

Abstract
The efficacy of steroids and immunosuppressive treatments in idiopathic nephrotic syndrome (INS) hints at the implication of immune cells in the pathophysiology of the disease. Toll-like receptor (TLR) dysfunctions are involved in many kidney diseases of immune origin, but remain little described in INS. We investigated the expression and function of TLRs in peripheral blood mononuclear cells (PBMC) of INS children, including 28 in relapse, 23 in remission and 40 controls. No child had any sign of infection, but a higher Epstein–Barr virus viral load was measured in the PBMC of relapsing patients. TLR-3 expression was increased in B cells only during INS remission. There was a negative correlation between proteinuria and TLR-3 expression in total and the main subsets of PBMC from INS patients. The expression of TLR-8 was also increased in both CD4+ T cells and B cells in INS remission. There was a negative correlation between proteinuria and TLR-8 expression in total PBMC, CD4+ T cells and B cells of INS patients. Nevertheless, TLR-3 and TLR-8 expression was normalized in all PBMC subsets in an additional group of 15 INS patients in remission with B cell repletion after rituximab therapy. Paradoxically, interferon (IFN) regulatory factor 3 transactivation was increased in PBMC of all INS patients. In-vitro secretion of IFN-α and interleukin 6 were increased spontaneously in PBMC of INS remission patients, whereas PBMC from all INS patients displayed an impaired IFN-α secretion after TLR-3 stimulation. Thus, TLR-3 pathway dysfunctions may be closely involved in INS pathogenesis.
Keywords: blood leucocytes, cytokines, immunoglobulins, proteinuria, steroid-sensitive nephrotic syndrome
Introduction
Idiopathic nephrotic syndrome (INS) accounts for 90% of all glomerular nephropathies in childhood, and is characterized by massive proteinuria and hypoalbuminaemia 1. The disease affects the kidney exclusively and is marked typically by the effacement of podocyte foot processes without glomerular deposit or inflammatory lesion. The clinical course of INS is largely dependent upon the response to steroids. Relapses are frequent after withdrawal of steroid treatment, with up to 60% of patients being steroid-dependent. Immunosuppressive treatments, such as cyclophosphamide and calcineurin inhibitors, are administered as a second-line treatment for INS to prevent relapses and to spare steroid usage 2. Rituximab (RTX), a chimeric human–mouse anti-CD20 monoclonal antibody, is also effective in the treatment of INS in preventing relapses in steroid-dependent patients 3,4.
The efficacy of immunosuppressive treatments indicates that INS may be associated with dysfunctions of the immune system 5. The main subsets of immune cells have all been linked to the pathogenesis of INS. The injection of CD34+ peripheral blood mononuclear cells (PBMC), but not CD34− PBMC, from relapsing INS patients induces albuminuria and podocyte foot effacement in immunodeficient mice 6. Immature CD34+ circulating cells are expanded during INS relapses 7, as well as several T cell populations such as memory T cells 8 and interleukin (IL)-17-producing CD4+ T cells, associated with a reduced number and activity of regulatory T cells 9,10. Numerous studies have concluded that INS patients present an imbalance between T helper type 1 (Th1) and Th2 responses, with a trend towards a greater Th2 response 11. Suppressor T cells also inhibit T–B cooperation, responsible for a decreased immunoglobulin (Ig) class-switch recombination 12. B cell involvement is suggested by the alteration of plasma IgG subclasses and IgM concentrations in INS relapse 13 by the achievement of INS remission after plasma Ig depletion 14,15, the decrease in B cell numbers during INS remission 7 and the rare but classic association of INS with Hodgkin lymphoma 16. Blood monocytes are also affected during INS relapse, marked by an increased production of proinflammatory IL-18 17 and a decreased production of IL-8 and tumour necrosis factor alpha (TNF-α), surprisingly associated with a down-regulation of CD14 expression 18. No global explanation for these anomalies has ever been put forward.
Infections appear to be involved in the onset of INS. Partial evidence for viral involvement was provided in the late 1960s by the detection of myxovirus-like particles in renal biopsies of patients suffering from INS 19. One prospective study in the 1980s showed that 70% of relapses were preceded by an upper respiratory tract infection with serology and viral cultures identifying respiratory syncytial virus, influenza virus, parinfluenza virus, varicella zoster virus and adenovirus as potential triggers of the relapses 20. In studies of INS relapses, upper respiratory viral infections are associated strongly with proteinuria 21 and steroid dependency 22,23. The prevalence and a recent infection or reactivation of Epstein–Barr virus (EBV) DNA was found to be higher in children at onset of INS compared to an age- and gender-matched population 24.
Recognition of pathogen-associated molecular patterns (PAMPs) by T cells bearing Toll-like receptors (TLRs) induces their activation and the secretion of proinflammatory cytokines 25. TLRs also induce B cell differentiation and Ig production via nuclear factor-kappa B (NF-κB) in a T cell-independent manner 26. Recognition of PAMPs by TLRs of monocytes, macrophages and dendritic cells triggers the NF-κB, activator protein 1 (AP-1) and interferon (IFN) pathways, resulting in the translocation of transcription factors and cytokine modulation leading to inflammatory responses via T cell stimulation 27. Some receptors for PAMPs are implicated more frequently in responses to infections during childhood, such as the extracellular TLR-2 activated by peptidoglycan from Gram+ bacteria and TLR-4 activated by lipopolysaccharides from Gram– bacteria, but also by the intracellular TLR-3, TLR-7/-8 and TLR-9 recognizing viral double-strand (ds)RNA, viral single-strand (ss)RNA and microbial cytosine–phosphate–guanine DNA, respectively 27,28.
Overall, we hypothesize that TLRs in PBMC might be involved in INS disease. The aim of this study was to investigate the expression and function of TLRs in PBMC of INS patients at various stages of the disease.
Materials and methods
Patients and samples
Twenty-eight patients during INS relapse, 23 patients in remission, 15 patients in remission after rituximab therapy (remission RTX+) and 40 control subjects were recruited prospectively at the Robert Debré Hospital in Paris. The study protocol was approved by the local ethics committee. Paediatric patients were enrolled following written informed consent from the parents. INS children were sampled if they were aged < 18 years, at least either 1 month from any steroid therapy or 3 months from immunosuppressive therapy. INS relapse and remission were defined by the French National Authority for Health, a proteinuria/creatininuria ratio > 0·25 g/mmol and a hypoalbuminaemia < 30 g/l, a proteinuria/creatininuria ratio < 0·02 g/mmol and albuminaemia > 30 g/l, respectively. Due to ethical paediatric reasons, biopsies were not performed in children aged < 11 years and in adolescents when they were steroid-sensitive and had no complication of INS. It is universally admitted that patients with steroid-sensitive nephrotic syndrome commonly display minimal change disease at renal histology. In our series of 66 patients with steroid-sensitive nephrotic syndrome, only nine patients had renal histology that consistently showed minimal change disease. Control subjects were aged < 18 years, had neither proteinuria nor a history of nephrotic syndrome, and they had never received steroids or immunosuppressive therapy. Urine analyses and blood cell counts were undertaken routinely in the laboratories of the Robert Debré Hospital in Paris. Ethylenediamine tetracetic acid venous peripheral blood was collected from patients and controls matched by age and gender. Plasma and PBMC were isolated after blood centrifugation using a density gradient (Ficoll Paque Premium; VWR, Fontenay-sous-Bois, France).
DNA extraction and EBV detection in PBMC
The QIAamp DNA Blood Mini Kit (Qiagen, Courtaboeuf, France) was used for DNA isolation of PBMC. Extracted DNA was quantified by spectrophotometry (NanoPhotometer Pearl; Implen, Sciencetec, Courtaboeuf, France). EBV DNA levels were measured by a commercial quantitative real-time polymerase chain reaction (PCR) kit (Diagenode, Seraing, Belgium), using CFX96 C1000T Thermal Cycle (Bio-Rad, Marnes-la-Coquette, France). EBV load was calculated as EBV copies per µg of extracted DNA from PBMC.
TLR protein expression in PBMC
Protein expression of TLRs was quantified in PBMC by flow cytometry. V450-conjugated CD4, allophycocyanin (APC)-conjugated CD8, V500-conjugated CD19 and APC-H7-conjugated CD14 antibodies (Becton Dickinson, Le Pont de Claix, France) identified CD4+ T, CD8+ T, CD19+ B lymphocyte subsets and CD14+ monocytes, respectively. Monoclonal antibodies against TLRs were obtained from Imgenex (Clinisciences, Nanterre, France), including anti-TLR-2/CD282 fluorescein isothiocyanate (FITC) conjugate (clone: TL2.1, product number: IMG-416C), anti-TLR-3/CD283 red–phycoerythrin (PE) conjugate (clone: 40C1285.6, product number: IMG-315D), anti-TLR-4/CD284 FITC conjugate (clone: HTA125, product number: IMG-417C), anti-TLR-8/CD288 PE conjugate (clone: 44C143, product number: IMG-321D) and anti-TLR-9/CD289 FITC conjugate (clone: 26C593.2, product number: IMG-305C). Isotype controls were from Becton Dickinson. PBMC were stained with anti-CD marker antibodies and anti-extracellular TLR-2 and TLR-4 for 30 min at 4°C. PBMC were also stained with anti-CD marker antibodies for 30 min at 4°C, permeabilized with the Cytofix/Cytoperm kit (Becton Dickinson) and incubated with anti-intracellular TLR-3, TLR-8 or TLR-9 antibodies for 30 min at 4°C. A total of 10 000 viable PBMC were collected using BD fluorescence activated cell sorter (FACS)Diva Software by flow cytometry (FACSCanto II; Becton Dickinson). Results were analysed using FlowJo software (TreeStar, Inc., Ashland, OR, USA). TLR expressions were calculated as the ratio of mean fluorescence intensity (MFI) of TLR/MFI of isotype control.
