

Abstract
Dietary energy restriction (DER) inhibits experimentally-induced mammary cancer, an effect accompanied by elevated levels of silent information regulator 2 (SIRT1), a class III histone deacetylase (HDAC). However, the effect of DER on targets of other classes of HDACs has not been reported, a highly relevant issue given evidence that HDAC induction favors the development of cancer and tumor growth. Experiments were conducted to determine if suberoylanilide hydroxamic acid (SAHA), a histone deacetylase inhibitor with broad activity, would affect the anti-cancer activity of DER. Female Sprague Dawley rats (n=30/group) were injected with 1-methyl-1-nitrosourea (50 mg/kg) at 21 days of age and 7 days thereafter were randomized to groups fed: 1) control diet (AIN-93G), 2) 0.1% SAHA (w/w), 3) 40% DER, or 4) 0.1% SAHA+40% DER. An additional group was fed 0.1% SAHA+40%DER for 5 weeks and released to control diet for 3 weeks. DER significantly reduced mammary cancer incidence, multiplicity, and cancer burden and prolonged cancer latency (P < 0.01). Cancer inhibition was maintained in SAHA+DER despite evidence that histone (H2ALys9, H2BLys5, and H4Lys5/8/12/16, but not H3Lys9 P < 0.001) and non-histone protein deacetylation (p53Lys373 and p53Lys382 P < 0.001), induced by DER, were reversed by SAHA. This indicates that DER’s inhibition of cancer is not dependent on HDAC induction. After releasing rats from DER+SAHA, cancer multiplicity remained lower than control (P < 0.05), consistent with apoptosis mediated cell deletion. These findings support further investigation of the hypothesis that HDAC induction by DER blunts its anti-carcinogenic impact.
Keywords: apoptosis, dietary energy restriction, histone deacetylase, mammary carcinogenesis, suberoylanilide hydroxamic acid
Introduction
Dietary energy restriction (DER), also referred to as caloric restriction, is a physiological inhibitor of the carcinogenic process in many model systems including those for breast cancer (1, 2). Recognizing that DER is a model for investigating weight gain prevention and is not a model by which to study the effects of weight loss (3), there is strong evidence that DER’s powerful effects are also operative in human populations in which preventing adult weight gain is associated with reduced lifetime risk for breast cancer (4–6). At the cellular level, DER acts by reducing the drive for the proliferation of transformed cells and by inducing cell death via apoptosis, effects that account, at least in part, for DER’s cancer inhibitory activity (7–9). The effect of DER on apoptosis induction is dominant but appears insufficient to result in the deletion of populations of premalignant cells that would render sustained protection against cancer in the absence of continuous treatment (10). Since apoptosis induction is one of a number of mechanisms that are targeted to kill cancer cells during therapy, the induction of apoptosis by DER, and its preferential deletion of transformed cells could conceivably offer a new approach to cancer prevention through early cure via elimination of pathologies before they become clinically detectable.
While an increasing amount of data is accumulating about the mechanisms that underlie the cancer inhibitory activity of DER, those investigations have uncovered two effects that could serve to limit DER’s protective activity (11). They are: induction of autophagy mediated by down regulation of mammalian target of rapamycin (mTOR) signaling (12), and induction of SIRT1, a class III histone deacetylase (HDAC) (13). The work reported herein focused on HDAC mediated effects of DER.
SIRT1 is a member of one of three classes of HDACs. While the induction of SIRT1 by DER has been widely reported and has been frequently linked to the effects of DER on longevity extension (14), emerging evidence indicates that induction of SIRT1 and other classes of HDACs are associated with pro-carcinogenic effects and enhanced tumor growth (15–17). Consequently, HDACs are currently targets for drug development (18–20). However, there is limited information about how DER affects overall HDAC activity or the acetylation of histone and/or non-histone protein targets of HDAC’s in mammary carcinoma. Because of the complex role that protein acetylation plays in carcinogenesis and the limited information available about energetics-driven changes in protein acetylation, the study reported herein was designed to assess effects of DER on site specific histone acetylation and on acetylation of p53 at sites known to play role in apoptosis induction (21–23). As a starting point for this work, the effect of DER was studied alone and in combination with suberoylanilide hydroxamic acid (SAHA), an HDAC inhibitor generally considered to target class I and II HDACs, but that has also been reported to inhibit SIRT1 gene transcription (24). An advantage of using SAHA was that its effect on chemically induced mammary carcinogenesis in the rat has been investigated, providing useful information about dose and route of administration (25, 26).
Materials and Methods
Chemicals and Reagents
Primary antibodies used in this study were anti-Bax, anti-Bcl 2 and XIAP from BD Biosciences (San Diego, CA); anti-pACCSer79/ACC, anti-pAktSer473/Akt, anti-pAMPKThr172/AMPK, anti-Apaf-1, anti-Cleaved Caspase-3, anti- p4EBP1Thr37/46/4EBP1, anti-E2F-1, anti-HDAC1, anti-pmTORSer2448/mTOR, anti-pP70S6KThr389/P70S6K, anti-PARP, anti-PI3Kp110, anti-pRaptorSer792/Raptor, anti-pRbSer780/Rb, anti-SIRT-1, anti-rabbit immunoglobulin-horseradish peroxidase-conjugated secondary antibody and LumiGLO reagent with peroxide were purchased from Cell Signaling Technology (Beverly, MA); anti-IGF-1Rα, anti-p53, anti-p21 and anti-mouse immunoglobulin-horseradish peroxidase-conjugated secondary antibody were from Santa Cruz (Santa Cruz, CA); mouse anti-β-actin primary antibody was obtained from Sigma Aldrich (St. Louis, MO); anti-acetyl-p53Lys373 and anti-acetyl-p53Lys373/382, anti- actyl-histone H2ALys9, anti- actyl-histone H2BLys5, anti-actyl-histone H3Lys9, and anti-actyl-histone H4Lys5,8,12,16 were from Millipore (Millipore, Billerica, MA). Carcinogen: 1-methyl-1-nitrosourea (MNU) was obtained (Ash Stevens, Detroit, MI) and stored at −80°C prior to use. The following kits and reagents were used to conduct the experiments: glucose-hexokinase liquid stable reagent (Thermo Fisher Scientific Inc., Waltham, MA); multiplex and signalplex kits for insulin, leptin, and insulin growth factor-1 as well as ELISA kit of adiponectin (Millipore, Billerica, MA); insulin growth factor binding protein 3 (IGFBP-3) ELISA kit (Mediagnost, Reutlingen, Germany); Suberoylanilide hydroxamic acid (SAHA) was purchased from ChemieTek (Indianapolis, IN). Commercially available reagents for determination of plasma glucose were purchased from Thermo Fisher Scientific Inc (Waltham, MA).
