

Abstract
The α4β2 neuronal nicotinic acetylcholine receptor (nAChR) plays a crucial role in nicotine addiction. These receptors are known to desensitize and up-regulate after chronic nicotine exposure, but the mechanism remains unknown. Currently, the structure and functional role of the intracellular domains of the nAChR are obscure. To study the effect of subunit phosphorylation on α4β2 nAChR function and expression, eleven residues located in the M3-M4 cytoplasmic loop were mutated to alanine and aspartic acid. Two-electrode voltage clamp and 125I-labeled epibatidine binding assays were performed on Xenopus oocytes to assess agonist activation and receptor expression. When ACh was used as an agonist, a decrease in receptor activation was observed for the majority of the mutations. When nicotine was used as an agonist, four mutations exhibited a statistically significant hypersensitivity to nicotine (S438D, S469A, Y576A, and S589A). Additionally, two mutations (S516D and T536A) that displayed normal activation with ACh displayed remarkable reductions in sensitivity to nicotine. Binding assays revealed a constitutive up-regulation in these two nicotine mutations with reduced nicotine sensitivity. These results suggest that consensus phosphorylation residues in the M3-M4 cytoplasmic loop of the α4 subunit play a crucial role in regulating α4β2 nAChR agonist selectivity and functional expression. Furthermore, these results suggest that disruption of specific interactions at PKC putative consensus sites can render α4β2 nAChRs almost insensitive to nicotine without substantial effects on normal AChR function. Therefore, these PKC consensus sites in the M3-M4 cytoplasmic loop of the α4 nAChR subunit could be a target for smoking cessation drugs.
Keywords: nicotinic acetylcholine receptor, nicotine, two-electrode voltage clamp, nicotine sensitivity, phosphorylation
1. Introduction
Neuronal nicotinic acetylcholine receptors (nAChRs) are transmitted gated ion channels that belong to a gene super family of homologous receptors including γ–aminobutyric acid (GABA), glycine, and serotonin receptors. The receptor is composed of five subunits with each having four transmembrane domains (M1-M4) and a large intracellular loop (C2). The genes for the subunits that have been cloned so far are divided into two subfamilies of nine α (α2-α10) and three β (β2-β4) subunits, and are expressed in the nervous system, cochlea, and a number of non-neuronal tissues (Gotti and Clementi, 2004; Hogg et al., 2003).
Neuronal nAChRs have a role in the mediation of tolerance and addiction to nicotine in chronic tobacco users and the symptoms of withdrawal experienced upon cessation of use (Benowitz, 1996). The complex activities of nicotine in the nervous system are due to its ability to mimic the activity of acetylcholine (ACh) on these receptors (Gaimarri et al., 2007). At the molecular level, chronic nicotine exposure differentially affects the number (up-regulation), subunit composition, stoichiometry, and functional status (desensitization and inactivation) of some nAChR subtypes, leaving others substantially unaffected (Gaimarri et al., 2007). Nicotine, the addictive component of tobacco, has a predominant effect in the brain mainly on the α4β2 nAChR, the most abundant subtype.
Desensitization and up-regulation of nAChRs is thought to involve phosphorylation mediated by various kinases (Fenster et al., 1999b). For instance, protein kinase A (PKA) (Madhok et al., 1994) and protein kinase C (PKC) (Eilers et al., 1997) are examples of kinases that have been studied in the context of nAChR desensitization and up-regulation. Specifically, studies suggest that in the continuous presence of nicotine, receptors would be driven into inactive/desensitized conformations (Lukas, 1991; Marks et al., 1993; Peng et al., 1994), a process likely influenced by the level of phosphorylation (Eilers et al., 1997; Gopalakrishnan et al., 1997). There is evidence that activators of PKA and PKC increase nAChR binding sites and synergistically enhance nicotine-induced receptor up-regulation (Gopalakrishnan et al., 1997). These findings are validated by in vitro and in vivo studies, which have demonstrated that α4 nAChR subunits are phosphorylated (Hsu et al., 1997; Nakayama et al., 1993a, 1993b) and, more specifically, that they are targets of PKA phosphorylation (Wecker et al., 2001). Studies using phosphopeptide mapping have provided evidence that residues S365, S472, and S491 of the rat α4 subunit, corresponding to positions S364, S471 and S490 on the current α4 NCBI reference sequence, are substrates for PKA phosphorylation (Guo and Wecker, 2002). In addition, a recent study identified two major substrate sites for PKA phosphorylation on the human α4 subunit, namely S467 and S362, which are homologues to rat α4 positions S471 and S364 used in the current study (Pollock et al., 2007).
Recent work from our laboratory has shown that two point mutations of a PKC phosphorylation residue, S336A and S336D, exhibit constitutive up-regulation when expressed in oocytes (López-Hernández et al., 2009). In addition, we found that both of these PKC mutations changed the ACh affinity but exhibited no change in nicotine affinity, suggesting that the properties of agonist binding for α4β2 channel activation have very distinct dynamic and/or structural requirements for ACh and nicotine (López-Hernández et al., 2009).
On the basis of these findings, we examined various consensus positions in the M3-M4 cytoplasmic loop of the α4 subunit (Fig. S1) in an attempt to dissect the potential role of this domain in the functional response of the α4β2 nAChR. This domain contains conserved consensus sites for various protein kinases (PKA, PKC, CKII, and TK) (Viseshakul et al., 1998) and has been recently mentioned as a possible target for allosteric sites (Taly et al., 2009). In the present study, eleven of these sites were mutated, to alanine and aspartic acid, to study the effect of α4 subunit phosphorylation on the α4β2 nAChR activation and expression. The rationale for using alanine and aspartic acid substitutions is that alanine impairs phosphorylation, whereas aspartic acid mimics phosphorylation of the protein (Arany et al., 2013). We used ACh and nicotine as agonists to test the functionality of the mutations as compared with the wild-type receptor. The present results reveal a novel allosteric linkage between the M3-M4 cytoplasmic loop of the α4 neuronal nAChR subunit that can regulate an agonist's selectivity and functional expression. We suggest that this domain could be considered as a structural target for the development of smoking cessation drugs given that point mutations at this domain can decreased nicotine sensitivity (Taly et al., 2009).
2. Materials and methods
2.1. Site-directed mutagenesis
Single point mutations were prepared using the Quickchange™ Site-Directed Mutagenesis Kit (Stratagene, La Jolla, CA). The template used for the PCR reaction was the pGEMHE vector with Rattus norvegicus cDNA coding for the α4 neuronal nAChR subunit. DNA was purified using a QIAprep spin miniprep kit (Quiagen, Germantown, MD) and then sequenced to confirm the incorporation of each mutation. A total of 11 residues in the cytoplasmic loop of the α4 subunit were chosen to be mutated: S364, T417, S438, S469, S471, S490, S504, S516, T536, Y576, and S589. Each residue was mutated to alanine (A) and aspartic acid (D).