Activation of NF-κB, AP-1 and interferon regulatory factor 3 (IRF3) pathways in PBMC
Nuclear extracts of 1·5 × 106 PBMC were prepared with the Nuclear Extract kit (Active Motif, La Hulpe, Belgium) and protein concentrations were quantified using the Pierce BCA Protein Assay Kit (Thermo Fisher Scientific, Courtaboeuf, France). NF-κB, AP-1 and IRF3 binding activities in nuclear extracts were measured with TransAM™ NF-κB, AP-1 and IRF3 transcription factor assay kits, respectively (Active Motif). Activation of NF-κB, AP-1 and IRF3 was calculated as the ratio of optical density (OD) per µg of proteins in nuclear extracts.
Ex-vivo induction of proinflammatory cytokines in PBMC by TLR agonists
PBMC were suspended in RPMI-1640 Glutamax culture medium (Life Technologies, Saint-Aubin, France) supplemented with 10% fetal calf serum (Biowest, Paris, France), 1% penicillin–streptomycin (Sigma-Aldrich); 5 × 105 PBMC per condition were then incubated with or without TLR agonists, including polyinosinic–polycytidylic acid (Poly I:C, TLR-3 agonist) at 40 µg/ml and Resiquimod (R848, TLR-7/-8 agonist) at 1 µg/ml (all from Cayla-Invivogen, Toulouse, France) at 37°C in a 5% CO2 atmosphere for 3 and 11 days to detect the production of proinflammatory cytokines and of immunoglobulins in supernatants, respectively.
Enzyme-linked immunosorbent assay (ELISA)
IFN-α production was measured in plasma and in supernatants using the VeiriKineTM human IFN-α serum sample ELISA kit (pbl interferon source, Piscataway, NJ, USA). Production of IL-6, IL-1β, IL-8, IL-13 and TNF-α was measured using the ELISA DuoSet kits (R&D Systems, Abingdon, UK). Production of IgM and IgG was measured using the ELISA quantification sets (Bethyl, Montgomery, TX, USA).
Statistical analyses
Quantitative data were first analysed by the non-parametric Kruskal–Wallis test, and if significant, the Mann–Whitney U-test was applied for between-group analyses. Qualitative data were analysed first using the χ2 test and if significant, by Fisher's exact test. Spearman's rank correlation test was used to assess the correlation between two variables. P < 0·05 was considered statistically significant. Results are presented as mean ± standard error of the mean (s.e.m.). Statistical analyses were performed using the GraphPad Prism version 5·0 software (La Jolla, CA, USA).
Results
Sixty-six INS children without steroid and/or immunosuppressive treatment at the time of sampling were divided into three groups, i.e. 28 in relapse, 23 in remission and 15 in remission RTX+. Forty non-nephrotic controls comprised children with urinary tract malformation (n = 26), mineral metabolism disorders (n = 8), isolated haematuria (n = 4) and lithiasis (n = 2). The clinical features of all children are presented in Table1. Patients in remission after RTX therapy had a more severe history of the disease than other patients in relapse and in remission, as underlined by their dependence upon steroid and immunosuppressive therapies, meaning that this group will be analysed separately from others. Only three INS relapsing patients and one control had a history of upper respiratory infection within 4 weeks of the blood sample being obtained. At the time of sampling, no child had any sign of infection. In agreement with these clinical observations, blood leucocyte subset counts were within the range of normal values and were similar in both patients and controls (P > 0·05; Supporting information, Table S1).
Table 1.
Clinical characteristics of patients with idiopathic nephrotic syndrome (INS) and controls.
| Control | Relapse | Remission | Remission RTX+ | P | |
|---|---|---|---|---|---|
| Number of patients (n) | 40 | 28 | 23 | 15 | n.a. |
| Gender ratio (male/female) | 27/13 | 18/10 | 14/9 | 10/5 | 0.96 |
| Age (years) | 9·9 (6·7–14·4) | 7·8 (3·6–17·4) | 9·7 (5·1–12·8) | 10·4 (7·6–16·5) | 0·06 |
| Proteinuria (g/mmol creatinine) | 0·012 (0·007–0·021)a | 0·397 (0·196–1·308)b | 0·010 (0·007–0·016)a | 0·009 (0·005–0·131)a | <0·0001 |
| Serum albumin (g/l) | 42·8 (41·5–45·0)a | 13·4 (8·1–32·6)b | 43·7(41·1–45·0)a | 42·0 (38·0–46·0)a | <0·0001 |
| eGFR (ml/min/1·73 m2) | 102 (86–110)a | 139 (113–153)b | 126 (105–130)c | 166 (145–187)d | <0·0001 |
| First flare or scare relapse (n) | n.a. | 16a | 9a | 0b | 0·001 |
| Steroid dependency (n) | n.a. | 12a | 14a | 15b | 0·001 |
| At the time of sampling | |||||
| Number of relapses since INS onset | n.a. | 1 (0–5)a | 3 (1–6)ab | 4 (3–12)b | 0·02 |
| Relapse-free period in INS (months) | n.a. | 26 (10–37) | 13 (8–25) | 11 (7–32) | 0·27 |
| Steroid-free period in INS (months) | n.a. | 13 (5–27) | 6 (3–15) | 9 (5–28) | 0·37 |
| History of previous therapies | |||||
| None (n) | 40a | 11b | 0c | 0c | <0·0001 |
| Prednisone (n) | 0a | 17b | 23c | 15c | <0·0001 |
| Rituximab (n) | 0a | 0a | 0a | 15b | <0·0001 |
| Other immunosuppressants (n) | 0a | 6b | 10b | 13c | <0·0001 |
Data are expressed as median (interquartile range). eGFR = estimated glomerular filtration rate for the children using the revised Schwartz formula (k-value = 0·413) updated in 2009 29; other immunosuppressants included mycophenolate mofetil, anti-calcineurin inhibitors and cyclophosphamide; RTX = rituximab therapy; n.a. = not applicable. When superscript letters are different (a, b, c, d), medians showed significant difference between groups, P < 0·05.
As EBV shows a high prevalence at onset of INS 24 and can activate various TLRs 30, EBV DNA copies were quantified in extracted DNA of PBMC from both cases and controls. EBV DNA load was detected in PBMC from 17 of 27 relapsing patients and from 16 of 35 controls, as well as from 10 of 20 patients in remission. Moreover, the EBV DNA load detected in PBMC was higher in relapsing patients than in controls, but not than in remission (Fig. 1a). Positive EBV DNA load detected in PBMC was not correlated with proteinuria in INS patients (Fig. 1b).
Figure 1.
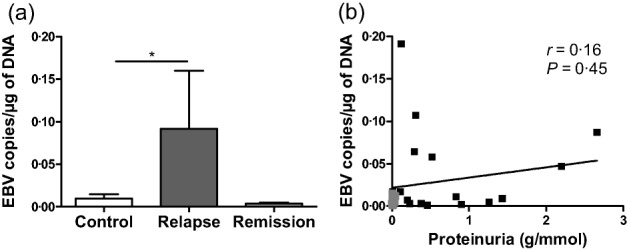
Epstein–Barr virus (EBV) load in peripheral blood mononuclear cells (PBMC) and lack of correlation with proteinuria. (a) Copies of EBV DNA were measured by quantitative real time polymerase chain reaction in extracted DNA from PBMC of 35 controls, 28 patients in relapse and 20 patients in remission; *P < 0·05. (b) Correlation between proteinuria (g/mmol creatinine) and samples with positive EBV DNA load from 16 patients in relapse (dark grey plots) and 10 patients in remission (light grey plots) using Spearman's rank correlation test, not significant.
Many viral primary infections, particularly those due to herpesvirus, are free of symptoms during childhood 31. As these infections appeared to be implicated in INS pathogenesis 24 we sought to characterize TLRs, which are able to recognize PAMPs. Specifically, we investigated the expression of TLRs in total PBMC and in PBMC subsets, including CD4+ and CD8+ T cells, CD14+ monocytes and CD19+ B cells from both cases and controls. The numbers of total PBMC, CD4+ and CD8+ T cells, CD14+ monocytes and CD19+ B cells were comparable between groups of INS patients and controls (P > 0·05; Supporting information, Table S2). Representative flow cytometry analyses for intracellular TLR-3 expression in total PBMC and PBMC subsets are given in Supporting information, Fig. S1. In total PBMC, patients are not different from controls, but TLR-3 expression was significantly higher in remission than in relapse (Fig. 2a). Consistently, TLR-3 expression in total PBMC was correlated negatively with proteinuria in INS patients (Fig. 2f). In remission, TLR-3 expression was significantly higher than in relapse in all studied PBMC subsets (Fig. 2b–e). In addition, TLR-3 expression in CD19+ B cells was higher in remission than in controls (Fig. 2e), whereas it remained at base level in CD4+ and CD8+ T cells and in CD14+ monocytes (Fig. 2b–d). TLR3 expression was reduced significantly in CD4+ and CD8+ T cells and in CD14+ monocytes in relapse compared to controls (Fig. 2b–d), whereas it remained at base level in CD19+ B cells (Fig. 2e). Negative correlations were significant between proteinuria and TLR-3 expression in all PBMC subsets studied (Fig. 2g–j).