Experimental Design
Female Sprague Dawley rats were obtained (Charles River, Wilmington, MA) at 20 days of age. At 21 days of age, rats were injected with 50 mg MNU/kg body weight, i.p., as previously described (27). Rats were individually housed in solid bottomed polycarbonate cages equipped with food cups. At 28 days of age, 1 week after carcinogen injection, rats were assigned by stratified randomization using body weight to one of five groups (30 rats/group): 1) ad libitum fed control (Ad Lib Ctl); 2) 0.1%SAHA (w/w); 3) 40%DER; 4) 40%DER + 0.1%SAHA (w/w); and 5) 40%DER + 0.1%SAHA (w/w)-Release. The approach used for feeding rats has been described in detail (7). Briefly, rats were ad libitum meal-fed with AIN-93G diet for Group 1 or 0.1% (w/w) dietary SAHA in AIN-93G diet for Group 2, and were restricted to 60% the amount of that control animals consumed in remaining groups (40%DER; 4) 40%DER + 0.1%SAHA (w/w); and 5) 40%DER + 0.1%SAHA (w/w)-Release) for six weeks. During the last three weeks of the experiment, the animals in Groups 1 – 4 were maintained on the same diet and fed in the same manner. The animals in the Release group (Group 5) were switched to AIN-93G diet and fed in the same manner as Group 1, i.e. they were released from 40% DER + 0.1% (w/w) SAHA diet.
Throughout the experiment, animal rooms were maintained at 22 ± 1°C with 50% relative humidity and a 12-h light/12-h dark cycle. Rats were weighed twice per week and were palpated for the detection of mammary tumors twice weekly starting from 21 days post carcinogen. At necropsy, rats were skinned and the skin to which mammary gland chains were attached was examined under translucent light for detectable mammary pathologies. All grossly detectable mammary gland pathologies were excised and prepared for histological classification according to published criteria (28, 29). Only confirmed mammary carcinomas are reported since they represented > 98% of the pathologies that were observed. The experimental protocols were reviewed and approved by the Institutional Animal Care and Use Committee and conducted according to the committee guidelines.
Blood collection and plasma biomarker analyses
Blood collection
Following an overnight fast, rats were euthanized via inhalation of gaseous carbon dioxide and blood was directly obtained from the retro-orbital sinus and gravity fed through heparinized capillary tubes (Fisher Scientific, Pittsburgh, PA) into EDTA coated tubes (Becton Dickinson, Franklin Lakes, NJ) for plasma that was isolated by centrifugation at 1000 × g for 10 min at room temperature.
Assessment of circulating molecules
Glucose was determined using a kit obtained from Thermo Fisher Scientific Inc. Insulin-like growth factor 1 (IGF-1), IGF binding protein 3 (IGFBP-3), adiponectin, insulin, and leptin, in plasma were determined using a commercially available ELISA or as previously described (30).
Western blotting
Forty mammary carcinomas (8 per group) were homogenized for Western blotting as described previously (11). The levels of p21, Bax, Bcl 2, XIAP, pACCSer79/ACC, pAktSer473/Akt, pAMPKThr172/AMPK, Apaf-1, Cleaved Caspase-3, p4EBP1Thr37/46/4EBP1, E2F-1, HDAC1, pmTORSer2448/mTOR, pP70S6KThr389/P70S6K, PARP, PI3Kp110, pRaptorSer792/Raptor, pRbSer780/Rb, SIRT-1, IGF-1Rα, p53, acetyl-p53Lys373, acetyl-p53Lys373/382, actyl-histone H2ALys9, actyl-histone H2BLys5, actyl-histone H3Lys9, and actyl-histone H4Lys5/8/12/16 and β-actin were determined using specific primary antibodies, followed by treatment with the appropriate peroxidase-conjugated secondary antibodies and visualized by LumiGLO reagent western blotting detection system. The chemiluminescence signal was captured using a ChemiDoc densitometer (Bio-Rad) and analyzed using Quantity One software (Bio-Rad). All Western blot signals were within a range where the signal was linearly related to the mass of protein and actin-normalized scanning density data were used for analysis.
Statistical Analyses
Differences among groups were evaluated as follows: incidence of mammary carcinomas by the Fischer exact text, the number of mammary carcinomas per rat (multiplicity) by ANOVA after square root transformation of tumor count data, and cancer burden and actin-normalized Western blot data by the nonparametric Kruskal-Wallis test with post hoc unpaired comparisons using the Dwass-Steel-Chritchlow-Fligner test (31–33). Cancer latency was evaluated by survival analysis using the Mantel-Haenszel method (34). Differences in final body weight and circulating analytes were evaluated by ANOVA with post hoc comparisons by the method of Tukey. Effects of treatment group on series of mechanistically interrelated variables were evaluated by multivariate analysis of variance (35). All analyses were performed using Systat statistical analysis software, version 13 (Systat Software, Inc., Chicago, IL). All P values are 2-sided and statistical significance was set a priori at P < 0.05.
Results
Body weight gain
While SAHA had no significant effect on rate of growth, DER slowed the rate of growth either alone or in combination with SAHA as intended, but SAHA+DER had no additional effect on growth in comparison to DER alone. Discontinuing treatment with SAHA+DER (the release group) resulted in rapid weight gain (Fig. 1A), with final body weights approaching that of the control group within 3 weeks from their release from the intervention.
Figure 1.
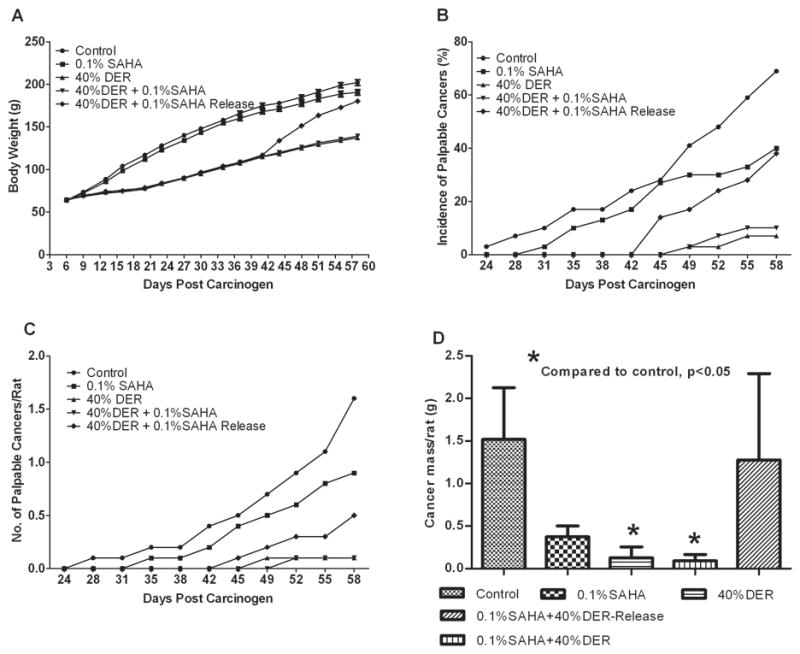
Effects of dietary suberoylanilide hydroxamic acid (SAHA) with dietary energy restriction (DER) on body weight gain and the carcinogenic response. (A) Body weight gain. (B) Incidence of palpable mammary cancer. (C) Number of palpable mammary carcinoma number per rat. (D) Tumor weight. For panels A and D, values are means ±SEM, n=30 rats/group.