2.2. In vitro synthesis of mRNA and oocyte microinjection
Each subunit encoding mRNA was synthesized in vitro from linearized pGEMHE plasmid templates of Rattus norvegicus cDNA coding for α4 and β2 nAChR subunits using the mMessage mMachine RNA transcription kit (Ambion, Austin, TX). mRNA mixtures of α4 and β2 subunits were prepared at 2 μg:3 μg ratio, and 32.2 nL of this mixture was microinjected into each oocyte. The mRNA mixture was microinjected, using a displacement injector (Drummond Instruments, Broomhall, PA), into stage V and VI oocytes that had been extracted, incubated in collagenase Type 1A (Sigma, St. Louis, MO), and defolliculated by manual dissection. The injected oocytes were incubated at 19 °C for 3-4 days in ND-96 medium supplemented with albumin, gentamicin, tetracycline, and theophyline. Electrophysiological experiments were performed after the third or fourth day of mRNA injection.
2.3. Electrophysiological characterization of α4β2 nAChRs
Oocytes injected with the mRNA transcripts of α4 and β2 subunits were characterized using a two-electrode voltage clamp. ACh- and nicotine-induced currents were recorded at 20 °C, 3-4 days after mRNA injection, with a GeneClamp 500B Amplifier (Axon Instruments, Foster City, CA). Electrodes were filled with 3M KCl and had a resistance of less than 5 megaohms. Impaled oocytes in the recording chamber were continuously perfused at a rate of 5 ml/min with MOR-2 buffer (115 mM NaCl, 2.5 mM KCl, 5 mM HEPES, 1 mM Na2HPO4, 0.2 mM CaCl2, 5 mM MgCl2, and 0.5 mM EGTA, pH 7.4). All the reagents used were purchased from Sigma-Aldrich, Co. (St. Louis, MO). For dose-response curves, each oocyte was held at a membrane potential of −70 mV. Membrane currents were digitized using the DigiData 1322A interface (Axon Instruments, Foster City, CA), filtered at 2 kHz during recording. The Clampex 10.0 software running on a Pentium 4-based computer was used for data acquisition. Data analysis was via Prism 3.0 (Graphpad Software, San Diego, CA). Dose-response data for the α4β2 combination were collected using seven ACh doses (0.1, 1, 3, 10, 30, 100, and a seventh concentration ranging from 300 to 1000 μM depending on the mutant) and seven nicotine concentrations (0.1, 1, 3, 10, 30, 100, 300 μM). The data were fitted using a one-component sigmoidal dose-response equation, Y = I/ImaxBottom + (I/ImaxTop – I/ImaxBottom)/(1 + 10^((LogEC50 – X) × HillSlope)) where X is the logarithm of concentration and Y is the response.
2.4. Epibatidine Binding Assays
125I-labeled epibatidine (PerkinElmer Life Sciences, Boston, MA) binding assays were performed to determine membrane expression of nAChR in oocytes. The oocytes were incubated in 50 pM 125I–labeled epibatidine with 5 mg/mL albumin serum bovine in MOR-2 without EGTA at room temperature for 2 h. Non-injected oocytes were also incubated in 125I–labeled epibatidine to measure nonspecific binding (PerkinElmer 2470 WIZARD Gamma Counter). Excess epibatidine was removed by washing each oocyte with 60 mL of MOR-2 without EGTA. A standard linear regression was obtained by plotting the counts per minute (CPM) against 125I-labeled epibatidine concentration (0.5-20 fmol).
2.5. Determination in potential changes in subunit stoichiometry of α4β2 nAChRs using A-85380
To determine whether the α4β2 mutations α4S469Aβ2, α4S471Dβ2, α4T536Aβ2 and α4Y576Aβ2 expressed in Xenopus laevis oocytes assemble in the α4(2):β2(3) or the α4(3):β2(2) stoichiometry, we estimated the efficacy of agonist A-85380 when compared to ACh 300 μM. The α4(2):β2(3) stoichiometry was favored by microinjecting α4β2 mRNA in a 1:10 ratio, and the α4(3):β2(2) by microinjecting in a 10:1 ratio. The efficacy of A-85380 in WTα4(2):β2(3) and WTα4(3):β2(1) was determined by comparing the macroscopic current elicited by A-85380 100 nM and ACh 300 μM in oocytes microinjected α4β2 mRNA in 1:10 and 10:1 ratios, respectively. For these experiments, we focused our attention on one mutation from each set (PKA, CKII, PKC, and TK) rather than performing a systematic assessment in all the mutations. In doing so, we selected mutations from each set that displayed interesting functional deviations from the WT, to explore the possibility that such changes arose from changes in subunit stoichiometry. Because this work contains quite a lot of data, we were inclined not to probe all mutations, but rather test only a functionally interesting representative from each set. The efficacy of A-85380 in α4S469Aβ2, α4S471Dβ2, α4T536Aβ2 and α4Y576Aβ2 was determined by comparing the macroscopic current elicited by A-85380 100 nM and ACh 300 μM in oocytes microinjected α4β2 mRNA in 2:3 ratio. The changes in stoichiometry in these four mutants were assessed by comparing the efficacy of A-85380 in each mutant compared to WTα4(2):β2(3) and WTα4(3):β2(2).
3. Results
3.1 Functional effects of mutations at PKA putative phosphorylation sites in the α4 subunit when using ACh as an agonist
Mutations α4S364A, α4S364D, α4S471D, and α4S490A resulted in receptors with a significant decrease in macroscopic peak current as compared with the wild-type (WT) receptor (Fig. 1). Mutations α4S471A and α4S490D resulted in non-functional receptors. Statistical analysis using an unpaired t test showed no significant difference in the ACh EC50 value between mutations α4S364A, α4S364D, α4S490A, and the WT α4β2 nAChR (2.55 ± 0.08, 2.71 ± 0.05, 1.51 ± 0.08, and 2.33 ± 0.03 μM, respectively). On the other hand, mutation α4S471D exhibited a significant EC50 increase (i.e. 53.60 ± 0.05 μM) as compared with the WT ACh EC50 value (Table 1).
Fig. 1.

Binding and functional characterization of mutations on PKA putative sites in the α4 subunit. Voltage-clamp recordings were used to determine the macroscopic response ofmutations α4S364Aβ2, α4S364Dβ2, α4S471Aβ2, α4S471Dβ2, α4S490Aβ2, α4S490Dβ2 andwild-type α4β2 nAChRs expressed in Xenopus laevis oocytes to several ACh and nicotineconcentrations. (A) Family of ACh-induced macroscopic currents. Calibration bars are shown for all family of currents, horizontal bars indicate time (5 s) and vertical bars indicate the inward current (500 nA). (B) Dose-response relationships obtained by voltage-clamp using ACh as an agonist. ACh dose-response curves were determined using seven ACh concentrations (0.1, 1, 3, 10, 30, 100, and a seventh concentration ranging from 300 to 1000 μM depending on the mutant). The responses were normalized to the maximum response (I/Imax). (C) Left panel, comparison of the macroscopic peak currents of all the mutations and wild-type receptor shown in nA. Right panel, results of the 125I-labeled epibatidine binding experiments performed in Xenopus laevis oocytes expressing the mutations α4S364Aβ2, α4S364Dβ2, α4S471Aβ2, α4S471Dβ2, α4S490Aβ2, α4S490Dβ2 and wild-type α4β2 nAChRs shown in fmoles (n=6-17) (*** p<0.0005, ** p<0.005).