Figure 2.
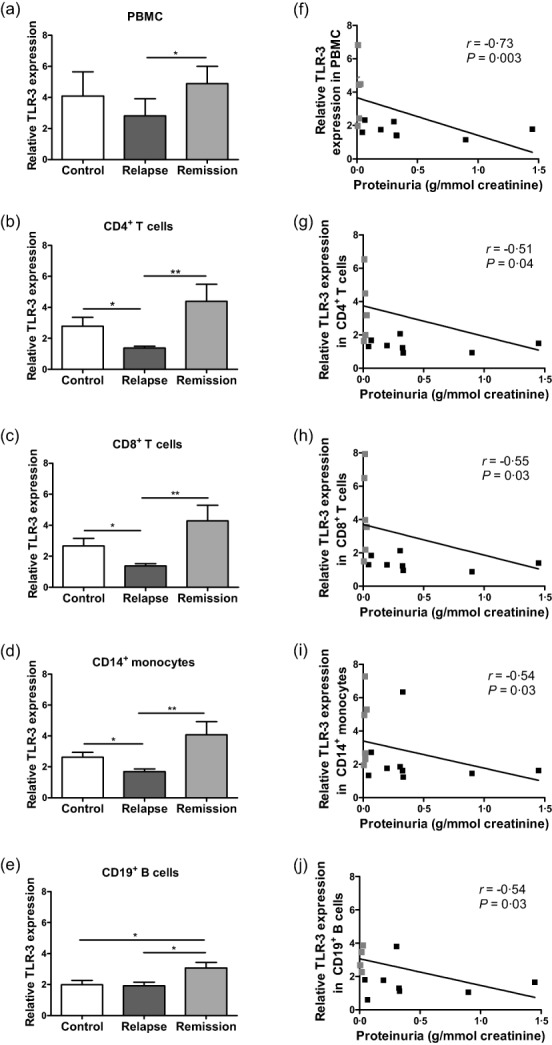
Expression of Toll-like receptor (TLR)−3 in peripheral blood mononuclear cells (PBMC) and correlation with proteinuria. TLR-3 expression was measured in (a) PBMC, (b) CD4+ T cells, (c) CD8+ T cells, (d) CD14+ monocytes and (e) CD19+ B cells by flow cytometry and results are expressed as mean fluorescence intensity (MFI) of TLR-3/MFI of isotype control in 10 controls, 9 patients in relapse and 6 patients in remission; *P < 0·05; **P < 0·01. Correlation between proteinuria (g/mmol creatinine) and TLR-3 expression in (f) total PBMC, (g) CD4+ T cells, (h) CD8+ T cells, (i) CD14+ monocytes and (j) CD19+ B cells from 9 patients in relapse (dark grey plots) and 6 patients in remission (light grey plots) using Spearman's rank correlation test.
Similar results were found for intracellular TLR-8 expression in PBMC. No difference was found between the two groups of INS patients and controls (Fig. 3a), but TLR-8 expression in total PBMC was correlated negatively with proteinuria in INS patients (Fig. 3f). In PBMC subsets, only CD4+ T cells and CD19+ B cells showed a significant increase of TLR-8 expression in remission compared to controls and relapsing patients (Fig. 3b,e), with a negative correlation with proteinuria in INS patients (Fig. 3g,j).
Figure 3.
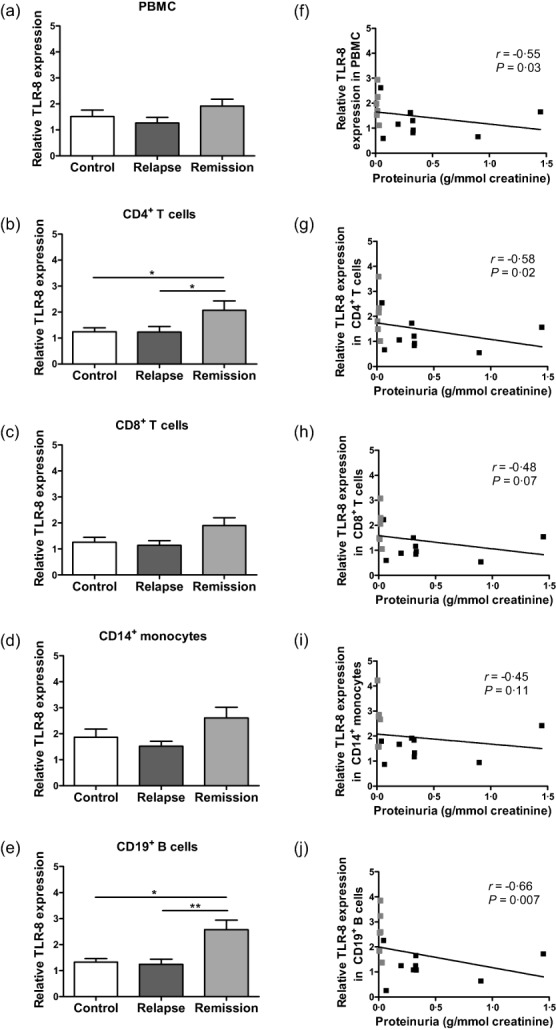
Expression of Toll-like receptor (TLR)-8 in peripheral blood mononuclear cells (PBMC) and correlation with proteinuria. TLR-8 expression was measured in (a) total PBMC, (b) CD4+ T cells, (c) CD8+ T cells, (d) CD14+ monocytes and (e) CD19+ B cells by flow cytometry and results are expressed as mean fluorescence intensity (MFI) of TLR/MFI of isotype control in 10 controls, 9 patients in relapse and 6 patients in remission; *P < 0·05; **P < 0·01. Correlation between proteinuria (g/mmol creatinine) and TLR-8 expression in (f) total PBMC, (g) CD4+ T cells, (h) CD8+ T cells, (i) CD14+ monocytes and (j) CD19+ B cells from 9 patients in relapse (dark grey plots) and 6 patients in remission (light grey plots) using Spearman's rank correlation test.
In contrast, no modification of the expression of extracellular TLR-2, TLR-4 and TLR-9 was detected in total PBMC as well as in all studied PBMC subsets between INS patients and controls (Table2). No correlation was found between proteinuria and the expression of TLR-2, TLR-4 and TLR-9 in total and subsets of PBMC from INS patients (Table2). Taken together, these data show that only TLRs recognizing RNA, i.e. TLR-3 linking dsRNA and TLR-8 linking ssRNA, are implied in both relapse and remission of INS and are related to proteinuria. Moreover, patients in remission RTX+ had shown a decrease of TLR-3 expression in monocytes and of TLR-8 expression in CD8+ T cells compared to patients in remission (Table3). In patients in remission RTX+, TLR-3 and TLR-8 expression of CD19+ B cells, as well as TLR-8 expression in CD4+ T cells, were decreased significantly compared to patients in remission to return to base level of controls (Table3).
Table 2.
Expression of Toll-like receptors (TLR)-2, -4 and -9 in peripheral blood mononuclear cells (PBMC) and correlation to proteinuria.
| Control | Relapse | Remission | P | Proteinuria | |
|---|---|---|---|---|---|
| Number of patients | 10 | 9 | 6 | n.a. | n.a. |
| Relative TLR-2 expression | |||||
| PBMC | 4·01 ± 0·59 | 4·13 ± 0·78 | 2·98 ± 0·27 | 0·33 | r = −0·26, P = 0·34 |
| CD4+ T cells | 1·50 ± 0·14 | 1·71 ± 0·16 | 1·39 ± 0·11 | 0·37 | r = 0·06, P = 0·84 |
| CD8+ T cells | 1·31 ± 0·10 | 1·35 ± 0·08 | 1·19 ± 0·06 | 0·49 | r = 0·18, P = 0·52 |
| CD14+ monocytes | 6·40 ± 1·10 | 7·88 ± 1·24 | 5·10 ± 0·47 | 0·30 | r = 0·28, P = 0·31 |
| CD19+ B cells | 2·94 ± 0·44 | 3·04 ± 0·37 | 2·31 ± 0·22 | 0·42 | r = 0·09, P = 0·75 |
| Relative TLR-4 expression | |||||
| PBMC | 2·82 ± 0·37 | 2·47 ± 0·24 | 2·81 ± 0·58 | 0·81 | r = −0·36, P = 0·19 |
| CD4+ T cells | 1·83 ± 0·24 | 1·75 ± 0·23 | 1·73 ± 0·30 | 0·89 | r = −0·37, P = 0,17 |
| CD8+ T cells | 1·40 ± 0·14 | 1·58 ± 0·18 | 1·49 ± 0·22 | 0·79 | r = −0·11, P = 0·69 |
| CD14+ monocytes | 3·06 ± 0·35 | 2·67 ± 0·20 | 3·51 ± 0·22 | 0·15 | r = −0·12, P = 0·67 |
| CD19+ B cells | 2·76 ± 0·38 | 2·54 ± 0·19 | 2·59 ± 0·51 | 0·96 | r = −0·13, P = 0·66 |
| Relative TLR-9 expression | |||||
| PBMC | 86·3 ± 19·2 | 133·7 ± 31·6 | 141 ± 41·8 | 0·54 | r = −0·13, P = 0·64 |
| CD4+ T cells | 136·4 ± 34·7 | 165·5 ± 42·3 | 186·4 ± 58·9 | 0·72 | r = −0·09, P = 0·75 |
| CD8+ T cells | 129·6 ± 29·8 | 192·9 ± 40·7 | 162·4 ± 38·1 | 0·75 | r = −0·06, P = 0·84 |
| CD14+ monocytes | 58·8 ± 10·4 | 88·8 ± 18·0 | 53·7 ± 7·9 | 0·62 | r = −0·35, P = 0·23 |
| CD19+ B cells | 115·3 ± 32·7 | 191·9 ± 27·5 | 244·5 ± 66·32 | 0·19 | r = −0·27, P = 0·32 |
TLR expression was measured in PBMC subsets by flow cytometry and results are expressed as mean fluorescence intensity (MFI) of TLR/MFI of isotype control, and no significant difference between groups was observed using the Kruskal–Wallis test, P > 0·05; r corresponds to Spearman's rank correlation test used to assess the correlation between TLR expression of total PBMC and each PBMC subset from idiopathic nephrotic syndrome (INS) patients and proteinuria (g/mmol creatinine), and no significant correlation was found; n.a. = not applicable.
