pP are parameters that are fitted to measured distributions. In other words, the probability that a P-sampled SS is found with S = log(Ω) is distributed binomially. By definition 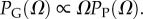 Taking normalization into account is enough to show that PG(Ω) has the same binomial form as equation (4.1), but with parameter pP replaced by
Taking normalization into account is enough to show that PG(Ω) has the same binomial form as equation (4.1), but with parameter pP replaced by 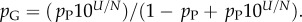 Fixing the parameters N, U and either pG or pP thus fixes both distributions.
Fixing the parameters N, U and either pG or pP thus fixes both distributions.
For L = 20, equation (4.1) with U = 10, N = 8.0 and pP = 0.55 describes the exact PP(Ω) from full enumeration very well, as can be seen in figure 1b. The related approximation for PG(Ω) performs slightly less well for G-sampled data for L = 20, but it still captures the main qualitative features.
For larger L, we determine the parameters as follows: first, we estimate U from the largest non-trivial NS size found. This method inevitably provides a lower bound U′ on the true maximum U. However, given the rather sharp upper bound generated by the binomial fit, we expect that the relative errors in U are quite small. For example, for L = 60, we used 20 000 samples to determine U′ = 30.56. From the binomial form, we estimate that U′ is within an error 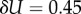 of the true U with 90% probability. Next, we calculate
of the true U with 90% probability. Next, we calculate 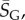 the G-sampled average of S = log(Ω), as well as the standard deviation
the G-sampled average of S = log(Ω), as well as the standard deviation 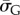 in S from G-sampled data. We can then determine the parameter
in S from G-sampled data. We can then determine the parameter 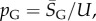 derived by taking the mean of q through equation (4.1). The parameter N can subsequently be extracted from the measured G-sampled standard deviation:
derived by taking the mean of q through equation (4.1). The parameter N can subsequently be extracted from the measured G-sampled standard deviation: 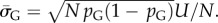 The P-sampled standard deviation is given by
The P-sampled standard deviation is given by 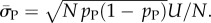 Because
Because 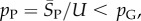 so also
so also 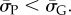 In this way, we obtained the binomial fits shown in figure 3 and the electronic supplementary material, figures S1 and S2. The close agreement between the sampled data and our fits for lengths up to L = 126 suggests that this procedure is fairly robust.
In this way, we obtained the binomial fits shown in figure 3 and the electronic supplementary material, figures S1 and S2. The close agreement between the sampled data and our fits for lengths up to L = 126 suggests that this procedure is fairly robust.
It is harder to find structures with small Ω, so that the PP(Ω) can only be partially sampled, especially for larger L. However, given how well our simple ansatz works for predicting PG(Ω), and given that for L = 20 RNA the binomial form works so well for the full range of structures, we expect the full PP(Ω) to be at least similar if not very close to equation (4.1). Further evidence for this form can also be extracted from combinatorial arguments for the distributions of stacks [30], which are correlated with log(Ω) (see below).
4.5. Rank plots
Analytic rank-plot functions are the cumulative density function of PP(Ω). For L = 20, all structures are known, so a rank plot can be directly made. For L > 20, the measured Ω of natural SSs was used to align points to a rank function calculated from the analytic binomial fit to PP(Ω).
4.6. Scaling forms as a function of L
We further used the lengths L = 30–100 to extract scaling forms for several properties as a function of L. Linear fits in L are shown for U, T, SG and N in the electronic supplementary material, figure S5. T is close to U, so that the relative difference between T and U decreases as L increases (electronic supplementary material, figure S5). A summary of the scaling forms for different properties in the large L limit can be found in table 1.
4.7. Probability PT to find the trivial structure
The probability of finding the trivial structure, PT, decreases exponentially with increasing L:
| 4.2 |
For example, for L = 20, we can directly measure 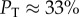 , whereas for L = 55 it has already dropped down to a mere 0.04%. This rapid decrease in PT justifies our decision to ignore the trivial structure in our fitting to PG(S) and PP(S).