Table 1.
Electrophysiological characterization of α4β2 nAChR mutations reveal different nicotine and ACh sensitivities
| Kinase | Mutation | ACh | Nicotine | ||||
|---|---|---|---|---|---|---|---|
| EC50 (μM) | Hill Coef. | Peak Current (nA) | EC50 (μM) | Hill Coef. | Peak Current (nA) | ||
| – | WT α4β2 | 2.33± 0.03 | 1.00 | 2110±243 | 4.21±0.41 | 1.80 | 387±66 |
| PKA | α4S364Aβ2 | 2.55±0.08 | 1.22 | 759±157*** | n/f | n/f | n/f |
| α4S364Dβ2 | 2.71±0.05 | 1.07 | 135±87*** | n/f | n/f | n/f | |
| α4S471Aβ2 | n/f | n/f | n/f | n/f | n/f | n/f | |
| α4S471Dβ2 | 53.60±0.05*** | 1.01 | 1039±139** | 5.02±0.62 | 2.00 | 142±313** | |
| α4S490Aβ2 | 1.51±0.08 | 0.91 | 200±115*** | n/f | n/f | n/f | |
| α4S490Dβ2 | n/e | n/e | n/e | n/e | n/e | n/e | |
| CKII | α4T417Aβ2 | 77.80±0.12*** | 0.76 | 1557±306 | 2.12±0.61* | 1.33 | 293±120 |
| α4T417Dβ2 | 46.09±0.05*** | 0.99 | 360±89*** | 2.11±0.32** | 1.78 | 582±255 | |
| α4S438Aβ2 | 11.00±0.19*** | 0.49 | 97±29*** | n/f | n/f | n/f | |
| α4S438Dβ2‡ | 3.96±0.09* | 0.68 | 1938±367 | 2.17±0.16** | 1.29 | 2156±475** | |
| α4S469Aβ2‡ | 43.59±0.09*** | 0.87 | 2222±413 | 3.37±0.20 | 1.84 | 1552±313** | |
| α4S469Dβ2 | n/f | n/f | n/f | n/f | n/f | n/f | |
| α4S504Aβ2 | n/f | n/f | n/f | n/f | n/f | n/f | |
| α4S504Dβ2 | n/f | n/f | n/f | n/f | n/f | n/f | |
| PKC | α4S516Aβ2 | 65.89±0.06*** | 1.04 | 1027±169*** | n/f | n/f | n/f |
| α4S516Dβ2¶ | 43.72±0.12*** | 0.79 | 3296±581 | 3.80±0.68 | 1.48 | 76±16*** | |
| α4T536Aβ2¶ | 38.25±0.11*** | 0.67 | 3345±129*** | 3.84±0.61 | 1.80 | 110±28*** | |
| α4T536Dβ2 | 85±17*** | 0.69 | 216±85*** | n/f | n/f | n/f | |
| α4S589Aβ2‡ | 77±10*** | 0.80 | 1644±192 | 4.67±0.56 | 1.61 | 1882±672* | |
| α4S589Dβ2 | n/f | n/f | n/f | n/f | n/f | n/f | |
| TK | α4Y576Aβ2‡ | 55±8*** | 0.91 | 4398±450*** | 3.76±0.60 | 1.65 | 874±221* |
| α4Y576Dβ2 | 35±7*** | 0.62 | 396±65*** | n/f | n/f | n/f | |
Mutations highlighted in green (‡) exhibit hypersensitivity to nicotine while those in red (¶) exhibit agonist selectivity evidenced by their remarkable reductions in sensitivity to nicotine. Legend: (n/f) non-functional; (n/e) no-expression; Error estimates are expressed as the mean ± SEM of 6-17 oocytes.
P<0.05
P<0.005
P<0.0005. Comparisons are relative to the WT.
3.2. Functional effects of mutations at PKA putative phosphorylation sites in the α4 subunit when using nicotine as an agonist
All of the PKA mutants were studied with the use of nicotine as an agonist. The only mutation that exhibited functional activation by nicotine was α4S471D (Fig. 2). This mutation exhibited a significant decrease in the macroscopic peak current as compared with that of the WT receptor. The nicotine EC50 values revealed no difference between the mutation and the WT receptor (Table 1). This was a remarkable observation as the α4S471D mutation shows a significant difference in ACh activation as compared with the WT; however, nicotine activation in this mutant was similar to that in the WT receptor. This kind of behavior was previously described in mutations of PKC residue S336 (López-Hernández et al., 2009).
Fig. 2.

Functional characterization of mutations on PKA putative sites in the α4 subunit with nicotine used as agonist. (A) Family of nicotine-induced macroscopic currents for mutation α4S471Dβ2 and the wild-type α4β2 nAChR. Calibration bars are shown for all family of currents, horizontal bars indicate time (5 s) and vertical bars indicate the inward current (500 nA). (B) Dose-response relationships obtained by voltage-clamp using nicotine as agonist. Nicotine dose-response curves were determined using seven nicotine concentrations (0.1, 1, 3, 10, 30, 100 and 300 M). The responses were normalized to the maximum response (I/Imax). (C) Comparison of the nicotine-induced macroscopic peak currents of the mutations and wild-type receptor shown in nA (n=6-17) (** p<0.005).
3.3. Effects on nAChR expression of mutations at PKA putative phosphorylation sites in the α4 subunit
125I-labeled epibatidine binding assays revealed the expression patterns of the various mutations of PKA residues in the α4 subunit (Fig. 1C, right panel). Statistical analysis using an unpaired t test revealed no significant changes among any of the mutations as compared with the WT nAChR. Although mutation α4S490D did not exhibit any function during the voltage-clamp experiments (Table 1), it was still subjected to the binding assays which revealed no expression for mutation α4S490D.
3.4. Functional effects of mutations at CKII putative phosphorylation sites in the α4 subunit when using ACh as an agonist
Voltage-clamp data analysis revealed significant decreases in the macroscopic peak currents for mutations α4T417D and α4S438A (Fig. 3). On the other hand, mutations α4T417A, α4S438D, and α4S469A showed no change in the macroscopic peak current (Table 1). At the same time, mutations α4S469D, α4S504A, and α4S504D resulted in non-functional receptors. The ACh EC50 values for all of the functional mutations of the CKII putative sites α4T417A, α4T417D, α4S438A, α4S438D, and α4S469A resulted in significant increases when compared with the WT nAChR (Table 1).
Fig. 3.

