

Abstract
Gut-derived bacterial lipopolysaccharides (LPS) play an essential role in inducing intestinal and systemic inflammatory responses and have been implicated as a pathogenic factor of necrotizing enterocolitis (NEC) and inflammatory bowel disease (IBD). The defective intestinal tight junction (TJ) barrier has been shown to be an important factor contributing to the development of intestinal inflammation. LPS, at physiological concentrations, cause an increase in intestinal tight junction permeability (TJP) via a TLR-4 dependent process; however the intracellular mechanisms that mediate LPS regulation of intestinal TJP remain unclear. The aim of this study was to investigate the adaptor proteins and the signaling interactions that mediate LPS modulation of intestinal TJ barrier using an in-vitro and in-vivo model system. LPS caused a TLR-4 dependent activation of membrane-associated adaptor protein FAK in Caco-2 monolayers. LPS caused an activation of both MyD88-dependent and –independent pathways. SiRNA silencing of MyD88 prevented LPS-induced increase in TJP. LPS caused a MyD88-dependent activation of IRAK4. TLR-4, FAK and MyD88 were co-localized. SiRNA silencing of TLR-4 inhibited TLR-4 associated FAK activation; and FAK knockdown prevented MyD88 activation. In-vivo studies also confirmed that LPS-induced increase in mouse intestinal permeability was associated with FAK and MyD88 activation; knockdown of intestinal epithelial FAK prevented LPS-induced increase in intestinal permeability. Additionally, high dose LPS-induced intestinal inflammation was also dependent on TLR-4/FAK/MyD88 signal-transduction axis. Our data show for the first time that LPS-induced increase in intestinal TJP and intestinal inflammation was regulated by TLR-4 dependent activation of FAK-MyD88-IRAK4 signaling pathway.
Keywords: Intestinal Permeability, Lipopolysaccharide, MyD88, focal adhesion kinase
Introduction
The intestinal epithelium provides a physical barrier that separates the many trillions of commensal bacteria in the intestinal lumen from the underlying lamina propria and the deeper intestinal layers(1, 2). Defective intestinal epithelial tight junction (TJ) barrier, characterized by an increase in intestinal permeability, has been shown to be an important pathogenic factor contributing to the development of inflammatory bowel disease (IBD) and necrotizing enterocolitis (NEC) by allowing paracellular permeation of luminal antigens that elicit and promote inflammatory response(1, 2). The importance of defective intestinal TJ barrier in the development and the prolongation of intestinal inflammation in IBD and NEC has been shown in clinical and animal studies(1–4). It is well-established that patients with IBD and NEC have a defective intestinal TJ barrier manifested by an increase in intestinal permeability (5, 6); and animal studies have shown that enhancement or protection of intestinal TJ barrier prevents the development of intestinal inflammation in animal models of IBD and NEC(7, 8).
Lipopolysaccharide (LPS), also referred to as endotoxin, is an important structural component of the outer membrane of gram negative bacteria. Normally, LPS concentrations are highest in the gut lumen and low or undetectable in the circulating plasma, as LPS in the gut lumen do not penetrate across the healthy intestinal epithelium(9, 10). However, in IBD and NEC, the defective or leaky TJ barrier allows paracellular permeation of LPS and other luminal antigens, which leads to the increase in the intestinal tissue and plasma concentration of LPS(11–13). The importance of LPS in the pathogenesis of NEC and IBD are highlighted by studies that show that the circulating levels of LPS are elevated in patients with these diseases. Similarly, animal models of NEC and IBD also have increased levels of LPS in the intestinal tissue and in the serum(14, 15). Additionally, the intestinal tissue expression of toll-like receptor-4 (TLR-4), the pattern recognition receptor which binds the LPS, is also markedly increased in IBD and NEC(16, 17). TLR-4 polymorphism is associated with increased risk of IBD and more extensive colonic involvement in UC(18). The increase in the intestinal tissue and circulating levels of LPS has been shown to exert deleterious effects on the intestinal epithelial cells, characterized by a reduction in intestinal barrier integrity. However, how the circulating LPS regulates the intestinal epithelial barrier remains unknown.
