

Abstract
The role of the macrophage in influenza virus infection is complex. Macrophages are critical for resolution of influenza virus infections but implicated in morbidity and mortality in severe infections. They can be infected with influenza virus and consequently macrophage infection is likely to have an impact on the host immune response. Macrophages display a range of functional phenotypes, from the prototypical pro-inflammatory classically activated cell to alternatively activated anti-inflammatory macrophages involved in immune regulation and wound healing. We were interested in how macrophages of different phenotype respond to influenza virus infection and therefore studied the infection of bone marrow-derived macrophages (BMDMs) of classical and alternative phenotype in vitro. Our results show that alternatively activated macrophages are more readily infected and killed by the virus than classically activated. Classically activated BMDMs express the pro-inflammatory markers inducible nitric oxide synthase (iNOS) and TNF-α, and TNF-α expression was further upregulated following infection. Alternatively activated macrophages express Arginase-1 and CD206; however, following infection, expression of these markers was downregulated whilst expression of iNOS and TNF-α was upregulated. Thus, infection can override the anti-inflammatory state of alternatively activated macrophages. Importantly, however, this results in lower levels of pro-inflammatory markers than those produced by classically activated cells. Our results showed that macrophage phenotype affects the inflammatory macrophage response following infection, and indicated that modulating the macrophage phenotype may provide a route to develop novel strategies to prevent and treat influenza virus infection.
Introduction
Influenza A viruses impose a considerable burden on human health. Seasonal influenza virus infections range from mild to life-threatening with the outcome dependent on both strain of virus and host response. They are associated with significant morbidity and mortality, particularly in the elderly and the very young. The emergence of highly pathogenic avian strains that can infect humans, albeit with a limited ability to spread human to human, presents an additional threat (Webster & Govorkova, 2014). A key feature of these strains is an overreactive immune response that fails to control the infection, resulting in excessive production of cytokines and chemokines, influx of immune cells, and severe immunopathology which contributes to high mortality (Cheung et al., 2002; Kobasa et al., 2007; Korteweg & Gu, 2008).
Influenza virus primarily infects epithelial cells in the respiratory tract but also has a well-recognized ability to infect macrophages (Rodgers & Mims, 1982; Tumpey et al., 2005). Macrophages play a central role in initiating and controlling the immune response to infections, and infection of these innate immune cells is highly likely to have consequences for the outcome of infection. It is clear, however, that the ability to infect and replicate in macrophages is dependent on both virus strain and origin of the macrophage, and that the results of infection are highly variable (Nicol & Dutia, 2014; Short et al., 2012). Infection of monocyte-derived macrophages in vitro with highly pathogenic viruses leads to production of higher levels of inflammatory cytokines and chemokines than are produced in response to infection with low-pathogenicity seasonal viruses (Cheung et al., 2002; Zhou et al., 2006), suggesting that infection of macrophages may be, at least in part, responsible for the increased pathogenicity of these viruses. In vivo studies, however, have shown that a virus strain that readily infects macrophages in vitro is less pathogenic in mice than a related virus that fails to infect macrophages (Tate et al., 2010). In this instance, depletion of macrophages increases the virulence, suggesting that macrophage infection can attenuate pathogenesis.
Macrophages are not, however, a single homogeneous population. They are highly pleiomorphic cells with a range of phenotypes and functions (Gordon & Taylor, 2005). At the extremes of the spectrum of macrophage phenotypes are the ‘classically activated’ or M1 macrophage, generally considered to be pro-inflammatory, producing TNF-α, IL-6, IL-1β and inducible nitric oxide synthase (iNOS), and the ‘alternatively activated’ or M2 macrophage which has upregulated expression of the macrophage mannose receptor CD206 and MHC class II, produces high levels of Arginase-1 (Arg-1) and endocytic function, and is considered to be associated with wound healing and repair. IFN-γ and TNF-α drive classical macrophage activation, whilst alternative macrophage activation is driven by the T-helper type 2 cytokines IL-4 and IL-13. Between these two extremes lies a range of subtly different phenotypes which orchestrate and regulate the immune response (Gordon, 2003).
We hypothesized that susceptibility and subsequent response of macrophages to influenza virus infection may depend on their phenotype, and that it would be possible to alter the extent of infection and thus influenza-associated pathology by manipulating macrophage phenotype. Here, we show that alternatively activated bone marrow-derived macrophages (BMDMs) are more susceptible to infection with the A/WSN/33 strain of influenza virus than classically activated BMDMs and are more readily killed by infection. Infection of alternatively activated BMDMs overrides their anti-inflammatory state, inducing a pro-inflammatory macrophage phenotype. However, infection of alternatively activated BMDMs results in production of lower levels of pro-inflammatory markers, including iNOS and TNF-α, than infection of classically activated BMDMs. Overall, our study supports the hypothesis that alternatively activated macrophages have a protective role in highly pathogenic virus infection.
Results
129Sv/Ev BMDMs can be infected with influenza virus strain A/WSN/33
Previous studies have reported that influenza viruses can infect macrophages with varying efficiency in a virus strain-dependent manner (Reading et al., 2000; Rodgers & Mims, 1981; Tate et al., 2010). We wished to investigate the ability of A/WSN/33 to infect macrophages from mice on the 129Sv/Ev background and therefore we derived macrophages from femurs of female 129Sv/Ev mice (6–8 weeks old) by culture for 7 days in medium containing macrophage colony-stimulating factor (M-CSF). FACS analysis showed that >95 % of the cells expressed the macrophage markers CD11b and F4/80 (data not shown), and therefore were of macrophage phenotype (Misharin et al., 2013). The BMDMs were infected at varying m.o.i. using viral titres determined on Madin-Darby canine kidney (MDCK) cells, incubated for various lengths of time and stained for viral antigen using a polyclonal antibody directed against purified H1N1 virus. No antigen-positive cells were detected at 1 h post-infection (p.i.) (Fig. 1b), but positive cells were detected from 6 h p.i. (Fig. 1c, d), indicating that viral protein synthesis was required for antibody staining. An m.o.i. 10 resulted in infection of ∼60 % of cells, but increasing the amount of input virus did not increase the percentage of cells infected further. Poisson distribution predicts that, if BMDMs were infected with the same efficiency as MDCK cells, an m.o.i. 5 should result in an infection rate of >99 % of cells. Thus, although BMDMs can be infected with A/WSN/33, they are less readily infected than MDCK cells.