Binding and functional characterization of mutations on CKII putative phosphorylation sites in the α4 subunit with ACh used as an agonist. Mutations α4T417Aβ2, α4T417Dβ2, α4S438Aβ2, α4S438Dβ2, α4S469Aβ2, α4S469Dβ2, α4S504Aβ2, and α4S504Dβ2 were expressed in Xenopus laevis oocytes. (A) Family of ACh-induced macroscopic currents. Calibration bars are shown for all family of currents, horizontal bars indicate time (5 s) and vertical bars indicate the inward current (500 nA). (B) Dose-response curves obtained by voltage-clamp using ACh as agonist. ACh dose-response curves were determined using seven ACh concentrations (0.1, 1, 3, 10, 30, 100, and a seventh concentration ranging from 300 to 1000 μM depending on the mutant). The responses were normalized to the maximum response (I/Imax). (C) Left panel. Comparison of the macroscopic peak currents of all the mutations compared with the wild-type receptor, shown in nA. Right panel. Results of the 125I-labeled epibatidine binding experiments performed in Xenopus laevis oocytes expressing the mutations α4T417Aβ2, α4T417Dβ2, α4S438Aβ2, α4S438Dβ2, α4S469Aβ2, α4S469Dβ2, α4S504Aβ2, and α4S504Dβ2 and wild-type α4β2 nAChRs, shown in fmoles (n=6-17) (* p<0.05, ***p<0.0001).
3.5. Functional effects of mutations at CKII putative phosphorylation sites in the α4 subunit when using nicotine as an agonist
When nicotine was used as agonist, we found that mutations α4S469D, α4S504A, and α4S504D also resulted in non-functional receptors. However, although mutation α4S438A was a functional receptor when ACh was used as agonist, it resulted in a non-functional receptor when nicotine was used as an agonist. Mutations of residue α4T417 exhibited no significant change in macroscopic current when compared with the WT receptor; however, their nicotine EC50 values decreased significantly (Fig. 4). These mutations displayed a higher potency for nicotine. Interestingly, mutations α4S438D and α4S469A exhibited significant increases in the macroscopic peak currents (2156 ± 475 and 1552 ± 313 nA, respectively) when compared with the WT receptor (387 ± 66 nA) (Table 1). The nicotine EC50 value for α4S438D exhibited a significant decrease whereas mutation α4S469A exhibited no change. These results suggest that mutations α4S438D and α4S469A enhance the sensitivity to nicotine.
Fig. 4.

Functional characterization of mutations on CKII putative phosphorylation sites in the α4 subunit with nicotine used as agonist. Voltage-clamp recordings were used to determine the macroscopic response to several nicotine concentrations of mutations of CKII putative sites and wild-type α4β2 nAChRs expressed in Xenopus laevis oocytes. (A) Family of nicotine-induced macroscopic currents for mutations α4T417Aβ2, α4T417Dβ2, α4S438Dβ2, α4S469Aβ2, and the wild-type α4β2 nAChR. Calibration bars are shown for all family of currents, horizontal bars indicate time (5 s) and vertical bars indicate the inward current (500 nA). (B) Dose-response relationships obtained by voltage-clamp experiments with nicotine used as an agonist. Nicotine dose-response curves were determined using seven nicotine concentrations (0.1, 1, 3, 10, 30, 100, and 300 μM). The responses were normalized to the maximum response (I/Imax). (C) Comparison of the nicotine-induced macroscopic peak currents of the mutations and wild-type receptor, shown in nA (n=6-17) (**p<0.005).
3.6. Effects on nAChR expression of mutations at CKII putative phosphorylation sites in the α4 subunit
125I-labeled epibatidine binding assays on mutations of the CKII residues revealed significant decreases in receptor expression for mutations α4T417D, α4S438D, and α4S469A (Fig. 3C, right panel). On the other hand, mutations α4T417A and α4S438A did not exhibit changes in expression when compared with the WT receptor. More interestingly, three mutations that resulted in non-functional receptors when using ACh and nicotine (α4S469D, α4S504A, and α4S504D) exhibited no change in receptor expression (Fig. 3C right panel, and Table 1).
3.7. Functional effects of mutations at PKC putative phosphorylation sites in the α4 subunit when using ACh as an agonist
The alanine substitution at position α4S516 displayed a significant decrease in macroscopic peak current (1027 ± 169 nA) (Fig. 5). Mutation α4T536A showed a significant increase in macroscopic peak current (3345 ± 129 nA), whereas mutation α4T536D showed a significant decrease in macroscopic peak current (216 ± 85 nA) when compared with the WT receptor (Table 1). Mutation α4S589A displayed a functional receptor with no change in macroscopic peak current (1644 ± 192 nA) when compared with the WT receptor. However, mutating the same residue to aspartic acid, α4S589D, resulted in a non-functional receptor (i.e., no significant current was detected). The five mutations that exhibited functional channels (α4S516A, α4S516D, α4T536A, α4T536D, and α4S589A) exhibited a significant increase in the ACh EC50 value as compared with the WT receptor (Table 1).
Fig. 5.

Binding and functional characterization of mutations on PKC putative phosphorylation sites in the α4 subunit with ACh used as an agonist. Mutations α4S516Aβ2, α4S516Dβ2, α4T536Aβ2, α4T536Dβ2, α4S589Aβ2, and α4S589Dβ2 were expressed in Xenopus laevis oocytes. (A) Family of ACh-induced macroscopic currents. Calibration bars are shown for all family of currents, horizontal bars indicate time (5 s) and vertical bars indicate the inward current (500 nA). (B) Dose-response curves obtained by voltage-clamp experiments with ACh used as an agonist. ACh dose-response curves were determined using seven ACh concentrations (0.1, 1, 3, 10, 30, 100, and a seventh concentration ranging from 300 to 1000 μM depending on the mutant). The responses were normalized to the maximum response (I/Imax). (C) Left panel. Comparison of the macroscopic peak currents of all the mutations compared to the wild-type receptor shown in nA. Right panel. Results of the 125I-labeled epibatidine binding experiments performed in Xenopus laevis oocytes expressing the mutations α4S516Aβ2, α4S516Dβ2, α4T536Aβ2, α4T536Dβ2, α4S589Aβ2, and α4S589Dβ2 and wild-type α4β2 nAChRs, shown in fmoles (n=6-17) (*p<0.05, **p<0.005, ***p<0.0005).
3.8. Functional effects of mutations at PKC putative phosphorylation sites in the α4 subunit when using nicotine as an agonist
We performed voltage-clamp experiments on the mutations of PKC putative sites, this time using nicotine as an agonist. Mutations α4S516A, α4T536D, and α4S589D exhibited no nicotine-induced currents, an unsurprising outcome given that their response to ACh was significantly decreased. However, mutations α4S516D and α4T536A exhibited extremely low nicotine-induced currents (Fig. 6). This was an interesting finding because both of these mutations exhibited either no change (α4S516D) or an increase (α4T536A) in the ACh-induced macroscopic peak current. On the other hand, mutation α4S589A displayed a significant increase in macroscopic peak current, while exhibiting no change in the nicotine EC50 value (Table 1).
Fig. 6.