Table 3.
Impact of rituximab (RTX) therapy in expression of Toll-like receptors (TLR)-3 and -8 in peripheral blood mononuclear cells (PBMC).
| Control | Remission | Remission RTX+ | P | |
|---|---|---|---|---|
| Number of patients | 10 | 6 | 8 | n.a. |
| Relative TLR-3 expression | ||||
| PBMC | 4·09 ± 1·56 | 4·89 ± 1·11 | 2·39 ± 0·33 | 0·12 |
| CD4+ T cells | 2·79 ± 0·57 | 4·39 ± 1·10 | 1·95 ± 0·31 | 0·10 |
| CD8+ T cells | 2·65 ± 0·48 | 4·28 ± 1·02 | 1·86 ± 0·28 | 0·06 |
| CD14+ monocytes | 2·63 ± 0·32ab | 4·07 ± 0·86a | 1·91 ± 0·17b | 0·04 |
| CD19+ B cells | 2·00 ± 0·26a | 3·25 ± 0·34b | 2·08 ± 0·59a | 0·04 |
| Relative TLR-8 expression | ||||
| PBMC | 1·51 ± 0·25 | 1·92 ± 0·26 | 1·24 ± 0·12 | 0·11 |
| CD4+ T cells | 1·24 ± 0·15a | 2·07 ± 0·36b | 1·17 ± 0·09a | 0·04 |
| CD8+ T cells | 1·26 ± 0·19ab | 1·90 ± 0·29a | 1·10 ± 0·11b | 0·04 |
| CD14+ monocytes | 1·87 ± 0·31 | 2·61 ± 0·40 | 1·53 ± 0·16 | 0·11 |
| CD19+ B cells | 1·32 ± 0·14a | 2·58 ± 0·37b | 1·36 ± 0·11a | 0·01 |
TLR expression was measured in PBMC subsets by flow cytometry and results are expressed as mean fluorescence intensity (MFI) of TLR/MFI of isotype control. When superscript letters are different (a, b), means showed significant difference between groups, P < 0·05; n.a. = not applicable.
TLR-3 activates the IRF3 leading to the induction of type I IFN by many immune cells, but also activates the alternative pathway of NF-κB and AP-1 leading to the production of cytokines in PBMC, as well as production of Igs by B cells 27. Activation of IRF3 was increased in nuclear extracts of PBMC during remission and also in relapse, compared to controls (Fig. 4a). Consistently, plasma IFN-α level was correlated with the nuclear transactivation of IRF3 in PBMC from INS patients (Fig. 4b). In standardized conditions of culture, to circumvent secondary changes induced by massive proteinuria in relapse, IFN-α secretion was higher in PBMC from patients in remission compared to controls, whereas it was similar between relapsing patients and controls (Fig. 4c). In-vitro stimulation of PBMC from controls by TLR-3 agonist induced a 10-fold increase of IFN-α secretion compared to unstimulated PBMC (Fig. 4c). However, PBMC from INS patients in remission did not increase their secretion of IFN-α in response to TLR-3 agonist stimulation (Fig. 4c). Similar results were found in PBMC from relapsing patients (Fig. 4c). Stimulation of PBMC with TLR-7/-8 agonists could also induce IFN-α production via IRF7 activation 27, as shown in PBMC from controls and relapsing patients (Fig. 4c). However, PBMC from INS patients in remission did not increase their secretion of IFN-α in response to TLR-7/-8 agonist stimulation (Fig. 4c). In contrast, NF-κB and AP-1 were not transactivated significantly in nuclear extracts of PBMC from all INS patients, either in relapse or in remission with and without RTX, compared to controls (Supporting information, Fig. S2). These data suggest that IFN-α secretion, which is dependent upon TLR-3 and IFR3, is activated specifically in PBMC of patients in remission and to a lesser extent in relapsing patients.
Figure 4.
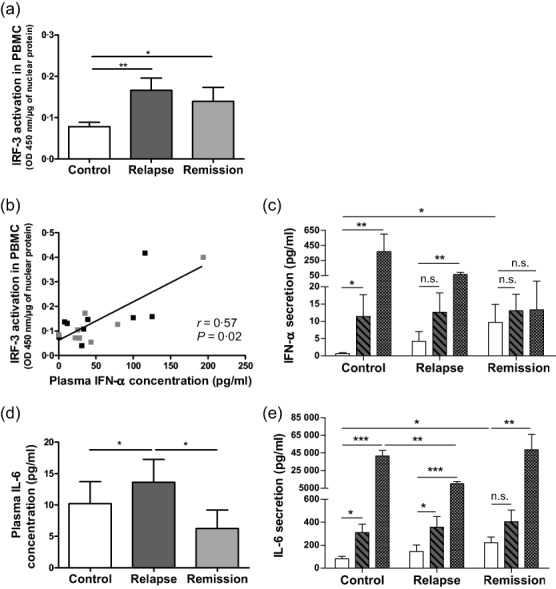
Transactivation of interferon regulatory transcription factor (IRF3) in peripheral blood mononuclear cells (PBMC) and production of interferon (IFN)-α and interleukin (IL)-6 in plasma, in unstimulated and in Toll-like receptor (TLR)-3 and TLR-7/-8-stimulated PBMC. (a) Activation of IRF3 was measured by enzyme-linked immunosorbent assay (ELISA) in nuclear extracts of PBMC from 14 controls, 13 patients in relapse and 13 patients in remission. (b) Correlation between plasma IFN-α and IRF3 activation in nuclear extracts of PBMC from 10 patients in relapse (dark grey plots) and 8 patients in remission (light grey plots) using Spearman's rank correlation test. (c) Secretion of IFN-α was measured using ELISA by day 3 in supernatants of cultured PBMC without stimulation (white bars) and after stimulation by Poly I:C (TLR-3 agonist, hatched bars) and by Resiquimod (TLR-7/-8 agonist, squared bars) in 15 controls, 16 patients in relapse and 13 patients in remission; *P < 0·05; ** P < 0·01; n.s. = not significant. (d) Plasma IL-6 production was measured using ELISA in 40 controls, 28 patients in relapse and 23 patients in remission. (e) Secretion of IL-6 was measured using ELISA by day 3 in supernatants of cultured PBMC without stimulation (white bars) and after stimulation by Poly I:C (TLR-3 agonist, hatched bars) and by Resiquimod (TLR-7/-8 agonist, squared bars) in 21 controls, 22 patients in relapse and 15 patients in remission; *P < 0·05; **P < 0·01; ***P < 0·001; n.s. = not significant.
Despite a significant increase of IRF3 activation in PBMC similar to other INS patients, plasma IFN-α concentration was lower in patients in remission RTX+ than in controls and in remission (Table4). Moreover, unstimulated PBMC from patients in remission RTX+ secreted low levels of IFN-α, compared to patients in remission (P < 0·05), to return to base level of controls (Table4). However, PBMC from INS patients in remission RTX+ did not increase their secretion of IFN-α in response to TLR-3 agonist stimulation, as described in other INS patients, but they were able to induce a high increase of IFN-α secretion in response to TLR-7/-8 agonist stimulation, contrary to patients in remission (Table4).
Table 4.