, whereas for L = 55 it has already dropped down to a mere 0.04%. This rapid decrease in PT justifies our decision to ignore the trivial structure in our fitting to PG(S) and PP(S).
4.8. The number of SS, NP, as a function of L
For L = 11−20, we used full enumerations [20] to calculate NP. For longer L, we used our analytic fit as follows: the mean (including trivial structure) of Ω is 4L/NP, so that 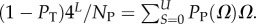 Given the binomial form of PP(Ω), the sum can be carried out analytically, from which it follows that
Given the binomial form of PP(Ω), the sum can be carried out analytically, from which it follows that 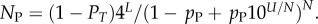 At each L, we evaluated N,PT,pP and U, and used this to evaluate NP. A simple linear form,
At each L, we evaluated N,PT,pP and U, and used this to evaluate NP. A simple linear form,
| 4.3 |
provides a good fit to the data (electronic supplementary material, figure S5).
4.9. Scaling forms for the Shannon entropy H and bias parameter β as a function of L
From the expression for the entropy derived above, it directly follows that 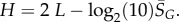 Using the fit derived above for SG, the entropy grows with L as
Using the fit derived above for SG, the entropy grows with L as 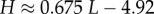 and the effective number of states scales as
and the effective number of states scales as 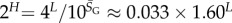 (this ignores the trivial structure, but the effect is very small for larger L). Note that one only has to find
(this ignores the trivial structure, but the effect is very small for larger L). Note that one only has to find 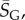 which is easily obtainable, to determine this important quantity. By combining with equation (4.3), we find that the bias parameter scales as
which is easily obtainable, to determine this important quantity. By combining with equation (4.3), we find that the bias parameter scales as 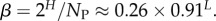
4.10. Sampling natural RNA from the fRNA database
To generate the distributions of functional ncRNA, we took all available sequences for each length studied (ranging from L = 40 to 126) from the non-coding functional RNA database (fRNAdb [21]). For L = 20, we took data from Drosophila melanogaster only, but this made up 77% of all L = 20 SSs in the database. For each sequence, we found the SS and used the NSSE to estimate its Ω. A small fraction of the natural RNA sequences contained non-standard nucleotide letters, e.g. N or R; such sequences were ignored, because the standard packages cannot treat them. Similarly, a small fraction of sequences were also discarded owing to the NSSE failing to converge (see electronic supplementary material). We checked that there were no repeated sequences in the database of natural RNA. Finally, in the electronic supplementary material, we provide a breakdown of the identity of the structures for L = 20, 55, 70 and 126 and also take curated subsets of the data to emphasize structures where the SS is known to be important. In figure 3 and the electronic supplementary material, figures S1 and S2, we show for L = 55 and L = 126 that the close correlation with PG(Ω) remains when data are curated.
4.11. Checking for codon bias
Genetic mutations are random in the sense that they do not arise to benefit an organism. Nevertheless, it is well known that in other ways mutations are not uniformly random [58]. One example (among several) is that transitions (pyramidines ↔ pyramidines or purines ↔ purines) are more frequent that transversions (purines ↔ pyramidines) [58]. For many of these biases, mutations can still effectively sample the whole space uniformly without preferring certain genotypes over others. Nevertheless, there are biases that lead, for example, to an excess of GC over AT base pairs, or vice versa in DNA [58]. To test for the effect of strong bias of this type, we generated Ω distributions with 30% GC (AU bias) and 70% GC (GC bias) content for different lengths. The overall effect becomes less pronounced for longer strands (electronic supplementary material, figure S4). Because natural DNA (and by extension RNA) can show biases for both larger and smaller GC content, we argue that this can to first order be ignored when comparing with natural datasets across many species, although the effects may be observable on datasets with large content bias.
