

Abstract
Foldback priming at DNA double-stranded breaks is one mechanism proposed to initiate palindromic gene amplification, a common feature of cancer cells. Here we show small (5–9 bp) inverted repeats drive the formation of large palindromic duplications, the major class of chromosomal rearrangements recovered from yeast cells lacking Sae2 or the Mre11 nuclease. RPA dysfunction increased the frequency of palindromic duplications in Sae2 or Mre11 nuclease-deficient cells by ~1000-fold, consistent with intra-strand annealing to create a hairpin-capped chromosome that is subsequently replicated to form a dicentric isochromosome. The palindromic duplications were frequently associated with duplication of a second chromosome region bounded by a repeated sequence and a telomere, suggesting the dicentric chromosome breaks and repairs by recombination between dispersed repeats to acquire a telomere. We propose secondary structures within single-stranded DNA are potent instigators of genome instability, and RPA and Mre11-Sae2 play important roles in preventing their formation and propagation, respectively.
Keywords: gene amplification, RPA, Sae2, Mre11, palindrome, inverted duplication
INTRODUCTION
Diverse and complex chromosome rearrangements, including inter-chromosomal translocations, deletions and gene amplifications, are often found in the genomes of cancer cells (Stephens et al., 2011). Intra-chromosomal gene amplifications are organized as direct or inverted (palindromic) duplications, and can confer a growth advantage to promote tumor initiation, metastatic dissemination or drug resistance resulting in treatment failure (Kitada and Yamasaki, 2007, 2008; Marotta et al., 2012; Slamon et al., 1987; Tanaka and Yao, 2009). Palindromic duplications have been identified in metastatic pancreatic cancer and ErbB-2 (HER2) positive breast cancers (Campbell et al., 2010; Marotta et al., 2012; Slamon et al., 1987; Waddell et al., 2015).
Mechanisms used to explain palindromic amplification commonly invoke a dicentric isochromosome that undergoes breakage-fusion-bridge (BFB) cycles (Marotta et al., 2013; Narayanan et al., 2006; Tanaka and Yao, 2009). A dicentric isochromosome can be formed by fusion of the sister chromatids produced by replication of a chromosome broken in the G1 phase of the cell cycle, or with dysfunctional telomeres (Fig S1). Asymmetric breakage of the dicentric chromosome during mitosis results in inverted duplication close to the breakpoint, further BFB cycles can result in extensive gene amplification. A role for inverted repeats was first proposed to explain the formation of short, linear palindromic chromosomes during nuclear differentiation of Tetrahymena (Yasuda and Yao, 1991). A 42 bp inverted repeat was shown to be essential for palindrome formation, and a subsequent study supported a model whereby annealing of the repeats after end resection from a double-strand break (DSB) creates a hairpin-capped molecule that is replicated to generate the palindromic duplication (Fig S1) (Butler et al., 1996). Short inverted repeats have also been shown to mediate palindromic gene amplification in yeast and mammalian cells (Butler et al., 1996; Maringele and Lydall, 2004; Marotta et al., 2013; Putnam et al., 2014; Rattray et al., 2005; Tanaka et al., 2002). In yeast the recovery of palindromic duplications is markedly increased in cells lacking Sae2 or expressing a nuclease-defective mutant form of Mre11 (Rattray et al., 2001; Rattray et al., 2005). Sae2 activates the Mre11-Rad50-Xrs2 (MRX) complex to cleave the 5′ strand internal to the DSB end to initiate 5′-3′ DNA end resection and promotes opening of hairpin-capped DNA ends (Cannavo and Cejka, 2014; Lengsfeld et al., 2007; Lobachev et al., 2002).
Long inverted repeats (>300 bp) function as fragile sites in yeast and stimulate gross chromosome rearrangements (GCRs), including dicentric/acentric chromosome formation, non-reciprocal translocation and gene amplification (Lemoine et al., 2005; Lobachev et al., 2002; Mizuno et al., 2009; Narayanan et al., 2006; Paek et al., 2009). Inverted repeats are thought to extrude to form a cruciform or hairpin structure that is cleaved at the base to create DSBs terminated by covalently closed hairpins; alternatively, intramolecular annealing of inverted repeats exposed by end resection of a DSB could create a hairpin-capped end (Darmon et al., 2010). Failure to resolve the hairpin-capped ends by MRX and Sae2 (or SbcCD in Escherichia coli) leads to the formation of acentric and dicentric palindromic molecules that can undergo further rearrangement (Darmon et al., 2010; Eykelenboom et al., 2008; Lobachev et al., 2002; Narayanan et al., 2006). Recombination-dependent replication fork restart within inverted repeats at a stalled replication fork can also result in dicentric/acentric palindromic chromosome rearrangements (Mizuno et al., 2009).
Conditions that favor annealing between short homologies are expected to increase foldback priming resulting in palindromic duplications. Replication protein A (RPA), the main eukaryotic single-stranded DNA (ssDNA) binding protein, removes secondary structure from ssDNA and prevents annealing between short oligonucleotides in vitro (Gibb et al., 2012; Sugiyama et al., 1998). Depletion of Rfa1 (RFA1/RPA1 encodes the largest subunit of the heterotrimeric RPA complex) from yeast cells results in the formation of foldback structures by annealing between short inverted repeats within the ssDNA formed by end resection at DSBs (Chen et al., 2013). Furthermore, expression of hypomorphic alleles of RFA1, which encode proteins with reduced DNA binding activity, greatly increases the frequency of microhomology-mediated end joining (MMEJ), suggesting that RPA binding to ssDNA prevents annealing between short homologies that can result in genome destabilization (Deng et al., 2014).
Because depletion of RPA from cells results in foldback structures at DSBs we were interested in whether rfa1 hypomorphic mutants would exhibit an increased frequency of palindromic duplication. We found that naturally occurring short inverted repeats (5–9 bp) stimulate formation of large (51–83 kb) palindromic duplications, which are the main class of GCRs recovered from sae2Δ and mre11-H125N mutants. Although palindromic duplications were not found among the GCRs from the rfa1-t33 mutant, the frequency of these events was increased ~1000-fold in the rfa1-t33 sae2Δ and rfa1-t33 mre11-H125N double mutants relative to the sae2Δ and mre11-H125N mutants. We also recovered rearrangements from the rfa1-t33 sae2Δ mutant that had a more than two-fold amplification, similar to amplifications observed in tumor cells.