Fig. 1. Influenza virus infection of 129Sv/Ev BMDMs. BMDMs cultured from femurs of female 129Sv/Ev mice were mock-infected or infected with A/WSN/33 and stained with antiserum to viral antigens. (a) Mock-infected cells, (b) 1 h p.i., m.o.i. 10, (c) 6 h p.i., m.o.i. 10 and (d) 48 h p.i., m.o.i. 10.

Effect of macrophage phenotype on infection
In order to produce polarized macrophage populations, we treated macrophages derived from WT 129Sv/Ev mice and mice on the same genetic background lacking the IFN-γ receptor (IFN-γR− / − ) with IFN-γ or IL-4 for 16 h, and measured levels of iNOS and Arg-1 by quantitative reverse transcription (qRT)-PCR and biochemical assay. Fig. 2 shows that treatment of both 129Sv/Ev and IFN-γR− / − BMDMs with IL-4 (Fig. 2a, b) for 16 h led to induction of Arg-1 mRNA and arginase activity, indicating that the macrophages had differentiated to an alternatively activated-like phenotype. Treatment of 129Sv/Ev BMDMs with IFN-γ resulted in production of iNOS mRNA and iNOS activity (Fig. 2c, d), confirming that these BMDMs had differentiated to a classical phenotype. IFN-γR− / − BMDMs cannot respond to IFN-γ and therefore were not able to produce a classical macrophage response, i.e. they did not upregulate iNOS upon IFN-γ treatment (Fig. 2c, d). We next infected the polarized macrophages with A/WSN/33 and stained with antibody to H1N1 virus (Fig. 3). Treatment of 129Sv/Ev and IFN-γR− / − BMDMs with IFN-γ or IL-4 and infection with virus did not affect the cell density (Fig. 3a–c, f–h). However, it was clear from 6 h p.i. that IL-4-treated macrophages were more readily infected than IFN-γ-treated macrophages (Fig. 3d, i). Similar results were observed at 48 h p.i. (Fig. 3e, j) and intervening time points. Thus, alternatively activated macrophages from both strains of mice were more readily infected than classically activated.
Fig. 2. Treatment of 129Sv/Ev BMDMs with IFN-γ or IL-4 leads to polarization of the macrophages to classical or alternative phenotypes. BMDMs derived from 129Sv/Ev and IFN-γR− / − mice were treated for 16 h with 1 ng IFN-γ ml− 1 or 4 ng IL-4 ml− 1. (a–c) Expression of Arg-1 (a) or iNOS (c) mRNA was measured by qRT-PCR, or by biochemical assay for Arg-1 activity (b) or iNOS activity (d).

Fig. 3. Alternatively activated macrophages are more readily infected with A/WSN/33 than classically activated macrophages. BMDMs derived from (a–e) 129Sv/Ev and (f–j) IFN-γ R− / − mice were cultured in medium containing M-CSF alone (a, f) or treated with IFN-γ (b, g) or IL-4 (c, h) for 16 h and infected with 10 p.f.u. A/WSN/33 per cell followed by staining with antiserum to virus antigens. The percentage of cells positive for antigen was quantified at 6 (d, i) and 48 h (e, j) *P < 0.05; ***P < 0.001.

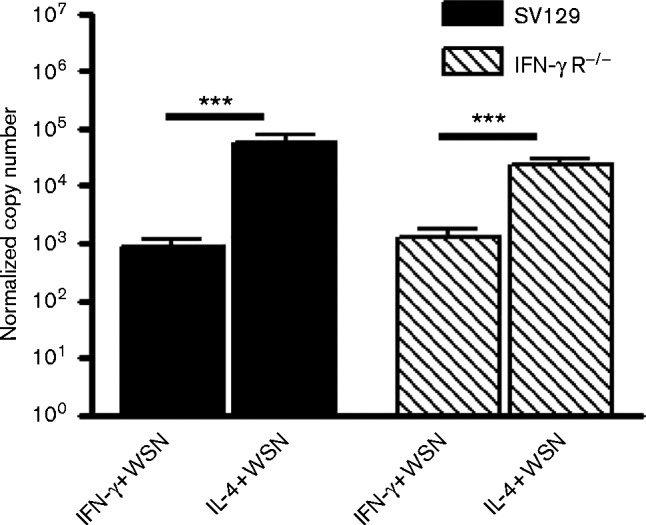
Consistent with the higher levels of viral antigen present in alternatively activated macrophages, qRT-PCR showed significantly higher amounts of viral M1 mRNA synthesized in these cells, indicating higher levels of viral infection (P < 0.001) (Fig. 4).
Fig. 4. Alternatively activated BMDMs produce more M1 mRNA than classically activated macrophages. BMDMs were activated with IFN-γ (classically activated) or IL-4 (alternatively active) and infected with 10 p.f.u. A/WSN/33 per cell (WSN). At 48 h p.i., M1 mRNA levels were measured by qRT-PCR. ***P < 0.001.