Impact of rituximab (RTX) therapy in transactivation of interferon regulatory factor 3 (IRF3) in peripheral blood mononuclear cells (PBMC) and in interferon (IFN)-α and interleukin (IL)-6 production in plasma, in unstimulated and in Toll-like receptor (TLR)-3- and TLR-7/-8-stimulated PBMC.
| Control | Remission | Remission RTX+ | P | |
|---|---|---|---|---|
| Number of patients | 14 | 13 | 13 | n.a. |
| IRF3 activation in PBMC | 0·08 ± 0·01a | 0·14 ± 0·03b | 0·23 ± 0·08b | 0·001 |
| Plasma IFN-α | 29·80 ± 8·73a | 53·36 ± 21·53a | 0·00 ± 0·00b | 0·0001 |
| IFN-α in unstimulated PBMC | 0·63 ± 0·28a | 9·69 ± 5·33b | 0·20 ± 0·13a | 0·02 |
| IFN-α in TLR-3-stimulated PBMC | 11·80 ± 6·70a | 9·10 ± 2·94a | 2·97 ± 2·97b | 0·01 |
| IFN-α in TLR-7/-8-stimulated PBMC | 397 ± 263ab | 13·34 ± 10·14a | 1078 ± 520b** | 0·03 |
| Number of patients | 40 | 23 | 15 | n.a. |
| Plasma IL-6 | 10·21 ± 3·51 | 6·26 ± 2·95 | 4·85 ± 2·75 | 0·79 |
| Number of patients | 21 | 15 | 10 | n.a. |
| IL-6 in unstimulated PBMC | 82·73 ± 20·28a | 222 ± 50b | 18·40 ± 6·95c | 0·002 |
| IL-6 in TLR-3-stimulated PBMC | 310 ± 72 | 405 ± 99 | 303 ± 132* | 0·55 |
| IL-6 in TLR-7/-8-stimulated PBMC | 41 505 ± 6399 | 48 627 ± 17 355 | 26 093 ± 8014* | 0·41 |
IRF3 activation in PBMC was expressed as optical density (OD) 450 nm per µg of nuclear protein and IFN-α and IL-6 concentration as pg/ml. Production of IFN-α and IL-6 by PBMC was measured in supernatant after 3 days of culture following or not stimulation with Poly I:C (TLR-3 agonist) and Resiquimod (TLR-7/-8 agonist). All the data were obtained by enzyme-linked immunosorbent assay (ELISA). When superscript letters are different (a, b, c), means showed significant difference between groups, P < 0·05. Mean with ** showed a significant difference (P < 0·001) in production of IFN-α in TLR-7/-8-stimulated PBMC compared to unstimulated PBMC in patients in remission RTX+. Means with * showed significant difference (P < 0·05) in production of IL-6 in TLR-3- and TLR-7/-8-stimulated PBMC compared to unstimulated PBMC in patients in remission RTX+; n.a. = not applicable.
Plasma concentration of IL-6 was higher during relapse than during remission or in controls (Fig. 4d), but was strikingly independent of NF-κB, AP1 or IRF3 activation in PBMC of INS patients (r = 0·10, P = 0·61; r = 0·07, P = 0·72 and r = −0·06, P = 0·77, respectively). In standardized in-vitro conditions and similar to IFN-α secretion, IL-6 secretion by PBMC from patients in remission was higher than in controls, while being similar between relapsing patients and controls (Fig. 4e). Stimulations of PBMC with TLR-3 agonist induced a 10-fold increase of IL-6 secretion in controls compared to unstimulated PBMC (Fig. 4e). TLR-3 agonist did not induce IL-6 production in PBMC of patients in remission (Fig. 4e). TLR-3 stimulation of PBMC from relapsing patients also induced production of IL-6 similar to control PBMC (Fig. 4e). Moreover, TLR-7/-8 agonists also stimulated IL-6 secretion by PBMC from patients in remission and relapse, as in controls (Fig. 4e). However, IL-6 secretion was lower in PBMC from relapsing patients compared to controls after TLR-7/-8 stimulation (Fig. 4e), showing that TLR-7/-8 is implicated in PBMC of relapsing patients for the secretion of IL-6. Nevertheless, plasma concentration of IL-6 was normal in patients in remission RTX+, but their unstimulated PBMC secreted significantly low levels of IL-6 compared to patients in remission and controls (Table4). However, PBMC from INS patients in remission RTX+ increase their secretion of IL-6 in response to TLR-3 and TLR-7/-8 agonist stimulation (Table4). Conversely, no difference between INS patients and controls has been demonstrated with the production of IL-1β, IL-8, IL-13 and TNF-α in plasma and in unstimulated PBMC (data not shown).
Furthermore, as TLR-3 and TLR-8 expression in B cells was different between patients in relapse and in remission, B cell functions were assessed through measurements of plasma and in-vitro immunoglobulin productions. Plasma IgM levels were increased in relapse compared to controls and remission (Fig. 5a, Table5), while spontaneous IgM secretion was not different between cultured PBMC of INS patients and controls (Fig. 5b, Table5). TLR-3 agonist stimulation of PBMC from controls and patients did not induce variations of IgM secretion compared to unstimulated PBMC (Fig. 5b, Table5). However, PBMC from patients in remission RTX+ secreted lower IgM levels compared to controls and patients in remission after TLR-3 stimulation (Table5). Conversely, TLR-7/-8 agonists stimulated IgM secretion by PBMC in controls and INS patients compared to unstimulated cultured PBMC (Fig. 5b, Table5). TLR-7/-8 agonist-induced stimulation of IgM secretion by PBMC was higher in relapse compared to remission or to controls (Fig. 5b).
Figure 5.
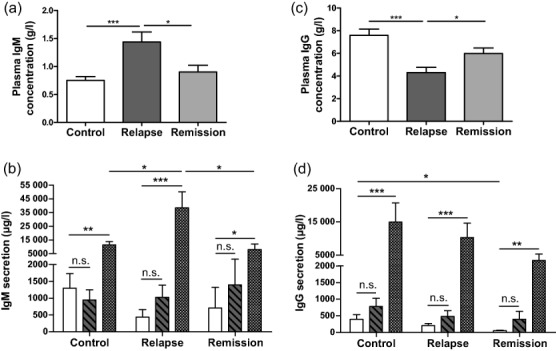
Immunoglobulin (Ig)M and IgG production in plasma, in unstimulated and in -Toll-like receptor (TLR)-3 and TLR-7/-8-stimulated peripheral blood mononuclear cells (PBMC). (a) Plasma IgM production was measured using enzyme-linked immunosorbent assay (ELISA) in 40 controls, 28 patients in relapse and 23 patients in remission. (b) Secretion of IgM was measured using ELISA by day 11 in supernatants of cultured PBMC without stimulation (white bars) and after stimulation by Poly I:C (TLR-3 agonist, hatched bars) and by Resiquimod (TLR-7/-8 agonist, squared bars) in 10 controls, 11 patients in relapse and 7 patients in remission. (c) Plasma IgG production was measured using ELISA in 40 controls, 28 patients in relapse and 23 patients in remission. (d) Secretion of IgG was measured using ELISA by day 11 in supernatants of cultured PBMC without stimulation (white bars) and after stimulation by Poly I:C (TLR-3 agonist, hatched bars) and by Resiquimod (TLR-7/-8 agonist, squared bars) in 10 controls, 11 patients in relapse and 7 patients in remission; *P < 0·05; **P < 0·01; ***P < 0·001; n.s. = not significant.
Table 5.
Impact of rituximab (RTX) therapy in immunoglobulin (Ig)M and IgG production in plasma, in unstimulated and in Toll-like receptor (TLR)-3- and TLR-7/-8-stimulated peripheral blood mononuclear cells (PBMC).
| Control | Remission | Remission RTX+ | P | |
|---|---|---|---|---|
| Number of patients | 40 | 23 | 15 | n.a. |
| Plasma IgM | 0·74 ± 0·07 | 0·90 ± 0·12 | 0·75 ± 0·17 | 0·39 |
| Plasma IgG | 7·61 ± 0·54 | 6·00 ± 0·48 | 7·48 ± 0·56 | 0·08 |
| Number of patients | 10 | 7 | 10 | n.a. |
| IgM in unstimulated PBMC | 1300 ± 433 | 709 ± 616 | 95 ± 12 | 0.14 |
| IgM in TLR-3-stimulated PBMC | 945 ± 306a | 1397 ± 681a | 100 ± 31b | 0·009 |
| IgM in TLR-7/-8-stimulated PBMC | 11 386 ± 2531 | 8058 ± 4078 | 5648 ± 1799* | 0·29 |
| IgG in unstimulated PBMC | 360 ± 131a | 56 ± 13b | 155 ± 42ab | 0·04 |
| IgG in TLR-3-stimulated PBMC | 778 ± 249 | 398 ± 234 | 166 ± 61 | 0·06 |
| IgG in TLR-7/-8-stimulated PBMC | 14 919 ± 5800a | 3385 ± 1298ab | 743 ± 203b* | 0·006 |
Plasma concentration of IgM and IgG was expressed as g/l. Production of IgM and IgG was expressed in µg/l and was measured by day 11 in supernatants of cultured PBMC following or not stimulation with Poly I:C (TLR-3 agonist) and Resiquimod (TLR-7/-8 agonist). All the data were obtained by enzyme-linked immunosorbent assay (ELISA). When superscript letters are different (a, b), means showed significant difference between groups, P < 0·05. Means with
showed significant difference in production of IgM (P < 0·05) or IgG (P < 0·001) in TLR-7/-8-stimulated PBMC compared to unstimulated PBMC in patients in remission RTX+. P < 0·05; n.a. = not applicable.