RESULTS
rfa1-t33 and sae2Δ synergistically increase the GCR rate
To examine genomic instability caused by rfa1-t33 and sae2Δ mutations we used a well-characterized assay that measures the accumulation of spontaneous GCRs on the left arm of chromosome V (Ch V) by simultaneous loss of the URA3 and CAN1 genes (Fig 1A) (Chen and Kolodner, 1999). This assay detects a broad spectrum of GCR events with the frequency indicative of general genome stability and distinct repair products recovered being reflective of the inherent nature of the genotype (Putnam et al., 2005). The rfa1-t33 mutant was chosen for this study because the RPAt33 mutant protein is partially defective for removal of secondary structure from ssDNA in vitro (Deng et al., 2014). The rfa1-t33 mutant showed a 205-fold increased rate of GCR accumulation compared to wild type, consistent with a previous study (Fig. 1B, Table 1) (Chen and Kolodner, 1999). Although loss of Sae2 resulted in only a 5-fold increase in the GCR rate, a synergistic increase was observed in the rfa1-t33 sae2Δ double mutant (1.86 × 10−6, 9000-fold elevation).
Figure 1. Increased GCR rate and an altered spectrum of events in sae2Δ derivatives.
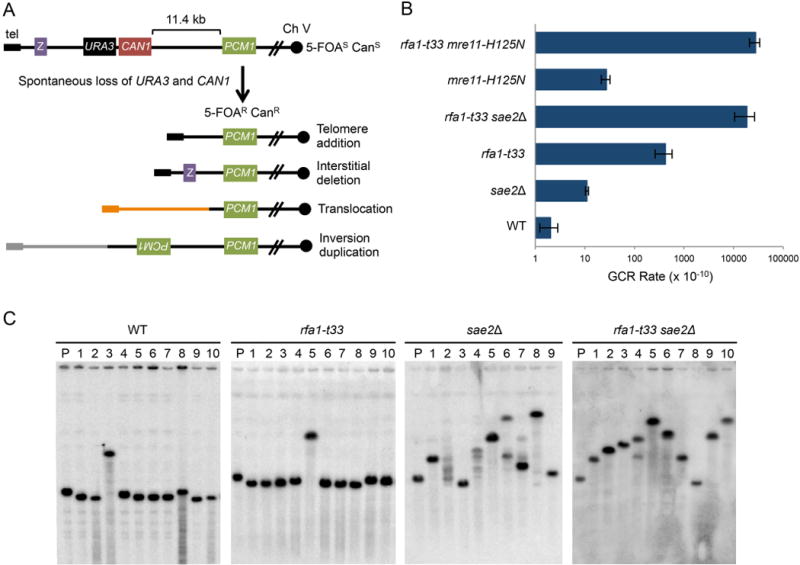
A. Schematic of the GCR assay and rearrangements. Z indicates a distal Ch V gene that would be retained after interstitial deletion and the solid circle denote the centromere. B. GCR rates for the indicated genotypes, WT denotes wild type. The rates shown are the average of three independent trials for WT, sae2Δ, mre11-H125N, rfa1-t33 mre11-H125N and five and seven-independent trials for rfa1-t33 and rfa1-t33 sae2Δ, respectively. Error bars indicate standard deviation. C. PFGE of WT, rfa1-t33, sae2Δ and rfa1-t33 sae2Δ GCR clones. The first lane of each blot (P) shows the parental un-rearranged Ch V. See also Figure S1.
Table 1.
Spectrum of GCR events
| Relevant genotype | CanR 5-FOAR mutation rate* | Isolates analyzed | Telomere addition | Deletion/translocation | Ch V left arm duplication (class I, class II) |
|---|---|---|---|---|---|
| WT | 2.04 [± 0.72] × 10−10 (1.0) | 121 | 9 | 1 | 1 (1, 0) |
| sae2Δ | 1.11 [± 0.08] × 10−9 (5.4) | 9 | 1 | 1 | 7 (4, 3) |
| mre11-H125N | 2.70 [± 0.46] × 10−9 (13.2) | 10 | 2 | 0 | 8 (6, 2) |
| rfa1-t33 | 4.20 [± 1.80 ] × 10−8 (205) | 14 | 7 | 7 | 0 (0, 0) |
| rfa1-t33 sae2Δ | 1.86 [± 1.10] × 10−6 (9079) | 10 | 1 | 0 | 9 (9, 0) |
| rfa1-t33 mre11-H125N | 2.75 [± 0.57] × 10−6 (13436) | 10 | 3 | 0 | 7 (3, 4) |
Rate of accumulating CanR 5-FOAR progeny. Numbers in brackets indicate standard deviation.
The number in parenthesis is the fold increase relative to wild type.
Class I: Duplication on Ch V and second homology dependent invasion
Class II: Duplication on Ch V only
One WT GCR clone contained a point mutation in the CAN1 gene and no discernable mutation at URA3. This clone was able to grow on 5-FOA containing and SC-URA media.
See also Figure S3.
The large increase in the frequency of GCRs in the rfa1-t33 mutant could be due to more initiating lesions, defective homologous recombination (HR) or an altered mode of repair. We found the GCR rate of the HR-defective rad51Δ mutant was increased by only 4-fold relative to wild type, and while the sae2Δ mutation did synergize with rad51Δ, the rate of GCRs was ~100-fold lower than the rfa1-t33 sae2Δ double mutant (Fig S2A). An increased number of spontaneous lesions would be expected to increase the rate of deletions between direct repeats. We found a small but significant increase in the rate of deletions in the rfa1-t33 mutant (p=0.05), consistent with more initiating lesions, while the rate of spontaneous Rad51-dependent gene conversion was comparable to wild-type cells (Fig S2B). Thus, the increased GCR rate of the rfa1-t33 mutant is not a consequence of impaired HR and is likely due to aberrant repair of replication-associated DNA breaks.