Survival of influenza virus-infected macrophages
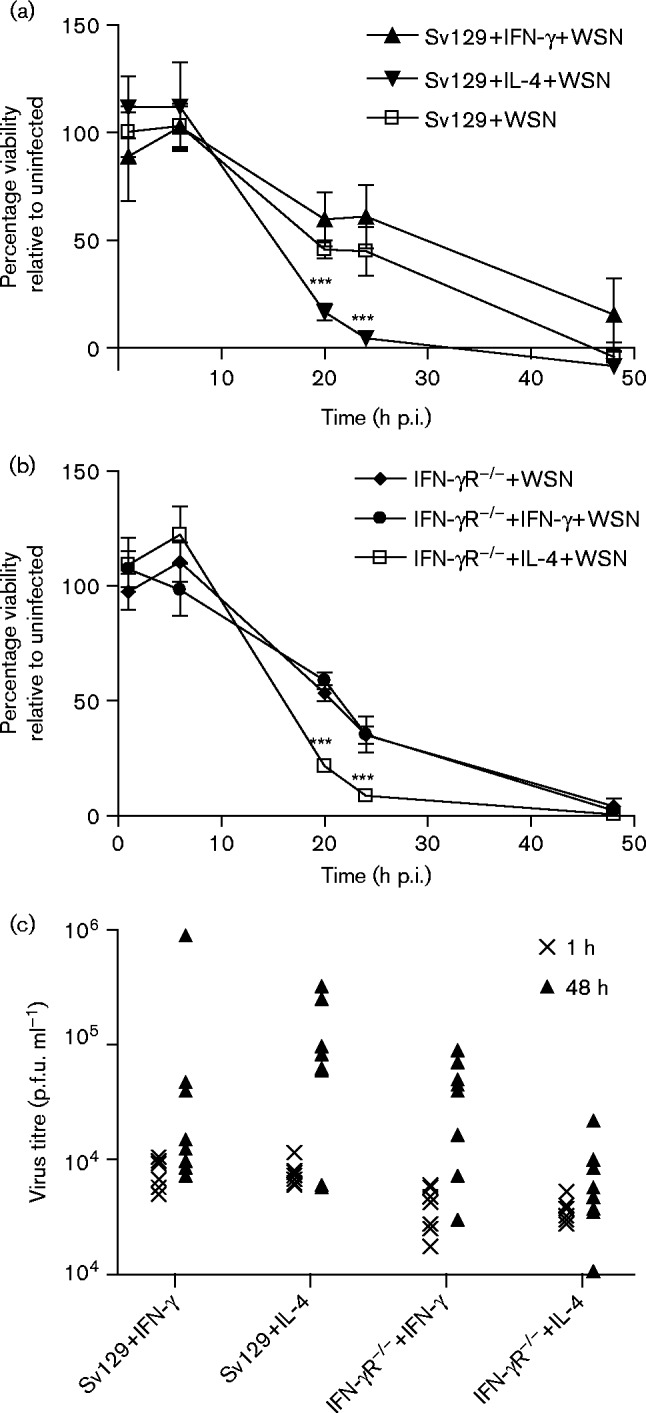
The survival of influenza virus-infected BMDMs was assessed by use of a CellTiter-Blue Viability Assay. Fig. 5(a, b) shows that by 20 h p.i., infection of alternatively activated macrophages resulted in a significantly lower rate of viability than seen in infected classically activated macrophages. Interestingly, 129Sv/Ev IFN-γ-treated macrophages show a trend towards higher viability than untreated BMDMs from the same mice (Fig. 5a). This difference was not apparent for the IFN-γR− / − BMDMs, which could not respond to IFN-γ, suggesting that classical activation conferred protection against virus induced cell death. These data suggested that IL-4 activation rendered BMDMs more permissive for A/WSN/33 and that once infected, these cells were more readily killed by the virus than classically activated cells. In order to determine whether higher levels of productive virus infection occurred in alternatively activated macrophages, we measured the infectious virus present in the cell supernatants (Fig. 5c). At 48 h p.i., the amount of virus in supernatants was higher than at 1 h p.i., indicating that there was a low-level replication in the BMDMs. However, the amount of virus recovered was less than the amount of input virus and we found no evidence that alternative activation resulted in production of higher levels of infectious virus by the BMDMs.
Fig. 5. Alternatively activated BMDMs are more susceptible to cell death following infection with A/WSN/33, but do not support higher levels of virus replication. BMDMs from 129Sv/Ev and IFN-γR− / − mice were untreated or activated with IFN-γ or IL-4 and infected with 10 p.f.u. A/WSN/33 per cell (WSN). Cell viability was measured by a CellTiter-Blue Viability Assay at various times after infection. (a) 129Sv/Ev BMDMs and (b) IFN-γR− / − BMDMs. ***P < 0.001. (c) Virus titres in cell supernatants at 1 and 48 h p.i. were measured by titration on MDCK cells. Results are representative of three independent experiments.
