

Abstract
Background
FNR homologues constitute an important class of transcription factors that control a wide range of anaerobic physiological functions in a number of bacterial species. Since FNR homologues are some of the most pervasive transcription factors, an understanding of their involvement in regulating anaerobic gene expression in different species sheds light on evolutionary similarity and differences. To address this question, we used a combination of high throughput RNA-Seq and ChIP-Seq analysis to define the extent of the FnrL regulon in Rhodobacter capsulatus and related our results to that of FnrL in Rhodobacter sphaeroides and FNR in Escherichia coli.
Results
Our RNA-seq results show that FnrL affects the expression of 807 genes, which accounts for over 20 % of the Rba. capsulatus genome. ChIP-seq results indicate that 42 of these genes are directly regulated by FnrL. Importantly, this includes genes involved in the synthesis of the anoxygenic photosystem. Similarly, FnrL in Rba. sphaeroides affects 24 % of its genome, however, only 171 genes are differentially expressed in common between two Rhodobacter species, suggesting significant divergence in regulation.
Conclusions
We show that FnrL in Rba. capsulatus activates photosynthesis while in Rba. sphaeroides FnrL regulation reported to involve repression of the photosystem. This analysis highlights important differences in transcriptional control of photosynthetic events and other metabolic processes controlled by FnrL orthologues in closely related Rhodobacter species. Furthermore, we also show that the E. coli FNR regulon has limited transcriptional overlap with the FnrL regulons from either Rhodobacter species.
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-015-2162-4) contains supplementary material, which is available to authorized users.
Keywords: Transcriptomics, Redox regulation, Global transcription factor binding
Background
The purple non-sulfur α-proteobacterium Rhodobacter capsulatus possesses a metabolically versatile metabolism that allows growth in a wide variety of environments. Much is known about its photosynthetic growth metabolism along with transcription factors that control anaerobic photosystem gene expression such as RegA, CrtJ, and AerR [1–5]. However, the redox responding transcription factor FnrL, which is a homologue of FNR (for fumarate nitrate reduction) from E. coli, has not been well characterized in Rba. capsulatus [5–7]. FnrL from Rba. capsulatus is reported to have a role in production of respiratory cytochromes but not in the production of the photosystem machinery [2, 5, 7, 8]. Beyond these observations, the involvement of FnrL in controlling anaerobic gene expression is unknown.
FNR from E. coli has a central role in controlling many changes in metabolism that occurs when these cells shift from aerobic to anaerobic growth conditions [6, 9]. FNR directly senses changes in oxygen tension via the presence of a redox sensitive 4Fe-4S cluster that is coordinated by four cysteines [10]. Under anaerobic conditions, the iron cluster is stable allowing FNR to form a dimer that binds to target DNA sequences [11, 12]. However, under aerobic conditions, this cluster becomes oxidized leading to its disassembly with a concomitant loss of FNR dimerization and ultimately loss of DNA binding activity [8, 11]. FnrL from Rhodobacter capsulatus, and its homolog in Rhodobacter sphaeroides, also contain four Fe coordinating cysteines as described for E. coli FNR, however their placement within the peptide sequence is different from FNR. This suggests that the coordination of the 4Fe-4S cluster may be altered and/or there exist dissimilarities in redox regulation and allosteric behavior between the FnrL homologs and FNR.
Analysis of the FNR regulon in E. coli has been well characterized most recently using a combination of the deep sequencing technologies; RNA-seq and chromatin immunoprecipitation sequencing (ChIP-seq) [6]. This recent study has established that the FNR regulon is quite large and complex and is responsible for controlling variety of genes that affect the ability to effectively grow under conditions of oxygen limitation. For example, FNR controls the expression of high oxygen affinity terminal oxidases and a DMSO reductase that uses DMSO as an alternative electron acceptor under anaerobiosis [6]. The FNR regulon not only includes genes whose expression are directly regulated by FNR, but also genes indirectly regulated by FNR via secondary regulation [6, 13]. The latter occurs when FNR directly controls the expression of a transcription factor that subsequently regulates expression of downstream genes either directly or through additional downstream transcription cascades. Analysis of the E. coli FNR regulon is further complicated by the observation that a number of FNR binding sites as defined by ChIP-seq occur near or within genes that do not exhibit a corresponding difference in expression upon deletion of FNR [6]. Thus, there appears to be a number of “silent” FNR binding sites that presumably are involved in control of gene expression under conditions that have not yet been tested. Additionally, these silent sites may have a role that does not affect transcription but instead have a role in providing chromosomal structural integrity. For example, FNR may have a yet to be defined nucleoid-associated role that would affect such processes as chromosome packing [14].
Both RNA-seq and ChIP-seq analysis of the Rba, sphaeroides FnrL regulon has recently been reported [18]. Their analysis indicated that FnrL is directly involved in regulating anaerobic respiration, tetrapyrrole biosynthesis and iron metabolism. However, there does not appear to be direct control of the photosynthetic structural proteins with overall photosynthetic events negatively regulated by FnrL. In contrast, detailed analysis of the Rba. capsulatus FnrL regulon has not been undertaken, but is necessary as there are key differences between the observed phenotypes of FnrL deletions in these species. For example, FnrL mutants in Rba. sphaeroides are unable to grow photosynthetically while a FnrL deletion mutant of Rba. capsulatus remains viable during photosynthetic growth [5, 7, 15–17]. To address these differences, we utilize a combination of ChIP-seq and RNA-seq analyses to provide a high-resolution description of the FnrL regulon in Rba. capsulatus. We have identified a large set of genes scattered throughout the genome involved in diverse metabolic pathways that are directly and indirectly regulated by FnrL. We present a global picture of the regulatory involvement of FnrL and also provide a detailed depiction of the photosynthetic events controlled by FnrL in Rba. capsulatus. For completeness, we compare the Rba. capsulatus FnrL regulon with the FnrL regulon from Rba. sphaeroides and the FNR regulon in E. coli [6, 18]. While the FnrL regulons from Rhodobacter species do share similarities, they differ significantly and are unambiguously different from the E. coli FNR regulon. Consequently, there is considerable plasticity in number and type of genes that constitute members of FNR regulons in different organisms.
Results and discussion
Identifying direct and indirect members of the FnrL regulon using comparative RNA-Seq and ChIP-Seq
We identified members of the FnrL regulon by performing RNA-seq transcriptome analysis of anaerobically (photosynthetically) grown wild-type versus ΔfnrL strains. Over 10 million (M) strand specific RNA-seq reads were collected per sample from three biological replicates. Differentially expressed genes (DEGs) from pair-wise comparison of wild type and ΔfnrL data sets were identified as those that had altered photosynthetic/aerobic changes in expression with a p-value ≤ 0.05. The motivation behind using a p-value cutoff of ≤ 0.05 was to make our results directly comparable to that of previously published E. coli and Rba. sphaeroides FNR/FnrL RNA-seq data sets that used a similar p-value of ≤ 0.05 [6, 18]. With a p-value cutoff of ≤ 0.05 we categorized 807 DEGs as members of the Rba. capsulatus FnrL regulon (Fig. 1, Table 1, Additional files 1 and 2: Table S1 and S2). This number of genes in the Rba. capsulatus FnrL regulon is comparable to that observed for the FnrL regulon from Rba. sphaeroides, which has 917 genes DEG’s with p-value ≤ 0.05 We also note that several FnrL ChIP-seq peaks containing well-defined FnrL binding consensus sequences are present upstream of DEGs with p-values between 0.05 and 0.1. These genes are noted in the ChIP-seq data set in Additional file 3: Table S3 and suggest that a p-value ≤ 0.05 at times acts as too stringent of a filter. Nevertheless we used the p value ≤ 0.05 as a cut off so as to be confident that the genes that are included in the FnrL regulon are not falsely identified and to be consistent with similar studies in other species.
Fig. 1.
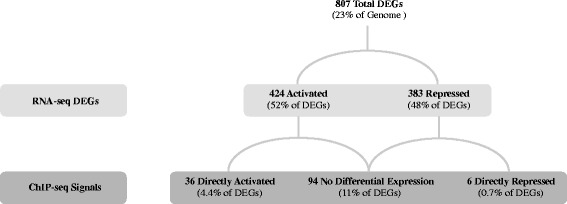
Total FnrL directly and indirectly controlled genes. Differentially expressed genes that are directly and indirectly activated by FnrL as determined from RNA-seq (green) and ChIP-seq (purple). No Differential Expression are instances where ChIP signal was observed without a corresponding RNA-seq expression
Table 1.