GCRs recovered from sae2Δ derivatives have an expanded chromosome V
To determine the spectrum of GCRs, we first examined Ch V by pulsed-field gel electrophoresis (PFGE) and Southern blot hybridization using a PCM1 probe, an essential gene on Ch V. We found that the majority of rearrangements in wild type and the rfa1-t33 mutant resulted in a Ch V species with a faster mobility than the parental Ch V (Fig 1C). In contrast, the majority of sae2Δ and rfa1-t33 sae2Δ GCR clones exhibited an expanded Ch V as evidenced by the slower mobility by PFGE (Fig 1C). Some sae2Δ GCR isolates displayed a smear of Ch V products indicating either a mixed population of repaired products or an unstable Ch V.
To better understand the types of GCRs in the rfa1-t33 and sae2Δ derivatives, we utilized a previously described method to identify the location of the breakpoint by overlap PCR using Ch V primers, and then attempted to sequence across the junction using an arbitrary PCR strategy (Fig S3) (Schmidt et al., 2006). We characterized 12 GCR isolates from wild-type cells and found that repair occurred primarily by telomere addition (Table 1). We were unable to identify the breakpoint junction by PCR of the GCR clone with an expanded Ch V; this event was further characterized by array-Comparative Genome Hybridization (aCGH, see below). Of 14 GCR clones analyzed from the rfa1-t33 mutant, we found that 7 were due to telomere addition, 2 events were due to translocations and 5 events were interstitial deletions mediated by microhomologies (Table 1). Telomere addition and interstitial deletion are consistent with a shorter Ch V species by PFGE, while the translocation events exhibited an expanded Ch V. The spectra of GCR events found for wild type and the rfa1-t33 mutant are consistent with a previous study (Chen and Kolodner, 1999).
Rearrangements in sae2Δ mutants result from inverted duplication of Ch V
Of the 9 GCR clones analyzed from the sae2Δ mutant, one resulted from telomere addition and another was due to a translocation mediated by microhomologies. For the remaining 7 events we identified the breakpoints but could not amplify the rearrangement junctions by PCR. Similarly, 1/10 GCR clones from the rfa1-t33 sae2Δ mutant was due to telomere addition and we failed to amplify junctions from the other 9 clones. Since previous studies had shown recovery of palindromic duplications from sae2Δ mutants (Putnam et al., 2014; Rattray et al., 2001; Rattray et al., 2005), it seemed likely that the failure to amplify junctions from the majority of sae2Δ and rfa1-t33 sae2Δ GCR clones was due to the presence of inverted duplications, which are difficult to amplify as a result of snapback structures formed during PCR.
Two strategies were used to determine the structure of the rearrangements. First, we physically mapped the region around the breakpoint of several GCR clones by restriction digestion of genomic DNA and hybridization with appropriate probes (Fig 2A). The distance between each restriction endonuclease site chosen and the breakpoint is defined as distance “X” and we reasoned that if a palindromic duplication had occurred we would recover DNA fragments of size 2X because the restriction site would be duplicated on the other side the breakpoint. We used several restriction endonucleases that were predicted to cleave at varying distances from the breakpoint and all showed a fragment of twice the expected size, consistent with a palindromic duplication. Interestingly, a shadow band of half the predicted size was observed by native gel electrophoresis (Fig 2A) and only a single band of the predicted size was seen by alkaline gel electrophoresis (data not shown) suggesting formation of snapback structures during DNA manipulation or extrusion of the cruciform in vivo.
Figure 2. Inversion duplications are recovered from sae2Δ derivatives after chromosomal rearrangement.
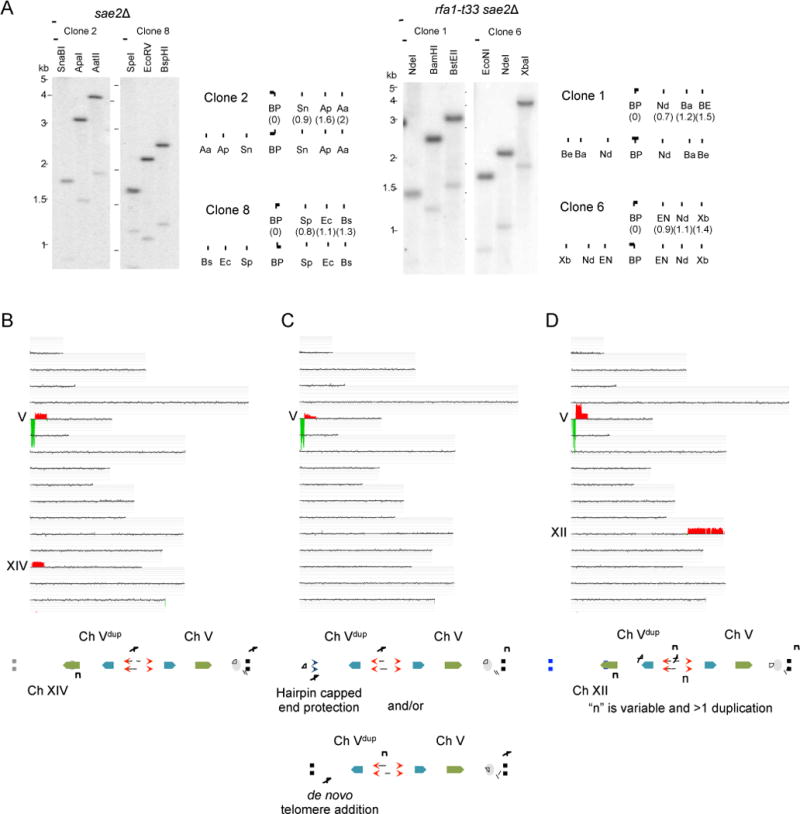
A. Physical analysis of the breakpoint region surrounding the DSB. Schematic shows location of the breakpoint (BP) and restriction enzyme recognition sites adjacent to the BP. For each pair of lines, the top line shows the sites for the control strain, and the bottom line shows the sites in the strain with the chromosome rearrangement. Abbreviations for restriction enzymes are as follows: SnaBI (Sn) ApaI (Ap), AatII (Aa), SpeI (Sp), EcoRV (Ec), BspHI (BS), NdeI (Nd), BamHI (Ba), BstEII (BE), EcoNI (EN), XbaI (Xb). B-D. aCGH analysis of GCR clones. B. Representative aCGH from a sae2Δ GCR clone indicating deletion of the terminal fragment of Ch V, duplication adjacent to the breakpoint on Ch V and an additional duplication to create non-reciprocal translocation. Each horizontal line represents each yeast chromosome (I–XVI) and the roman numerals to the left indicate the chromosomes with alterations. Green below the line indicates sequence loss and red above the line shows sequence gain. C. Representative aCGH from a sae2Δ GCR clone showing duplication adjacent to the break on Ch V only. D. Representative aCGH from a rfa1-t33 sae2Δ GCR clone with a higher order amplification adjacent to the breakpoint. Schematics to show the rearranged Ch V in each of the aCGH plots are shown below the plots. Solid green indicates a Ty-related or tRNA element. Blue represents gene X within the duplicated region. See also Figure S2.