Cytokine response to influenza virus infection
In order to assess the effect of influenza virus infection on macrophage phenotype, we analysed the expression of phenotypic markers in infected/polarized BMDMs. iNOS, TNF-α and IL-12p40 were chosen as markers of classically activated macrophages. iNOS is associated with inflammatory macrophage responses, and the secreted cytokines TNF-α and IL-12p40 contribute to macrophage-driven inflammation. In addition to Arg-1, which is highly expressed in alternatively activated macrophages (Fig. 2), we measured expression of the mannose receptor CD206, which is upregulated on alternatively activated macrophages (Gordon, 2003). BMDMs were treated with IFN-γ or IL-4 and infected with A/WSN/33 at m.o.i. 10. Cells were harvested at 48 h p.i. and the expression of markers was measured by qRT-PCR. Treatment of 129Sv/Ev macrophages with IFN-γ upregulated iNOS by >104-fold and no further upregulation was induced by infection with virus (Fig. 6a). Similar results were found for IL-12p40 (data not shown). In contrast, TNF-α expression in these macrophages, although elevated by IFN-γ alone, was significantly higher in cells which had been infected with virus (P < 0.05; Fig. 6e). Thus, virus infection could drive expression of this pro-inflammatory cytokine. Arg-1 and CD206 expression in the IFN-γ-treated 129Sv/Ev macrophages was low and virus infection did not alter this (Fig. 6c, g). As expected, IFN-γ treatment of macrophages grown from IFN-γR− / − mice did not result in significant changes in expression of the classical markers iNOS and TNF-α (Fig. 6a, e). Infection of IFN-γR− / − macrophages with influenza virus upregulated iNOS expression (Fig. 6a), indicating the virus alone could switch on expression of this inflammatory marker. TNF-α expression was clearly completely dependent on the IFN-γ activity as there was no change in expression following virus infection of the IFN-γR− / − macrophages (Fig. 6e). Similarly, there was no significant change in IL-12p40 expression (data not shown). Thus, infection of classically activated macrophages resulted in increased expression of TNF-α, enhancing the pro-inflammatory state of these macrophages. In the absence of IFN-γ responsiveness, infection led to enhanced expression of iNOS but did not induce synthesis of secreted pro-inflammatory cytokines (TNF-α and IL-12), indicating that these macrophages did not enter a pro-inflammatory state.
Fig. 6. Expression of pro- and anti-inflammatory markers in classically and alternatively activated macrophages infected with influenza virus A/WSN/33. BMDMs were treated with 1 ng IFN-γ ml− 1 or 4 ng IL-4 ml− 1 for 16 h and then infected with 10 p.f.u. A/WSN/33 per cell (WSN). Cells were harvested at 48 h p.i. and expression of cellular markers was monitored by qRT-PCR. Results are representative of three independent experiments. (a, b) iNOS, (c, d) Arg-1, (e, f) TNF-α and (g, h) CD206. *P < 0.05; **P < 0.005; ***P < 0.001.

Alternative activation of BMDMs from both WT 129Sv/Ev and IFN-γR− / − mice resulted in upregulation of Arg-1 and CD206 compared with untreated BMDMs (Fig. 6d, h). Classical markers were low or undetectable in these cells (Fig. 6b, f). However, infection of alternatively activated 129Sv/Ev BMDMs induced expression of iNOS and TNF-α, indicating that virus infection could override the alternative, anti-inflammatory state of these macrophages, producing a pro-inflammatory state within the cells (iNOS) and leading to secretion of pro-inflammatory cytokines (Fig. 6b, f). Similarly, iNOS was significantly upregulated following infection of alternatively activated IFN-γR− / − BMDMs (Fig. 6b; P < 0.001), but levels were lower than those produced in alternatively activated 129Sv/Ev BMDMs. Again, expression of TNF-α was completely dependent on responsiveness to IFN-γ (Fig. 6f). Infection of alternatively activated WT 129Sv/Ev and IFN-γR− / − BMDMs led to decreased expression of Arg-1 and CD206 (Fig. 6d, h). Thus, infection of alternatively activated macrophages led to downregulation of alternative markers and induction of the pro-inflammatory mediators iNOS and TNF-α. However, the levels of these cytokines produced by alternatively activated macrophages were lower than those produced by classically activated macrophages. Whilst infection could clearly override the anti-inflammatory state, infection of alternatively activated macrophages led to lower levels of pro-inflammatory cytokine production than those observed following infection of classically activated macrophages.
Discussion
Our results show that alternatively activated macrophages are more susceptible to infection with A/WSN/33 than classically activated cells. A higher percentage of cells express viral antigens and higher levels of M1 mRNA are produced in alternatively activated cells than in classically activated cells. Similarly, Hoeve et al. (2012) showed that following infection with the H3N2 virus Udorn, a significantly higher number of human monocyte-derived macrophages with anti-inflammatory characteristics contained viral antigen than those with the pro-inflammatory phenotype. The effect of IL-4 on uptake of antigens by macrophages is dependent on both antigen and pathway. Treatment of macrophages with IL-4 leads to increased uptake of soluble antigen as well as increased mannose receptor-dependent uptake of antigen (Montaner et al., 1999; Raveh et al., 1998). However, alternative activation of macrophages with IL-4 has been shown to impair phagocytosis of bacteria and microbial particles (Varin et al., 2010). The higher levels of M1 mRNA in IL-4-treated cells argue that the presence of virus antigen in cells is not simply due to increased phagocytosis of viral antigens, but rather is due to increased infection of these macrophages. Influenza viruses usually enter cells by endocytosis following initial binding of the haemagglutinin to sialic acids on the cell surface (Matlin et al., 1981; Skehel & Wiley, 2000). However, there is evidence that influenza A virus can use other cell surface molecules, including the macrophage mannose receptor CD206, and macrophage galectin-type lectins DC-SIGN and L-SIGN, to bind to and enter macrophages (Londrigan et al., 2011; Reading et al., 2000; Upham et al., 2010). Alternatively activated macrophages express a different range of cell surface proteins to those found on classically activated macrophages. For example, CD206 is more highly expressed on the surface of alternatively activated macrophages than on classically activated macrophages and is indeed considered a marker for alternative activation (Gordon, 2003; Stein et al., 1992). At this point, further work is required to understand the mechanisms by which alternatively activated macrophages are more readily infected, but this may have important implications for influenza virus pathogenesis.