Carcinogenic Response
SAHA numerically suppressed the carcinogenic response in comparison to the control group but the effects were not statistically significant after adjustment for multiple comparisons (Fig. 1B, C, and D). Cancer incidence was reduced 17.1%, cancer multiplicity was 36.5% lower, cancer burden was decreased by 74.9%, and cancer latency was extended 9.6%. The carcinogenic response was significantly inhibited by DER or DER + SAHA. In comparison to the control group, cancer incidence was reduced 57.2% or 42.8%, cancer multiplicity was 84.9% or 73.8% lower, cancer burden was decreased 91.6% or 91.0%, and cancer latency was extended 23.1% or 21.2%, respectively. When the animals in DER + SAHA-Release group were ad libitum fed control diet for three weeks after six-weeks of feeding the DER + SAHA experimental diet, inhibition of the carcinogenic response was reversed, but cancer multiplicity remained lower than observed in the control group (P < 0.05).
Acetylation histones and p53
Acetylation of histones in mammary carcinomas was significantly increased by SAHA (SAHA vs. control or DER, P < 0.05, multivariate Hotelling P < 0.001), but was decreased by DER (DER vs. control or SAHA, P < 0.05, multivariate Hotelling P < 0.001), although DER did not affect acetylation of histone H3 (Table 1, representative Western blots in Supplementary Fig. S1). SAHA in combination with DER appeared to block DER mediated histone deacetylation with values being similar to those observed in the control group.
Table 1.
Effect of dietary SAHA and/or dietary energy restriction on HDAC and histone and p53 acetylation in mammary carcinomas
| Dietary treatment | Ad Lib Ctl | 0.1% SAHA | 40%DER | 40%DER +0.1%SAHA | 40%DER +0.1% SAHA-Release | Overall P |
|---|---|---|---|---|---|---|
| p53Lys373 | 146 ± 8a | 170 ± 2b | 115 ± 4c | 163 ± 6ab | 152 ± 6ab | < 0.0001 |
| p53Lys373/382 | 338 ± 11a | 425 ± 8b | 300 ± 9c | 352 ± 6a | 327 ± 5ac | < 0.0001 |
| P53 | 109 ± 6 | 119 ± 4 | 108 ± 2 | 113 ± 4 | 108 ± 3 | 0.286 |
| H2ALys9 | 310 ± 15a | 425 ± 23b | 265 ± 10a | 316 ± 14a | 328 ± 16a | < 0.0001 |
| H2BLys5 | 245 ± 14a | 282 ± 14a | 166 ± 11b | 194 ± 12b | 165 ± 12b | < 0.0001 |
| H3Lys9 | 138 ± 7a | 248 ± 14b | 139 ± 8a | 147 ± 6a | 149 ± 8a | < 0.0001 |
| H4Lys5/8/12/16 | 433 ± 22a | 719 ± 41b | 365 ± 22a | 620 ± 40b | 439 ± 24a | < 0.0001 |
| SIRT1 | 82 ± 3a | 61 ± 1b | 110 ± 5c | 90 ± 4a | 84 ± 4a | < 0.0001 |
| HDAC1 | 305 ± 3a | 232 ± 3b | 298 ± 7a | 236 ± 4b | 307 ± 4a | < 0.0001 |
Values are means ± SEM (n=8). Actin-normalized western blot data, which are semi-quantitative estimates of protein expression, were analyzed by Kruskall-Wallis rank test. Different superscripts within the same row are statistically significant among different groups (P < 0.05). Ratio, the ratio of phospho-protein (arbitrary units of optical density) to non-phospho-protein (arbitrary units of optical density). Ad Lib, ad libitum; Ctl, control; Ad Lib, ad libitum; Ctl, control; SAHA, suberoylanilide hydroxamic acid; DER, dietary energy restriction. H2A, actyl-histone H2A; H2B, actyl-histone H2B; H3, actyl-histone H3; H4, actyl-histone H4; HDAC, histone deacetylase; SIRT-1, sirtuin 1.
Mammary carcinomas from rats released from SAHA+DER also had levels of histone acetylation similar to the control group with the exception of H2B which remained low (P < 0.05). Similarly, acetyl-p53Lys373 and acetyl- p53Lys373/382 were significantly increased in carcinomas from SAHA treated rats, decreased in DER carcinomas and DER mediated deacetylation was restored by SAHA. Acetylation of both sites was similar in carcinomas from rats released from SAHA+DER and control carcinomas.
Protein levels of SIRT1 and HDAC1 were also determined (Table 1). SAHA decreased levels of both proteins (SAHA vs. control or DER, p < 0.05); whereas, DER increased SIRT1(DER vs. control or SAHA, P < 0.05) and had no effect on HDAC1. SAHA diminished SIRT1 induction by DER and DER had no effect on the SAHA mediated reduction in HDAC1. Levels of both proteins were similar in the SAHA+DER release and control groups.
Apoptosis and cell cycle regulation
Patterns of acetylation of histones and non-histone proteins such as p53 affect many cellular processes. The focus of this study was on apoptosis. Intervention effects on two indicators of apoptosis are shown (Table 2, representative Western blots in Supplementary Fig. 2S). Both SAHA and DER increased levels of cleaved caspase-3 and PARP cleavage reflected by the ratio of PARP89 to PARP 116 (SAHA or DER vs. control, P < 0.05). While these effects were observed in the SAHA+DER group, the level of cleaved caspase 3 was lower than in DER alone (SAHA+DER vs. DER, P < 0.05) and the level of cleaved PARP was higher than in either DER or SAHA alone (SAHA+DER vs. SAHA or DER, P < 0.05). Cleaved caspase-3 in the SAHA+DER release group was similar to that observed in the control group but the level of cleaved PARP remained elevated in carcinomas from the Release group. Effects on cellular machinery involved in regulating apoptosis were also investigated. Apaf-1 was elevated by SAHA, DER, and SAHA+DER as well as the SAHA+DER release group compared to the control (P < 0.05). The widely assessed indicator of apoptosis mediated by the intrinsic pathway, i.e. the ratio of BAX/BCL-2 was also elevated by SAHA or DER and almost 2-fold further elevated in the SAHA+DER group compared to the control (P < 0.05), and it remained elevated in the SAHA+DER release group. The effect on the ratio was primarily accounted for by suppression of the level of cellular BCL-2 in the intervention groups. Levels of XIAP, an inhibitor of caspase activity, were suppressed by SAHA or DER compared to the control (P < 0.05) but unaffected in the combined treatment or release groups.