Second, we turned to aCGH to determine the size of the palindromic duplications. aCGH confirmed loss of distal sequence on Ch V and also indicated a duplication of sequence immediately adjacent to the break point; the breakpoints were consistent with the PCR mapping results (Fig 2B, 2C and 2D). One clone from the wild-type strain (8%), 7/9 (78%) sae2Δ clones and 9/10 (90%) rfa1-t33 sae2Δ clones analyzed had inversion duplications. Three of the 7 inversion duplications recovered from the sae2Δ mutant exhibited a “fading” duplication (class II): sequences immediately adjacent to the breakpoint were present at 2X, but exhibited a gradient of 2X to 1X copy number over a ~70 kb region (Fig 2C); thus giving rise to variable Ch V sizes by PFGE (Fig 1C). Passage of a single class II inversion duplication clone and isolation of single colonies confirmed that the Ch V remains unstable through many generations but some single colonies isolated did stabilize to form a discrete-sized Ch V (data not shown). The chromosome end may be protecting essential genes by formation of palindromes, as reported previously for survival of recombination and telomerase defective cells (Maringele and Lydall, 2004), and some of these may eventually be stabilized by de novo telomere addition.
Inversion duplications require a secondary invasion to stabilize the chromosome
Many of the GCR clones with an inversion duplication of Ch V had a duplication of another genomic region from the telomere to an internal site (Class I) (Fig 2B), suggesting that a secondary event is required to form a stable chromosome by acquisition of a telomere (Pennaneach and Kolodner, 2009). Four of the seven inversion duplications analyzed from the sae2Δ mutant and the single event from wild type were composed of sequences from the break point to the Ty1-containing ura3-52 locus (located 76–83 kb away) and were associated with a duplication of another chromosome arm bounded by a Ty1-related element (Ty or delta element) and a telomere. All of the inversion duplications found in the rfa1-t33 sae2Δ mutant contained a duplication of another genomic region. Eight of the rfa1-t33 sae2Δ clones had an inverted duplication to the ura3-52 locus associated with a secondary duplication initiated at a Ty1-related element, the remaining clone had an inverted duplication up to a serine tRNA (51 kb) associated with a duplication from an identical serine tRNA present on Ch IV to the telomere. GCRs found in other studies frequently involve repetitive elements, such as delta sequences, but tRNAs have rarely been observed at breakpoint junctions (Fischer et al., 2000; Lemoine et al., 2005; Mieczkowski et al., 2006; Narayanan et al., 2006; Paek et al., 2009). The serine tRNA is only 84 nucleotides in length, considerably shorter than delta (330 bp) and Ty elements (6 kb). The spectra of GCRs recovered from wild type and mutant cells are summarized in Figure 3A.
Figure 3. Summary of GCR data and model for palindromic gene duplication.
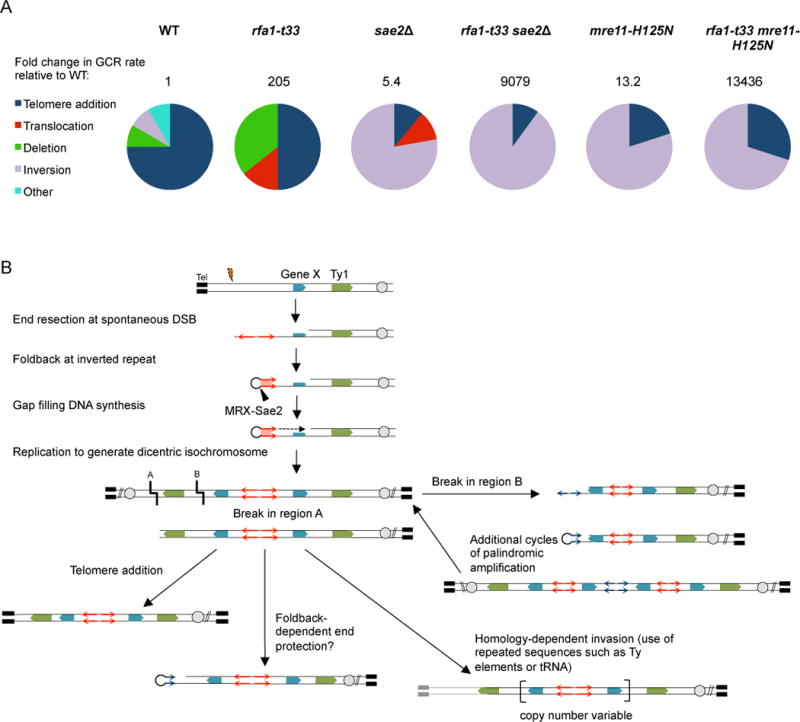
A. The increase in the GCR rate for all of the mutants relative to wild type and the spectrum of events recovered from each genotype are shown. B. A spontaneous DSB centromere distal to Gene X initiates rearrangement. A foldback in the 3′ single-stranded DNA tail formed by end resection primes DNA synthesis and the 3′ end is ligated to the resected 5′ end to form a hairpin-capped end. Replication results in a dicentric isochromosome that is broken at mitosis and undergoes additional cycles of foldback priming, telomere addition or recombination between repeated sequences. A foldback might also stabilize the end to prevent degradation and checkpoint activation. Grey circles represent centromeres and thick black lines denote telomeres. See also Figures S3, S4 and Table S1.