Most of the published studies used high pharmacologic concentrations of LPS (1–50 μg/ml) to show that LPS causes rapid cell death and apoptosis in various cell types, but these studies do not provide accurate depiction of biological activity of physiological concentrations of LPS(19, 20). Previous studies from our laboratory showed that LPS at physiologically and clinically relevant concentrations (0–10 ng/ml) do not cause intestinal epithelial cell death, but causes a selective increase in intestinal TJ permeability in-vitro and in-vivo by inducing enterocyte membrane expression of TLR-4 and CD14(21). LPS stimulation of mammalian cells occurs through a series of interactions with several membrane-associated proteins including the LPS binding protein (LBP), CD14, MD-2 and TLR-4(22). Upon ligation of TLR-4 with LPS, signaling cascades are activated which result in the production of innate effector responses as well as the initiation of an adaptive immune response. Adaptor proteins are recruited to the TLR-4 receptor complex via TIR-TIR interactions. TLR-4 signaling has been divided into MyD88-dependent and MyD88-independent or TRIF-dependent pathways(22). Focal adhesion kinase (FAK) plays a key role in focal adhesion dynamics and signal transduction process(23). MyD88 has been shown to be involved in FAK-regulated protein I/II-induced cytokine release in mice macrophages(24). The mechanisms of how LPS regulates TLR-4 signal transduction and the involvement of adaptor proteins remain unclear. The major aim of this study was to examine the role of adaptor proteins FAK and MyD88 in LPS-induced increase in intestinal epithelial TJ permeability, using filter-grown Caco-2 intestinal epithelial monolayers and mouse re-cycling intestinal perfusion as in-vitro and in-vivo models of intestinal epithelium. Our data show for the first time that the LPS-induced increase in intestinal epithelial TJ permeability is mediated by the activation of TLR-4/FAK/MyD88 signal transduction axis; moreover, our data also show that high dose LPS-induced inflammation and mucosal damage are also dependent on TLR-4/FAK/MyD88 axis activation.
Materials and Methods
Reagents
DMEM, trypsin, FBS, glutamine, penicillin, streptomycin, PBS, HRP-conjugated secondary antibodies for Western blot analysis were purchased from Invitrogen Life Technologies (Carlsbad, CA). siRNA of FAK, TRIF, TRAM, MyD88 and transfection reagents were from Dharmacon (Lafayette, CO). LPS (O111:B4) and focal adhesion kinase (FAK) inhibitor PF-573228 (3,4-dihydro-6-[[4-[[[3-(methyl-sulfonyl)phenyl]methyl]amino]-5-(trifluoromethyl)-2-pyrimidinyl]amino]-2(1H)-quinolinone) (PF-228) were purchased from Sigma-Aldrich (St. Louis, MO). All other chemicals were of reagent grade and were purchased from Sigma-Aldrich (St. Louis, MO), VWR (Aurora, CO), or Fisher Scientific (Pittsburgh, PA).
Cell culture
Caco-2 cells (passage 20) were purchased from the American Type Culture Collection (Manassas, VA) and maintained at 37°C in a culture medium composed of DMEM with 4.5 mg/ml glucose, 50 U/ml penicillin, 50 U/ml streptomycin, 4 mM glutamine, 25 mM HEPES, and 10% FBS as previously described. Caco-2 cells were used between passages 22 and 28 in this study. The cells were kept at 37°C in a 5% CO2 environment. For growth on filters, high-density cells (1 × 105 cells) were plated on Transwell filters with 0.4 μm pore (Corning Incorporated, Corning, NY) and monitored regularly by visualization with an inverted microscope (Eclipse TS100/100-F, Nikon, Melville, NY) and by epithelial resistance measurements.
Assessment of protein expression by western blot analysis
Protein expression from Caco-2 cells and mouse tissue was assessed by western blot as previously described. Cells and mouse tissue were lysed with lysis buffer (50 mM Tris•HCl (pH 7.5), 150 mM NaCl, 500 μM NaF, 2 mM EDTA, 100 μM vanadate, 100 μM PMSF, 1 μg/ml leupeptin, 1 μg/ml pepstatin A, 40 mM paranitrophenyl phosphate, 1 μg/ml aprotinin, and 1% Triton X-100) on ice for 30 min. The lysates were centrifuged at 10,000 g for 10 min in an Eppendorf Centrifuge (5417R, Hauppauge, NY) to obtain a clear lysate. The supernatant was collected and protein concentration was determined using the Bio-Rad Protein Assay kit (Bio-Rad Laboratories, Hercules, CA). Laemmli gel loading buffer (Bio-Rad Laboratories) was added to the lysate containing 10 – 20 μg of protein and boiled at 100°C for 7 min, after which proteins were separated on an SDS-PAGE gel. Proteins from the gel were transferred to the membrane (Trans-Blot Transfer Medium, Nitrocellulose Membrane; Bio-Rad) overnight. The membrane was incubated for 2 h in blocking solution (5% dry milk in TBS-Tween 20 buffer). The membrane was then incubated with antibody in blocking solution. After a wash in TBS-1% Tween buffer, the membrane was incubated in secondary antibody and developed using the Santa Cruz Western Blotting Luminol Reagents (Santa Cruz Biotechnology, Santa Cruz, CA) on the Kodak BioMax MS film (Fisher Scientific, Pittsburgh, PA). The films were exposed between 5 s to 10 min.