FnrL directly regulated genes based on ChIP-seq signal with corresponding RNA-seq expression change that also contain a consensus binding sequence
| Locus ID | Gene Name | Description | Recognition Sequence | Enrichment | Regulation | Fold Change |
|---|---|---|---|---|---|---|
| COG C: Energy production and conversion | ||||||
| RCC01157 | ccoN | cbb 3-type cytochrome c oxidase subunit I | TTGATCAAGGTCAAb | 25 | + | 1.55 |
| RCC01157 | ccoN | cbb 3-type cytochrome c oxidase subunit I | ATGATGTCGATCAAa | 25 | + | 1.55 |
| RCC00728 | NnrU family protein | NnrU family protein | CTGCCGCAGATCAAa | 4 | + | 1.47 |
| RCC00732 | sdhD | succinate dehydrogenase | ATGATGAGCGTCAAb | 3 | + | 1.41 |
| RCC00022 | Oxidoreductase | oxidoreductase | ATGATTTACCGCAAa | 5 | + | 1.38 |
| COG E: Amino acid transport and metabolism | ||||||
| RCC01724 | speB1 | agmatinase | TTGATCTGCGTCAAb | 10 | + | 1.33 |
| COG F: Nucleotide transport and metabolism | ||||||
| RCC00400 | pyrB | aspartate carbamoyltransferase | CTGACGCAGATCAAa | 10 | + | 1.47 |
| COG G: Carbohydrate transport and metabolism | ||||||
| RCC00731 | sdhC | succinate dehydrogenase, cytochrome b556 subunit | ATGATGAGCGTCAAa | 3 | + | 1.53 |
| COG I: Lipid transport and metabolism | ||||||
| RCC00480 | rpsU | 30S ribosomal protein S21 | CTGATGCAACTCAAb | 4 | + | 1.57 |
| COG J: Translation, ribisomal tructure and biogenesis | ||||||
| RCC01495 | fusA | translation elongation factor G | TTGGCATGGGTCAAb | 17 | + | 3.48 |
| COG L: Replication, recombination and repair | ||||||
| RCC03240 | ATPase AAA | K01144 exodeoxyribonuclease V | ATGCGCCAGATCAAa | 4 | - | -1.34 |
| RCC02193 | DNA-3-methyladenine glycosylase II | K01247 DNA-3-methyladenine glycosylase II | ATGACGCGGATCAAa | 4 | - | –1.97 |
| COG M: Cell wall/membrane/envelope biogenesis | ||||||
| RCC02479 | lipoprotein | lipoprotein | CTGATGCAGCGCAAb | 13 | + | 1.42 |
| COG N: Cell motility | ||||||
| RCC00481 | mcpI | methyl-accepting chemotaxis protein McpI | CTGACCGAGATCAAa | 4 | - | –1.53 |
| RCC03524 | flagellar FlaF family protein | K06602 flagellar protein FlaF | CTGATCGACATCAAa | 4 | - | –1.87 |
| RCC03523 | flbT | flagellin synthesis repressor protein FlbT | CTGATCGACATCAAb | 4 | - | –2.14 |
| COG O: Post-translational modification, protein turnover, and chaperones | ||||||
| RCC01156 | UspA domain-containing protein | UspA domain-containing protein | TTGACGCGGATCAAb | 26 | + | 5.21 |
| RCC01723 | ccpA | cytochrome-c peroxidase | TTGATCTGCGTCAAb | 10 | + | 3.81 |
| RCC02829 | hypothetical protein | K03699 putative hemolysin | TTGACCCTCGTCAAa | 6 | - | –1.64 |
| COG R: General function prediction only | ||||||
| RCC02684 | polyphosphate kinase 2 | polyphosphate kinase 2 | TTGATGCGTGTCAAb | 14 | + | 2.25 |
| RCC02665 | hemolysin-type calcium-binding | hemolysin-type calcium-binding | ATGACCGGCGTCAAa | 9 | + | 1.46 |
| COG S: Function unknown | ||||||
| RCC00435 | hypothetical protein | hypothetical protein | CTGACCCAGATCAAb | 21 | + | 9.19 |
| RCC00901 | hypothetical protein | hypothetical protein | TTGACACGGGTCAAb | 10 | + | 6.87 |
| RCC00747 | hypothetical protein | hypothetical protein | ATGACGCAGATCAAb | 5 | + | 3.78 |
| RCC00424 | hypothetical protein | hypothetical protein | ATGATTCAGATCAAb | 20 | + | 3.51 |
| RCC02321 | hypothetical protein | hypothetical protein | ATGATCCGGATCAAb | 26 | + | 2.17 |
| RCC02988 | hypothetical protein | hypothetical protein | TTGACCCAGATCAAb | 8 | - | –1.41 |
| RCC01027 | hypothetical protein | hypothetical protein | TTGACCAAGGTCAAb | 6 | - | –1.64 |
| COG T: Signal transduction mechanisms | ||||||
| RCC02849 | dorS | DMSO/TMAO-sensor hybrid histidine kinase | TTGATCGGGATCAAa | 26 | + | 4.53 |
| RCC02590 | dksA | DnaK suppressor protein | TTGATTCAGGTCAAb | 28 | + | 1.79 |
| COG X: Photosynthesis | ||||||
| RCC00667 | aerR | regulatory CrtJ antirepressor AerR | ATGCTCGAGTTCAAb | 8 | + | 1.39 |
| RCC00666 | bchF | 2-vinyl bacteriochlorophyllide hydratase | ATGACATGGGTCAAb | 8 | + | 1.39 |
Recognition sequences were determined using MEME server
aSequence is found within the coding region of the gene
bSequence is found in the upstream intergenic or promoter region
We determined which DEGs are directly controlled by FnrL by identifying FnrL binding sites in vivo using ChIP-seq analysis. Our ChIP-seq results provided near-complete representation of the entire genome with significant peaks called that exhibited a false discovery rate (FDR) cutoff of 5 % (corresponding to an unadjusted p value <1E-5) using the MACS package. In making our results comparable to datasets available for E. coli and Rba. sphaeroides, we present FDR values with a cutoff of 5 %. As shown in Additional file 1: Table S1 we identified 82 ChIP-seq peaks that were above this significance threshold. These peaks were found primarily within the intergenic regions where 47 ChIP sites (57 %) are enriched in promoter regions and of these 28 show a corresponding differential expression. Using chi-squared test it was determined that this exhibits statistical enrichment for promoters since intergenic regions only make up 9.19 % of Rba. capsulatus’ genome. Furthermore, we also identified peaks that were located within a gene next to neighboring genes that exhibited differential gene expression in the ΔfnrL strain (12 cases). We also found 34 called FnrL ChIP-seq peaks that did not exhibit an alteration in neighboring gene expression (Additional file 1: Table S1). It is difficult to reconcile the possibility that the latter category represents false positives on the basis of excellent enrichment coupled with a clear FnrL recognition sequence; rather, it may signal that FnrL bound to these location either has long range expression effects that are not being recognized or that additional auxiliary regulatory factors supersede the activity of FnrL. Furthermore, since only the photosynthetic state was investigated, these binding sites may be important in gene regulation during other growth states such as dark anaerobic or microaerobic growth or under nutrient limiting conditions.
A consensus FnrL recognition sequence was obtained using the MEME server from called ChIP-seq sites (Fig. 2). The derived sequence (T/C/A)TGA-N6-TCAA has second and third positions that were invariably TG while the 12th and 13th positions were invariably CA. The first position was somewhat variable with T, C, or A accounting for 37, 34 and 24 %, respectively, whereas the 14th position was an A at a frequency of 90 %. As shown in Fig. 2, the derived FnrL consensus sequence is highly similar to consensus sequences derived from similar studies from Rba. sphaeroides and E. coli. Variants of the Rba. capsulatus FnrL recognition sequence were identified by MEME in 69 out of 82 called ChIP-seq sites (Additional file 1: Table S1) with potential FnrL binding recognition sequences also manually found in ChIP peaks where no consensus sequence was identified by MEME. These manually identified potential recognition sequences are not listed in Additional file 1: Table S1 since flanking TTG/CAA sequences are common throughout the genome.
Fig. 2.
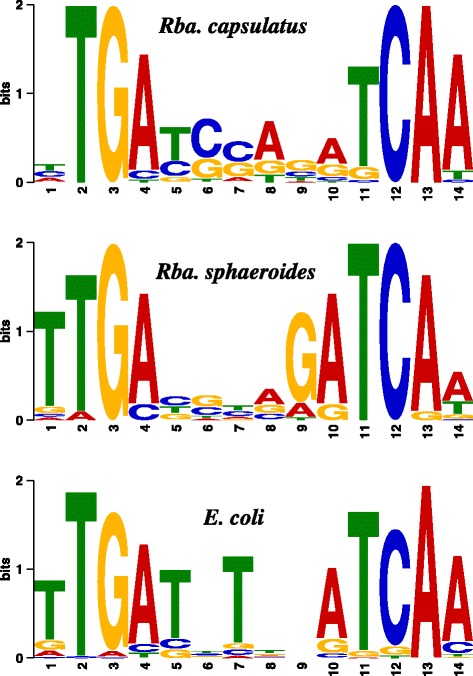
DNA binding motifs of FnrL/FNR orthologues. DNA consensus binding site was determined using MEME server from ChIP enriched sequences.