Functional characterization of mutations on PKC putative phosphorylation sites in the α4 subunit with nicotine used as an agonist. Voltage-clamp recordings were used to determine the macroscopic response to several nicotine concentrations of mutations of PKC putative sites and wild-type α4β2 nAChRs expressed in Xenopus laevis oocytes. (A) Family of nicotine-induced macroscopic currents for the mutations and the wild-type α4β2 nAChR. Calibration bars are shown for all family of currents, horizontal bars indicate time (5 s) and vertical bars indicate the inward current (500 nA). (B) Dose-response relationships obtained by voltage-clamp experiments with nicotine used as an agonist. Nicotine dose-response curves were determined using seven nicotine concentrations (0.1, 1, 3, 10, 30, 100, and 300 μM). The responses were normalized to the maximum response (I/Imax). (C) Comparison of the nicotine-induced macroscopic peak currents, shown in nA (n=6-17) (*p<0.05, ***p<0.0005).
3.9. Effects on nAChR expression of mutations at PKC putative phosphorylation sites in the α4 subunit
The binding assays, using 125I-labeled epibatidine, to measure receptor expression showed significant increases in receptor expression when compared with the WT receptor. Mutations α4S516D, and α4T536A showed significant increases in receptor expression when compared with the WT α4β2 nAChR (Fig. 5C, right panel). These mutations resulted in receptors that are constitutively up-regulated. Mutation α4S589D displayed a behavior similar to that seen previously in mutations α4S504A and α4S504D; it showed no signs of ACh- and nicotine-induced macroscopic currents (Table 1). However, binding assays revealed no change in receptor expression for mutation α4S589D when compared with the WT receptor (Fig. 5C, right panel).
3.10. Functional effects of mutations of a TK putative phosphorylation site in the α4 subunit when using ACh as an agonist
The alanine substitution in the TK putative site α4Y576 exhibited a significant increase in the macroscopic peak current (4398 ±450 nA), and the aspartic acid substitution resulted in a significant decrease in macroscopic peak current (396 ± 65 nA) when compared with the α4β2 nAChR (Fig. 7). The ACh EC50 values for α4Y576A (55 ± 8 μM) and for α4Y576D (35 ± 7 μM) increased significantly when compared with the WT receptor, which has an EC50 value of 2.33 ± 0.03 μM (Table 1). These increases in ACh EC50 values have also been seen in other mutations.
Fig. 7.

Binding and functional characterization of mutations on the TK putative phosphorylation site in the α4 subunit with ACh used as an agonist. Mutations α4Y576Aβ2 and α4Y576Dβ2 were expressed in Xenopus laevis oocytes. (A) Family of ACh-induced macroscopic currents. Calibration bars are shown for all family of currents, horizontal bars indicate time (5 s) and vertical bars indicate the inward current (500 nA). (B) Dose-response curves obtained by voltage-clamp experiments with ACh used as an agonist. ACh dose-response curves were determined using seven ACh concentrations (0.1, 1, 3, 10, 30, 100, and a seventh concentration ranging from 300 to 1000 μM depending on the mutant). The responses were normalized to the maximum response (I/Imax). (C) Left panel. Comparison of the macroscopic peak currents of all the mutations compared with the wild-type receptor, shown in nA. Right panel. Results of the 125I-labeled epibatidine binding experiments performed in Xenopus laevis oocytes expressing the mutations α4Y576Aβ2 and α4Y576Dβ2 and wild-type α4β2 nAChRs, shown in fmoles (n=6-17) (*p<0.05, ***p<0.0005).
3.11. Functional effects of mutations of a TK putative phosphorylation site in the α4 subunit when using nicotine as an agonist
Mutation α4Y576A exhibited a significant increase in the macroscopic peak current whereas mutation α4Y576D exhibited no significant nicotine-induced current, resulting in a nonfunctional channel (Fig. 8). Since mutation α4Y576D exhibited a significant decrease in the ACh-induced macroscopic peak current, it was not surprising that there was no significant nicotine-induced current. The nicotine EC50 value for mutation α4Y576A revealed no significant difference when compared with the WT receptor (Table 1). Our data indicate that mutation α4Y576A exhibits an enhanced sensitivity to nicotine.
Fig. 8.

Functional characterization of mutations on the TK putative phosphorylation site in the α4 subunit with nicotine used as an agonist. Voltage-clamp recordings were used to determine the macroscopic response to several nicotine concentrations of mutations of TK putative sites and wild-type α4β2 nAChRs expressed in Xenopus laevis oocytes. (A) Family of nicotine-induced macroscopic currents for mutation α4Y576Aβ2 and the wild-type α4β2 nAChR. Calibration bars are shown for all family of currents, horizontal bars indicate time (5 s) and vertical bars indicate the inward current (500 nA). (B) Dose-response relationships obtained by voltage-clamp experiments with nicotine used as an agonist. Nicotine dose-response curves were determined using seven nicotine concentrations (0.1, 1, 3, 10, 30, 100, and 300 μM). The responses were normalized to the maximum response (I/Imax). (C) Comparison of the nicotine-induced macroscopic peak currents, shown in nA (n=6-17) (*p<0.05).
3.12. Effects on nAChR expression of mutations of TK putative phosphorylation sites in the α4 subunit
The results of the binding assays performed on mutations α4Y576A and α4Y576D revealed that only mutation α4Y576D decreased receptor expression significantly, whereas mutation α4Y576A saw no change in receptor expression when compared with the α4β2 nAChR (Fig. 7C, right panel).
4. Discussion
4.1. Functional implications of PKA consensus phosphorylation residues in the α4 subunit: Possible inhibitory role for PKA residues
PKA mutants that expressed functional receptors displayed significant decreases in macroscopic current, whereas receptor expression was unchanged. These results indicate that assembly of the α4β2 nAChR was unaffected by PKA mutations while receptor function was significantly decreased. Also, when ACh was used in mutation α4S364A, it displayed faster decay rates as compared with WT. This result suggests that a residue far away from the binding site of the α4 subunit can affect agonist association and channel activation and inactivation kinetics via a complex network of allosteric interactions.
The mutant α4S490D resulted in no measurable expression levels suggesting that phosphorylation of this residue shuts down α4β2 nAChR expression completely, by affecting trafficking or assembly. Thus, PKA residues may play an inhibitory role in vivo since the mutations mentioned above resulted in functional inhibition and, in one case, complete shut down of α4β2 nAChR expression.
4.2. Functional implications of CKII consensus phosphorylation residues in the α4 subunit: Possible modulatory role for CKII residues
Residues α4T417, α4S438, α4S469, and α4S504 may be phosphorylated by CKII due to their amino acid sequence (Viseshakul et al., 1998), but they have not been extensively studied. We found that mutations in two of these positions, namely α4S438D and α4S469A, displayed significant increases in their nicotine-induced functional responses, whereas α4S438A and α4S469D exhibited no functional response to nicotine. These results suggest that a single mutation in the α4 intracellular domain can enhance nicotine sensitivity, and that consensus positions in the M3-M4 intracellular domain can influence the allosteric properties of α4β2. Furthermore, because α4S469A exhibited decreased potency for ACh but retained the same potency for nicotine as the WT receptor further demonstrates that nicotine and ACh have different requirements for channel activation. Since α4S469A exhibited an increase in peak currents to nicotine, this mutation could be a novel model for the study of nicotine tolerance. Interestingly, α4S469A results from the substitution of only one nucleotide and could, therefore, occur in vivo via a single nucleotide polymorphism (SNP) of CHRNA4 (Kim et al., 2003).