Co-immunoprecipitation analysis
Co-immunoprecipitation analysis was performed using Dynabeads Protein G (Life Technologies AS, Norway). After LPS treatment, Caco-2 cells were lysed in immunoprecipitation buffer (20 mM Tris•HCl (pH 7.5), 0.5 M NaCl, 1% Triton X-100, 50 mM NaF, 2 mM EDTA, 1 mM Na3VO4, 0.1% sodium dodecyl sulfate, 0.5% Nonidet P-40 and 10% glycerol (vol/vol)), containing protease inhibitor cocktail (Roche, Mannheim, Germany). After 4 cycles of freeze-thaw and sonication, lysates were centrifuged at 12,000 g for 10 min. 1.5 mg Dynabeads were incubated with 10 μg TLR-4 antibody for 10 min at room temperature. After washing with washing buffer (PBS pH7.4 with 0.02% Tween-20), 200 μl sample lysates were added into the beads-Ab complex, and incubated with rotation overnight at 4°C. After washing the Dynabeads-Ab-Ag complex 3 times with 200 μl washing buffer, 20 μl elution buffer (50 mM Glycine pH2.8) and 20 μl SDS sample buffer were added, and heated for 10 min at 95°C. Immunoprecipitates were separated by SDS-PAGE and further analyzed by Western blot using anti-FAK and anti-MyD88 rabbit polyclonal Abs (Abcams).
Determination of epithelial monolayer resistance and paracellular permeability
Caco-2 transepithelial electrical resistance (TER) was measured by using an epithelial voltohmeter (World Precision Instruments) as previously reported. Both apical and basolateral sides of the epithelium were bathed with buffer solution. Electrical resistance was measured until similar values were recorded on three consecutive measurements. Caco-2 paracellular permeability was determined by using an established paracellular marker inulin(21). Known concentrations of permeability marker (10 μM) and its radioactive tracer were added to the apical solution (25). Low concentrations of permeability markers were used to ensure that negligible osmotic or concentration gradient was introduced.
siRNA transfection
Caco-2 monolayers were transiently transfected with siRNA using DharmaFect transfection reagent (Lafayette, Co) as previously described (26). Briefly, cells (1 × 105/filter) were seeded into a 12-well transwell plate and grown to confluency. Caco-2 monolayers were then washed with PBS twice and 0.5 ml Accell medium (Thermo Scientific, Lafayette, CO) was added to the apical compartment of each filter and 1.5 ml was added to the basolateral compartment of each filter. siRNA (5 ng) of interest and DharmaFect reagent (2 μl) were preincubated in Accell medium. After 5 min of incubation, the two solutions were mixed, and the mixture was added to the apical compartment of each filter. For LPS experiments, Caco-2 cells were transfected with siRNA for 24 h prior to the LPS treatment. The Caco-2 TJ barrier assessments were carried out at the end of day 5 of LPS treatment.
IRAK4 activity measurement
IRAK4 activity was measured using IRAK4 Kinase Enzyme System and ADP-Glo Kinase Assay (Promega, Madison, WI) with modifications. IRAK4 kinase was immunoprecipitated using Dynabeads Protein G as described above. After immunoprecipitation, IRAK4 kinase was eluted with 20 μl Elution buffer (50 mM Glycine pH2.8). For the functional assay, the pH of the eluate was adjusted by adding 20 μl of 1 M Tris (pH7.5). The final pH of the eluate was 7.5. In 96-well plate, the 25 μl total reaction volume included 5 μl 5X kinase buffer, 10 μl enzyme, and 10 μl substrate/ATP mix (2.5 μg MBP protein, 50 μM ATP, and 50 μM DTT). The kinase reaction was incubated at room temperature for 60 min. Then 25 μl ADP-Glo Reagent was added. Followed by incubation at room temperature for 40 min, 50 μl Kinase Detection Reagent was added. After incubation at room temperature for 30 min, the luminescence was recorded with 0.5 s Integration time using PerkinElmer Multilabel Counter.