We also screened the Rba. capsulatus genome for additional FnrL sites with Virtual Footprint using FnrL recognition sequences identified from ChIP-seq peaks [19]. Our motivation for this stemmed from the fact that technical limitations exist that likely limit effective in vivo crosslinking of FnrL and/or immunoprecipitation of crosslinked DNA segments thus prohibiting our ability to identify all sites that are bound with FnrL. For example, we utilized formaldehyde as a crosslinker as it is typically used for ChIP-seq analysis. However, formaldehyde is known to form an ineffective adduct with B-form double stranded DNA and is thought to only be an effective crosslinker in cases where DNA binding proteins have perturbed or melted the DNA structure to allow formaldehyde to interact with the amine group of adenine [20]. Therefore, it is conceivable that FnrL bound to some sites may be ineffectively crosslinked with formaldehyde. Consequently the additional screening for potential FnrL sites using the MEME identified recognition sequences not surprisingly resulted in the identification of 332 additional potential FnrL recognition sites for a total of 414 possible sites in the genome. These additional sites were subsequently analyzed for their location relative to FnrL dependent differential gene expression. From this analysis, we were able to determine that an additional 77 genes are likely under direct control of FnrL as evidenced by the presence of a putative FnrL recognition site near a differentially expressed gene (Additional file 4: Table S4). Note that even thought some of these additional genes are likely directly regulated by FnrL they have remained in the “indirectly regulated” category (Additional file 2: Table S2) as it will require additional experimentation to determine which of genes are indeed under direct control by FnrL.
COG assignment of the FnrL regulon members
To address the role of members of the FnrL regulon in controlling anaerobic physiology, we placed individual genes into different “Clusters Of Orthologous Groups” (COGs) (as categorized in Additional file 5: Table S5. Inspection of the bar chart in Fig. 3 shows that the largest set of genes directly controlled by FnrL are in the category “Function Unknown”, which, accounts for 27 % of the genes in this regulon. This underscores that the role of many gene products in microbial physiology remain to be discovered. The largest COG categories that have a defined function are “Amino Acid Transport and Metabolism” and “Energy Production and Conversion”. These major COG categories highlight that FnrL has a role in controlling the energy metabolism of these cells. Another major category is “Signal transduction” of which more genes are repressed than activated. Signal transduction, along with the COG category “Transcription”, underscores that FnrL is an overarching global regulator that indirectly regulates a large number of genes.
Fig. 3.
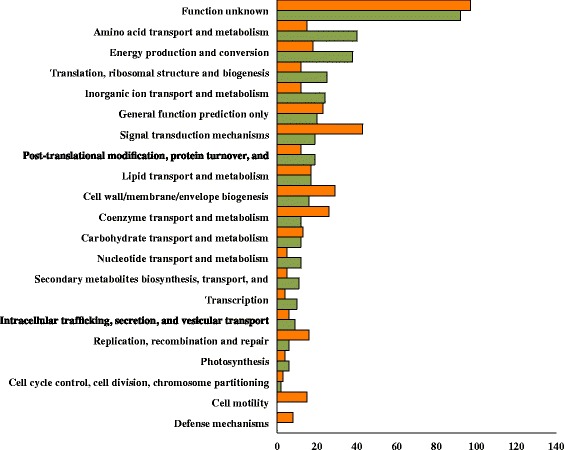
Rba. capsulatus genes clustered based on orthologous groups/functions. All FnrL directly and indirectly controlled genes clustered based on orthologous groups with orange representing repressed and green activated gene counts. COGs were determined using eggNOG server
FnrL regulates a variety of transcription factors and signal transduction components
Analysis of regulatory proteins that are directly regulated by FnrL shows that MerR (rcc03147) and TetR (rcc03059) transcription factor family members are directly repressed by FnrL (Additional file 4: Table S4). There is also a ChIP-seq identified FnrL binding site located directly upstream of a BadM/Rf2 family regulator (Additional file 1: Table S1). FnrL also directly regulates several two-component signal transduction components. For example, FnrL binds upstream of three sensor histidine kinases coded by rcc03452, rcc02198, and RegB2 (rcc01026). RegB2 is divergently transcribed from its cognate response regulator partner RegA2 so FnrL may control expression of both signaling components with the caveat that no affect of deleting FnrL was observed on RegB2 and RegA2 expression under the assayed growth conditions. The physiological role of RegB2, RegA2 is unknown, but they do share some degree of similarity (28 and 44 %) to RegB/RegA system, which is a well-characterized redox response system in Rba. capsulatus.
A two-component histidine kinase (rcc02198) is also a direct member of the FnrL regulon with its presumed cognate transcription response regulator (rcc02197) immediately upstream. These regulators are next to a propanediol gene cluster and may have a function in propanediol metabolism. The DNA binding site is located in the intergenic region of rcc02198-rcc02199 thus only rcc02198 is counted in the direct FnrL regulon. The ChIP-seq peak is located 185 bp upstream of the histidine kinase coding region with a corresponding 2-fold difference in transcription expression (Fig. 4c). We also observed that expression of DorS is induced 4-fold by FnrL with the presence of a ChIP-seq peak upstream of DorS, which is required for activation of the torCAD operon that codes for the DMSO/TMAO reductase system. It has been reported that a deletion of FnrL leads to a defect in utilizing DMSO as a terminal electron acceptor [16].
Fig. 4.
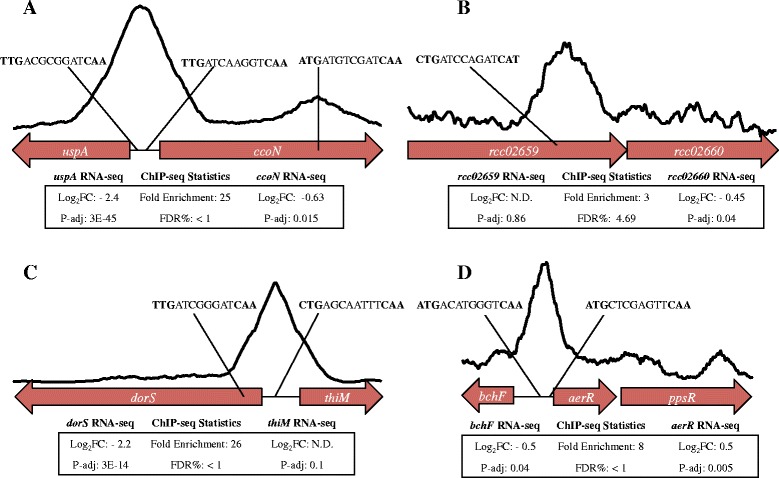
Selected ChIP and RNA-seq signal profile and statistics. Selected FnrL ChIP-seq signals of, A, cytochrome cbb 3 promoter region, B, ABC transporters rcc02659/rcc02660 with low enrichment but one with an FnrL binding site and a corresponding differential expression based on RNA-seq, C, promoter region of DMSO histidine kinase for DMSO reductase induction, D, bacteriochlorophyll biosynthesis bchF and CrtJ anti-repressor aerR for photosynthetic induction
Finally, FnrL also directly activates several genes that control synthesis and or hydrolysis of di-c-GMP (rcc02540, rcc01110 and rcc00783), which is often involved in regulating motility and biofilm biosynthesis suggesting that FnrL also has a role in controlling these processes [21].
FnrL is a direct controller of anaerobic respiration and photosynthesis
Cytochrome cbb3 (ccoNOQP) appears to be under direct control of FnrL. A ChIP-seq peak was found containing an FnrL binding sequence 100 bp upstream of the ccoN start codon and a second recognition site within the ccoN gene (Fig. 4a). RNA-Seq indicates that FnrL up-regulates expression of the ccoNOQP operon 1.5-fold under photosynthetic conditions. This is peculiar since this operon is repressed by several additional redox regulators such as by RegA [5, 7, 22]. One explanation might be that significant FnrL activation of the divergently transcribed neighbor uspA, overpowers FnrL repression of ccoNOQP. The second FnrL binding site located within the ccoN gene may be used for regulation of downstream cytochrome biogenesis proteins ccoGHIS since FnrL represses this second downstream operon. To this point, it is likely that the actual protein content of assembled cytochrome cbb3 is lower even with higher RNA transcription levels of ccoNOQP.