Interestingly, alternatively activated macrophages are more readily killed by infection with A/WSN/33 than classically activated macrophages. Influenza virus infection leads to cell death; hence, it is likely that the difference reflects the level of infection. qRT-PCR data show that at 48 h p.i. there is up to 100-fold more M1 mRNA in alternatively activated cultures than in classically activated. This, together with the fact that a higher percentage of alternatively activated macrophages is infected, is consistent with the higher level of cell death found in alternatively activated cultures. It is notable, however, that the apparently more permissive state of alternatively activated macrophages did not lead to production of higher levels of infectious virus than are found in classically activated cells. Although there is evidence for productive influenza virus infections in human macrophages (Hoeve et al., 2012; Perrone et al., 2008; van Riel et al., 2011; Yu et al., 2011), a number of publications have reported that influenza virus infection is abortive in murine macrophages (Rodgers & Mims, 1981; Tate et al., 2010, 2011). Our data show that whilst there is some replication of A/WSN/33 in murine BMDMs, the ability to produce infectious virus is not related to the activation state of the macrophage. The ease with which alternatively activated macrophages become infected has important implications, i.e. manipulation of phenotype in vivo may allow macrophages to act as a sink for viruses.
Infection of both classically and alternatively activated 129Sv/Ev macrophages resulted in upregulation of TNF-α, a cytokine associated with severe influenza virus infections in vivo. Macrophages can produce IFN-γ (Gessani & Belardelli, 1998; Schroder et al., 2004), and therefore it is likely that autocrine production of this cytokine contributes to the ability of alternatively activated macrophages to override the IL-4 response and produce an inflammatory response. Infection also resulted in upregulation of iNOS and IL-12p40 in alternatively activated 129Sv/Ev macrophages. Type I IFNs and IL-1β can induce synthesis of iNOS (Gao et al., 1998; Geller et al., 1995), and it is likely these cytokines together with TNF-α and IFN-γ are involved in induction of iNOS and IL-12p40 following virus infection (Drapier et al., 1988; Farrell & Blake, 1996; Ma et al., 1996). The upregulation of pro-inflammatory markers together with the downregulation of Arg-1 and CD206 expression demonstrates that infection results in a switch in cell phenotype. However, although both classes of macrophage produced a pro-inflammatory response following virus infection, alternatively activated macrophages produced lower levels of pro-inflammatory markers than classically activated macrophages. Thus, the data support the hypothesis that manipulation of the macrophage phenotype could have an impact on influenza virus pathogenesis.
IFN-γR− / − macrophages were significantly compromised in pro-inflammatory responses and TNF-α production was severely limited. Influenza virus infection did induce iNOS and IL-12p40 in these macrophages, most likely due to the action of type I IFNs and IL-1β, but levels were 1000-fold lower than in WT classically activated infected macrophages. Non-activated IFN-γR− / − BMDMs have significantly higher expression of CD206 than 129Sv/Ev BMDMs. Given that CD206 has been shown to act as a receptor for influenza virus, it is interesting to speculate that this may play a role in the increased susceptibility to infection found in these macrophages. Overall, these data provide evidence that IFN-γ responsiveness is critical to the macrophage response to influenza virus infection and highlight the role of IFN-γ as a critical cytokine in pathogenesis of influenza virus infections.
We chose to investigate the infection of BMDMs and successfully demonstrated that alternatively activated macrophages and those which cannot effectively mount a classical response (IFN-γR− / − ) are more susceptible to influenza virus infection. Whilst these studies were carried out with BMDMs rather than alveolar macrophages, they provide important clues for understanding the function of macrophages in control of influenza virus infections. Wang et al. (2013) recently showed that alternatively activated alveolar macrophages can protect against lethal challenge in a mouse model. Their study did not address macrophage infection, but our data would suggest that infection of alternatively activated macrophages per se is likely to play a role in this protective effect.
Macrophages clearly play a critical role in influenza virus infection. Depletion of macrophages in animal models leads to exacerbation of infection, indicating their importance in an effective host response to infection (Tate et al., 2011; Tumpey et al., 2005). Similarly, transfer of macrophages accounts for the protective effect of prior infection with a herpesvirus (Saito et al., 2013). However, they are major producers of inflammatory cytokines and hence have been implicated in the development of the severe pathology associated with fatal infections (Cheung et al., 2002; Perrone et al., 2008). Our study shows that macrophages of different phenotypes respond very differently to influenza virus infection. Given the diversity and plasticity of macrophages, understanding the interaction between macrophage phenotype and virus infection is likely to be fundamental to the development of novel strategies to prevent and treat severe influenza virus infections.
Methods
Cell culture
L929 murine fibroblasts were grown in tissue culture flasks (Nunc) in RPMI supplemented with 10 % FCS, 100 U penicillin ml− 1, 100 μg streptomycin ml− 1 and 2 mM l-glutamine (Invitrogen). Supernatant from confluent cultures was pooled, clarified by centrifugation at 8000 g and stored in aliquots at − 20 °C as a source of M-CSF for BMDMs.
MDCK cells were grown in Dulbecco's modified Eagle's medium (DMEM; Invitrogen) supplemented as for RPMI.
Virus growth and assay
A/WSN/33 was propagated on MDCK cells at m.o.i. 0.001 for 48 h before harvest of supernatant containing the virus. Supernatant was clarified by centrifugation at 3000 g and stored in aliquots at − 80 °C. Titre was determined by plaque assay as described previously (Nicol et al., 2012).