Table 2.
Effect of dietary SAHA and/or dietary energy restriction on indicators of cell proliferation and apoptosis in mammary carcinomas
| Dietary treatment | Ad Lib Ctl | 0.1% SAHA | 40%DER | 40%DER +0.1%SAHA | 40%DER +0.1% SAHA-Release | Overall P |
|---|---|---|---|---|---|---|
| Cell proliferation | ||||||
| E2F1 | 130 ± 5a | 115 ± 6ac | 85 ± 5b | 114 ± 5ac | 105 ± 2c | < 0.0001 |
| RbSer780 ratio | 0.90 ± 0.02a | 0.84 ± 0.01ac | 0.68 ± 0.01b | 0.62 ± 0.01b | 0.79 ± 0.02c | < 0.0001 |
| p21 | 128 ± 5a | 139 ± 3ac | 157 ± 3bc | 164 ± 5b | 144 ± 3c | < 0.0001 |
|
| ||||||
| Apoptosis | ||||||
| Cleaved caspase-3 | 111 ± 5a | 318 ± 11bc | 357 ± 16b | 291 ± 10c | 110 ± 6a | < 0.0001 |
| PARP89 | 50 ± 2a | 59 ± 2bc | 57 ± 3ac | 66 ± 2b | 56 ± 2ac | < 0.0001 |
| PARP116 | 49 ± 3a | 41 ± 2ab | 41 ± 2ab | 38 ± 1b | 40 ± 2b | 0.006 |
| PARP89/116 ratio | 1.02 ± 0.03a | 1.43 ± 0.03b | 1.40 ± 0.03b | 1.74 ± 0.03c | 1.42 ± 0.03b | < 0.0001 |
| Apaf-1 | 152 ± 4a | 202 ± 7b | 230 ± 7c | 203 ± 6b | 185 ± 7b | < 0.0001 |
| Bax | 72 ± 2 | 90 ± 3 | 88 ± 3 | 110 ± 4 | 94 ± 3 | 0.258 |
| Bcl-2 | 216 ± 5 | 163 ± 3 | 161 ± 3 | 114 ± 3 | 154 ± 4 | 0.382 |
| Bax/Bcl-2 ratio | 0.33 ± 0.01a | 0.55 ± 0.02b | 0.55 ± 0.02b | 0.97 ± 0.04c | 0.61 ± 0.02b | < 0.0001 |
| XIAP | 1755 ± 29a | 1485 ± 24b | 1405 ± 38b | 1642 ± 30a | 1741 ± 30a | < 0.0001 |
Values are means ± SEM (n=8). Actin-normalized western blot data, which are semi-quantitative estimates of protein expression, were analyzed by Kruskall-Wallis rank test. Different superscripts within the same row are statistically significant among different groups (P < 0.05). Ratio, the ratio of phospho-protein (arbitrary units of optical density) to non-phospho-protein (arbitrary units of optical density). Ad Lib, ad libitum; Ctl, control; Ad Lib, ad libitum; Ctl, control; SAHA, suberoylanilide hydroxamic acid; DER, dietary energy restriction. Apaf-1, apoptosis protease-activating factor-1; Bax, Bcl (B cell lymphoma)-associated X; Bcl, B cell leukemia oncogene; E2F1, transcription factor family including E2F- and DP-like subunits; PARP, Poly (ADP-ribose) polymerase; Retinoblastoma, Rb; XIAP, X-linked inhibitor of apoptosis protein.
Given that effects on cell proliferation are generally observed to occur concomitantly with apoptosis, and that alterations in acetylation are known to impact this process, three commonly affected molecular components of the cell cycle apparatus were assessed (Table 2). DER had the greatest effects in reducing the phosphorylation of Rb and levels of unbound E2F1 and in inducing p21 compared to the control (P < 0.05). SAHA was without effect and tended to blunt the effects of DER on these molecular determinants when given in combination. Levels of these proteins in the release group were intermediate to those observed in the SAHA or DER group.
mTOR-related signaling
HDACs are induced by cellular stress and DER is a recognized energy stressor. Therefore, it was decided that one of the primary networks that responds to energetic stress should be evaluated. DER induced the same pattern of mTOR-related regulation (Table 3, representative Western blots in Supplementary Fig. S3) as previously reported (11). Specifically, compared to the control, DER induced upregulation of AMPK-activated protein kinase and its direct targets, acetyl CoA carboxylase (ACC) and Raptor, and it suppressed multiple elements in the insulin/IGF-1 signaling cascade that also regulate mTOR. Specifically, DER downregulated IGF recetor 1 (IGF1Rα), PI-3 kinase subunit p110 (PIK3CA), activated protein kinase B (Akt) and the site on mTOR that it phosphorylates. Moreover, DER down regulated two downstream targets of activated mTOR, p70 S6K and 4EBP1. Of note, because it has not been previously reported, SAHA alone induced the same pattern of mTOR related regulation as DER. Although the effects were not as robust as observed in response to DER, they were generally statistically significant. This was confirmed by multivariate regression of all parameters (Table 3) with multivariate Hotelling statistics P < 0.001 for both DER and SAHA. While SAHA reversed the effects of DER on the acetylation of protein targets (Table 2), the effects of SAHA+DER on mTOR-related signaling were essentially identical to the effects of DER alone. In the SAHA-DER release group, the pattern of regulation was similar to that observed in the control group.
Table 3.