Even though the ΔfnrL strain is capable of photosynthetic growth, it appears that FnrL is directly involved in regulating photosynthesis in this species. This conclusion is supported by spectral analysis of anaerobically grown ΔfnrL mutant strain of Rba. capsulatus which exhibits a clear reduction in photosystem spectral components relative to that observed with wild type cells (Fig. 5). A mechanism for this reduction in pigment synthesis is revealed by the presence of an FnrL ChIP-seq peak containing a FnrL recognition sequence in the intergenic region between the divergently transcribed bacteriochlorophyll biosynthesis gene bchF and the bacteriochlorophyll regulator aerR (Fig. 4d). Two potential FnrL binding sites were identified within the bchF-aerR intergenic region with both sites exhibiting good similarity to the consensus sequence. AerR is a cobalamin binding anti-repressor of the bacteriochlorophyll/carotenoid/light harvesting repressor CrtJ and thus the 2-fold activation of AerR expression by FnrL would relieve repression by CrtJ (Fig. 6) [1]. Furthermore this RNA–seq data is validated by a previous in vivo expression study using lacZ reporter plasmids which showed that AerR expression increases 2-fold under anaerobic conditions [7, 23].
Fig. 5.
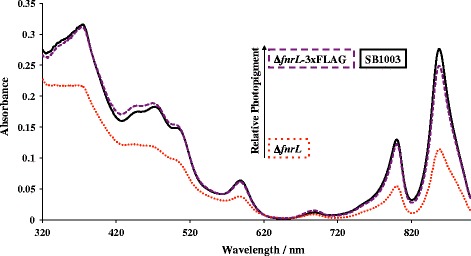
fnrL mutant in Rba. capsulatus show a reduction in photopigments. Photopigments from whole cell extracts of Rba. capsulatus shown in solid black, fnrL mutant in dotted red and fnrL mutant complemented with 3xFLAG tag shown in dashed purple
Fig. 6.
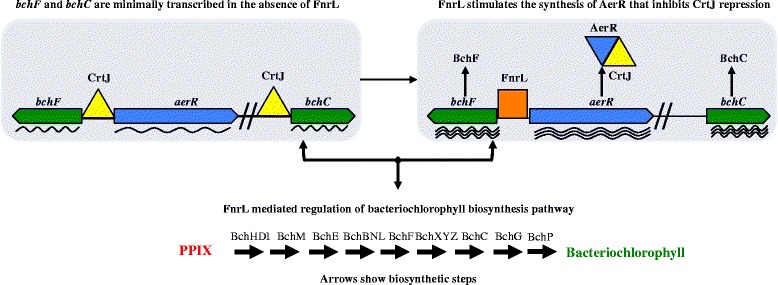
Schematic of Bacteriochlorophyll regulation. Repressor and antirepressor CrtJ/AerR system is shown as yellow/blue triangles, FnrL as an orange square, genes as chevrons and biosynthetic steps are shown as straight arrows. Wavy arrows indicate mRNA transcripts
We have also identified FnrL binding sites in the puc and puf light harvesting and reaction center operons (Additional file 1: Table S1). Specifically, there is a FnrL site that overlaps with the translational start site of pucA as well as a second site located 250 bp downstream of the start codon of pucC. The expression of pucB and pucDE up-regulated by FnrL indicating one or both of these sites may indeed be involved in activation of puc operon expression. There is also a ChIP-seq peak that spans the genetic space of pufLM with an FnrL binding sequence within pufM (42 bp upstream of the pufX start codon). RNA-sequencing show that pufLM is also up-regulated.
FnrL has a limited but suppressing role in motility
A number of flagellar, chemotaxis, aerotaxis and gas vesicle genes are either directly or indirectly repressed by FnrL (Additional files 1 and 2: Table S1 and S2). Many structural flagellar genes are located, in large part, in five operons. RNA-seq and ChIP-seq results indicate that FnrL directly represses a 5-gene operon (rcc03522- rcc03525) that codes for an unknown function flagellar protein, FlbT, FlaF, and FlaA (flagellin protein needed for synthesis of the flagella filament). A ChIP-seq peak was observed that spans this operon with a consensus FnrL binding site located 42 bp upstream of the FlbT start codon (Table 1).
In addition to flagellar structural proteins, FnrL also represses cheA1 that codes for chemotaxis signal transduction protein, a number of methyl-accepting chemotaxis receptors (rcc00644, rcc02611 rcc02887, rcc02139, and rcc01667), two aerotaxis receptors (rcc02075 and rcc03176) and several gas vesicle proteins (rcc01054 and rcc01056) (Table 1, Additional files 1 and 2: Table S1, and S2). One possible explanation for FnrL repression of motility may be that there is selective pressure to suppress motility under anaerobic photosynthetic growth conditions where light driven energy production is not limiting. Under photosynthetic growth conditions these metabolically diverse cells are very capable of directly synthesizing all essential cellular metabolites and likely not as reliant on chemotaxis. Repression of these motility components by FnrL would be relieved in the presence of oxygen that would disrupt the DNA binding activity of FnrL. This would allow the cell to synthesize components needed to either aerotax to areas with increasing oxygen content or increase their buoyancy so that they can rapidly “float” in an aquatic environment towards an oxygen source.
FnrL’s role in anaerobic carbon metabolism
FnrL is not directly involved in glycolysis or gluconeogenesis; however, there are two of steps in glycolysis/gluconeogenesis that are indirectly activated such as phosphopyruvate hydratase (rcc01715) and glyceraldehyde-3-phosphate dehydrogenases (rcc02160). However, tricarboxylic acid cycle (TCA) genes are directly activated by FnrL (also called the Krebs cycle and the citric acid cycle) (Additional file 2: Table S2). Of the TCA genes, succinate dehydrogenase is directly activated by FnrL and contains a consensus binding sequence 26 bp upstream of the sdhD start codon and within sdhC coding region. Succinate dehydrogenase in turn provides reducing power to ubiquinone to drive cytochrome bc1 (petABC) complex that is indirectly activated (Fig. 7).
Fig. 7.
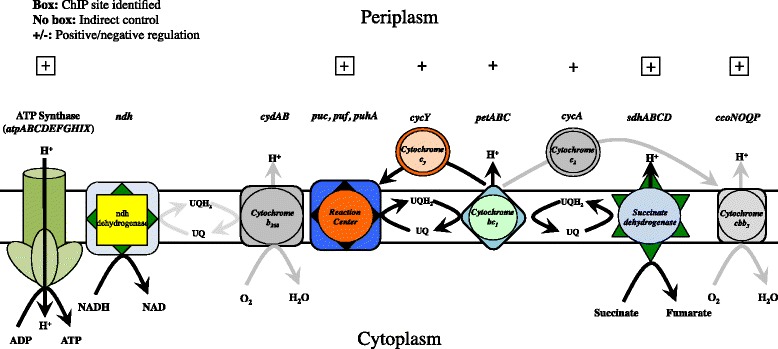
Schematic of Rba. capsulatus respiration. Boxes represent direct control by FnrL and activation/repression is shown as +/−
Rba. capsulatus contains two forms of RuBisCO where form I is coded by cbbLS and form II is coded by cbbM. Form I and II cbb operons are regulated by related LysR family transcription factors CbbRI and CbbRII, respectively. FnrL does not control these regulators, but deletion of fnrL causes the, expression of cbbLS to be reduced.
Regulation of tetrapyrrole biosynthesis and iron transport by FnrL
The common trunk of the tetrapyrrole pathway from δ-aminolevulinic acid to uroporphyrinogen III is used for cobalamin, heme and bacteriochlorophyll biosynthesis [5, 7, 24]. There is indirect activation of hemA expression (Additional file 2: Table S2) with possible direct activation of ferrochelatase (hemH) expression with a predicted FnrL binding site that shows good similarity to the FnrL consensus recognition sequence. While there is no detectable FnrL binding site in the intergenic region between divergently transcribed hemB and rcc01809 genes, there is a ChIP-seq peak with an FnrL recognition sequence located within rcc01809. This suggests that the promoter for hemB may be within the rcc01809 coding sequence. Interestingly, FnrL has an indirect role in repressing cobalamin (cob gene) synthesis (Additional file 2: Table S2). We hypothesize that the cell attenuates cobalamin biosynthesis in order to divert intermediates for the biosynthesis of PPIX and bacteriochlorophyll (unpublished observation).
We did not find any direct regulation of FnrL on siderophore or iron transport genes. Iron is an essential component of heme as well as the redox responding cofactor in FnrL and we were surprised to find a limited direct role of FnrL in iron transport. We did observe that FnrL does indirectly repress a siderophore ABC transporter (rcc02116), a FeoA family protein (rcc02028), a Fe(III) type ABC transporter (rcc02579) and FeoA2 that codes for a ferrous iron transporter (rcc00091) (Additional file 2: Table S2). One of the highest enriched (21-fold) sites was found in one uncharacterized set of genes (rcc3401-rcc3402) the first of which is a band 7/SPFH family protein thought to be the core of an ion channel while the second is a hypothetical protein that shares 24 % identity to a membrane protease found to be important for virulence in P. gingivalis W83 [25]. These two genes are typically found in an operon and appear to form the foundation of an ion channel. The role of this gene cluster is unclear in Rba. capsulatus, but it may be used for acquiring or sensing depleting ions including iron. Indeed it has been found that a knockout of homologous gene cluster in S. oneidensis shows a strong effect on iron metabolism with the disruption leading to a decrease in intracellular iron which affected proteins involved in respiratory chain that utilize iron [26].