4.3. Functional implications of PKC consensus phosphorylation residues in the α4 subunit: PKC residues may regulate agonist activation and up-regulation
It is known that PKC phosphorylates the α4 subunit (Wecker et al., 2001), and studies using mutations of residues phosphorylated by PKC have demonstrated how phosphorylation affects receptor desensitization and up-regulation by chronic nicotine treatment (Fenster et al., 1999a). Two of the PKC mutations, α4S516D and α4T536A, resulted in an increase in receptor expression; no other kinase residue mutations exhibited this kind of behavior. Moreover, these two mutations exhibited either normal (α4S516D) or increased (α4T536A) functional activation by ACh. However, their response to nicotine was extremely reduced as compared with the WT. Moreover, the peak current elicited by a nicotine concentration found in a chronic smoker (i.e. 0.1 μM) was almost undetected (i.e. 5 nA for α4S516D and 6 nA for α4T536A). This finding is remarkable: two novel mutations, far from the binding site, that can influence agonist activation. These two mutations produced α4β2 nAChRs that can discriminate for nicotine activation without major effects on ACh activation. These mutations could provide a novel perspective for new therapies for smoking cessation—targeting a residue that can make α4β2 nAChRs insensitive to nicotine without major effects on normal AChR function. At the same time, mutation α4S589A exhibited an enhanced sensitivity to nicotine as evidenced by a significant increase in macroscopic peak current. This result is noteworthy in that the identification of a position along the α4 subunit cytoplasmic loop that influences nicotine sensitivity in the α4β2 receptor suggests that this domain could have a role in the design of novel smoking cessation drugs.
Binding assays revealed no change in receptor expression for mutation α4S589D when compared with WT. This result suggests that a negative charge at this position completely shuts down receptor function, regardless of receptor expression levels.
4.4. Functional implications of the TK consensus phosphorylation residue in the α4 subunit: Possible role for regulating nicotine sensitivity and expression
Residue α4Y576 is proposed to be phosphorylated by tyrosine kinase (Viseshakul et al., 1998). The present data suggest that TK sites may play a role in regulating receptor expression and nicotine sensitivity. We base this assumption on the fact that mutation α4Y576A exhibits an increase in the peak current to ACh and nicotine. Mutation α4Y576D exhibits a decrease in the functional activation by ACh, fails to respond to nicotine, and exhibits a decrease in receptor expression. These results suggest that phosphorylation at the α4Y576 consensus position down-regulates both receptor function and expression and support our hypothesis for a role for this TK site in regulating agonist sensitivity and receptor expression.
4.5. Changes in ACh potency in the S469A, S471D, T536A and Y576A mutations does not involve alterations in α4β2 nAChRs subunit stoichiometry
The decrease in potency for ACh exhibited by the majority of the mutations suggests potential changes in α4β2 nAChR stoichiometry. Previous studies have shown that the α4(3):β2(2) stoichiometry has a characteristic lower affinity for ACh whereas the α4(2): β2(3) stoichiometry exhibits a higher affinity for ACh (López-Hernández et al., 2004; Moroni et al., 2006). Moreover, evidence suggests that phosphorylation of S467 (S471 in the present study) on free α4 subunits prior to their association with β2 subunits has long-lasting consequences, increasing the stability of the α4 subunits (Pollock et al., 2009). This increase in stability may result in an enhanced expression of α4β2 in the low affinity α4(3):β2(2) configuration (Moroni et al., 2006; Nelson et al., 2003). However, as Fig. 9 suggests, the observed decrease in ACh potency cannot be explained by alterations in the α4(2):β2(3) stoichiometry. The efficacy of agonist A-85380 has been reported to be significantly reduced in the α4(3):β2(2) stoichiometry (Zwart et al., 2006). The efficacy of A-85380 in α4β2 mutations S469A, S471D, T536A, and Y576A indicate that these are assembled according to the α4(2):β2(3) stoichiometry.
Fig. 9.
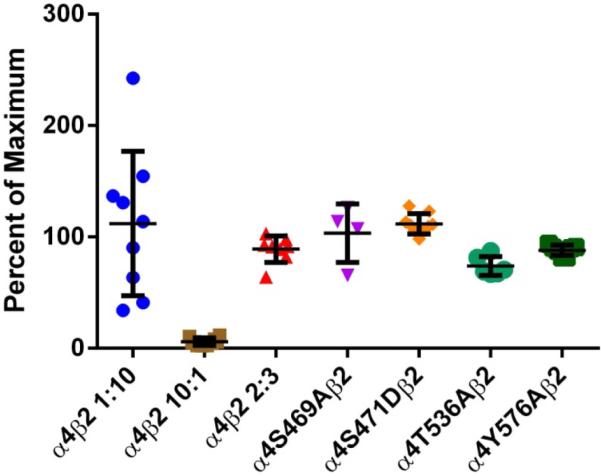
Efficacy of α4β2 agonist A-85380 for stoichiometry determination. Oocytes were injected with α4β2 mRNA at either a ratio of 10:1, 1:10 or 2:3 for WTα4β2 and in a 2:3 ratio for α4 mutants α4S469Aβ2, α4S471Dβ2, α4T536Aβ2 and α4Y576Aβ2. Current was measure for 300 μM ACh and 100 nM A-85380. Efficacy was measured as the percent of maximum when compared to ACh. The efficacy was significantly lower in oocytes injected at a ratio of 10:1 than oocytes injected at a ratio of 1:10 (***p<0.0001). Oocytes injected at a 2:3 ratio show no significant difference from those injected at a 1:10 ratio. The four mutations show no significant difference from the WT injected 2:3 or 1:10 ratio, suggesting that the receptors maintain the same stoichiometry.
Researchers are mostly in the dark about the structure and functional role of the intracellular domain of the AChR. The present data suggest a role for the cytoplasmic loop of the α4 subunit in α4β2 function, expression, and agonist selectivity. Point mutations at consensus sites of the α4 nAChR subunit have remarkable effects on α4β2 function and expression (Table 1). More specifically, these mutations affect the activation properties of ACh and nicotine; however, it is difficult to confirm a role for phosphorylation from the mutations’ effects alone. Since our data show that targeting residues S516 and T536 can render α4β2 nAChRs almost insensitive to nicotine without major effects on normal AChR function, we propose that PKC could be a feasible target for smoking cessation drugs. Therefore, this study provides the framework for further experiments with membrane-permeant cAMP analogs, antagonists, or other potential modulators of phosphorylation. Alternatively, it is possible that the mutations characterized in this study, besides affecting phosphorylation states, could also disrupt key point-to-point interactions essential for secondary structure and/or signal transduction. Twelve mutant receptors (S471A, S471D, T417A, S438A, S469D, S504A, S504D, S516A, T536D, S589A, S589D, and Y576A) exhibited significant changes in potency for ACh without affecting receptor expression levels.