Inversion duplications are mediated by short inverted repeats
Inversion duplications can be formed by end joining between two replicated broken sister chromatids or result from intra-strand annealing between short inverted repeats to form a foldback structure (Marotta et al., 2013). To determine the sequence at the breakpoint of the inversion duplications we treated the genomic DNA with sodium bisulfite to deaminate the cytosines and thus disrupt the palindrome sufficiently to allow for PCR amplification and DNA sequencing (Rattray, 2004). We found 5–9 bp inverted repeats separated by 3–12 bp spacer sequences present at the center of the inverted duplications (Table S1). Breakpoints were distributed throughout the 11 kb region between CAN1 and PCM1 with some clustering around the CAN1 locus (Table S1), in agreement with a previous study (Putnam et al., 2005). These data support the hypothesis that inversion duplications in sae2Δ derivatives initiate by intrastrand annealing at short inverted repeats followed by gap filling and ligation to create a hairpin-capped end. Replication of the hairpin-capped chromosome would yield a dicentric chromosome that could be broken at mitosis and the end healed by a secondary recombination event or telomere addition (Fig 3B).
Higher order amplifications observed in sae2Δ rfa1-t33 GCRs
Gene amplifications observed in cancers often have more than a single genomic duplication, some genes are amplified many fold (Kitada and Yamasaki, 2007, 2008; Marotta et al., 2012; Neiman et al., 2008; Tanaka and Yao, 2009). Interestingly, 3/9 GCRs with an inverted duplication recovered from the rfa1-t33 sae2Δ mutant contained a >2-fold amplification of the region adjacent to the break site (Fig 2D, Fig S3). The clone shown in Figure 2D is estimated to have 7 copies of the 49 kb region adjacent to the breakpoint by aCGH hybridization and qPCR. Based on the predicted size of Ch V, and the intensity of hybridization with the PCM1 probe, the amplification is intrachromosomal. This finding suggests more than one round of palindromic gene amplification and BFB cycles occurred to generate a higher order amplification (Fig 3B).
Inversion duplications are the primary class of GCRs recovered from mre11-H125N derivatives
Several studies have shown that loss of the Mre11 nuclease via the mre11-D56N or mre11-H125N mutations results in stabilization of hairpin-capped ends and palindromic duplications (Chen et al., 2013; Lobachev et al., 2002; Moreau et al., 1999; Rattray et al., 2001). However, palindromic duplications were not recovered from the mre11-H125N mutant using a variation of the GCR assay used here, even though they were identified as the main class of GCRs in the sae2Δ mutant (Putnam et al., 2014). Because the rfa1-t33 mutation enhances recovery of palindromic duplication in the sae2Δ background we expected a similar outcome in the absence of the Mre11 nuclease. The rate of GCRs was increased by 13 and 13000-fold in the mre11-H125N and mre11-H125N rfa1-t33 mutants, respectively, relative to wild type, similar to the increases found for the sae2Δ derivatives (Fig 1B, Table 1). Like the sae2Δ derivatives, most of the mre11-H125N and mre11-H125N rfa1-t33 clones exhibited an expanded Ch V by PFGE and an inverted duplication adjacent to the breakpoint (Fig S4). These data support the hypothesis that the Mre11 nuclease, in conjunction with Sae2, cleaves foldback structures to prevent palindromic duplications. There are two possible explanations for the discrepancy between our results and those of Putnam et al (2014). First, we used a slightly different GCR assay, and second, we created a knock-in allele of mre11-H125N instead of expressing the mutant allele from a plasmid in an mre11Δ strain.
DISCUSSION
Foldback priming at resected DSBs is one of the mechanisms proposed to drive palindromic gene duplication (Tanaka and Yao, 2009). Inverted duplications are a rare class of GCRs in wild-type cells, but are the major class of events recovered from sae2Δ and mre11-H125N mutants, and the frequency of their formation is increased by 50 to 130-fold relative to wild type. Although RPA plays an important role in preventing annealing between microhomologies that can lead to foldback structures (Chen et al., 2013; Deng et al., 2014), no inverted duplications were found among the 14 GCR events analyzed from the rfa1-t33 hypormorphic mutant. However, in the context of Sae2 or Mre11 nuclease deficiency, rfa1-t33 caused a >1,000-fold increase in the rate of inversion duplications. These data are consistent with the model that RPA normally removes foldback structures and when they do occasionally arise the Mre11 endonuclease and Sae2 efficiently cleave them to prevent formation of inverted duplications.
Palindromic duplications are a major threat to genomic stability because they act as fragile sites and stimulate further amplification and chromosome rearrangements (Lemoine et al., 2005; Narayanan et al., 2006; Tanaka and Yao, 2009). Therefore, the timely removal of foldbacks is essential to preserve genome integrity and could be one of the main cellular functions for the Mre11 nuclease and Sae2. CtIP (the functional ortholog of Sae2 in vertebrates) and the Mre11 nuclease are required for recombination induced at a hairpin-forming sequence in human cells, and SbcCD, the ortholog of the MR complex, destabilizes palindromes in E. coli suggesting hairpin cleavage is evolutionarily conserved (Eykelenboom et al., 2008; Wang et al., 2014).
Of the palindromic duplications observed in sae2Δ and mre11-H125N mutants, half were stabilized by a secondary rearrangement to acquire a telomere. We propose the dicentric isochromosome generated by replication of the hairpin-capped Ch V is broken between the two centromeres in the vicinity of the Ty1 element at the ura3 locus (Lopez et al., 2015), then the Ty or delta element is used for recombination with a Ty1 or delta element elsewhere in the genome (Fig 3B). Chromosome rearrangements mediated by Ty or delta elements have been reported in many other yeast studies, in particular, to stabilize dicentric chromosomes (Lemoine et al., 2005; Mieczkowski et al., 2006; Narayanan et al., 2006; Pennaneach and Kolodner, 2009; Surosky and Tye, 1985). Although we found a high frequency of GCRs in the rfa1-t33 sae2Δ and rfa1-t33 mre11-H125N mutants this is likely to be an underestimate of the global genome instability in these cells. The GCR assay used only detects events occurring in a ~30 kb region of Ch V and many of the secondary recombination events would not generate a viable product.