Plasma IgG concentration was lower in relapse than in controls (Fig. 5c), while spontaneous IgG secretion was normal in cultured PBMC in relapse (Fig. 5d). In contrast, plasma IgG had a similar level in remission and in controls (Fig. 5c), whereas IgG production was impaired severely in cultured PBMC in remission (Fig. 5d). Conversely concentration of IgG was normal in plasma and in unstimulated PBMC of patients in remission RTX+ compared to patients in remission and controls (Table5). TLR-3 agonists did not increase IgG production in all INS patients or controls compared to unstimulated PBMC (Fig. 5d, Table5). TLR-7/-8 stimulation of PBMC from controls and all INS patients similarly stimulated the secretion of IgG compared to unstimulated PBMC (Fig. 5d, Table5).
Taken together, these data obtained with cultured PBMC suggest impairment in IgG secretion by B cells in remission. Additionally, TLR-7/-8 stimulation induced exacerbated B cell response in relapse via IgM secretion.
Discussion
Interactions between TLRs and environmental agonists derived from either exogenous or commensal microbes or endogenous sources may impact kidney disorders 32,33. However, few studies have assessed the role of TLRs from PBMC of patients suffering from kidney diseases of immune origin. In this study, PBMC of INS patients showed a complex set of alterations affecting the expression and function of TLR-3, recognizing dsRNA and, to a lesser extent, of TLR-8, recognizing ssRNA.
Paradoxically, our results relate mainly to patients in remission of proteinuria: (1) increased TLR-3 expression in B cells and increased TLR-8 expression in both CD4+ T cells and B cells of patients in remission versus controls; interestingly, TLR-3 and TLR-8 expression levels were fully restored in main PBMC subsets of those patients in remission who were treated previously by rituximab; (2) increased TLR-3 expression in total and main subsets of PBMC and increased TLR-8 expression in CD4+ T cells and B cells in remission versus relapse with a negative correlation to proteinuria in INS patients; (3) increased transactivation of IRF3, which is mainly under the control of TLR-3, in PBMC from all INS patients versus controls; and (4) increased IFN-α and IL-6 production in cultured PBMC of patients in remission versus controls; interestingly, spontaneous in-vitro IFN-α production by PBMC from these patients in remission RTX+ was also restored to the level of controls.
Altogether, these results suggest that the TLR-3 pathway was activated constitutively in PBMC of INS patients in remission. Our results differ from previous reports showing that TLR-3 expression was similar between INS patients and controls 34 and that IRF3 was not activated in nucleus of PBMC from INS patients, whereas transactivation of NF-κB and AP-1 was increased in INS relapsing patients 35,36. Previous reports mixed children and adults among INS patients who were with and without treatment 34–36. However, ongoing steroid therapy in patients is known to directly alter the viability of immune cells, avoiding the possibility of functional analysis ex vivo 37. Consistently, our patients were sampled only at a distance of any steroids and immunosuppressive treatments to prevent any direct toxic effects. Consequently, results obtained in PBMC from our patients in remission represent only the state of recovery.
The intracellular TLR-3 and TLR-8 are usually stimulated by specific agonists, i.e. dsRNAs and ssRNAs, respectively, which can originate from any microbial agent able to enter into the cell 27. TLR-3 and TLR-8, as well as caspase activation and recruitment domains (CARD) helicases (retinoic acid-inducible gene 1, i.e. RIG-1, and melanoma differentiation-associated protein 5) recognizing RNA species released into the cytoplasm in a variety of cell types, can coordinate anti-viral programmes via IFN-α induction in a TLR-dependent and -independent manner 27. Intracellular RNAs might also originate from the normal metabolism of nucleic acids in the cell; however, TLRs do not recognize endogenous RNAs, except those released from necrotic cells, as a result of tissue inflammation 38. In fact, INS patients do not display any tissue or kidney inflammation 1. Usually, RNA viruses induce an increase of TLR-3 expression and of IFN-α and IL-6 secretion leading to viral elimination 39,40. Consequently, TLR-3 and TLR-8 over-expression might suggest that PBMC and specifically B cells are reacting to a latent microbial agent during remission. Consistently, IRF3 as well as IFN-α and IL-6 production were also activated constitutively in PBMC from INS patients during remission, also suggesting latent infection. In addition, a PBMC-impaired response to TLR-3 stimulation may reflect the viral immunomodulatory effect of foreign dsRNA in remission. Interestingly, viral infections or recurrences are linked closely to the triggering of both the first manifestations and relapses of the disease 20. A recent work has shown that herpesviruses and especially EBV were associated significantly with the first manifestation of INS 24. EBV is the most emblematic DNA virus, capable of latency in immune cells, with a limited production of viral proteins and viral dsRNAs named EBERs that are effectively recognized by TLR-3 41,42.
Our results regarding TLR-8 showed an up-regulation of expression and a reverse correlation to proteinuria, similar to TLR-3, but limited to CD4+ T cells and in B cells in remission. The significance of these results is not clear, while TLR-8 recognizes ssRNA and usually activates the production of proinflammatory cytokines through NF-κB and AP-1 pathways and the production of IFN-α through IRF7 43. Unfortunately, the literature on TLR-8 expression and functions in disease is scarce, and does not help to explain this issue.
Relapses are marked by a massive proteinuria. During relapse, TLR-3 expression is decreased in T cells and in monocytes but not in B cells, while TLR-8 expression, as well as those of all other studied TLRs, is unchanged in the total and main subsets of PBMC. This dissociation between TLR expression suggests that the decrease of TLR-3 in relapse cannot be attributed to the general dysregulation of protein metabolism secondary to massive proteinuria. Paradoxically, PBMC from patients in relapse displayed a similar IRF3 stimulation and IFN-α secretion to patients in remission. An explanation might be that TLR-3 is not the only receptor of foreign dsRNAs in the cells. Indeed, CARD helicases also play this critical role in vivo for defence against RNA from viruses in cytoplasm of the cells, where they recognized dsRNAs from viruses entering directly into cytoplasm or dsRNAs synthesized during active viral replication 44. After recognition of dsRNAs, CARD helicases also activate IRF3 and increase IFN-α release. Interactions between TLR-3 and CARD helicases have not been well characterized until now, but they can act in synergy or in opposition. In accord with the high viral load of EBV in relapsing patients and with the probable replication of other viruses at this stage of the disease, it can be reasonably postulated that CARD helicases may be activated in relapse and enhanced IRF3 activation and INF-α release, whereas TLR-3 is down-regulated in these patients. TLR-3 expression is highly variable between individuals, and might determine the response of the immune system to chronic viral infection. A decrease of TLR-3 expression in macrophages derived from PBMC was shown recently in patients infected chronically by hepatitis C virus (HCV), whereas patients who cleared the virus spontaneously had a higher expression of TLR-3, inducing the production of IFN 45. Additionally, herpesvirus 8 decreased TLR-3 mRNA expression and inhibited the anti-viral IFN response, which could explain the viral latency in patients with Kaposi sarcoma 46. The decrease of TLR-3 expression observed in PBMC of relapsing patients might also reflect an inappropriate response to viral infections, similar to those described above with the inefficient viral clearance of hepatitis C virus (HCV) or by the development of malignancy in Kaposi sarcoma, and be the base for a chronic relapsing course.
Globally, PBMC from all INS patients were unable to be restimulated in vitro by TLR-3 agonist, meaning that IFN-α production remained at the same level as unstimulated PBMC. Indeed, in-vivo viral-sensitized PBMC are already known to be unable to be restimulated in vitro with TLR agonists, as demonstrated in PBMC from patients infected by human immunodeficiency virus 47 and HCV 48. A decrease of IL-6 and IFN-α was also observed after TLR-3 and TLR-7/-8 stimulation of PBMC from HCV post-liver transplants with rapid fibrosis compared to low fibroses indicating the impairment of HCV immune system control 49. These observations favour the hypothesis of an inappropriate immune response against a virus in all INS patients, whether in remission or in relapse. Moreover, in-vitro IL-6 secretion was normal or decreased, as it was synthesized only by PBMC, whereas the increase of plasma IL-6 concentration from patients in relapse may more reflect the global synthesis of IL-6 by the non-immune organs that are able to secrete IL-6, i.e. kidneys and liver, in response to the stress of glomerular barrier injury and severe hypoproteinaemia, respectively 50–52.
B cell behaviour in relapse is quite different from other PBMC subsets, while TLR-3 and TLR-8 expression is normal. B cells are also highly sensitive to TLR-7/-8 stimulation in vitro that leads to an abnormal increased IgM production. The TLR-7/-8 agonist induces memory B cells to produce IgM and IgG, as well as naive human B cells to differentiate into IgM- and IgG-producing cells in the absence of B cell receptor cross-linking and CD40–CD40L interaction 53. The efficient in-vitro stimulation of TLR-7/-8 inducing IgG and IgM secretion, respectively, confirms that B cells are operational during relapse.
Clinically, the effect of rituximab in preventing relapses in steroid-dependent patients 3,4 argues strongly for the involvement of B cells in the primary mechanisms of the disease. An original result of this study is the normalization of the expression of TLR-3 and TLR-8 in all PBMC subsets, not only in B cells. At the functional level, decreased in-vitro IgG secretion in remission reveals a defect in IgG synthesis that is consistent with previous clinical observations 13,54,55. Interestingly, in our study, not only IgG synthesis but also the production of IL-6 and IFN-α by PBMC were normalized in remission following B cell recovery after RTX therapy. In contrast, both IRF3 transactivation and the lack of restimulation of IFN-α production by TLR-3 agonist remained unchanged in PBMC from INS patients after RTX compared to all INS patients. Finally, the lack of restimulation of IFN-α production by TLR-3 agonist as well as the remaining IRF3 transactivation are a hallmark of all INS patients, whatever the stage of the disease and previous therapies.