We previously proposed a model that is consistent with an allosteric site for ACh activation, whereas nicotine activation displayed a biphasic curve (López-Hernández et al., 2004; Vibat et al., 1995). In the present study, seven mutations displaced dose-response curves to higher ACh concentrations while displaying WT-like biphasic profiles for nicotine activation. Therefore, all of these mutations produced a negative allosteric effect on ACh activation without affecting the biphasic curve for nicotine activation. Furthermore, three other mutations showed only minor changes in the nicotine profile.
Overall, the effects that we observed in this study are fairly complex, for example, we observed decrease in peak response with both A and D mutations. In this particular case it is very likely that the mutation is not really mimicking a phosphorylation. Nonetheless, these results are consistent with previous reports of biphasic nicotine profiles for α4β2 nAChRs (López-Hernández et al., 2004; Vibat et al., 1995). Most important, we discovered mutations that can provide novel perspectives for the design of nicotine smoking cessation medications that do not act directly or indirectly on the agonist's binding site. Currently, we are designing a α4T536A knock-in mutant construct for the development of a transgenic mouse line with reduced nicotine sensitivity to be used in future studies (patent pending). Overall, these findings provide a new perspective for developing smoking cessation drugs that can definitely cause changes in the pharmacology of nicotinic receptors.
Supplementary Material
HIGHLIGHTS.
α4β2 nAChR phosphorylation plays crucial role in agonist selectivity and expression
Eleven residues in M3-M4 cytoplasmic loop were mutated to alanine and aspartic acid
Two-electrode voltage clamp and [125I]epibatidine binding assays were performed
Four α4β2 nAChR mutations exhibited significant hypersensitivity to nicotine
Two α4β2 nAChR mutations displayed remarkable reductions in sensitivity to nicotine
Acknowledgements
This work was supported by NIH Grant SNRP-U54N54301. Nilza M. Biaggi-Labiosa was sponsored by the Research Initiative for Scientific Enhancement (RISE) (NIH grant 2 R25 GM061151), the Puerto Rico Industrial Development Company (PRIDCO) and the José Trías Monge Scholarship. Daniel Caballero-Rivera was sponsored by the Research Initiative for Scientific Enhancement (RISE) Program (NIH grant 2 R25 GM061151), the Puerto Rico Industrial Development Company (PRIDCO) and the UPR Golf Tournament Fellowship.
ABBREVIATIONS
- nAChR
nicotinic acetylcholine receptor
- ACh
acetylcholine
- PKA
protein kinase A
- PKB
protein kinase B
- PKC
protein kinase C
- CKII
casein kinase II
- TK
tyrosine kinase
- HEPES
N-(2-hydroxyethyl)piperazine-N’-2-ethanesulfonic acid
- CPM
counts per minute
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Authorship Contributions
Participated in research design: Biaggi-Labiosa, Caballero-Rivera, Báez-Pagán, Lasalde-Dominicci
Conducted experiments: Biaggi-Labiosa, Avilés-Pagán, Caballero-Rivera
Performed data analysis: Biaggi-Labiosa, Avilés-Pagán, Caballero-Rivera
Wrote or contributed to the writing of the manuscript: Biaggi-Labiosa, Avilés-Pagán, Caballero-Rivera, Báez-Pagán, Lasalde-Dominicci
References
- Arany I, Clark J, Reed DK, Juncos LA. Chronic nicotine exposure augments renal oxidative stress and injury through transcriptional activation of p66shc. Nephrol. Dial. Transplant. 2013 Jun;28(6):1417–1425. doi: 10.1093/ndt/gfs596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benowitz NL. Pharmacology of nicotine: addiction and therapeutics. Annu. Rev. Pharmacol. Toxicol. 1996;36:597–613. doi: 10.1146/annurev.pa.36.040196.003121. doi:10.1146/annurev.pa.36.040196.003121. [DOI] [PubMed] [Google Scholar]
- Eilers H, Schaeffer E, Bickler PE, Forsayeth JR. Functional deactivation of the major neuronal nicotinic receptor caused by nicotine and a protein kinase C-dependent mechanism. Mol. Pharmacol. 1997;52:1105–1112. [PubMed] [Google Scholar]
- Fenster CP, Beckman ML, Parker JC, Sheffield EB, Whitworth TL, Quick MW, Lester RA. Regulation of alpha4beta2 nicotinic receptor desensitization by calcium and protein kinase C. Mol. Pharmacol. 1999a;55:432–443. [PubMed] [Google Scholar]
- Fenster CP, Whitworth TL, Sheffield EB, Quick MW, Lester RA. Upregulation of surface alpha4beta2 nicotinic receptors is initiated by receptor desensitization after chronic exposure to nicotine. J. Neurosci. Off. J. Soc. Neurosci. 1999b;19:4804–4814. doi: 10.1523/JNEUROSCI.19-12-04804.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaimarri A, Moretti M, Riganti L, Zanardi A, Clementi F, Gotti C. Regulation of neuronal nicotinic receptor traffic and expression. Brain Res. Rev. 2007;55:134–143. doi: 10.1016/j.brainresrev.2007.02.005. doi:10.1016/j.brainresrev.2007.02.005. [DOI] [PubMed] [Google Scholar]
- Gopalakrishnan M, Molinari EJ, Sullivan JP. Regulation of human alpha4beta2 neuronal nicotinic acetylcholine receptors by cholinergic channel ligands and second messenger pathways. Mol. Pharmacol. 1997;52:524–534. [PubMed] [Google Scholar]
- Gotti C, Clementi F. Neuronal nicotinic receptors: from structure to pathology. Prog. Neurobiol. 2004;74:363–396. doi: 10.1016/j.pneurobio.2004.09.006. doi:10.1016/j.pneurobio.2004.09.006. [DOI] [PubMed] [Google Scholar]
- Guo X, Wecker L. Identification of three cAMP-dependent protein kinase (PKA) phosphorylation sites within the major intracellular domain of neuronal nicotinic receptor alpha4 subunits. J. Neurochem. 2002;82:439–447. doi: 10.1046/j.1471-4159.2002.01027.x. [DOI] [PubMed] [Google Scholar]