Macrophage isolation, activation and infection
129Sv/Ev and IFN-γR− / − mice on the 129Sv/Ev background (Huang et al., 1993) were purchased from B & K Universal and bred in-house. Femurs from female mice aged 6–8 weeks old were removed and cleaned in alcohol, and the bone marrow flushed out with supplemented RPMI, using a 25G needle and syringe. Bone marrow cells were plated onto 100 mm bacteriological dishes (Sterilin) in 50 % supplemented RPMI : 50 % L929 fibroblast conditioned media (complete macrophage media). Cultures were initially set up by seeding cells from one femur in each plate. BMDMs were passaged on day 4 by the following method: medium was removed and retained, and the adherent cells were incubated with Dulbecco's PBS (D-PBS; Life Technologies) for 5 min, then detached from the plastic by washing vigorously with D-PBS using an 18G needle and syringe, and recovered by centrifugation at 8000 g. Cells from each plate were then reseeded into two new plates in complete macrophage medium supplemented with 5 ml original medium. On day 7, BMDMs were harvested, counted and seeded into 100 mm dishes, 96-well plates or onto eight-well glass chamber slides (BD Falcon) at 1 × 105 cells ml− 1. BMDMs were activated with either 1 ng IFN-γ ml− 1 or 4 ng IL-4 ml− 1 (Peprotech) in complete macrophage media for 16 h prior to infection with A/WSN/33. Activating medium was removed from activated BMDMs and kept at 37 °C whilst infection was carried out. BMDMs were infected at m.o.i. 10 by incubation with virus diluted in serum-free DMEM for 1 h. The inoculum was then removed and activating media replaced onto the cells to allow continued exposure to cytokine. After 48 h, medium was removed for assay of iNOS and Arg-1; the cells were washed by incubation in Ca2+/Mg+-free PBS (Invitrogen) for 5 min, flushed from the dishes as described above, counted and pelleted by centrifugation at 1500 g. Cell pellets were stored at − 80 °C.
Cell viability assays
Activated and non-activated BMDMs in 96-well plates were infected at m.o.i. 10 and, at appropriate time points, cell viability was measured using a CellTiter-Blue Cell Viability Assay (Promega). Samples were assayed in triplicate.
Immunostaining
Chamber slides were washed with PBS and fixed for 30 min with 4 % (w/v) paraformaldehyde. After fixation, slides were washed with PBS and either stained immediately or stored at 4 °C until required. Before staining, stored slides were washed with PBS and blocked for 30 min at room temperature with CAS-Block (Invitrogen). After extensive washing, slides were probed with 1 : 500 dilution of polyclonal goat anti-influenza A H1N1 strain USSR antibody (AbD Serotec) in CAS-Block. After 30 min incubation at room temperature, slides were washed with PBS and bound antibody was detected by incubation for 30 min with rabbit anti-goat/sheep Alexa Fluor 488-conjugated secondary antibody, diluted 1 : 1000 in CAS-Block (Invitrogen). Unbound conjugate was removed by washing with PBS, and the slides were counterstained with DAPI and mounted in Prolong Gold (Life Technologies) mounting medium.
iNOS assays
Active iNOS was determined by the Greiss reagent bioassay, which results in production of nitrite and a colour change from colourless to pink in the presence of enzyme. An aliquot of 100 μl Greiss reagent, 5.8 % (v/v) H3PO4, 1 % (w/v) sulphanilamide and 0.1 % (w/v) N-(1-naphthyl) ethylenediamine dihydrochloride was added to 100 μl BMDM supernatant or 100 μl sodium nitrite standard (Sigma) and the A540 determined.
Arg-1 bioassay
Bioactive Arg-1 was measured by conversion of l-arginine to urea as follows. First, 1 × 105 BMDMs were plated onto 96-well flat-bottomed plates (Nunc), washed with PBS and lysed with 0.1 % Triton-X (Sigma). The lysate was then removed to sterile 1.5 ml tubes. After addition of 100 μl 25 mM Tris/HCl and 20 μl 10 mM MnCl2 tubes were incubated at 56 °C for 10 min. Then, 100 μl of each sample was transferred to fresh tubes and incubated with 100 μl 0.5 M l-arginine for 2 h. During this time a standard dilution series of urea was made. Following the incubation step, 800 μl 10 % (v/v) sulphuric/30 % (v/v) phosphoric acid solution was added along with 40 μl isonitropropiophenone, mixed by vortexing and incubated at 95 °C for 30 min. Once cooled, samples and standards were placed in a 96-well plate and the A540 determined.
qRT- PCR
RNA was extracted from frozen BMDMs using an RNeasy Minikit and QIAshredders (Qiagen), as per the manufacturer's guidelines. Genomic DNA was removed by treatment with a DNA-free kit (Ambion) according to the manufacturer's instructions. RNA (1–2 μg) was reverse transcribed to cDNA with Superscript III (Invitrogen). cDNA was routinely diluted 1 : 20 for qRT-PCR analysis. Primers were designed as follows for each gene of interest, along with reference genes succinate dehydrogenase A and calnexin. These reference genes were chosen from a panel of 12 housekeeping genes (Quantace), which were tested to determine the genes with the most stable expression in BMDMs. Optimal amplification conditions were determined for each gene of interest to ensure >95 % efficiency of single products. qRT-PCR was carried out using a Rotorgene 3000 cycler (Qiagen).
Primer sets (forward/reverse) were: calnexin, TTAGTTGACCAGTCTGTTG/CCTTTCATCCCAATCTTCAG; succinate dehydrogenase A, GCTCCTACTGATGAAACCTG/AACTCAATCCCTTACAGCAA; iNOS, TGCTACTGAGACAGGGAAG/GACAGTCTCCATTCCCAA; TNF-α, CACCACCATCAAGGACTCAA/GACAGAGGCAACCTGACCAC; IL-12p40, GGAAGCACGGCAGCAGAATA/TTGAGGGAGAAGTAGGAATGG; M1, CTCTCTATCGTCCCGTCAGG/GAGCGTGAACACAAATCCTA.
Each gene of interest was normalized to the reference genes using Genex software (MultiD) and relative expression of infected to mock controls was calculated.
Statistical methods
Statistical analysis of the differences in the various parameters of interest (the percentage of macrophages infected, normalized copy number, percentage survival and gene expression) following classical and alternative activation of macrophages in both 129Sv/Ev and IFN-γR− / − mice was performed using standard linear-mixed effect models (Pinheiro & Bates, 2004), with individual mouse entered as the random effect to take account of the repeated measures taken from them. All statistical analyses were carried out on log10-transformed data to normalize the residuals and were performed in r (version 3.1.1; The R Foundation for Statistical Computing), using the package nlme (version 3.1-117).