4.12. Calculating P-distribution of stacks
We used a linear relationship between log(Ω) and K (electronic supplementary material, figure S6) to transform PP(Ω) into an estimated P-sampled distribution of stacks, as shown in figure 4a, and used this to obtain the P-sampled average  We also adapted analytic results from [30] based on combinatorics (see the electronic supplementary material) to calculate estimates for the P-sampled distribution of K and for
We also adapted analytic results from [30] based on combinatorics (see the electronic supplementary material) to calculate estimates for the P-sampled distribution of K and for  The close agreement shown in figure 4a,b, between the two independent methods for estimating the number of stacks from P-sampling increases our confidence in our binomial fits to the full P-sampled distribution.
The close agreement shown in figure 4a,b, between the two independent methods for estimating the number of stacks from P-sampling increases our confidence in our binomial fits to the full P-sampled distribution.
4.13. Calculating the robustness distribution
A strong positive correlation between mutational robustness and Ω is well established in the literature [6]. For figure 6, the robustness for G-sampled data (taken from 105 random sequences) and natural data (taken from the 504 natural sequences for L = 55) was calculated by folding all 3L strands within one point mutation and calculating the fraction r that generates the same SS. Error bars come from our binning procedure. For the P-sampled distribution, such a sequence robustness cannot be defined, so instead the robustness per sequence was first averaged over a set of sequences for each SS to estimate a mean robustness (phenotype robustness) per SS. We next generated a cubic fit to the mean r per SS versus logΩ. The fit was constrained to have r = 0 at Ω = 1 and had R2 = 0.93. This was then combined with PP(Ω) to generate an estimate of the r distribution for P-sampled data. Because we do not have many data points at small Ω, the fit is partially an extrapolation. Further error may arise from the partial sampling of the phenotype robustness. Nevertheless, we do not expect this procedure to lead to a significant difference in the qualitative comparison between P- and G-sampled data for robustness, given that the two are so different.
Acknowledgements
We acknowledge discussions with S.E. Ahnert, F.Q. Camargo, J.P.K. Doye, J.L. England, T.M.A. Fink, W. Haerty, I.G. Johnston, D.C. Lahti, A.C. Love, O. Martin, T.C.B. McLeish, T.E. Ouldridge, C.P. Ponting, F. Randisi and P. Sulc.
Competing interests
We declare we have no competing interests.
Funding
K.D. acknowledges funding from the EPSRC/EP/G03706X/1.
References
- 1.Gould SJ. 1989. Wonderful life: the Burgess Shale and the nature of history. New York, NY: W. W. Norton & Co. [Google Scholar]
- 2.Morris S. 2003. Life’s solution: inevitable humans in a lonely universe. Cambridge, UK: Cambridge University Press. [Google Scholar]
- 3.McGhee GR. 2006. The geometry of evolution: adaptive landscapes and theoretical morphospaces. Cambridge, UK: Cambridge University Press. [Google Scholar]
- 4.Salisbury FB. 1969. Natural selection and the complexity of the gene. Nature 224, 342–343. ( 10.1038/224342a0) [DOI] [PubMed] [Google Scholar]
- 5.Maynard Smith J. 1970. Natural selection and the concept of a protein space. Nature 225, 563–564. ( 10.1038/225563a0) [DOI] [PubMed] [Google Scholar]