In-vivo determination of mouse intestinal permeability
The Laboratory Animal Care and Use Committee at the University of New Mexico approved all experimental protocols. Male C57BL/6 mice, TLR-4 knock out and MyD88 knockout mice (9–10 wk) were purchased from Jackson Laboratories (Bar Harbor, ME). The mice were kept two per cage in a temperature-controlled room at 25°C with a 12:12 h light-dark cycle. Diet and drinking water were provided ad libitum.
LPS effect on intestinal permeability in an in-vivo mouse model system was determined using a re-cycling intestinal perfusion method as previously described. Mice were injected with varying concentrations of LPS intraperitoneally (i.p.) every 24 h for up to 5 days of experimental period. A 6 cm segment of mouse small intestine was isolated and cannulated with a small-diameter plastic tube (in an anesthetized mouse maintained in 1% isoflurane in oxygen) and continuously perfused with 5 ml Krebs-phosphate saline buffer for a 2 h perfusion period. An external recirculating pump (Econo Pump, Bio-Rad) was used to recirculate the perfusate at a constant flow rate (0.75 ml/min). The body temperature of mouse was maintained at 37°C with a temperature-controlled warming blanket. The intestinal permeability was assessed by measuring flux rate of paracellular probe, Texas Red-labeled dextran (MW = 10,000 g/mol). Water absorption was determined by using a non-absorbable marker sodium ferrocyanide or by measuring the difference between initial and final volume of the perfusate.
In-vivo transfection of FAK siRNA
The effect of FAK siRNA on mouse small intestinal TJ permeability was determined using the perfusion model described previously. In these studies, mice were fasted for 24 h prior to the surgery. With the abdominal cavity open, a 6-cm segment of mouse small intestine was isolated. The transfection solution (0.5 ml), consisting of FAK siRNA (2.5 nmol) or scramble nontarget siRNA and tansfecting agent Lipofectamine (50 μl), was injected through a 33-gauge needle into the lumen of the small intestine, and the small intestine was cannulated for 1 h. The small intestine was then placed back into the abdominal cavity, and the abdominal cavity was closed with sutures. The FAK siRNA transfection was performed at day 0. After 5 days LPS (0.1 mg/kg body weight, i.p.) treatment, the intestinal permeability was measured using a re-cycling intestinal perfusion method described above. The surgery had no effect on the food intake and the body weight of the animals during the experimental period.
High dose LPS-induced small intestinal inflammation in mice
To study the effect of high dose LPS on mice small intestine, mice were injected with high dose LPS intraperitoneally (i.p. 1 mg/Kg body weight), and intestinal permeability was measured at 3 h and 24 h using a re-cycling intestinal perfusion method as described above. After the procedure, small intestine tissue was collected for western blot analysis and histological examination. After high dose LPS administration for 24 h, mice body weights were weighted using a digital balance, and the clinical disease activity index was observed and recorded, including ruffled fur, decrease in movement and avoidance behavior, loose stools, and bloody stools.
Statistical analysis
Results are expressed as means ± SE, and analyzed using Student’s t-tests for unpaired data (GraphPad Prism 5.00 for Windows; GraphPad Software). A p value of ≤ 0.05 was used to indicate statistical significance. All experiments were carried out in triplicates or quadruplicates and repeated at a minimum of three times to ensure reproducibility.
Results
LPS-induced increase in Caco-2 intestinal epithelial TJ permeability is mediated by FAK activation
Previous studies have shown that LPS at very high pharmacologic concentrations (50 μg/ml) cause rapid cell death in various cell types studied, including intestinal and immune cells. We previously showed that LPS, at physiological and clinically relevant concentrations (0.1–1 ng/ml), caused a selective increase in intestinal TJ permeability by inducing enterocyte membrane expression and localization of TLR-4 without causing cell death. LPS (0.3 ng/ml) basolateral (but not apical) treatment caused a time-dependent increase in TLR-4 protein expression in filter-grown Caco-2 monolayers (Supplemental Fig. 1), which correlated with the time course of the drop in Caco-2 TER (Fig. 1B).