Acknowledgements
This work was funded by a Scottish Higher Education Funding Council grant to the Interdisciplinary Centre for Human and Avian Influenza Research (ICHAIR), and a joint ICHAIR–BBSRC Studentship to G. M. C.
References
- Cheung C.Y., Poon L.L.M., Lau A.S., Luk W., Lau Y.L., Shortridge K.F., Gordon S., Guan Y., Peiris J.S.M. (2002). Induction of proinflammatory cytokines in human macrophages by influenza A (H5N1) viruses: a mechanism for the unusual severity of human disease? Lancet 360 1831–1837 10.1016/S0140-6736(02)11772-7 . [DOI] [PubMed] [Google Scholar]
- Drapier J.C., Wietzerbin J., Hibbs J.B., Jr. (1988). Interferon-gamma and tumor necrosis factor induce the l-arginine-dependent cytotoxic effector mechanism in murine macrophages Eur J Immunol 18 1587–1592 10.1002/eji.1830181018 . [DOI] [PubMed] [Google Scholar]
- Farrell A.J., Blake D.R. (1996). Nitric oxide Ann Rheum Dis 55 7–20 10.1136/ard.55.1.7 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J.J., Filla M.B., Fultz M.J., Vogel S.N., Russell S.W., Murphy W.J. (1998). Autocrine/paracrine IFN-alphabeta mediates the lipopolysaccharide-induced activation of transcription factor Stat1alpha in mouse macrophages: pivotal role of Stat1alpha in induction of the inducible nitric oxide synthase gene J Immunol 161 4803–4810 . [PubMed] [Google Scholar]
- Geller D.A., de Vera M.E., Russell D.A., Shapiro R.A., Nussler A.K., Simmons R.L., Billiar T.R. (1995). A central role for IL-1 beta in the in vitro and in vivo regulation of hepatic inducible nitric oxide synthase. IL-1 beta induces hepatic nitric oxide synthesis J Immunol 155 4890–4898 . [PubMed] [Google Scholar]
- Gessani S., Belardelli F. (1998). IFN-gamma expression in macrophages and its possible biological significance Cytokine Growth Factor Rev 9 117–123 10.1016/S1359-6101(98)00007-0 . [DOI] [PubMed] [Google Scholar]
- Gordon S. (2003). Alternative activation of macrophages Nat Rev Immunol 3 23–35 10.1038/nri978 . [DOI] [PubMed] [Google Scholar]
- Gordon S., Taylor P.R. (2005). Monocyte and macrophage heterogeneity Nat Rev Immunol 5 953–964 10.1038/nri1733 . [DOI] [PubMed] [Google Scholar]
- Hoeve M.A., Nash A.A., Jackson D., Randall R.E., Dransfield I. (2012). Influenza virus A infection of human monocyte and macrophage subpopulations reveals increased susceptibility associated with cell differentiation PLoS One 7 e29443 10.1371/journal.pone.0029443 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang S., Hendriks W., Althage A., Hemmi S., Bluethmann H., Kamijo R., Vilcek J., Zinkernagel R.M., Aguet M. (1993). Immune response in mice that lack the interferon-gamma receptor Science 259 1742–1745 10.1126/science.8456301 . [DOI] [PubMed] [Google Scholar]
- Kobasa D., Jones S.M., Shinya K., Kash J.C., Copps J., Ebihara H., Hatta Y., Kim J.H., Halfmann P., other authors (2007). Aberrant innate immune response in lethal infection of macaques with the 1918 influenza virus Nature 445 319–323 10.1038/nature05495 . [DOI] [PubMed] [Google Scholar]
- Korteweg C., Gu J. (2008). Pathology, molecular biology, and pathogenesis of avian influenza A (H5N1) infection in humans Am J Pathol 172 1155–1170 10.2353/ajpath.2008.070791 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Londrigan S.L., Turville S.G., Tate M.D., Deng Y.M., Brooks A.G., Reading P.C. (2011). N-linked glycosylation facilitates sialic acid-independent attachment and entry of influenza A viruses into cells expressing DC-SIGN or L-SIGN J Virol 85 2990–3000 10.1128/JVI.01705-10 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma X., Chow J.M., Gri G., Carra G., Gerosa F., Wolf S.F., Dzialo R., Trinchieri G. (1996). The interleukin 12 p40 gene promoter is primed by interferon gamma in monocytic cells J Exp Med 183 147–157 10.1084/jem.183.1.147 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matlin K.S., Reggio H., Helenius A., Simons K. (1981). Infectious entry pathway of influenza virus in a canine kidney cell line J Cell Biol 91 601–613 10.1083/jcb.91.3.601 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misharin A.V., Morales-Nebreda L., Mutlu G.M., Budinger G.R., Perlman H. (2013). Flow cytometric analysis of macrophages and dendritic cell subsets in the mouse lung Am J Respir Cell Mol Biol 49 503–510 10.1165/rcmb.2013-0086MA . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montaner L.J., da Silva R.P., Sun J., Sutterwala S., Hollinshead M., Vaux D., Gordon S. (1999). Type 1 and type 2 cytokine regulation of macrophage endocytosis: differential activation by IL-4/IL-13 as opposed to IFN-gamma or IL-10 J Immunol 162 4606–4613 . [PubMed] [Google Scholar]
- Nicol M.Q., Dutia B.M. (2014). The role of macrophages in influenza A virus infection Future Virol 9 847–862 10.2217/fvl.14.65. [DOI] [Google Scholar]
- Nicol M.Q., Ligertwood Y., Bacon M.N., Dutia B.M., Nash A.A. (2012). A novel family of peptides with potent activity against influenza A viruses J Gen Virol 93 980–986 10.1099/vir.0.038679-0 . [DOI] [PubMed] [Google Scholar]