- 6.Wagner A. 2005. Robustness and evolvability in living systems. Princeton, NJ: Princeton University Press. [Google Scholar]
- 7.Mattick JS, Makunin IV. 2006. Non-coding RNA. Hum. Mol. Genet. 15(Suppl 1), R17–R29. ( 10.1093/hmg/ddl046) [DOI] [PubMed] [Google Scholar]
- 8.Carothers J, Oestreich S, Davis J, Szostak J. 2004. Informational complexity and functional activity of RNA structures. J. Am. Chem. Soc. 126, 5130–5137. ( 10.1021/ja031504a) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bailor MH, Mustoe AM III, Brooks CL, Al-Hashimi HM. 2011. Topological constraints: using RNA secondary structure to model 3d conformation, folding pathways, and dynamic adaptation. Curr. Opin. Struct. Biol. 21, 296–305. ( 10.1016/j.sbi.2011.03.009) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hofacker I, Fontana W, Stadler P, Bonhoeffer L, Tacker M, Schuster P. 1994. Fast folding and comparison of RNA secondary structures. Chem. Mon. 125, 167–188. ( 10.1007/BF00818163) [DOI] [Google Scholar]
- 11.Zuker M, Mathews D, Turner D. 1999. Algorithms and thermodynamics for RNA secondary structure prediction: a practical guide. RNA Biochem. Biotechnol. 70, 11–44. ( 10.1007/978-94-011-4485-8_2) [DOI] [Google Scholar]
- 12.Schuster P, Fontana W, Stadler P, Hofacker I. 1994. From sequences to shapes and back: a case study in RNA secondary structures. Proc. R. Soc. Lond. B 255, 279–284. ( 10.1098/rspb.1994.0040) [DOI] [PubMed] [Google Scholar]
- 13.Fontana W, Konings DA, Stadler PF, Schuster P. 1993. Statistics of RNA secondary structures. Biopolymers 33, 1389–1404. ( 10.1002/bip.360330909) [DOI] [PubMed] [Google Scholar]
- 14.Schultes EA, Spasic A, Mohanty U, Bartel DP. 2005. Compact and ordered collapse of randomly generated RNA sequences. Nat. Struct. Mol. Biol. 12, 1130–1136. ( 10.1038/nsmb1014) [DOI] [PubMed] [Google Scholar]
- 15.Schuster P. 2006. Prediction of RNA secondary structures: from theory to models and real molecules. Rep. Prog. Phys. 69, 1419 ( 10.1088/0034-4885/69/5/R04) [DOI] [Google Scholar]
- 16.Smit S, Yarus M, Knight R. 2006. Natural selection is not required to explain universal compositional patterns in rRNA secondary structure categories. RNA 12, 1–14. ( 10.1261/rna.2183806) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cowperthwaite M, Economo E, Harcombe W, Miller E, Meyers L. 2008. The ascent of the abundant: how mutational networks constrain evolution. PLoS Comput. Biol. 4, e1000110 ( 10.1371/journal.pcbi.1000110) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jorg T, Martin O, Wagner A. 2008. Neutral network sizes of biological RNA molecules can be computed and are not atypically small. BMC Bioinformatics 9, 464 ( 10.1186/1471-2105-9-464) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Aguirre J, Buldú JM, Stich M, Manrubia SC. 2011. Topological structure of the space of phenotypes: the case of RNA neutral networks. PLoS ONE 6, e26324 ( 10.1371/journal.pone.0026324) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schaper S, Louis AA. 2014. The arrival of the frequent: how bias in genotype–phenotype maps can steer populations to local optima. PLoS ONE 9, e86635 ( 10.1371/journal.pone.0086635) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kin T, et al. 2007. FRNAdb: a platform for mining/annotating functional RNA candidates from non-coding RNA sequences. Nucleic Acids Res. 35(Suppl 1), D145–D148. ( 10.1093/nar/gkl837) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.de Vries H. 1904. Species and varieties, their origin by mutation. Amsterdam, The Netherlands: The Open Court Publishing Company. [Google Scholar]