Comparison of FNR/FnrL differentially expressed genes in Rba. capsulatus, Rba. sphaeroides, and E. coli
The number of genes that encompass the Rba. capsulatus FnrL regulon (807 genes) is similar to the number of genes reported for the Rba. sphaeroides FnrL regulon (917 genes) [6, 18]. However, analysis for congruence shows that only 171 genes are differentially expressed in common (Tables 2 and 3 in Additional file 6: Table S6). This means that 78 and 81 % of the genes in the Rba. capsulatus and Rba. sphaeroides FnrL regulons, respectively, are uniquely regulated by FnrL in these photosynthetic species [18]. Among the 171 commonly regulated genes, 52 are convergently activated and 36 are convergently repressed with 83 exhibiting differences in regards to activation versus repression. Divergent roles of FnrL in these species is also highlighted by the fact that only 9 FnrL ChIP-seq peaks are located in common positions relative to a common downstream gene out of the 82 FnrL peaks in Rba. capsulatus and 28 FnrL peaks in Rba. sphaeroides (Additional file 7: Table S7).
Table 2.
Comparison of selected genes directly controlled by FnrL in Rba. capsulatus and Rba. sphaeroides
| Locus IDa | Locus IDb | Gene Name | Function | Regulationc | FnrL Recognition Sequence |
|---|---|---|---|---|---|
| Unique to Rhodobacter sphaeroides | |||||
| RSP0820 | RCC00426 | cytochrome b 561 | electron transporting and shutting | + | TTGATGCGGATCAA |
| RSP2984 | RCC00147 | hemA | 5-aminolevulinate synthase | + | TTGATAAGGATCAA |
| RSP0317 | RCC00151 | hemN | Coproporphyrinogen III oxidase | + | TTGCGCAGGATCAA |
| RSP1819 | RCC00091 | feoA | ferrous iron transport protein | + | TTGACGCGGATCAA |
| RSP1949 | RCC01601 | Iron/Sulfur | FeS assembly SUF system protein | + | GTGATCTGCATCAA |
| RSP0100 | RCC01517 | nuoA | NADH dehydrogenase | + | CTGATGCAGATCAA |
| Unique to Rhodobacter capsulatus | |||||
| RSP0278 | RCC02532 | pucC | light harvesting protein | + | CTGATCGGCTTCAA |
| RSP0284 | RCC00666 | bchF | 2-vinyl bacteriochlorophyllide hydratase | + | ATGACATGGGTCAA |
| RSP0283 | RCC00667 | aerR/ppaA | regulatory protein PpaA | + | ATGCTCGAGTTCAA |
| RSP1149 | RCC01729 | oxidoreductase | oxidoreductase | + | ATGATCCAAGTCAT |
| Directly activated in both organisms | |||||
| RSP0775 | RCC02479 | cytochrome c | peroxidase | + | CTGATGCAGCGCAA |
| RSP Recognition sequence | TTGACGCAGATCAG | ||||
| RSP0696 | RCC01157 | ccoN | cbb3-type cytochrome c oxidase subunit I | + | TTGATCAAGGTCAA |
| RSP Recognition sequence | TTGATCCTCATCAA | ||||
| RSP3044 | RCC02849 | dorS | DMSO/TMAO-sensor hybrid histidine kinase | + | TTGATCGGGATCAA |
| RSP Recognition sequence | TTGACGTCAATCAA | ||||
| RSP0166 | RCC02590 | dksA2 | DnaK suppressor protein | + | TTGATTCAGGTCAA |
| RSP Recognition sequence | TTGATGCAGGTCAA | ||||
| RSP2247 | RCC01495 | fusA | translation elongation factor G | + | TTGGCATGGGTCAA |
| RSP Recognition sequence | TTGATTCAGGTCAA | ||||
| RSP0697 | RCC01156 | uspA | universal heath shock protein | + | TTGACGCGGATCAA |
| RSP Recognition sequence | TTGATCCATGTCAA | ||||
| RSP2337 | RCC01723 | ccpA1 | cytochrome-c peroxidase | + | TTGATCTGCGTCAA |
| RSP Recognition sequence | TTGATCTGCGTCAT | ||||
| RSP0698 | RCC02493 | fnrL | CRP/FNR family transcriptional regulator | + | TTGTCCCAAATCAA |
| RSP Recognition sequence | TTGATTCAGATCAA | ||||
| RSP0467 | RCC01733 | ubiD | 3-octaprenyl-4-hydroxybenzoate carboxy-lyase | N.D. | TTGATCAATATCAA |
| RSP Recognition sequence | TTGATGTAGGTCAA | ||||
N.D No differential gene expression based on RNA-seq (Not Determined)
RSP Recognition sequence Rba. sphaeroides recognition sequence
a Rba. sphaeroides
b Rba. capsulatus
c(+/-) indicate activation/repression by FnrL
The large number of uniquely regulated genes in these two Rhodobacter species indicates that FnrL has adopted dissimilar regulatory roles. This conclusion is highlighted by divergent roles of FnrL in regards to the regulation of tetrapyrrole biosynthesis and photosystems. For example, FnrL directly activates hemA in Rba. sphaeroides but not in Rba. capsulatus. Bacteriochlorophyll genes bchM, bchJ, bchO, and bchD are also convergently repressed by both species while bchC, bchE and bchF are activated in Rba. capsulatus and repressed in Rba. sphaeroides. Furthermore, an FnrL ChIP signal is observed in the light harvesting complex pufALM operon from Rba. capsulatus which is positively regulated by FnrL, but not in Rba. sphaeroides where this operon appears to be negatively regulated by FnrL [18]. This difference also extends to downstream secondary photosystem regulators. Specifically, we found an FnrL ChIP signal in the Rba. capsulatus promoter region of AerR which is a photosystem regulator that functions as an antirepressor of the bch/crt repressor CrtJ [1–5]. In Rba. sphaeroides the control of this downstream regulator by FnrL does not appear to exist [18]. These differences signal that there is significant variation in the role of FnrL for the control of photosystem synthesis between these species.
Some notable similarities do, however, exist between these Rhodobacter species. For example, FnrL directly ctivates DMSO reductase and cbb3 cytochrome oxidases and has direct negative effects on cobalamin biosynthesis in both of these species (Table 2). Furthermore, both organisms use FnrL to indirectly activate cbbLS (Calvin-Benson-Bassham cycle).
Searching for convergence of FnrL/FNR regulons across genera we observed that there is only a handful of examples where the E. coli FNR regulon shows congruence with either of the Rhodobacter regulons. For example, the DMSO reductase system and uspA (universal stress protein) is directly activated by FnrL/FNR in all three species (Additional files 8, 9 and 10: Tables S8, S9, S10) [6]. Similarly, the fadBA (fatty acid metabolism) operon is repressed in all three species though in all cases this repression appears to be indirect. The E. coli and Rba. capsulatus FNR/FnrL orthologues also directly control nrdD (anaerobic ribonucleoside reductase) but this does not appear to be the case in Rba. sphaeroides. These results clearly demonstrate that there exist considerable divergence in function of FNR/FnrL orthologues from distant and more closely related bacteria.
Conclusions
This study shows that genes constituting the FnrL regulon from Rba. capsulatus are remarkably dissimilar from the published FnrL regulon from Rba. sphaeroides. Indeed only 9 genes in these two photosynthetic species have FnrL binding sites upstream from common targets. This dissimilarity is striking given that these organisms share similar anoxygenic photosynthetic physiologies and therefore presumably face similar challenges in controlling energy balance (redox poise) in response to light, oxygen, and nutrient availability. The fact that these FnrL orthologues exhibit high sequence identity (Fig. 8) and utilize similar target sequences (Fig. 2), and yet control many different target genes, indicates that there is significant evolutionary drift in the location of transcription factor recognition sequences even among related species that occupy similar environmental niches (Fig. 9).
Fig. 8.