- Perrone L.A., Plowden J.K., García-Sastre A., Katz J.M., Tumpey T.M. (2008). H5N1 and 1918 pandemic influenza virus infection results in early and excessive infiltration of macrophages and neutrophils in the lungs of mice PLoS Pathog 4 e1000115 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinheiro J.C., Bates D.M. (2004). Mixed-Effects Models in S and S-PLUS 3rd Edition Springer; New York. [Google Scholar]
- Raveh D., Kruskal B.A., Farland J., Ezekowitz R.A. (1998). Th1 and Th2 cytokines cooperate to stimulate mannose-receptor-mediated phagocytosis J Leukoc Biol 64 108–113 . [PubMed] [Google Scholar]
- Reading P.C., Miller J.L., Anders E.M. (2000). Involvement of the mannose receptor in infection of macrophages by influenza virus J Virol 74 5190–5197 10.1128/JVI.74.11.5190-5197.2000 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodgers B., Mims C.A. (1981). Interaction of influenza virus with mouse macrophages Infect Immun 31 751–757 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodgers B.C., Mims C.A. (1982). Influenza virus replication in human alveolar macrophages J Med Virol 9 177–184 10.1002/jmv.1890090304 . [DOI] [PubMed] [Google Scholar]
- Saito F., Ito T., Connett J.M., Schaller M.A., Carson W.F., IV, Hogaboam C.M., Rochford R., Kunkel S.L. (2013). MHV68 latency modulates the host immune response to influenza A virus Inflammation 36 1295–1303 10.1007/s10753-013-9668-1 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroder K., Hertzog P.J., Ravasi T., Hume D.A. (2004). Interferon-gamma: an overview of signals, mechanisms and functions J Leukoc Biol 75 163–189 10.1189/jlb.0603252 . [DOI] [PubMed] [Google Scholar]
- Short K.R., Brooks A.G., Reading P.C., Londrigan S.L. (2012). The fate of influenza A virus after infection of human macrophages and dendritic cells J Gen Virol 93 2315–2325 10.1099/vir.0.045021-0 . [DOI] [PubMed] [Google Scholar]
- Skehel J.J., Wiley D.C. (2000). Receptor binding and membrane fusion in virus entry: the influenza hemagglutinin Annu Rev Biochem 69 531–569 10.1146/annurev.biochem.69.1.531 . [DOI] [PubMed] [Google Scholar]
- Stein M., Keshav S., Harris N., Gordon S. (1992). Interleukin 4 potently enhances murine macrophage mannose receptor activity: a marker of alternative immunologic macrophage activation J Exp Med 176 287–292 10.1084/jem.176.1.287 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tate M.D., Pickett D.L., van Rooijen N., Brooks A.G., Reading P.C. (2010). Critical role of airway macrophages in modulating disease severity during influenza virus infection of mice J Virol 84 7569–7580 10.1128/JVI.00291-10 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tate M.D., Schilter H.C., Brooks A.G., Reading P.C. (2011). Responses of mouse airway epithelial cells and alveolar macrophages to virulent and avirulent strains of influenza A virus Viral Immunol 24 77–88 10.1089/vim.2010.0118 . [DOI] [PubMed] [Google Scholar]
- Tumpey T.M., García-Sastre A., Taubenberger J.K., Palese P., Swayne D.E., Pantin-Jackwood M.J., Schultz-Cherry S., Solórzano A., Van Rooijen N., other authors (2005). Pathogenicity of influenza viruses with genes from the 1918 pandemic virus: functional roles of alveolar macrophages and neutrophils in limiting virus replication and mortality in mice J Virol 79 14933–14944 10.1128/JVI.79.23.14933-14944.2005 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Upham J.P., Pickett D., Irimura T., Anders E.M., Reading P.C. (2010). Macrophage receptors for influenza A virus: role of the macrophage galactose-type lectin and mannose receptor in viral entry J Virol 84 3730–3737 10.1128/JVI.02148-09 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Riel D., Leijten L.M., van der Eerden M., Hoogsteden H.C., Boven L.A., Lambrecht B.N., Osterhaus A.D., Kuiken T. (2011). Highly pathogenic avian influenza virus H5N1 infects alveolar macrophages without virus production or excessive TNF-alpha induction PLoS Pathog 7 e1002099 10.1371/journal.ppat.1002099 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varin A., Mukhopadhyay S., Herbein G., Gordon S. (2010). Alternative activation of macrophages by IL-4 impairs phagocytosis of pathogens but potentiates microbial-induced signalling and cytokine secretion Blood 115 353–362 10.1182/blood-2009-08-236711 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J., Li F., Sun R., Gao X., Wei H., Li L.J., Tian Z. (2013). Bacterial colonization dampens influenza-mediated acute lung injury via induction of M2 alveolar macrophages Nat Commun 4 2106 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster R.G., Govorkova E.A. (2014). Continuing challenges in influenza Ann N Y Acad Sci 1323 115–139 10.1111/nyas.12462 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu W.C., Chan R.W., Wang J., Travanty E.A., Nicholls J.M., Peiris J.S., Mason R.J., Chan M.C. (2011). Viral replication and innate host responses in primary human alveolar epithelial cells and alveolar macrophages infected with influenza H5N1 and H1N1 viruses J Virol 85 6844–6855 10.1128/JVI.02200-10 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J., Law H.K., Cheung C.Y., Ng I.H., Peiris J.S., Lau Y.L. (2006). Differential expression of chemokines and their receptors in adult and neonatal macrophages infected with human or avian influenza viruses J Infect Dis 194 61–70 10.1086/504690 . [DOI] [PMC free article] [PubMed] [Google Scholar]