Effect of dietary SAHA and/or dietary energy restriction on regulatory nodes that affect the mTOR-related signaling in mammary carcinomas
| Dietary treatment | Ad Lib Ctl | 0.1% SAHA | 40%DER | 40%DER +0.1%SAHA | 40%DER +0.1% SAHA-Release | Overall P |
|---|---|---|---|---|---|---|
| IGF-1R | 121 ± 5a | 105 ± 6ab | 93 ± 7b | 95 ± 6bc | 117 ± 5ac | <0.0001 |
| PI3Kp110 | 90 ± 5a | 84 ± 3a | 49 ± 3b | 71 ± 5c | 86 ± 6a | <0.0001 |
| AktSer473 ratio | 1.73 ± 0.03a | 1.55 ± 0.01b | 1.39 ± 0.02c | 1.30 ± 0.02c | 1.61 ± 0.02b | <0.0001 |
| AMPKThr172 ratio | 0.106 ± 0.006a | 0.182 ± 0.004b | 0.203 ± 0.003c | 0.144 ± 0.004d | 0.116 ± 0.005a | <0.0001 |
| ACCSer79 ratio | 1.62 ± 0.04a | 1.91 ± 0.04b | 2.48 ± 0.06c | 2.10 ± 0.08b | 1.50 ± 0.04a | <0.0001 |
| RaptorSer792 ratio | 0.131 ± 0.002a | 0.160 ± 0.004bc | 0.168 ± 0.006b | 0.153 ± 0.002ac | 0.141 ± 0.002a | <0.0001 |
| mTORSer2448 ratio | 0.65 ± 0.01a | 0.58 ± 0.04a | 0.27 ± 0.03b | 0.44 ± 0.03c | 0.59 ± 0.04a | <0.0001 |
| P70S6KThr389 ratio | 1.51 ± 0.04a | 1.26 ± 0.03b | 0.83 ± 0.05c | 0.98 ± 0.04c | 1.24 ± 0.06b | <0.0001 |
| 4EBP1Thr37/46 ratio | 1.38 ± 0.04a | 1.03 ± 0.06b | 0.95 ± 0.04b | 0.97 ± 0.04b | 1.23 ± 0.06a | <0.0001 |
Values are means ± SEM (n=8). Actin-normalized western blot data, which are semi-quantitative estimates of protein expression, were analyzed by Kruskall-Wallis rank test. Different superscripts within the same row are statistically significant among different groups (P < 0.05). Ratio, the ratio of phospho-protein (arbitrary units of optical density) to non-phospho-protein (arbitrary units of optical density). Ad Lib, ad libitum; Ctl, control; Ad Lib, ad libitum; Ctl, control; SAHA, suberoylanilide hydroxamic acid; DER, dietary energy restriction. ACC, acetyl-CoA carboxylase; Akt, protein kinase B; AMPK, adenosine monophosphate-activated protein kinase; 4EBP1, eukaryotic translation initiation factor 4E-binding protein 1; Hif-1α, hypoxia-inducible factor-1α; IGF-1R, insulin growth factor 1 receptor; mTOR, mammalian target of rapamycin; P70S6K, 70-kDa ribosomal protein S6 kinase; PI3Kp110, Phosphoinositide 3-kinase p110; Raptor, regulatory associated protein of mTOR.
Systemic Factors
Given the unexpected effects of SAHA on mTOR-related signaling, plasma was assessed for circulating factors known to regulate various nodes of this signaling network. Compared to the control, SAHA alone had no significant effects on IGF-1, IGFBP3, insulin, glucose, or leptin, although it did induce a marked increase in adiponectin (Table 4). On the other hand, these same parameters, other than adiponectin, were significantly lower in the plasma of DER and SAHA+DER rats in comparison to the control group and adiponectin was elevated. The values in the SAHA+DER release group for these plasma analytes were similar to the values observed in the control group.
Table 4.
Effect of dietary SAHA and/or dietary energy restriction on plasma analytes
| Dietary treatment | Ad Lib Ctl | 0.1%SAHA | 40%DER | 40%DER +0.1%SAHA | 40%DER +0.1%SAHA-Release | Overall P |
|---|---|---|---|---|---|---|
| IGF-1 (ng/ml) | 370 ± 9a,b | 333 ± 8a | 225 ± 9c | 248 ±6c | 388 ± 17b | < 0.0001 |
| IGFBP-3 (ng/ml) | 122 ± 4ab | 120 ± 4ab | 113 ±5a | 126 ± 3ab | 135 ± 5b | 0.0092 |
| IGF-1/IGFBP-3 | 3.06 ± 0.05a | 2.83 ± 0.08a | 2.01 ± 0.05b | 2.00 ± 0.06b | 2.91 ± 0.11a | < 0.0001 |
| Insulin (pg/ml) | 1295 ± 40a | 1286 ± 59a | 997 ± 21b | 1060 ± 38a | 1217 ± 55a | < 0.0001 |
| Glucose (mg/dl) | 148 ± 4 | 141 ± 5 | 127 ± 5 | 133 ± 7 | 134 ± 7 | 0.1129 |
| Adiponectin (μg/ml) | 17.4 ± 0.8a | 26.3 ± 0.9b | 21.3 ± 0.8c | 24.9 ± 0.9b | 20.2 ± 0.7ac | 0.019 |
| Leptin (pg/ml) | 2011 ± 56a | 2058 ± 63a | 1113 ± 37b | 1355 ± 69c | 1879 ± 55a | < 0.0001 |
Values are means ± SEM (n=30). Data were analyzed by one-way ANOVA with Bonferroni Post Hoc Test. Values within a row with different superscripts are statistically different from each other (P < 0.05). Ad Lib, ad libitum; Ctl, control; Ad Lib, ad libitum; Ctl, control; SAHA, suberoylanilide hydroxamic acid; DER, dietary energy restriction.
Discussion
Investigation of the role of protein acetylation and deacetylation in the prevention and control of cancer is a rapidly expanding field and not surprisingly, there are many contradictions in what has been reported. Nonetheless, it is clear that changes in acetylation patterns occur in response to cellular stresses including those associated with the development of cancer and with DER (13). Other than the consistent observation that chronic DER induces SIRT1, limited additional information exists about the role of DER-mediated HDAC induction in accounting for DER’s powerful, physiologic inhibition of cancer, especially in target organs such as the breast.
Carcinogenic response and acetylation
A growing body of literature indicates that protein deacetylation, at least in certain contexts and involving specific protein acetylation sites, promotes several steps in the carcinogenic process. If this is in fact the case, it creates the expectation that inhibition of HDAC activity would protect against cancer. Dietary administration of SAHA, a class I and II HDAC inhibitor that also inhibits SIRT1 gene transcription, at a concentration that had no effect on animal growth rate (Table 1, Fig. 1A), reduced all aspect of the carcinogenic response although the effects were not statistically significant when adjusted for multiple comparisons. This finding is consistent with other reports in similar model systems for breast cancer (25, 26). And SAHA increased the acetylation of the proteins selected for assessment (Table 1). On the other hand, DER, which robustly inhibited all the criteria by which the carcinogenic response was assessed (Table 1, Fig. 1), induced the deacetylation of the same proteins whose acetylation SAHA increased (Table 1). When SAHA and DER were combined, protein acetylation was restored to the level observed in carcinomas from control animals, but inhibition of carcinogenesis was essentially the same as observed in the DER only group. These data are consistent with at least one of three explanations: the protective activity of DER is independent of its effect on HDAC activity, the protective effect of DER under the conditions of the experiment is so dominant based on other mechanisms that changes in anticancer activity associated with DER and SAHA mediated effects on acetylation were of no consequence, or the acetylation sites assessed are not those critical to elucidating the effects of DER. While the effects on cancer incidence of releasing rats from combined treatment with SAHA and DER support the notion that DER’s anticancer activity is independent of effects on protein acetylation, the cancer multiplicity data in which cancers per rat remained 2.5 times lower than in the control group, leave open the question of whether blocking the effects of DER on protein deacetylation might result in apoptosis-mediated deletion of transformed cells and sustainable protection against some aspects of carcinogenesis in the absence of continuous treatment, a phenomenon that we refer to as early cure which is a type of cancer prevention that has been sought since the term chemoprevention was coined (36, 37).