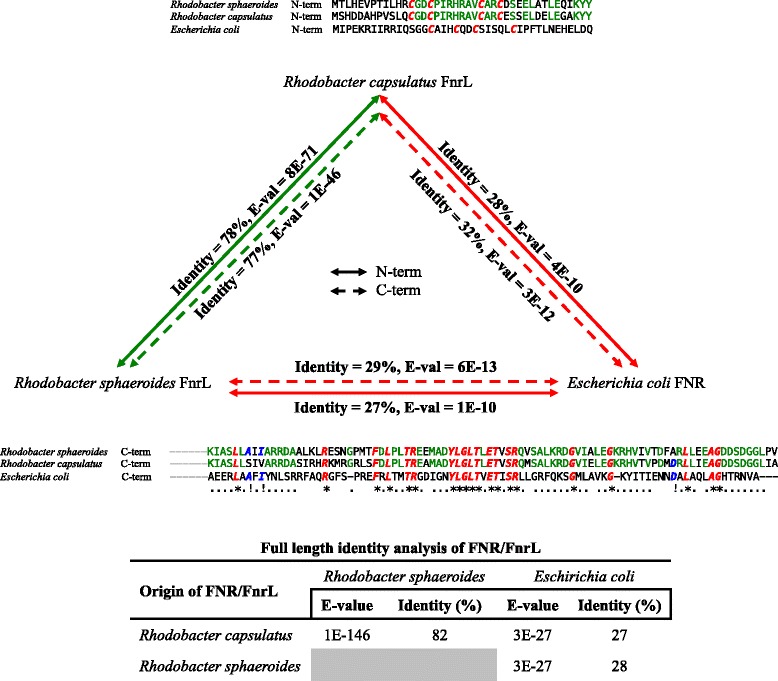
Comparison of FNR/FnrL homologues. Similarities of FnrL from Rba. capsulatus and Rba. sphaeroides and their differences to FNR from E. coli. Fe/S motif was taken from the N-terminus (solid) and HTH domain was taken from the C-terminus (dashed). Red colored amino acids denote critical residues, green denote similarities between Rhodobacter species, blue denote similarities between E. coli and Rhodobacter species, grey are unique to each organism and redundantly represented by ‘.’, ‘*’ and ‘!’
Fig. 9.

Phylogenetic relatedness of Rhodobacter species. The evolutionary history was inferred using the Neighbor-Joining method [42]. The bootstrap consensus tree inferred from 1000 replicates [43] is taken to represent the evolutionary history of the taxa analyzed [43]. The evolutionary distances were computed using the Maximum Composite Likelihood method [44] and are in the units of the number of base substitutions per site. The analysis involved 19 16S rRNA sequences. All positions containing gaps and missing data were eliminated. There were a total of 1325 positions in the final dataset. Evolutionary analyses were conducted in MEGA6 [45].
It is informative to note similarities and differences that exist between these Rhodobacter FnrL regulons as this can highlight areas of conservation that may apply to a broad spectrum of alpha-proteobacteria. For example, iron transport is controlled by FnrL in Rba. sphaeroides but not in Rba. capsulatus (Table 2) [27, 28]. Differences also exist for heme synthesis where FnrL from Rba. sphaeroides directly controls hemA, hemN and hemZ while FnrL in Rba. capsulatus is not directly involved in heme biosynthesis with the possible exception of hemH. We also note that numerous cobalamin biosynthesis genes are indirectly down-regulated by FnrL in both Rhodobacter species. This may not be an intuitive result since cobalamin is needed for anaerobic biosynthesis of bacteriochlorophyll where BchE uses cobalamin as its cofactor [29]. However, both Rhodobacter species undergo an extensive increase in bacteriochlorophyll biosynthesis (>100-fold) when they are grown anaerobically and yet both species show FnrL mediated repression of the cobalamin pathway.
In regards to the FNR regulon from E. coli [6], this species does not possess the ability to undergo photosynthesis and anaerobically relies on fermentative growth. Consequently, member of the E. coli FNR regulon are quite divergent from that of the Rba. capsulatus and Rba. sphaeroides FnrL regulons. Indeed despite the large number of genes that constitute the FNR/FnrL regulons from these species, we only found a few instances where all three organisms have direct orthologues that share the same direct FNR/FnrL control; the DMSO reductase system and the universal stress protein uspA. Although all three species do not share direct cytochrome oxidase orthologues, all three organisms do use FnrL/FNR to control the expression of oxygen utilizing terminal respiratory chain components [13, 16, 30, 31].
Finally, an example of metabolic divergence of E. coli from Rhodobacter species is highlighted by the direct involvement of E. coli FNR in regulating glycolysis while in the Rhodobacter species FnrL is not directly involved. Logically, in a non-photosynthetic organism such as E. coli it makes sense to direct phosphoenolpyruvate for either aerobic or anaerobic growth by an oxygen sensing transcriptional factor while it appears that both Rhodobacter species have adopted alternate modes of glycolytic routing mechanisms [6]. FnrL’s from Rba. capsulatus and Rba. sphaeroides are also indirectly involved in cobalamin repression while E. coli does not undertake de novo cobalamin biosynthesis and instead must go through a cobinamide intermediate [32].
The divergences observed with the FnrL/FNR regulons from Rba. capsulatus, Rba. sphaeroides and E. coli highlights the fact that analysis of transcription factor regulons must be experimentally derived on an individual basis as corollary regulatory events clearly differ between closely related organisms. This divergence can occur even among highly homologous transcription factor orthologs that bind to similar recognition sequences.
Methods
Strains, media, and growth conditions
The Rba. capsulatus parental strain SB1003, and its ΔfnrL derivative have previously been described [16]. These strains were routinely grown in peptone/yeast extract (PY) either in liquid or on agar plates with liquid media supplemented with MgCl2 and MgSO4 to a final concentration of 2 mM. Biological replicate strains were first grown semi-aerobically overnight as a 5 ml PY culture in culture tubes at 34 °C shaking at 200 rpm. Subsequently, these cultures were transferred and grown anaerobically in screw-cap vials overnight at 34 °C with four 75 W light bulbs after which the cells were subcultured to an optical density of 0.03 and spectrally monitored until harvesting at OD660 ~ 0.3. The optical density in the anaerobic vials was checked using Unico 1100 RS Spectrophotometer.
RNA isolation, validation, and sequencing (RNA-Seq)
After cultures reached OD660 ~ 0.3 the cultures were harvested by placing immediately into an ice/water bath and then transferred into 2 mL Eppendorf tubes, centrifuged at 6000 rpm for 3 min at 4 °C. The entire 2 mL cell pellet was then used for extracting total RNA using a Bioline Isolate II RNA extraction kit. Briefly, the bacterial pellet was dissolved in 100 μL of TE (10 mM Tris–HCl, 1 mM EDTA, pH 8) buffer containing 10 mg/mL lysozyme and incubated for 3 min at room temperature. After isolation of total RNA the DNA was removed by addition of 1 unit of Turbo DNAse and further incubated for 30 min at 37 °C. A cleanup step was performed with Zymogen Direct-zol RNA extraction kit according to manufacturers instructions. To check for residual DNA, qRT-PCR of the rpoZ housekeeping gene was performed with and without reverse transcriptase.
Total RNA was submitted to the University of Wisconsin-Madison Biotechnology Center where it was verified for purity and integrity with a NanoDrop2000 Spectrophotometer and Agilent 2100 BioAnalyzer, respectively. Samples that met Illumina sample input guidelines were prepared according the TruSeq® Stranded Total RNA Sample Preparation Guide (15031048 E) using the Illumina TruSeq® Stranded Total RNA kit (Illumina Inc., San Diego, California, USA) with minor modifications. For each library preparation, 2 μg of total RNA was reduced of ribosomal RNA using the EpiCentre RiboZero™ rRNA Removal (Bacteria) kit (EpiCentre Inc., Madison, WI, USA) as directed. Subsequently, each rRNA-depleted sample was fragmented using divalent cations under elevated temperature. The fragmented RNA was synthesized into first strand cDNA using SuperScript II Reverse Transcriptase (Invitrogen, Carlsbad, California, USA) combined with Actinomycin D and random primers followed by second strand synthesis using Second Strand Marking Master Mix. The blunt-ended double-stranded cDNA was purified by paramagnetic beads (Agencourt AMPure XP beads (Beckman Coulter, Indianapolis IN, USA). The cDNA products were incubated with A-Tailing Mix to add an ‘A’ base (Adenine) to the 3′ end of the blunt DNA fragments followed by ligation to Illumina adapters, which have a single ‘T’ base (Thymine) overhang at their 3′end. The adapter-ligated products were purified by paramagnetic beads. Adapter ligated DNA was then amplified in a Linker Mediated PCR reaction (LM-PCR) for 10 cycles using the PCR Master Mix and PCR Primer Cocktail and purified by paramagnetic beads. Quality and quantity of the finished libraries were assessed using an Agilent DNA1000 chip (Agilent Technologies, Inc., Santa Clara, CA, USA) and Qubit® dsDNA HS Assay Kit (Invitrogen, Carlsbad, California, USA), respectively and standardized to 2 μM. Cluster generation was performed using standard Cluster Kits (v3) and the Illumina Cluster Station. Single 100 bp sequencing was performed, using standard SBS chemistry (v3) on an Illumina HiSeq2000 sequencer. Images were analyzed using the standard Illumina Pipeline, version 1.8.2.