Short inverted repeats were identified by DNA sequencing at the breakpoints of the palindromic duplications characterized in sae2Δ GCR clones. The inverted repeats vary in size from 5 to 9 bp with 1–2 bp mismatches and are separated by 2–12 bp. Half of the 12 breakpoints sequenced utilized two sequences, which are the longest if mismatches are included, whereas the other 6 are unique. All of the palindromic junctions analyzed from sae2Δ and rad50Δ mutants in a previous study were formed at inverted repeats of 4–6 bp separated by 2–8 bp (Rattray et al., 2005). In contrast, the breakpoints of inverted duplications from tel1Δ mutants had inverted repeats of similar sizes to those identified in this study, but with longer spacers (25–44 bp) (Putnam et al., 2014). We speculate that Mre11 and Sae2 recognize and/or cleave foldbacks with short spacers; perhaps other structure-selective nucleases regulated by Tel1 can cleave long ssDNA loops.
Based on the properties of Mre11, Sae2 and RPAt33, intrastrand annealing to form a foldback is a logical mechanism to explain the palindromic duplication rearrangements. We speculate the initiating event is a resected DSB, but cannot exclude the possibility of annealing between the leading nascent strand and lagging strand template at a stalled replication fork linking the leading to the lagging strand (Fig S5), as suggested in previous studies (Brewer et al., 2011; Paek et al., 2009). Fork reversal could then lead to a hairpin-capped end, a substrate for MRX-Sae2 cleavage. It is unlikely that inversion duplications are created by MMEJ between replicated broken sister-chromosomes because MMEJ occurs at a high frequency in the rfa1-t33 mutant, yet we do not observe inversion duplications in the single mutant. Furthermore, an MMEJ mechanism would not explain why palindromic duplications are preferentially recovered from sae2Δ and mre11-H125N mutants. We argue that it is the hairpin opening activity of Mre11 and Sae2 that prevents formation of inversion duplications.
There are several arguments that our observations are relevant to the mechanisms of carcinogenesis. First, as described in the Introduction, palindromic insertions are a common type of genetic rearrangement in certain classes of tumors. For example, 16% of the chromosome rearrangements in metastatic pancreatic tumors are palindromic duplications compared to 2% of chromosome rearrangements in breast tumors (Campbell et al., 2010). Second, germline mutations in NBN (encoding Nbs1/Xrs2) result in the cancer-prone Nijmegen Breakage Syndrome (Thompson and Schild, 2002) and mutations in RAD50 are associated with an increased risk of familial breast cancer (Walsh and King, 2007). Third, mutations in RPA and MRE11A genes are found in various classes of sporadic tumors. For example, biallelic mutations of RPA1 were found in a pancreatic cancer study (Waddell et al., 2015). We examined data from the Broad Institute Cancer Genome Atlas Genome Data Analysis Center (http://gdac.broadinstitute.org/) for significant over-representation of mutations in RPA1-3, MRE11A, NBN, RAD50 and RBBP8/CtIP in various types of tumors. Both MRE11A and RPA3 were significantly over-represented (p values of 0.016 and 0.009, respectively) among colon adenocarcinomas (Broad Institute TCGA Genome Data Analysis Center (2015): Analysis Overview for Colon Adenocarcinoma (Primary solid tumor cohort) - 02 April 2015. Broad Institute of MIT and Harvard. doi:10.7908/C14Q7T1K), and RPA3 was over-represented (p=0.008) among colorectal adenocarcinomas (Broad Institute TCGA Genome Data Analysis Center (2015): Analysis Overview for Colorectal Adenocarcinoma (Primary solid tumor cohort) -02 April 2015. Broad Institute of MIT and Harvard. doi:10.7908/C12N5184). A more convincing association between our yeast observations and cancer data would be a demonstration that tumors with mutations in RPA1-3, and/or members of the MRN complex are associated with higher frequencies of palindromic duplications than observed in tumors with other types of mutations. To our knowledge, such an association has not yet been made.
EXPERIMENTAL PROCEDURES & METHODS
Yeast strain construction and cell culture were performed using standard methods. Fluctuation assays to determine the rate of GCRs, PCR mapping and amplification of the junctions were performed as previously described (Putnam and Kolodner, 2010). For physical analysis of inversion duplications, 3 μg of genomic DNA was digested with 20 units of the indicated restriction endonucleases, separated by gel electrophoresis and transferred to Biobond-Plus nylon membrane (Sigma) for hybridization. Samples for PFGE were prepared and analyzed following a published protocol (Argueso et al., 2008). Genomic DNA was extracted from agarose plugs and labeled for microarray hybridization as previously described (Zhang et al., 2013). For bisulfite sequencing, genomic DNA was first treated with the EpiMark Bisulfite Conversion kit (New England Biolabs) and then PCR amplified using EpiMark Hot Start Taq DNA Polymerase (New England Biolabs) according to the manufacturers instructions. Detailed information regarding methodology and associated references are available in the Supplemental Experimental Procedures.