Protein acetylation and tumor size homeostasis
In an effort to gain more insight about the alternatives posed in the previous subsection about the effects of DER on the carcinogenic response, attention was directed to cellular processes that regulate tumor growth and that are markedly affected by DER as previously reported (8, 9), specifically apoptosis and cell growth. While effects on elements of the cell cycle machinery involved with the G1/S transition were apparent and consistent with previously reported effects of DER, the outcome measures of apoptosis varied in a manner consistent with the unmasking of the apoptotic potential of DER by SAHA (Table 2). Levels of cleaved PARP and the ratio of BAX/BCL2, indicators of apoptosis induction and the pro-apoptotic potential of the environment, respectively, were markedly elevated in the SAHA+DER group in comparison to the DER group (Table 2). This is consistent with the changes in acetylation patterns reported (Table 1) and the effects of HDAC inhibitors on p53 and BCL2 (38–40). It is noteworthy that cleaved caspase 3 did not follow this pattern of induction. Whether this is due to higher levels of activated caspase 3 inhibitor, XIAP, which was induced in the SAHA+DER group, remains to be determined. If in fact, HDAC inhibitors do permit greater induction of apoptosis by DER, which is consistent with the cancer multiplicity data in the SAHA+DER release group, it will be critical to determine the basis of selectivity in deleting transformed foci of cells.
Metabolic regulation
Patterns of protein acetylation are known to be involved in regulating many aspects of cellular function including energy metabolism with a particularly strong causal linkage existing class III HDACs, the sirtuins, which are activated by NAD+. Presumably, DER induces SIRT1 due to its effect on the NAD/NADH ratio and the energy charge of the cell, although this has not been investigated in cancer models, and many argue that DER mediates effects on systemic factors and that it does not directly impact energy metabolism in peripheral tissues such as the mammary gland (41). Those arguments aside, DER induced higher levels of SIRT1 in mammary carcinoma without affecting cellular content of HDAC1 (Table 1). Hence, a direct effect of DER in the target tissue is apparent and might signal that energy metabolism is perturbed in these carcinomas. Consistent with published work, SAHA was observed to reduce cellular levels of SIRT1 as well as HDAC1, which in itself would be expected to reduce protein deacetylation.
The clear indication from the literature is that HDACs, at least class III, may play a role as intracellular energy sensors (13). Given our published evidence that a major site of DER’s impact on cancer is via highly conserved intracellular energy sensors and that the mTOR-related network of proteins is involved in mediating DERs anticancer activity, the effects of the various interventions on mTOR-related signaling was assessed. SAHA downregulated mTOR activity at each node that was studied (Table 3). While the magnitude of the effect was not as robust as DER, the effects were nonetheless statistically significant. Such activity has not previously been attributed to SAHA, but patterns of acetylation have been noted to affect mTOR activity, although not in mammary carcinomas (42). What was striking about the effects of SAHA is that it suppressed activated Akt in the absence of any effect on plasma levels of fasting insulin, IGF-1, IGFBP3 or glucose (Table 4). SAHA also induce activation of AMP activated protein kinase, an effect that might be associated with its induction of plasma adiponection levels, since adiponectin is known to activate AMPK. Thus, SAHA and perhaps other HDAC inhibitors may represent a new category of energy restriction mimetic agent (43). DER induced effects on both circulating factors and mTOR related signaling were consistent with previous reports. However, there was little evidence that SAHA+DER altered circulating factors or the activity of mTOR-related signaling nodes that would suggest combinatorial effects on this network, a network that is misregulated in the majority of human breast cancers (44–47).
Limitations
A limitation of the work reported is the number of acetylation sites and HDACs that were accessed. Nonetheless, the findings provide valuable information that can be used to guide future experiments. Another limitation was the duration of time that the SAHA+DER group was monitored following release from treatment since it could be argued that what was observed was due to a delay in tumor emergence rather than the deletion of transformed foci of mammary epithelial cells.
Translational significance
The identification of short term intervention strategies that can render long term protection against cancer would meet a critical public health objective. Given the episodic nature and high prevalence of dieting in the human population (48), efforts to identify agents that could be coupled with dieting to induce deletion of transformed foci of cells from a tissue could have high clinical impact. The evidence reported herein, particularly with regard to indicators of apoptosis and the machinery that regulate the intrinsic pathway of apoptosis induction, indicate that further investigation of HDAC inhibitors in combination with DER are warranted. A focus on inhibitors of Class III HDACs is of interest given their involvement in the cellular stress response, apoptosis, and energy metabolism.
Supplementary Material
Acknowledgments
Grant Support
This work was supported in part by United States Public Health Services Grant CA52626 from the National Cancer Institute.
The authors thank Amanda Blasingame and Elizabeth Neil for their expert technical assistance.
Abbreviations
- 4EBP1
eukaryotic translation initiation factor 4E-binding protein 1
- ACC
acetyl-CoA carboxylase
- Akt
protein kinase B
- AMPK
adenosine monophosphate-activated protein kinase
- Apaf-1
apoptosis protease-activating factor-1
- Bax
Bcl (B cell lymphoma)-associated X
- Bcl
B cell leukemia oncogene
- CCD
charge-coupled device
- DSCF
Dwass-Steel-Chritchlow-Fligner
- DTT
dithiothreitol
- DER
dietary energy restriction
- E2F1
transcription factor family including E2F- and DP-like subunits
- EDTA
Ethylene diamine tetraacetic acid
- EGTA
ethylene glycol tetraacetic acid
- H2A
actyl-histone H2A
- H2B
actyl-histone H2B
- H3
actyl-histone H3
- H4
actyl-histone H4
- HDAC
histone deacetylase
- IGF-1
insulin-like growth factor 1
- IGFBP-3
insulin-like growth factor binding protein 3
- IGF-1R
insulin growth factor 1 receptor
- MNU
N-methyl-N-nitrosourea
- mTOR
mammalian target of rapamycin
- P70S6K
70-kDa ribosomal protein S6 kinase
- PARP
Poly (ADP-ribose) polymerase
- PI3Kp110
Phosphoinositide 3-kinase p110
- PPAR
peroxisome proliferator-activated receptor
- Raptor
regulatory associated protein of mTOR
- Rb
retinoblastoma
- SAHA
suberoylanilide hydroxamic acid
- SDS-PAGE
sodium dodecyl sulfate-polyacrylamide gradient gel electrophoresis
- SIRT-1
sirtuin 1
- XIAP
X-linked inhibitor of apoptosis protein
Footnotes
Disclosure of Potential Conflicts of Interest: There are no conflicts of interest.