In conclusion, changes in innate immunity are numerous and complex in INS patients, affecting the cellular and humoral balance of specific immunity, both in relapse and remission. Those changes in TLR-3, as well as the lack of restimulation of IFN-α by TLR-3 agonist, might be the link between different stages of INS and the indirect mark of an exposure to external environmental factors, especially viral agents.
Acknowledgments
A.J. was supported by postdoctoral fellowships from the Association des Malades d'un Syndrome Néphrotique, the Société de Néphrologie and the Association des Séminaires de Robert Debré. L.D. was supported by a master fellowship and N.H. was supported by a doctoral fellowship, both from the Fondation pour la Recherche Médicale.
Disclosure
All authors declare no conflicts of interest.
Supporting Information
Additional Supporting information may be found in the online version of this article at the publisher's web site:
Fig. S1. Toll-like receptor (TLR)-3 expression in peripheral blood mononuclear cells (PBMC). Representative dot-plot analyses of flow cytometry showed the respective gating for PBMC, lymphocytes and monocytes, followed by representative histograms for CD4+ T cells, CD8+ T cells and CD19+ B cells in the lymphocyte gate and CD14+ monocytes in the monocyte gate. Then, representative histograms of intracellular TLR-3 expression were shown in total PBMC and in each PBMC subset from controls and idiopathic nephrotic syndrome (INS) patients in relapse and remission. TLR-3 expression and isotype controls are represented by the white and the grey layers, respectively.
Fig. S2. Transactivation of nuclear factor (NF)-κB and AP-1 in peripheral blood mononuclear cells (PBMC). Activation of (a) NF-κB and (b) AP-1 was measured by enzyme-linked immunosorbent assay (ELISA) in nuclear extracts of PBMC from 14 controls, 13 patients in relapse, 13 patients in remission and 13 patients in remission after rituximab therapy (remission RTX+). No significant difference was found between the four groups.
Table S1. Blood cell count of patients with idiopathic nephrotic syndrome and controls.
Table S2. Number of cells analysed by flow cytometry in isolated peripheral blood mononuclear cells (PBMC) subsets of patients with idiopathic nephrotic syndrome and controls.
References
- Eddy AA, Symons JM. Nephrotic syndrome in childhood. Lancet. 2003;362:629–39. doi: 10.1016/S0140-6736(03)14184-0. [DOI] [PubMed] [Google Scholar]
- Fujinaga S, Endo A, Ohtomo Y, Ohtsuka Y, Shimizu T. Uncertainty in management of childhood-onset idiopathic nephrotic syndrome: is the long-term prognosis really favorable? Pediatr Nephrol. 2013;28:2235–8. doi: 10.1007/s00467-013-2553-1. [DOI] [PubMed] [Google Scholar]
- Iijima K, Sako M, Nozu K, et al. Rituximab for childhood-onset, complicated, frequently relapsing nephrotic syndrome or steroid-dependent nephrotic syndrome: a multicentre, double-blind, randomised, placebo-controlled trial. Lancet. 2014;384:1273–81. doi: 10.1016/S0140-6736(14)60541-9. [DOI] [PubMed] [Google Scholar]
- Ravani P, Rossi R, Bonanni A, et al. Rituximab in children with steroid-dependent nephrotic syndrome: a multicenter, open-label, noninferiority, randomized controlled trial. J Am Soc Nephrol. 2015;26:2259–66. doi: 10.1681/ASN.2014080799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deschenes G, Doucet A. Free immunoglobulin light chains: a role in minimal change disease. Biosci Hypotheses. 2009;2:135–42. [Google Scholar]
- Sellier-Leclerc AL, Duval A, Riveron S, et al. A humanized mouse model of idiopathic nephrotic syndrome suggests a pathogenic role for immature cells. J Am Soc Nephrol. 2007;18:2732–9. doi: 10.1681/ASN.2006121346. [DOI] [PubMed] [Google Scholar]
- Lapillonne H, Leclerc A, Ulinski T, et al. Stem cell mobilization in idiopathic steroid-sensitive nephrotic syndrome. Pediatr Nephrol. 2008;23:1251–6. doi: 10.1007/s00467-008-0793-2. [DOI] [PubMed] [Google Scholar]
- Yan K, Nakahara K, Awa S, et al. The increase of memory T cell subsets in children with idiopathic nephrotic syndrome. Nephron. 1998;79:274–8. doi: 10.1159/000045049. [DOI] [PubMed] [Google Scholar]
- Araya C, Diaz L, Wasserfall C, et al. T regulatory cell function in idiopathic minimal lesion nephrotic syndrome. Pediatr Nephrol. 2009;24:1691–8. doi: 10.1007/s00467-009-1214-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao XS, Yang XQ, Zhao XD, et al. The prevalence of Th17 cells and FOXP3 regulate T cells (Treg) in children with primary nephrotic syndrome. Pediatr Nephrol. 2009;24:1683–90. doi: 10.1007/s00467-009-1194-x. [DOI] [PubMed] [Google Scholar]
- Pereira Wde F, Brito-Melo GE, Guimaraes FT, Carvalho TG, Mateo EC, Simoes e Silva AC. The role of the immune system in idiopathic nephrotic syndrome: a review of clinical and experimental studies. Inflamm Res. 2014;63:1–12. doi: 10.1007/s00011-013-0672-6. [DOI] [PubMed] [Google Scholar]
- Chen CH, Hsieh KH, Lee PP. Enhanced suppressor T cell activity resulting in increased IgM and decreased IgG productions in children with minimal change nephrotic syndrome. Int J Pediatr Nephrol. 1987;8:75–80. [PubMed] [Google Scholar]
- Kemper MJ, Altrogge H, Ganschow R, Muller-Wiefel DE. Serum levels of immunoglobulins and IgG subclasses in steroid sensitive nephrotic syndrome. Pediatr Nephrol. 2002;17:413–7. doi: 10.1007/s00467-001-0817-7. [DOI] [PubMed] [Google Scholar]
- Dantal J, Bigot E, Bogers W, et al. Effect of plasma protein adsorption on protein excretion in kidney-transplant recipients with recurrent nephrotic syndrome. N Engl J Med. 1994;330:7–14. doi: 10.1056/NEJM199401063300102. [DOI] [PubMed] [Google Scholar]
- Dantal J, Godfrin Y, Koll R, et al. Antihuman immunoglobulin affinity immunoadsorption strongly decreases proteinuria in patients with relapsing nephrotic syndrome. J Am Soc Nephrol. 1998;9:1709–15. doi: 10.1681/ASN.V991709. [DOI] [PubMed] [Google Scholar]
- Stephan JL, Deschenes G, Perel Y, et al. Nephrotic syndrome and Hodgkin disease in children: a report of five cases. Eur J Pediatr. 1997;156:239–42. doi: 10.1007/s004310050592. [DOI] [PubMed] [Google Scholar]
- Matsumoto K, Kanmatsuse K. Augmented interleukin-18 production by peripheral blood monocytes in patients with minimal-change nephrotic syndrome. Am J Nephrol. 2001;21:20–7. doi: 10.1159/000046214. [DOI] [PubMed] [Google Scholar]
- Chen SP, Cheung W, Heng CK, Jordan SC, Yap HK. Childhood nephrotic syndrome in relapse is associated with down-regulation of monocyte CD14 expression and lipopolysaccharide-induced tumour necrosis factor-alpha production. Clin Exp Immunol. 2003;134:111–9. doi: 10.1046/j.1365-2249.2003.02252.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duffy JL. Myxovirus-like particles in lipoid nephrosis. N Engl J Med. 1969;281:562–3. doi: 10.1056/NEJM196909042811018. [DOI] [PubMed] [Google Scholar]
- MacDonald NE, Wolfish N, McLaine P, Phipps P, Rossier E. Role of respiratory viruses in exacerbations of primary nephrotic syndrome. J Pediatr. 1986;108:378–82. doi: 10.1016/s0022-3476(86)80876-9. [DOI] [PubMed] [Google Scholar]
- Toyabe S, Nakamizo M, Uchiyama M, Akazawa K. Circannual variation in the onset and relapse of steroid-sensitive nephrotic syndrome. Pediatr Nephrol. 2005;20:470–3. doi: 10.1007/s00467-004-1780-x. [DOI] [PubMed] [Google Scholar]
- Abeyagunawardena AS, Trompeter RS. Increasing the dose of prednisolone during viral infections reduces the risk of relapse in nephrotic syndrome: a randomised controlled trial. Arch Dis Child. 2008;93:226–8. doi: 10.1136/adc.2007.116079. [DOI] [PubMed] [Google Scholar]
- Yap HK, Han EJ, Heng CK, Gong WK. Risk factors for steroid dependency in children with idiopathic nephrotic syndrome. Pediatr Nephrol. 2001;16:1049–52. doi: 10.1007/s004670100024. [DOI] [PubMed] [Google Scholar]
- Dossier C, Sellier-Leclerc AL, Rousseau A, et al. Prevalence of herpesviruses at onset of idiopathic nephrotic syndrome. Pediatr Nephrol. 2014;29:2325–31. doi: 10.1007/s00467-014-2860-1. [DOI] [PubMed] [Google Scholar]
- Mills KH. TLR-dependent T cell activation in autoimmunity. Nat Rev Immunol. 2011;11:807–22. doi: 10.1038/nri3095. [DOI] [PubMed] [Google Scholar]