- 23.Wagner A. 2014. Arrival of the fittest: solving evolution’s greatest puzzle. New York, NY: Penguin. [Google Scholar]
- 24.Mezard M, Montanari A. 2009. Information, physics, and computation. New York, NY: Oxford University Press. [Google Scholar]
- 25.Salehi-Ashtiani K, Szostak J. 2001. In vitro evolution suggests multiple origins for the hammerhead ribozyme. Nature 414, 82–83. ( 10.1038/35102081) [DOI] [PubMed] [Google Scholar]
- 26.Hammann C, Luptak A, Perreault J, de la Peña M. 2012. The ubiquitous hammerhead ribozyme. RNA 18, 871–885. ( 10.1261/rna.031401.111) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gan HH, Pasquali S, Schlick T. 2003. Exploring the repertoire of RNA secondary motifs using graph theory; implications for RNA design. Nucleic Acids Res. 31, 2926–2943. ( 10.1093/nar/gkg365) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Athavale SS, Spicer B, Chen IA. 2014. Experimental fitness landscapes to understand the molecular evolution of RNA-based life. Curr. Opin. Chem. Biol. 22, 35–39. ( 10.1016/j.cbpa.2014.09.008) [DOI] [PubMed] [Google Scholar]
- 29.Andronescu M, Bereg V, Hoos HH, Condon A. 2008. RNA STRAND: the RNA secondary structure and statistical analysis database. BMC Bioinformatics 9, 340 ( 10.1186/1471-2105-9-340) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hofacker IL, Schuster P, Stadler PF. 1998. Combinatorics of RNA secondary structures. Discrete Appl. Math. 88, 207–237. ( 10.1016/S0166-218X(98)00073-0) [DOI] [Google Scholar]
- 31.Greenbury SF, Ahnert SE, Louis AA.2015. Genetic correlations greatly increase mutational robustness and can both reduce and enhance evolvability. (http://arxiv.org/abs/1505.07821. )
- 32.Yampolsky L, Stoltzfus A. 2001. Bias in the introduction of variation as an orienting factor in evolution. Evol. Dev. 3, 73–83. ( 10.1046/j.1525-142x.2001.003002073.x) [DOI] [PubMed] [Google Scholar]
- 33.Amundson R. 2005. The changing role of the embryo in evolutionary thought: roots of evo-devo. Cambridge, UK: Cambridge University Press. [Google Scholar]
- 34.Laland K, Uller T, Feldman M, Sterelny K, Müller GB, Moczek A, Jablonka E, Odling-Smee J. 2014. Does evolutionary theory need a rethink? Yes, urgently. Nature 514, 161–164. ( 10.1038/514161a) [DOI] [PubMed] [Google Scholar]
- 35.Wray GA, Hoekstra HE, Futuyma D, Lenski R, Mackay T, Schluter D, Strassman J. 2014. Does evolutionary theory need a rethink? No, all is well. Nature 514, 161–164. [DOI] [PubMed] [Google Scholar]
- 36.Losos JB. 2011. Convergence, adaptation, and constraint. Evolution 65, 1827–1840. ( 10.1111/j.1558-5646.2011.01289.x) [DOI] [PubMed] [Google Scholar]
- 37.Scotland RW. 2011. What is parallelism? Evol. Dev. 13, 214–227. ( 10.1111/j.1525-142X.2011.00471.x) [DOI] [PubMed] [Google Scholar]
- 38.Pearce T. 2011. Convergence and parallelism in evolution: a neo-Gouldian account. Br. J. Philos. Sci. 63, 429–448. ( 10.1093/bjps/axr046) [DOI] [Google Scholar]
- 39.Powell R. 2012. Convergent evolution and the limits of natural selection. Eur. J. Philos. Sci. 2, 355–373. ( 10.1007/s13194-012-0047-9) [DOI] [Google Scholar]
- 40.Weismann A. 1893. The all-sufficiency of natural selection: a reply to Herbert Spencer. Contemp. Rev. 64, 309–338. [Google Scholar]
- 41.Morris SC. 2008. The deep structure of biology: is convergence sufficiently ubiquitous to give a directional signal. West Conshohocken, PA: Templeton Foundation Press. [Google Scholar]