Conflict of Interest Statement: None.
References
- 1.Thompson HJ, McTiernan A. Weight cycling and cancer: weighing the evidence of intermittent caloric restriction and cancer risk. Cancer Prev Res (Phila) 2011;4:1736–42. doi: 10.1158/1940-6207.CAPR-11-0133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hursting SD, Smith SM, Lashinger LM, Harvey AE, Perkins SN. Calories and carcinogenesis: lessons learned from 30 years of calorie restriction research. Carcinogenesis. 2010;31:83–9. doi: 10.1093/carcin/bgp280. [DOI] [PubMed] [Google Scholar]
- 3.Thompson HJ, Jiang W, Zhu Z. Energetics and Cancer: Exploring a Road Less Traveled. In: McTiernan A, editor. Physical Activity, Dietary Calorie Restriction, and Cancer. 1. Springer Science + Business Media, LLC; 2011. pp. 55–67. [Google Scholar]
- 4.Thompson HJ, McTiernan A. Weight cycling and cancer: weighing the evidence of intermittent caloric restriction and cancer risk. Cancer Prev Res (Phila) 2011;4:1736–42. doi: 10.1158/1940-6207.CAPR-11-0133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.International Agency for Research on Cancer. Physical Activity, Weight Control, and Cancer. [8] Lyon: IARC Press; 2002. [Google Scholar]; Vainio H, Bianchin IF. Cancer Prevention Handbook. [Google Scholar]
- 6.World Cancer Research Fund, American Institute for Cancer Research. Food, Nutrition, Physical Activity, and the Prevention of Cancer: a Global Perspective. Washington, DC: AICR; 2007. [Google Scholar]
- 7.Zhu Z, Haegele AD, Thompson HJ. Effect of caloric restriction on pre-malignant and malignant stages of mammary carcinogenesis. Carcinogenesis. 1997;18:1007–12. doi: 10.1093/carcin/18.5.1007. [DOI] [PubMed] [Google Scholar]
- 8.Jiang W, Zhu Z, Thompson HJ. Effect of energy restriction on cell cycle machinery in 1-methyl-1-nitrosourea-induced mammary carcinomas in rats. Cancer Res. 2003;63:1228–34. [PubMed] [Google Scholar]
- 9.Thompson HJ, Zhu Z, Jiang W. Identification of the apoptosis activation cascade induced in mammary carcinomas by energy restriction. Cancer Res. 2004;64:1541–5. doi: 10.1158/0008-5472.can-03-3108. [DOI] [PubMed] [Google Scholar]
- 10.Zhu Z, Jiang W, McGinley J, Wolfe P, Thompson HJ. Effects of dietary energy repletion and IGF-1 infusion on the inhibition of mammary carcinogenesis by dietary energy restriction. Mol Carcinog. 2005;42:170–6. doi: 10.1002/mc.20071. [DOI] [PubMed] [Google Scholar]
- 11.Jiang W, Zhu Z, Thompson HJ. Dietary energy restriction modulates the activity of AMP-activated protein kinase, Akt, and mammalian target of rapamycin in mammary carcinomas, mammary gland, and liver. Cancer Res. 2008;68:5492–9. doi: 10.1158/0008-5472.CAN-07-6721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Alers S, Loffler AS, Wesselborg S, Stork B. Role of AMPK-mTOR-Ulk1/2 in the regulation of autophagy: cross talk, shortcuts, and feedbacks. Mol Cell Biol. 2012;32:2–11. doi: 10.1128/MCB.06159-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kwon HS, Ott M. The ups and downs of SIRT1. Trends Biochem Sci. 2008;33:517–25. doi: 10.1016/j.tibs.2008.08.001. [DOI] [PubMed] [Google Scholar]
- 14.Baur JA, Chen D, Chini EN, Chua K, Cohen HY, de CR, et al. Dietary restriction: standing up for sirtuins. Science. 2010;329:1012–3. doi: 10.1126/science.329.5995.1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Marks PA, Richon VM, Rifkind RA. Histone deacetylase inhibitors: inducers of differentiation or apoptosis of transformed cells. J Natl Cancer Inst. 2000;92:1210–6. doi: 10.1093/jnci/92.15.1210. [DOI] [PubMed] [Google Scholar]
- 16.Liu T, Liu PY, Marshall GM. The critical role of the class III histone deacetylase SIRT1 in cancer. Cancer Res. 2009;69:1702–5. doi: 10.1158/0008-5472.CAN-08-3365. [DOI] [PubMed] [Google Scholar]
- 17.Liu T, Kuljaca S, Tee A, Marshall GM. Histone deacetylase inhibitors: multifunctional anticancer agents. Cancer Treat Rev. 2006;32:157–65. doi: 10.1016/j.ctrv.2005.12.006. [DOI] [PubMed] [Google Scholar]
- 18.Baur JA, Ungvari Z, Minor RK, Le Couteur DG, de CR. Are sirtuins viable targets for improving healthspan and lifespan? Nat Rev Drug Discov. 2012;11:443–61. doi: 10.1038/nrd3738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ma X, Ezzeldin HH, Diasio RB. Histone deacetylase inhibitors: current status and overview of recent clinical trials. Drugs. 2009;69:1911–34. doi: 10.2165/11315680-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 20.Stunkel W, Campbell RM. Sirtuin 1 (SIRT1): the misunderstood HDAC. J Biomol Screen. 2011;16:1153–69. doi: 10.1177/1087057111422103. [DOI] [PubMed] [Google Scholar]
- 21.Marks PA, Richon VM, Rifkind RA. Histone deacetylase inhibitors: inducers of differentiation or apoptosis of transformed cells. J Natl Cancer Inst. 2000;92:1210–6. doi: 10.1093/jnci/92.15.1210. [DOI] [PubMed] [Google Scholar]
- 22.Cellai C, Balliu M, Laurenzana A, Guandalini L, Matucci R, Miniati D, et al. The new low-toxic histone deacetylase inhibitor S-(2) induces apoptosis in various acute myeloid leukaemia cells. J Cell Mol Med. 2012;16:1758–65. doi: 10.1111/j.1582-4934.2011.01464.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Condorelli F, Gnemmi I, Vallario A, Genazzani AA, Canonico PL. Inhibitors of histone deacetylase (HDAC) restore the p53 pathway in neuroblastoma cells. Br J Pharmacol. 2008;153:657–68. doi: 10.1038/sj.bjp.0707608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kyrylenko S, Kyrylenko O, Suuronen T, Salminen A. Differential regulation of the Sir2 histone deacetylase gene family by inhibitors of class I and II histone deacetylases. Cellular and Molecular Life Sciences. 2003;60:1990–7. doi: 10.1007/s00018-003-3090-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cohen LA, Amin S, Marks PA, Rifkind RA, Desai D, Richon VM. Chemoprevention of carcinogen-induced mammary tumorigenesis by the hybrid polar cytodifferentiation agent, suberanilohydroxamic acid (SAHA) Anticancer Res. 1999;19:4999–5005. [PubMed] [Google Scholar]