- Hogg RC, Raggenbass M, Bertrand D. Nicotinic acetylcholine receptors: from structure to brain function. Rev. Physiol. Biochem. Pharmacol. 2003;147:1–46. doi: 10.1007/s10254-003-0005-1. doi:10.1007/s10254-003-0005-1. [DOI] [PubMed] [Google Scholar]
- Hsu YN, Edwards SC, Wecker L. Nicotine enhances the cyclic AMP-dependent protein kinase-mediated phosphorylation of alpha4 subunits of neuronal nicotinic receptors. J. Neurochem. 1997;69:2427–2431. doi: 10.1046/j.1471-4159.1997.69062427.x. [DOI] [PubMed] [Google Scholar]
- Kim H, Flanagin BA, Qin C, Macdonald RL, Stitzel JA. The mouse Chrna4 A529T polymorphism alters the ratio of high to low affinity alpha 4 beta 2 nAChRs. Neuropharmacology. 2003;45:345–354. doi: 10.1016/s0028-3908(03)00167-9. [DOI] [PubMed] [Google Scholar]
- López-Hernández GY, Biaggi-Labiosa NM, Torres-Cintrón A, Ortiz-Acevedo A, Lasalde-Dominicci JA. Contribution of position alpha4S336 on functional expression and up-regulation of alpha4beta2 neuronal nicotinic receptors. Cell. Mol. Neurobiol. 2009;29:41–53. doi: 10.1007/s10571-008-9293-y. doi:10.1007/s10571-008-9293-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- López-Hernández GY, Sánchez-Padilla J, Ortiz-Acevedo A, Lizardi-Ortiz J, Salas-Vincenty J, Rojas LV, Lasalde-Dominicci JA. Nicotine-induced up-regulation and desensitization of alpha4beta2 neuronal nicotinic receptors depend on subunit ratio. J. Biol. Chem. 2004;279:38007–38015. doi: 10.1074/jbc.M403537200. doi:10.1074/jbc.M403537200. [DOI] [PubMed] [Google Scholar]
- Lukas RJ. Effects of chronic nicotinic ligand exposure on functional activity of nicotinic acetylcholine receptors expressed by cells of the PC12 rat pheochromocytoma or the TE671/RD human clonal line. J. Neurochem. 1991;56:1134–1145. doi: 10.1111/j.1471-4159.1991.tb11403.x. [DOI] [PubMed] [Google Scholar]
- Madhok TC, Beyer HS, Sharp BM. Protein kinase A regulates nicotinic cholinergic receptors and subunit messenger ribonucleic acids in PC 12 cells. Endocrinology. 1994;134:91–96. doi: 10.1210/endo.134.1.8275974. doi:10.1210/endo.134.1.8275974. [DOI] [PubMed] [Google Scholar]
- Marks MJ, Grady SR, Collins AC. Downregulation of nicotinic receptor function after chronic nicotine infusion. J. Pharmacol. Exp. Ther. 1993;266:1268–1276. [PubMed] [Google Scholar]
- Moroni M, Zwart R, Sher E, Cassels BK, Bermudez I. alpha4beta2 nicotinic receptors with high and low acetylcholine sensitivity: pharmacology, stoichiometry, and sensitivity to long-term exposure to nicotine. Mol. Pharmacol. 2006;70:755–768. doi: 10.1124/mol.106.023044. doi:10.1124/mol.106.023044. [DOI] [PubMed] [Google Scholar]
- Nakayama H, Okuda H, Nakashima T. In vitro phosphorylation of rat brain nicotinic acetylcholine receptor by cAMP-dependent protein kinase. Ann. N. Y. Acad. Sci. 1993a;707:439–442. doi: 10.1111/j.1749-6632.1993.tb38092.x. [DOI] [PubMed] [Google Scholar]
- Nakayama H, Okuda H, Nakashima T. Phosphorylation of rat brain nicotinic acetylcholine receptor by cAMP-dependent protein kinase in vitro. Brain Res. Mol. Brain Res. 1993b;20:171–177. doi: 10.1016/0169-328x(93)90123-7. [DOI] [PubMed] [Google Scholar]
- Nelson ME, Kuryatov A, Choi CH, Zhou Y, Lindstrom J. Alternate stoichiometries of alpha4beta2 nicotinic acetylcholine receptors. Mol. Pharmacol. 2003;63:332–341. doi: 10.1124/mol.63.2.332. [DOI] [PubMed] [Google Scholar]
- Peng X, Gerzanich V, Anand R, Whiting PJ, Lindstrom J. Nicotine-induced increase in neuronal nicotinic receptors results from a decrease in the rate of receptor turnover. Mol. Pharmacol. 1994;46:523–530. [PubMed] [Google Scholar]
- Pollock VV, Pastoor T, Katnik C, Cuevas J, Wecker L. Cyclic AMP-dependent protein kinase A and protein kinase C phosphorylate alpha4beta2 nicotinic receptor subunits at distinct stages of receptor formation and maturation. Neuroscience. 2009;158:1311–1325. doi: 10.1016/j.neuroscience.2008.11.032. doi:10.1016/j.neuroscience.2008.11.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollock VV, Pastoor TE, Wecker L. Cyclic AMP-dependent protein kinase (PKA) phosphorylates Ser362 and 467 and protein kinase C phosphorylates Ser550 within the M3/M4 cytoplasmic domain of human nicotinic receptor alpha4 subunits. J. Neurochem. 2007;103:456–466. doi: 10.1111/j.1471-4159.2007.04853.x. doi:10.1111/j.1471-4159.2007.04853.x. [DOI] [PubMed] [Google Scholar]
- Taly A, Corringer P-J, Guedin D, Lestage P, Changeux J-P. Nicotinic receptors: allosteric transitions and therapeutic targets in the nervous system. Nat. Rev. Drug Discov. 2009;8:733–750. doi: 10.1038/nrd2927. doi:10.1038/nrd2927. [DOI] [PubMed] [Google Scholar]
- Vibat CR, Lasalde JA, McNamee MG, Ochoa EL. Differential desensitization properties of rat neuronal nicotinic acetylcholine receptor subunit combinations expressed in Xenopus laevis oocytes. Cell. Mol. Neurobiol. 1995;15:411–425. doi: 10.1007/BF02071877. [DOI] [PubMed] [Google Scholar]
- Viseshakul N, Figl A, Lytle C, Cohen BN. The alpha4 subunit of rat alpha4beta2 nicotinic receptors is phosphorylated in vivo. Brain Res. Mol. Brain Res. 1998;59:100–104. doi: 10.1016/s0169-328x(98)00128-4. [DOI] [PubMed] [Google Scholar]
- Wecker L, Guo X, Rycerz AM, Edwards SC. Cyclic AMP-dependent protein kinase (PKA) and protein kinase C phosphorylate sites in the amino acid sequence corresponding to the M3/M4 cytoplasmic domain of alpha4 neuronal nicotinic receptor subunits. J. Neurochem. 2001;76:711–720. doi: 10.1046/j.1471-4159.2001.00041.x. [DOI] [PubMed] [Google Scholar]
- Zwart R, Broad LM, Xi Q, Lee M, Moroni M, Bermudez I, Sher E. 5-I A-85380 and TC-2559 differentially activate heterologously expressed alpha4beta2 nicotinic receptors. Eur. J. Pharmacol. 2006;539:10–17. doi: 10.1016/j.ejphar.2006.03.077. doi:10.1016/j.ejphar.2006.03.077. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.