Supplementary Material
Acknowledgments
We thank R. Kolodner for gifts of yeast strains and plasmids, J. L. Argueso for advice on PFGE and D. Gordenin for advice on looking for cancer-associated mutations. This study was supported by grants from the National Institutes of Health (R01 GM041784 and P01 CA174653 to L.S.S, and R01 GM24110 and GM52319 to T.D.P.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Argueso JL, Westmoreland J, Mieczkowski PA, Gawel M, Petes TD, Resnick MA. Double-strand breaks associated with repetitive DNA can reshape the genome. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:11845–11850. doi: 10.1073/pnas.0804529105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brewer BJ, Payen C, Raghuraman MK, Dunham MJ. Origin-dependent inverted-repeat amplification: a replication-based model for generating palindromic amplicons. PLoS Genet. 2011;7:e1002016. doi: 10.1371/journal.pgen.1002016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler DK, Yasuda LE, Yao MC. Induction of large DNA palindrome formation in yeast: implications for gene amplification and genome stability in eukaryotes. Cell. 1996;87:1115–1122. doi: 10.1016/s0092-8674(00)81805-x. [DOI] [PubMed] [Google Scholar]
- Campbell PJ, Yachida S, Mudie LJ, Stephens PJ, Pleasance ED, Stebbings LA, Morsberger LA, Latimer C, McLaren S, Lin ML, et al. The patterns and dynamics of genomic instability in metastatic pancreatic cancer. Nature. 2010;467:1109–1113. doi: 10.1038/nature09460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannavo E, Cejka P. Sae2 promotes dsDNA endonuclease activity within Mre11-Rad50-Xrs2 to resect DNA breaks. Nature. 2014;514:122–125. doi: 10.1038/nature13771. [DOI] [PubMed] [Google Scholar]
- Chen C, Kolodner RD. Gross chromosomal rearrangements in Saccharomyces cerevisiae replication and recombination defective mutants. Nat Genet. 1999;23:81–85. doi: 10.1038/12687. [DOI] [PubMed] [Google Scholar]
- Chen H, Lisby M, Symington LS. RPA coordinates DNA end resection and prevents formation of DNA hairpins. Mol Cell. 2013;50:589–600. doi: 10.1016/j.molcel.2013.04.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darmon E, Eykelenboom JK, Lincker F, Jones LH, White M, Okely E, Blackwood JK, Leach DR. E. coli SbcCD and RecA control chromosomal rearrangement induced by an interrupted palindrome. Mol Cell. 2010;39:59–70. doi: 10.1016/j.molcel.2010.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng SK, Gibb B, de Almeida MJ, Greene EC, Symington LS. RPA antagonizes microhomology-mediated repair of DNA double-strand breaks. Nat Struct Mol Biol. 2014;21:405–412. doi: 10.1038/nsmb.2786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eykelenboom JK, Blackwood JK, Okely E, Leach DR. SbcCD causes a double-strand break at a DNA palindrome in the Escherichia coli chromosome. Mol Cell. 2008;29:644–651. doi: 10.1016/j.molcel.2007.12.020. [DOI] [PubMed] [Google Scholar]
- Fischer G, James SA, Roberts IN, Oliver SG, Louis EJ. Chromosomal evolution in Saccharomyces. Nature. 2000;405:451–454. doi: 10.1038/35013058. [DOI] [PubMed] [Google Scholar]
- Gibb B, Silverstein TD, Finkelstein IJ, Greene EC. Single-stranded DNA curtains for real-time single-molecule visualization of protein-nucleic acid interactions. Anal Chem. 2012;84:7607–7612. doi: 10.1021/ac302117z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitada K, Yamasaki T. The MDR1/ABCB1 regional amplification in large inverted repeats with asymmetric sequences and microhomologies at the junction sites. Cancer Genet Cytogenet. 2007;178:120–127. doi: 10.1016/j.cancergencyto.2007.06.014. [DOI] [PubMed] [Google Scholar]
- Kitada K, Yamasaki T. The complicated copy number alterations in chromosome 7 of a lung cancer cell line is explained by a model based on repeated breakage-fusion-bridge cycles. Cancer Genet Cytogenet. 2008;185:11–19. doi: 10.1016/j.cancergencyto.2008.04.005. [DOI] [PubMed] [Google Scholar]
- Lemoine FJ, Degtyareva NP, Lobachev K, Petes TD. Chromosomal translocations in yeast induced by low levels of DNA polymerase a model for chromosome fragile sites. Cell. 2005;120:587–598. doi: 10.1016/j.cell.2004.12.039. [DOI] [PubMed] [Google Scholar]
- Lengsfeld BM, Rattray AJ, Bhaskara V, Ghirlando R, Paull TT. Sae2 is an endonuclease that processes hairpin DNA cooperatively with the Mre11/Rad50/Xrs2 complex. Mol Cell. 2007;28:638–651. doi: 10.1016/j.molcel.2007.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobachev KS, Gordenin DA, Resnick MA. The Mre11 complex is required for repair of hairpin-capped double-strand breaks and prevention of chromosome rearrangements. Cell. 2002;108:183–193. doi: 10.1016/s0092-8674(02)00614-1. [DOI] [PubMed] [Google Scholar]
- Lopez V, Barinova N, Onishi M, Pobiega S, Pringle JR, Dubrana K, Marcand S. Cytokinesis breaks dicentric chromosomes preferentially at pericentromeric regions and telomere fusions. Genes Dev. 2015;29:322–336. doi: 10.1101/gad.254664.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maringele L, Lydall D. Telomerase- and recombination-independent immortalization of budding yeast. Genes Dev. 2004;18:2663–2675. doi: 10.1101/gad.316504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marotta M, Chen X, Inoshita A, Stephens R, Budd GT, Crowe JP, Lyons J, Kondratova A, Tubbs R, Tanaka H. A common copy-number breakpoint of ERBB2 amplification in breast cancer colocalizes with a complex block of segmental duplications. Breast Cancer Res. 2012;14:R150. doi: 10.1186/bcr3362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marotta M, Chen X, Watanabe T, Faber PW, Diede SJ, Tapscott S, Tubbs R, Kondratova A, Stephens R, Tanaka H. Homology-mediated end-capping as a primary step of sister chromatid fusion in the breakage-fusion-bridge cycles. Nucleic acids research. 2013;41:9732–9740. doi: 10.1093/nar/gkt762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mieczkowski PA, Lemoine FJ, Petes TD. Recombination between retrotransposons as a source of chromosome rearrangements in the yeast Saccharomyces cerevisiae. DNA Repair (Amst) 2006;5:1010–1020. doi: 10.1016/j.dnarep.2006.05.027. [DOI] [PubMed] [Google Scholar]