- 42.Mayr E. 1961. Cause and effect in biology. Science (New York, NY) 134, 1501–1506. ( 10.1126/science.134.3489.1501) [DOI] [PubMed] [Google Scholar]
- 43.Laland KN, Sterelny K, Odling-Smee J, Hoppitt W, Uller T. 2011. Cause and effect in biology revisited: is Mayr's proximate-ultimate dichotomy still useful? Science 334, 1512–1516. ( 10.1126/science.1210879) [DOI] [PubMed] [Google Scholar]
- 44.Scholl R, Pigliucci M. 2014. The proximate–ultimate distinction and evolutionary developmental biology: causal irrelevance versus explanatory abstraction. Biol. Philos. 30, 653–670. ( 10.1007/s10539-014-9427-1) [DOI] [Google Scholar]
- 45.Schwenk K, Wagner GP. 2004. The relativism of constraints on phenotypic evolution. In Phenotypic integration: studying the ecology and evolution of complex phenotypes (eds Pigliucci M, Preston K), pp. 390–408. Oxford, UK: Oxford University Press. [Google Scholar]
- 46.Gould S, Lewontin R. 1979. The spandrels of San Marco and the Panglossian paradigm: a critique of the adaptationist programme. Proc. R. Soc. Lond. B 205, 581–598. ( 10.1098/rspb.1979.0086) [DOI] [PubMed] [Google Scholar]
- 47.Gilbert SF. 2000. Developmental biology. Sunderland, MA: Sinauer Associates, Inc. [Google Scholar]
- 48.Li H, Helling R, Tang C, Wingreen N. 1996. Emergence of preferred structures in a simple model of protein folding. Science 273, 666–669. ( 10.1126/science.273.5275.666) [DOI] [PubMed] [Google Scholar]
- 49.England J, Shakhnovich E. 2003. Structural determinant of protein designability. Phys. Rev. Lett. 90, 218101 ( 10.1103/PhysRevLett.90.218101) [DOI] [PubMed] [Google Scholar]
- 50.Greenbury SF, Johnston IG, Louis AA, Ahnert SE. 2014. A tractable genotype–phenotype map modelling the self-assembly of protein quaternary structure. J. R. Soc. Interface 11, 20140249 ( 10.1098/rsif.2014.0249) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nochomovitz Y, Li H. 2006. Highly designable phenotypes and mutational buffers emerge from a systematic mapping between network topology and dynamic output. Proc. Natl Acad. Sci. USA 103, 4180–4185. ( 10.1073/pnas.0507032103) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Raman K, Wagner A. 2011. Evolvability and robustness in a complex signalling circuit. Mol. Biosyst. 7, 1081–1092. [DOI] [PubMed] [Google Scholar]
- 53.Borenstein E, Krakauer D. 2008. An end to endless forms: epistasis, phenotype distribution bias, and nonuniform evolution. PLoS Comput. Biol. 4, e1000202 ( 10.1371/journal.pcbi.1000202) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gerber S. 2014. Not all roads can be taken: development induces anisotropic accessibility in morphospace. Evol. Dev. 16, 373–381. ( 10.1111/ede.12098) [DOI] [PubMed] [Google Scholar]
- 55.Mathews DH, Disney MD, Childs JL, Schroeder SJ, Zuker M, Turner DH. 2004. Incorporating chemical modification constraints into a dynamic programming algorithm for prediction of RNA secondary structure. Proc. Natl Acad. Sci. USA 101, 7287–7292. ( 10.1073/pnas.0401799101) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Reuter JS, Mathews DH. 2010. Rnastructure: software for RNA secondary structure prediction and analysis. BMC Bioinformatics 11, 129 ( 10.1186/1471-2105-11-129) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gardner PP, Giegerich R. 2004. A comprehensive comparison of comparative RNA structure prediction approaches. BMC Bioinformatics 5, 140 ( 10.1186/1471-2105-5-140) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Li W, Graur D. 1999. Fundamentals of molecular evolution. Sunderland, MA: Sinauer. [Google Scholar]