Construction and sequencing of ChIP libraries (ChIP-Seq)
A plasmid expressing a FnrL 3xFLAG Tag with an isopropyl β-D-1-thiogalactopyranoside (IPTG) inducible lac promoter was constructed with the following reverse primer ctaGCTAGCttaCTTGTCATCGTCATCCTTGTAGTCGATGTCATGATCTTTATAATCACCGTCATGGTCTTTGTAGTCggatc containing NheI restricted site and forward primer acatGCATGCGGTTCATCCCCGATTGCGCCAG containing SphI restriction site and cloned into pSRK (complementation plasmid containing gentamycin resistance marker to produce pSRK-FnrL. This expression plasmid is described in detail in the following reference [33]. pSRK-FnrL was subsequently mated into Rba. capsulatus using S17-1 E. coli mating strain with complementation checked by growing cells anaerobically with 50 mM DMSO in the presence of 1.0 mM IPTG. FnrL mutants fail to utilize DMSO as a terminal electron acceptor due to their inability to express sufficient amounts of DMSO reductase [16] and also have reduced levels of photopigments (Fig. 5). The FnrL deletion strain complemented with pSRK-FnrL was subsequently able to restore growth on DMSO and to resort wild type photopigment levels (Fig. 5) identical to that of wild type cells.
Photosynthetically grown FnrL-3xFLAG complemented cells were treated with 37 % formaldehyde to a final concentration of 1 % for 15 min at room temperature. Crosslinking with formaldehyde quenched by the addition of Tris–HCl pH 8.2 to a final concentration of 500 mM for 5 min at room temperature after which the cells were harvested by centrifugation. The cells were washed with 40 mL TBS buffer and resuspended in 4 mL buffer composed of 50 mM Tris pH 7.5, 150 mM NaCl, 1 mM EDTA, 1 % Triton X100. After disruption by French press lysis, the DNA was sheared three times by sonication using a small tip sonicator with 15-W power output. Protein bound to DNA was then reverse crosslinked by heating to 65 °C overnight with concurrent removal of contaminating RNA by the addition of 1 μg of RNAse A per 100 μL sample. Immunoprecipitation was performed according to manufacturers instruction using ANTI-FALG® M2 Affinity Gel (Cat. Number A2220).
Purified immunoprecipitated and input DNA was submitted to the University of Wisconsin-Madison Biotechnology Center for library construction and sequence analysis. DNA concentration and sizing were verified using the Qubit® dsDNA HS Assay Kit (Invitrogen, Carlsbad, California, USA) and Agilent DNAHS chip (Agilent Technologies, Inc., Santa Clara, CA, USA), respectively. Samples that met the Illumina sample input guidelines were prepared according the TruSeq® ChIP Sample Preparation kit (Illumina Inc., San Diego, California, USA) with minor modifications. Libraries were size selected for an average size of 350 bp using SPRI-based bead selection. Quality and quantity of the finished libraries were assessed using an Agilent DNA1000 chip and Qubit® dsDNA HS Assay Kit, respectively with DNA concentration standardized to 2 μM. Cluster generation was performed using standard Cluster Kits (v3) and the Illumina Cluster Station. Single 100 bp sequencing was performed, using standard SBS chemistry (v3) on an Illumina HiSeq2000 sequencer. Images were analyzed using the standard Illumina Pipeline, version 1.8.2.
Data pre-processing, computer software and data analysis for RNA-sequencing and ChIP-sequencing
All computations were performed on a custom built computer running Ubuntu 13.10 equipped with Asus Z9PE-D8 WS motherboard, 2 x Intel Xeon E5-2630 V2 CPU, 128 GB DDR3-1600 RAM. Each fastq file was checked for good quality using FastQC and trimmed of low quality sequences using Trimmomatic program using a sliding window of 5:25 and a minimum length of 40. The reads were aligned to the genome using Bowtie2 [34] mapped individual genes using HTSeq-count [35]. Raw counts generated from HTSeq-count program were used to generate differentially expressed genes with DESeq2 package in R [36, 37]. Default parameters with noted exceptions were used for Trimmomatic, Bowtie2 and HTSeq-count programs.
For processing ChIP-seq, a pipeline consisting of Trimmomatic with a sliding window of 5:25 and a minimum length of 40 was used to trim poor quality reads, Bowtie2 to align the reads to the SB1003 reference genome, MACS to determine significantly enriched sites, and MEME for binding sequence extraction using default parameters [38]. All packages are available for download via github and/or bioconductor [33–35, 38–40]. Raw sequence data from our RNA-seq and ChIP-seq analysis can be accessed via NCBI Sequence Read Archive server under the accession number (PRJNA274121).
Cross-species orthologous analysis
Orthologues of Rba. capsulatus in Rba. sphaeroides and E. coli were found using OMA web server accessible at http://http://www.omabrowser.org [41]. Data sets for Rba. sphaeroides and E. coli used for differential gene expression comparison were taken directly from published results and presented in congruent style [6, 18].
Spectral scans of SB1003 and FnrL
Wild-type Rba. capsulatus, fnrL mutant and fnrL mutant complemented with 3xFLAG tag were grown in PY medium until OD660 nm reached 0.3 and 2 mL of each genotype was harvested and centrifuged to collect the pellet. The pellet was dissolved in buffer containing 20 mM Tris and 150 mM NaCl and sonicated three times using power of 9 W for duration of 15 s. The samples were clarified by centrifugation and the spectra were recorded.
Acknowledgements
We thank members of the Bauer research group for helpful discussions on RNA-Seq and ChIP-Seq data analysis and the University of Wisconsin-Madison Biotechnology Center for library construction and sequence analysis. This study was funded by a National Institutes of Health grant GM 040941 awarded to CEB.
Additional files
Genes containing ChIP-seq signal with corresponding RNA-seq expression. (XLSX 40 kb)
A table of genes indirectly controlled by FnrL in Rba. capsulatus. (DOCX 228 kb)
FnrL ChIP-seq signal with corresponding p-value > 0.05 based on RNA-seq expression change. (DOCX 66 kb)
FnrL predicted binding site based on PWM with corresponding RNA-seq expression change. (DOCX 165 kb)
Clusters of orthologous groups definitions. (DOCX 66 kb)
Orthologues of Rba. capsulatus in Rba. sphaeroides and their differential expression. (XLSX 280 kb)
Orthologues of Rba. capsulatus in Rba. sphaeroides and presence of FnrL ChIP signals. (XLSX 67 kb)
Orthologues of Rba. capsulatus in E. coli and their differential expression. (XLSX 85 kb)
Orthologues of Rba. capsulatus in E. coli and presence of FnrL/FNR ChIP signals. (XLSX 57 kb)
Orthologues of Rba.spaeroides in E. coli and their differential expression. (XLSX 63 kb)
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
Conceived and designed the experiments: YK, CB. Performed the experiments: YK. Analyzed the data: YK, CB. Contributed reagents/materials/analysis tools: YK. Wrote the paper: YK, CB. Provided funding and space: YK. Both authors read and approved the final manuscript.