- 26.Cohen LA, Marks PA, Rifkind RA, Amin S, Desai D, Pittman B, et al. Suberoylanilide hydroxamic acid (SAHA), a histone deacetylase inhibitor, suppresses the growth of carcinogen-induced mammary tumors. Anticancer Res. 2002;22:1497–504. [PubMed] [Google Scholar]
- 27.Thompson HJ, McGinley JN, Rothhammer K, Singh M. Rapid induction of mammary intraductal proliferations, ductal carcinoma in situ and carcinomas by the injection of sexually immature female rats with 1-methyl-1-nitrosourea. Carcinogenesis. 1995;16:2407–11. doi: 10.1093/carcin/16.10.2407. [DOI] [PubMed] [Google Scholar]
- 28.Singh M, McGinley JN, Thompson HJ. A comparison of the histopathology of premalignant and malignant mammary gland lesions induced in sexually immature rats with those occurring in the human. Lab Invest. 2000;80:221–31. doi: 10.1038/labinvest.3780025. [DOI] [PubMed] [Google Scholar]
- 29.Thompson HJ, Singh M, McGinley J. Classification of premalignant and malignant lesions developing in the rat mammary gland after injection of sexually immature rats with 1-methyl-1-nitrosourea. J Mammary Gland Biol Neoplasia. 2000;5:201–10. doi: 10.1023/a:1026495322596. [DOI] [PubMed] [Google Scholar]
- 30.Zhu Z, Jiang W, McGinley JN, Prokopczyk B, Richie JP, Jr, El BK, et al. Mammary gland density predicts the cancer inhibitory activity of the N-3 to N-6 ratio of dietary fat. Cancer Prev Res (Phila) 2011;4:1675–85. doi: 10.1158/1940-6207.CAPR-11-0175. [DOI] [PubMed] [Google Scholar]
- 31.Snedecor GW, Cochran WG. Statistical Methods. 8. Ames, IA: Iowa State University Press; 1989. [Google Scholar]
- 32.Sokal RR, Rohlf FJ. Biometry the principles and practice of statistics in biological research. 3. New York: W.H. Freeman; 1995. [Google Scholar]
- 33.Huh MH, Critchlow DE, Verducci JS, Kiecolt-Glaser J, Glaser R, Malarkey WB. A symmetric analysis of paired rankings with application to temporal patterns of hormonal concentration. Biometrics. 1995;51:1361–71. [PubMed] [Google Scholar]
- 34.Collett D. Modelling Survival Data in Medical Research. London: Chapman and Hall; 1994. [Google Scholar]
- 35.Morrison DF. Multivariate Statistical Methods. 3. New York: McGraw-Hill Publishing Co; 1990. [Google Scholar]
- 36.Sporn MB. Approaches to prevention of epithelial cancer during the preneoplastic period. Cancer Res. 1976;36:2699–702. [PubMed] [Google Scholar]
- 37.Sporn MB, Dunlop NM, Newton DL, Smith JM. Prevention of chemical carcinogenesis by vitamin A and its synthetic analogs (retinoids) Fed Proc. 1976;35:1332–8. [PubMed] [Google Scholar]
- 38.Li D, Marchenko ND, Moll UM. SAHA shows preferential cytotoxicity in mutant p53 cancer cells by destabilizing mutant p53 through inhibition of the HDAC6-Hsp90 chaperone axis. Cell Death Differ. 2011;18:1904–13. doi: 10.1038/cdd.2011.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fortson WS, Kayarthodi S, Fujimura Y, Xu H, Matthews R, Grizzle WE, et al. Histone deacetylase inhibitors, valproic acid and trichostatin-A induce apoptosis and affect acetylation status of p53 in ERG-positive prostate cancer cells. Int J Oncol. 2011;39:111–9. doi: 10.3892/ijo.2011.1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Uo T, Veenstra TD, Morrison RS. Histone deacetylase inhibitors prevent p53-dependent and p53-independent Bax-mediated neuronal apoptosis through two distinct mechanisms. J Neurosci. 2009;29:2824–32. doi: 10.1523/JNEUROSCI.6186-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pollak M. Do cancer cells care if their host is hungry? Cell Metab. 2009;9:401–3. doi: 10.1016/j.cmet.2009.04.006. [DOI] [PubMed] [Google Scholar]
- 42.Chu F, Chou P, Mirkin BL, Mousa SA, Rebbaa A. Cellular conditioning with trichostatin A enhances the anti-stress response through up-regulation of HDAC4 and down-regulation of the IGF/Akt pathway. Aging Cell. 2008;7:516–25. doi: 10.1111/j.1474-9726.2008.00403.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhu Z, Jiang W, McGinley JN, Thompson HJ. 2-Deoxyglucose as an energy restriction mimetic agent: effects on mammary carcinogenesis and on mammary tumor cell growth in vitro. Cancer Res. 2005;65:7023–30. doi: 10.1158/0008-5472.CAN-05-0453. [DOI] [PubMed] [Google Scholar]
- 44.Efeyan A, Sabatini DM. mTOR and cancer: many loops in one pathway. Curr Opin Cell Biol. 2010;22:169–76. doi: 10.1016/j.ceb.2009.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zoncu R, Efeyan A, Sabatini DM. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol. 2011;12:21–35. doi: 10.1038/nrm3025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.McAuliffe PF, Meric-Bernstam F, Mills GB, Gonzalez-Angulo AM. Deciphering the role of PI3K/Akt/mTOR pathway in breast cancer biology and pathogenesis. Clin Breast Cancer. 2010;10(Suppl 3):S59–S65. doi: 10.3816/CBC.2010.s.013. [DOI] [PubMed] [Google Scholar]
- 47.She QB, Chandarlapaty S, Ye Q, Lobo J, Haskell KM, Leander KR, et al. Breast Tumor Cells with PI3K Mutation or HER2 Amplification Are Selectively Addicted to Akt Signaling. PLoS ONE. 2008;3:e3065. doi: 10.1371/journal.pone.0003065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Keel PK, Baxter MG, Heatherton TF, Joiner TE., Jr A 20-year longitudinal study of body weight, dieting, and eating disorder symptoms. J Abnorm Psychol. 2007;116:422–32. doi: 10.1037/0021-843X.116.2.422. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.