- Mizuno K, Lambert S, Baldacci G, Murray JM, Carr AM. Nearby inverted repeats fuse to generate acentric and dicentric palindromic chromosomes by a replication template exchange mechanism. Genes Dev. 2009;23:2876–2886. doi: 10.1101/gad.1863009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreau S, Ferguson JR, Symington LS. The nuclease activity of Mre11 is required for meiosis but not for mating type switching, end joining, or telomere maintenance. Molecular and cellular biology. 1999;19:556–566. doi: 10.1128/mcb.19.1.556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayanan V, Mieczkowski PA, Kim HM, Petes TD, Lobachev KS. The pattern of gene amplification is determined by the chromosomal location of hairpin-capped breaks. Cell. 2006;125:1283–1296. doi: 10.1016/j.cell.2006.04.042. [DOI] [PubMed] [Google Scholar]
- Neiman PE, Elsaesser K, Loring G, Kimmel R. Myc oncogene-induced genomic instability: DNA palindromes in bursal lymphomagenesis. PLoS Genet. 2008;4:e1000132. doi: 10.1371/journal.pgen.1000132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paek AL, Kaochar S, Jones H, Elezaby A, Shanks L, Weinert T. Fusion of nearby inverted repeats by a replication-based mechanism leads to formation of dicentric and acentric chromosomes that cause genome instability in budding yeast. Genes Dev. 2009;23:2861–2875. doi: 10.1101/gad.1862709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennaneach V, Kolodner RD. Stabilization of dicentric translocations through secondary rearrangements mediated by multiple mechanisms in S. cerevisiae. PLoS One. 2009;4:e6389. doi: 10.1371/journal.pone.0006389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putnam CD, Kolodner RD. Determination of gross chromosomal rearrangement rates. Cold Spring Harb Protoc. 20102010 doi: 10.1101/pdb.prot5492. pdb prot5492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putnam CD, Pallis K, Hayes TK, Kolodner RD. DNA repair pathway selection caused by defects in TEL1, SAE2, and de novo telomere addition generates specific chromosomal rearrangement signatures. PLoS Genet. 2014;10:e1004277. doi: 10.1371/journal.pgen.1004277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putnam CD, Pennaneach V, Kolodner RD. Saccharomyces cerevisiae as a model system to define the chromosomal instability phenotype. Molecular and cellular biology. 2005;25:7226–7238. doi: 10.1128/MCB.25.16.7226-7238.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rattray AJ. A method for cloning and sequencing long palindromic DNA junctions. Nucleic acids research. 2004;32:e155. doi: 10.1093/nar/gnh143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rattray AJ, McGill CB, Shafer BK, Strathern JN. Fidelity of mitotic double-strand-break repair in Saccharomyces cerevisiae: a role for SAE2/COM1. Genetics. 2001;158:109–122. doi: 10.1093/genetics/158.1.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rattray AJ, Shafer BK, Neelam B, Strathern JN. A mechanism of palindromic gene amplification in Saccharomyces cerevisiae. Genes Dev. 2005;19:1390–1399. doi: 10.1101/gad.1315805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt KH, Pennaneach V, Putnam CD, Kolodner RD. Analysis of grosschromosomal rearrangements in Saccharomyces cerevisiae. Methods Enzymol. 2006;409:462–476. doi: 10.1016/S0076-6879(05)09027-0. [DOI] [PubMed] [Google Scholar]
- Slamon DJ, Clark GM, Wong SG, Levin WJ, Ullrich A, McGuire WL. Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science. 1987;235:177–182. doi: 10.1126/science.3798106. [DOI] [PubMed] [Google Scholar]
- Stephens PJ, Greenman CD, Fu B, Yang F, Bignell GR, Mudie LJ, Pleasance ED, Lau KW, Beare D, Stebbings LA, et al. Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell. 2011;144:27–40. doi: 10.1016/j.cell.2010.11.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugiyama T, New JH, Kowalczykowski SC. DNA annealing by RAD52 protein is stimulated by specific interaction with the complex of replication protein A and single-stranded DNA. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:6049–6054. doi: 10.1073/pnas.95.11.6049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surosky RT, Tye BK. Resolution of dicentric chromosomes by Ty-mediated recombination in yeast. Genetics. 1985;110:397–419. doi: 10.1093/genetics/110.3.397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka H, Tapscott SJ, Trask BJ, Yao MC. Short inverted repeats initiate gene amplification through the formation of a large DNA palindrome in mammalian cells. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:8772–8777. doi: 10.1073/pnas.132275999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka H, Yao MC. Palindromic gene amplification--an evolutionarily conserved role for DNA inverted repeats in the genome. Nat Rev Cancer. 2009;9:216–224. doi: 10.1038/nrc2591. [DOI] [PubMed] [Google Scholar]
- Thompson LH, Schild D. Recombinational DNA repair and human disease. Mutation research. 2002;509:49–78. doi: 10.1016/s0027-5107(02)00224-5. [DOI] [PubMed] [Google Scholar]
- Waddell N, Pajic M, Patch AM, Chang DK, Kassahn KS, Bailey P, Johns AL, Miller D, Nones K, Quek K, et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature. 2015;518:495–501. doi: 10.1038/nature14169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh T, King MC. Ten genes for inherited breast cancer. Cancer cell. 2007;11:103–105. doi: 10.1016/j.ccr.2007.01.010. [DOI] [PubMed] [Google Scholar]
- Wang H, Li Y, Truong LN, Shi LZ, Hwang PY, He J, Do J, Cho MJ, Li H, Negrete A, et al. CtIP maintains stability at common fragile sites and inverted repeats by end resection-independent endonuclease activity. Mol Cell. 2014;54:1012–1021. doi: 10.1016/j.molcel.2014.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuda LF, Yao MC. Short inverted repeats at a free end signal large palindromic DNA formation in Tetrahymena. Cell. 1991;67:505–516. doi: 10.1016/0092-8674(91)90525-4. [DOI] [PubMed] [Google Scholar]
- Zhang H, Zeidler AF, Song W, Puccia CM, Malc E, Greenwell PW, Mieczkowski PA, Petes TD, Argueso JL. Gene copy-number variation in haploid and diploid strains of the yeast Saccharomyces cerevisiae. Genetics. 2013;193:785–801. doi: 10.1534/genetics.112.146522. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.