- Bekeredjian-Ding I, Jego G. Toll-like receptors – sentries in the B-cell response. Immunology. 2009;128:311–23. doi: 10.1111/j.1365-2567.2009.03173.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawai T, Akira S. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity. 2011;34:637–50. doi: 10.1016/j.immuni.2011.05.006. [DOI] [PubMed] [Google Scholar]
- Arkwright PD, Abinun M. Recently identified factors predisposing children to infectious diseases. Curr Opin Infect Dis. 2008;21:217–22. doi: 10.1097/QCO.0b013e3282fa1824. [DOI] [PubMed] [Google Scholar]
- Schwartz GJ, Munoz A, Schneider MF, et al. New equations to estimate GFR in children with CKD. J Am Soc Nephrol. 2009;20:629–37. doi: 10.1681/ASN.2008030287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Gent M, Braem SG, de Jong A, et al. Epstein–Barr virus large tegument protein BPLF1 contributes to innate immune evasion through interference with toll-like receptor signaling. PLOS Pathog. 2014;10:e1003960. doi: 10.1371/journal.ppat.1003960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prendergast AJ, Klenerman P, Goulder PJ. The impact of differential antiviral immunity in children and adults. Nat Rev Immunol. 2012;12:636–48. doi: 10.1038/nri3277. [DOI] [PubMed] [Google Scholar]
- Anders HJ. Toll-like receptors and danger signaling in kidney injury. J Am Soc Nephrol. 2010;21:1270–4. doi: 10.1681/ASN.2010030233. [DOI] [PubMed] [Google Scholar]
- Smith KD. Toll-like receptors in kidney disease. Curr Opin Nephrol Hypertens. 2009;18:189–96. doi: 10.1097/MNH.0b013e32832a1d5f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coppo R, Camilla R, Amore A, et al. Toll-like receptor 4 expression is increased in circulating mononuclear cells of patients with immunoglobulin A nephropathy. Clin Exp Immunol. 2010;159:73–81. doi: 10.1111/j.1365-2249.2009.04045.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Audard V, Pawlak A, Candelier M, Lang P, Sahali D. Upregulation of nuclear factor-related kappa B suggests a disorder of transcriptional regulation in minimal change nephrotic syndrome. PLOS ONE. 2012;7:e30523. doi: 10.1371/journal.pone.0030523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahali D, Pawlak A, Le Gouvello S, et al. Transcriptional and post-transcriptional alterations of IkappaBalpha in active minimal-change nephrotic syndrome. J Am Soc Nephrol. 2001;12:1648–58. doi: 10.1681/ASN.V1281648. [DOI] [PubMed] [Google Scholar]
- Rozkova D, Horvath R, Bartunkova J, Spisek R. Glucocorticoids severely impair differentiation and antigen presenting function of dendritic cells despite upregulation of Toll-like receptors. Clin Immunol. 2006;120:260–71. doi: 10.1016/j.clim.2006.04.567. [DOI] [PubMed] [Google Scholar]
- Yu L, Wang L, Chen S. Endogenous toll-like receptor ligands and their biological significance. J Cell Mol Med. 2010;14:2592–603. doi: 10.1111/j.1582-4934.2010.01127.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato K, Ishikawa T, Okumura A, et al. Expression of Toll-like receptors in chronic hepatitis C virus infection. J Gastroenterol Hepatol. 2007;22:1627–32. doi: 10.1111/j.1440-1746.2006.04783.x. [DOI] [PubMed] [Google Scholar]
- Vercammen E, Staal J, Beyaert R. Sensing of viral infection and activation of innate immunity by toll-like receptor 3. Clin Microbiol Rev. 2008;21:13–25. doi: 10.1128/CMR.00022-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen JI. Epstein–Barr virus infection. N Engl J Med. 2000;343:481–92. doi: 10.1056/NEJM200008173430707. [DOI] [PubMed] [Google Scholar]
- Iwakiri D. Epstein–Barr virus-encoded RNAs: key molecules in viral pathogenesis. Cancers (Basel) 2014;6:1615–30. doi: 10.3390/cancers6031615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cervantes JL, Weinerman B, Basole C, Salazar JC. TLR8: the forgotten relative revindicated. Cell Mol Immunol. 2012;9:434–8. doi: 10.1038/cmi.2012.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MS, Kim YJ. Signaling pathways downstream of pattern-recognition receptors and their cross talk. Annu Rev Biochem. 2007;76:447–80. doi: 10.1146/annurev.biochem.76.060605.122847. [DOI] [PubMed] [Google Scholar]
- Qian F, Bolen CR, Jing C, et al. Impaired toll-like receptor 3-mediated immune responses from macrophages of patients chronically infected with hepatitis C virus. Clin Vaccine Immunol. 2013;20:146–55. doi: 10.1128/CVI.00530-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs SR, Gregory SM, West JA, et al. The viral interferon regulatory factors of Kaposi's sarcoma-associated herpesvirus differ in their inhibition of interferon activation mediated by toll-like receptor 3. J Virol. 2013;87:798–806. doi: 10.1128/JVI.01851-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lester RT, Yao XD, Ball TB, et al. Toll-like receptor expression and responsiveness are increased in viraemic HIV-1 infection. AIDS. 2008;22:685–94. doi: 10.1097/QAD.0b013e3282f4de35. [DOI] [PubMed] [Google Scholar]
- Imran M, Waheed Y, Manzoor S, et al. Interaction of hepatitis C virus proteins with pattern recognition receptors. Virol J. 2012;9:126. doi: 10.1186/1743-422X-9-126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howell J, Sawhney R, Skinner N, et al. Toll-like receptor 3 and 7/8 function is impaired in hepatitis C rapid fibrosis progression post-liver transplantation. Am J Transplant. 2013;13:943–53. doi: 10.1111/ajt.12165. [DOI] [PubMed] [Google Scholar]
- Batal I, De Serres SA, Mfarrej BG, et al. Glomerular inflammation correlates with endothelial injury and with IL-6 and IL-1beta secretion in the peripheral blood. Transplantation. 2014;97:1034–42. doi: 10.1097/01.TP.0000441096.22471.36. [DOI] [PubMed] [Google Scholar]
- de Sain-Van Der Velden MG, de Meer K, Kulik W, et al. Nephrotic proteinuria has no net effect on total body protein synthesis: measurements with (13)C valine. Am J Kidney Dis. 2000;35:1149–54. doi: 10.1016/s0272-6386(00)70053-9. [DOI] [PubMed] [Google Scholar]
- de Sain-van der Velden MG, Kaysen GA, de Meer K, et al. Proportionate increase of fibrinogen and albumin synthesis in nephrotic patients: measurements with stable isotopes. Kidney Int. 1998;53:181–8. doi: 10.1046/j.1523-1755.1998.00729.x. [DOI] [PubMed] [Google Scholar]
- Glaum MC, Narula S, Song D, et al. Toll-like receptor 7-induced naive human B-cell differentiation and immunoglobulin production. J Allergy Clin Immunol. 2009;123:224–30 e4. doi: 10.1016/j.jaci.2008.09.018. [DOI] [PubMed] [Google Scholar]
- Bazzi C, Petrini C, Rizza V, et al . Urinary excretion of IgG and alpha(1)-microglobulin predicts clinical course better than extent of proteinuria in membranous nephropathy. Am J Kidney Dis. 2001;38:240–8. doi: 10.1053/ajkd.2001.26080. [DOI] [PubMed] [Google Scholar]
- Roy RR, Roy E, Rahman MH, Hossain MM. Serum immunoglobulin G, M and IgG:IgM ratio as predictors for outcome of childhood nephrotic syndrome. World J Pediatr. 2009;5:127–31. doi: 10.1007/s12519-009-0025-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Toll-like receptor (TLR)-3 expression in peripheral blood mononuclear cells (PBMC). Representative dot-plot analyses of flow cytometry showed the respective gating for PBMC, lymphocytes and monocytes, followed by representative histograms for CD4+ T cells, CD8+ T cells and CD19+ B cells in the lymphocyte gate and CD14+ monocytes in the monocyte gate. Then, representative histograms of intracellular TLR-3 expression were shown in total PBMC and in each PBMC subset from controls and idiopathic nephrotic syndrome (INS) patients in relapse and remission. TLR-3 expression and isotype controls are represented by the white and the grey layers, respectively.
Fig. S2. Transactivation of nuclear factor (NF)-κB and AP-1 in peripheral blood mononuclear cells (PBMC). Activation of (a) NF-κB and (b) AP-1 was measured by enzyme-linked immunosorbent assay (ELISA) in nuclear extracts of PBMC from 14 controls, 13 patients in relapse, 13 patients in remission and 13 patients in remission after rituximab therapy (remission RTX+). No significant difference was found between the four groups.
Table S1. Blood cell count of patients with idiopathic nephrotic syndrome and controls.
Table S2. Number of cells analysed by flow cytometry in isolated peripheral blood mononuclear cells (PBMC) subsets of patients with idiopathic nephrotic syndrome and controls.