References
- 1.Cheng Z, Li K, Hammad LA, Karty JA, Bauer CE. Vitamin B12 regulates photosystem gene expression via the CrtJ antirepressor AerR in Rhodobacter capsulatus. Mol Microbiol. 2014;91(4):649–64. doi: 10.1111/mmi.12491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Smart JL, Willett JW, Bauer CE. Regulation of hem gene expression in Rhodobacter capsulatus by redox and photosystem regulators RegA, CrtJ, FnrL, and AerR. J Mol Biol. 2004;342(4):1171–86. doi: 10.1016/j.jmb.2004.08.007. [DOI] [PubMed] [Google Scholar]
- 3.Elsen S, Swem LR, Swem DL, Bauer CE. RegB/RegA, a highly conserved redox-responding global two-component regulatory system. Microbiol Mol Biol Rev. 2004;68(2):263–79. doi: 10.1128/MMBR.68.2.263-279.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wu J, Bauer CE. RegB/RegA, a global redox-responding two-component system. Bacterial Signal Transduction: Networks and Drug Targets. 2008;631:131–48. doi: 10.1007/978-0-387-78885-2_9. [DOI] [PubMed] [Google Scholar]
- 5.Yin L, Bauer CE. Controlling the delicate balance of tetrapyrrole biosynthesis. Philos Trans R Soc Lond B Biol Sci. 2013;368(1622):20120262. doi: 10.1098/rstb.2012.0262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Myers KS, Yan H, Ong IM, Chung D, Liang K, Tran F, et al. Genome-scale analysis of Escherichia coli FNR reveals complex features of transcription factor binding. PLoS Genet. 2013;9(6):e1003565. doi: 10.1371/journal.pgen.1003565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Swem DL, Bauer CE. Coordination of ubiquinol oxidase and cytochrome cbb3 oxidase expression by multiple regulators in Rhodobacter capsulatus. J Bacteriol. 2002;184(10):2815–20. doi: 10.1128/JB.184.10.2815-2820.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Khoroshilova N, Popescu C, Munck E, Beinert H, Kiley PJ. Iron-sulfur cluster disassembly in the FNR protein of Escherichia coli by O2: [4Fe-4S] to [2Fe-2S] conversion with loss of biological activity. Proc Natl Acad Sci U S A. 1997;94(12):6087–92. doi: 10.1073/pnas.94.12.6087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Spiro S. The FNR, family of transcriptional regulators. Antonie Van Leeuwenhoek. 1994;66(1–3):23–36. doi: 10.1007/BF00871630. [DOI] [PubMed] [Google Scholar]
- 10.Fleischhacker AS, Kiley PJ. Iron-containing transcription factors and their roles as sensors. Curr Opin Chem Biol. 2011;15(2):335–41. doi: 10.1016/j.cbpa.2011.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kiley PJ, Beinert H. Oxygen sensing by the global regulator, FNR: the role of the iron-sulfur cluster. FEMS Microbiol Rev. 1998;22(5):341–52. doi: 10.1111/j.1574-6976.1998.tb00375.x. [DOI] [PubMed] [Google Scholar]
- 12.Peuser V, Remes B, Klug G. Role of the Irr protein in the regulation of iron metabolism in Rhodobacter sphaeroides. PLoS One. 2012;7(8):e42231. doi: 10.1371/journal.pone.0042231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zannoni D, Schoepp-Cothenet B, Hosler J. Respiration and Respiratory Complexes. In: Hunter CN, Daldal F, Thurnauer MC, Beatty JT, editors. The Purple Photosynthetic Bacteria. Dordrecht: Springer Netherlands; 2009. pp. 537–61. [Google Scholar]
- 14.Dame RT. The role of nucleoid-associated proteins in the organization and compaction of bacterial chromatin. Mol Microbiol. 2005;56(4):858–70. doi: 10.1111/j.1365-2958.2005.04598.x. [DOI] [PubMed] [Google Scholar]
- 15.Zeilstra-Ryalls JH, Kaplan S. Aerobic and anaerobic regulation in Rhodobacter sphaeroides 2.4.1: the role of the fnrL gene. J Bacteriol. 1995;177(22):6422–31. doi: 10.1128/jb.177.22.6422-6431.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zeilstra-Ryalls JH, Gabbert K, Mouncey NJ, Kaplan S, Kranz RG. Analysis of the fnrL gene and its function in Rhodobacter capsulatus. J Bacteriol. 1997;179(23):7264–73. doi: 10.1128/jb.179.23.7264-7273.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zeilstra-Ryalls JH, Kaplan S. Role of the fnrL gene in photosystem gene expression and photosynthetic growth of Rhodobacter sphaeroides 2.4.1. J Bacteriol. 1998;180(6):1496–503. doi: 10.1128/jb.180.6.1496-1503.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Imam S, Noguera DR, Donohue TJ. Global analysis of photosynthesis transcriptional regulatory networks. PLoS Genet. 2014;10(12):e1004837. doi: 10.1371/journal.pgen.1004837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Munch R, Hiller K, Grote A, Scheer M, Klein J, Schobert M, et al. Virtual Footprint and PRODORIC: an integrative framework for regulon prediction in prokaryotes. Bioinformatics. 2005;21(22):4187–9. doi: 10.1093/bioinformatics/bti635. [DOI] [PubMed] [Google Scholar]
- 20.McGhee JD, von Hippel PH. Formaldehyde as a probe of DNA structure. r. Mechanism of the initial reaction of Formaldehyde with DNA. Biochemistry. 1977;16(15):3276–93. doi: 10.1021/bi00634a002. [DOI] [PubMed] [Google Scholar]
- 21.Boyd CD, O’Toole GA. Second messenger regulation of biofilm formation: breakthroughs in understanding c-di-GMP effector systems. Annu Rev Cell Dev Biol. 2012;28:439–62. doi: 10.1146/annurev-cellbio-101011-155705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Swem LR, Elsen S, Bird TH, Swem DL, Koch HG, Myllykallio H, et al. The RegB/RegA two-component regulatory system controls synthesis of photosynthesis and respiratory electron transfer components in Rhodobacter capsulatus. J Mol Biol. 2001;309(1):121–38. doi: 10.1006/jmbi.2001.4652. [DOI] [PubMed] [Google Scholar]
- 23.Dong C, Elsen S, Swem LR, Bauer CE. AerR, a second aerobic repressor of photosynthesis gene expression in Rhodobacter capsulatus. J Bacteriol. 2002;184(10):2805–14. doi: 10.1128/JB.184.10.2805-2814.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zappa S, Li K, Bauer CE. The tetrapyrrole biosynthetic pathway and its regulation in Rhodobacter capsulatus. Adv Exp Med Biol. 2010;675:229–50. doi: 10.1007/978-1-4419-1528-3_13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Walters S, Rodrigues P, Belanger M, Whitlock J, Progulske-Fox A. Analysis of a band 7/MEC-2 family gene of Porphyromonas gingivalis. J Dent Res. 2009;88(1):34–8. doi: 10.1177/0022034508328381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gao WM, Liu YQ, Giometti CS, Tollaksen SL, Khare T, Wu LY. Knock-out of SO1377 gene, which encodes the member of a conserved hypothetical bacterial protein family COG2268, results in alteration of iron metabolism, increased spontaneous mutation and hydrogen peroxide sensitivity in Shewanella oneidensis MR-1. Bmc Genomics. 2006;7:Artn 76. doi: 10.1186/1471-2164-7-76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Darie S, Gunsalus RP. Effect of heme and oxygen availability on hemA gene expression in Escherichia coli: role of the fnr, arcA, and himA gene products. J Bacteriol. 1994;176(17):5270–6. doi: 10.1128/jb.176.17.5270-5276.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Niehaus F, Hantke K, Unden G. Iron content and FNR-dependent gene regulation in Escherichia coli. FEMS Microbiol Lett. 1991;68(3):319–23. doi: 10.1111/j.1574-6968.1991.tb04617.x. [DOI] [PubMed] [Google Scholar]
- 29.Gough SP, Petersen BO, Duus JO. Anaerobic chlorophyll isocyclic ring formation in Rhodobacter capsulatus requires a cobalamin cofactor. Proc Natl Acad Sci U S A. 2000;97(12):6908–13. doi: 10.1073/pnas.97.12.6908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gunsalus RP. Control of electron flow in Escherichia coli: coordinated transcription of respiratory pathway genes. J Bacteriol. 1992;174(22):7069–74. doi: 10.1128/jb.174.22.7069-7074.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mouncey NJ, Kaplan S. Cascade regulation of dimethyl sulfoxide reductase (dor) gene expression in the facultative phototroph Rhodobacter sphaeroides 2.4.1 T. J Bacteriol. 1998;180(11):2924–30. doi: 10.1128/jb.180.11.2924-2930.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lawrence JG, Roth JR. The cobalamin (coenzyme B12) biosynthetic genes of Escherichia coli. J Bacteriol. 1995;177(22):6371–80. doi: 10.1128/jb.177.22.6371-6380.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Khan SR, Gaines J, Roop RM, 2nd, Farrand SK. Broad-host-range expression vectors with tightly regulated promoters and their use to examine the influence of TraR and TraM expression on Ti plasmid quorum sensing. Appl Environ Microbiol. 2008;74(16):5053–62. doi: 10.1128/AEM.01098-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9(4):357–9. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Anders S, Pyl PT, Huber W. HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31(2):166–9. doi: 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Robles JA, Qureshi SE, Stephen SJ, Wilson SR, Burden CJ, Taylor JM. Efficient experimental design and analysis strategies for the detection of differential expression using RNA-Sequencing. BMC Genomics. 2012;13:484. doi: 10.1186/1471-2164-13-484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bailey TL, Boden M, Buske FA, Frith M, Grant CE, Clementi L, et al. MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res. 2009;37(Web Server issue):W202–8. doi: 10.1093/nar/gkp335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, et al. Model-based analysis of ChIP-Seq (MACS) Genome Biol. 2008;9(9):R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30(15):2114–20. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Altenhoff AM, Skunca N, Glover N, Train CM, Sueki A, Pilizota I, et al. The OMA orthology database in 2015: function predictions, better plant support, synteny view and other improvements. Nucleic Acids Res. 2015;43(Database issue):D240–9. doi: 10.1093/nar/gku1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Felsenstein J. Confidence - Limits on phylogenies - An Approach using the bootstrap. Evolution. 1985;39(4):783–91. doi: 10.2307/2408678. [DOI] [PubMed] [Google Scholar]
- 43.Saitou N, Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol. 1987;4(4):406–25. doi: 10.1093/oxfordjournals.molbev.a040454. [DOI] [PubMed] [Google Scholar]
- 44.Tamura K, Nei M, Kumar S. Prospects for inferring very large phylogenies by using the neighbor-joining method. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(30):11030–5. doi: 10.1073/pnas.0404206101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: Molecular Evolutionary Genetics Analysis Version 6.0. Molecular Biology and Evolution. 2013;30(12):2725–9. doi: 10.1093/molbev/mst197. [DOI] [PMC free article] [PubMed] [Google Scholar]