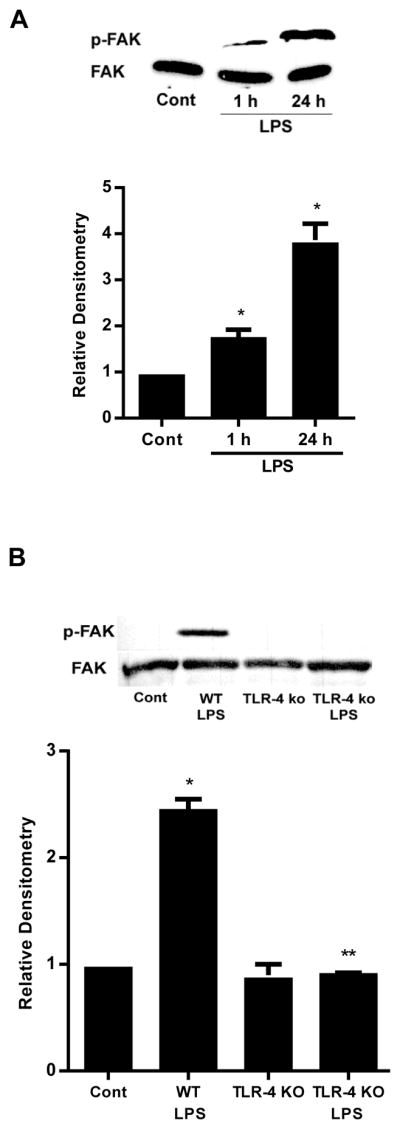
Figure 1.

The involvement of FAK in LPS-induced increase in Caco-2 permeability. (A). Time-course effect of LPS (0.3 ng/ml) on phospho-FAK in Caco-2 monolayers. The densitometry analysis indicates relative levels of phospho-FAK reveled time dependent significant increase. (B). Time-course effect of LPS (0.3 ng/ml) on Caco-2 TER. The effect of LPS (0.3 ng/ml) on Caco-2 TER was measured over a 5-day experimental period. The mean TER for control Caco-2 monolayers was 512 ± 15 Ω • cm2. (C). The siRNA FAK transfection resulted in a near-complete depletion of FAK expression as assessed by Western blot analysis and Densitometry analysis. The Western blot analysis was performed 72 h after siRNA FAK transfection. (D). The siRNA-induced silencing of FAK prevented LPS-induced increase in Caco-2 inulin flux. (E). The siRNA-induced silencing of FAK prevented LPS-induced drop in TER in Caco-2 monolayers. The mean TER for control Caco-2 monolayers was 520 ± 6 Ω • cm2. Inhibition of FAK function by inhibitor PF-228 prevented LPS-induced increase in Caco-2 inulin flux (F) and drop in TER (G). The mean TER for control Caco-2 monolayers was 485 ± 13 Ω • cm2. n=4. *, p<0.0001 vs control. **, p<0.0001 vs LPS treatment.
FAK is an adaptor protein that is concentrated in the focal adhesion and has been implicated in TLR-4 proinflammatory response. We hypothesized that FAK may play a regulatory role in initiating TLR-4 signal transduction process. To test this hypothesis, we first assessed the expression and activation of FAK by Western blot analysis. As shown in Fig. 1A, there was a time-dependent increase in FAK activation as evidenced by FAK tyrosine (Y397) phosphorylation (responsible for the signal transduction process), (27) following the LPS treatment; however, the total expression of FAK was not affected by the LPS treatment, indicating that LPS causes a time-dependent activation of FAK but not FAK expression. The LPS-induced increase in FAK phosphorylation correlated with the time course of the drop in Caco-2 TER (Fig. 1B). To determine the role of FAK in LPS-induced increase in intestinal permeability, FAK was selectively knockdown by siRNA (Fig. 1C). The selective knock-down of FAK by siRNA transfection resulted in complete inhibition of LPS-induced increase in mucosal-to-serosal flux of inulin in Caco-2 cells (Fig. 1D) and drop in Caco-2 TER (Fig. 1E), suggesting that FAK was necessary for the increase in Caco-2 TJ permeability. To further investigate the requirement of FAK activation on LPS-induced loss of barrier function in Caco-2 monolayers, we used a specific FAK inhibitor, PF-228, to evaluate the role of FAK activation. Recent studies have shown that PF-228 selectively inhibits the activation of FAK. Caco-2 cells were pretreated with PF-228 for 1 h prior to the LPS treatment. PF-228 (10 μM) treatment by itself did not cause significant change in Caco-2 inulin flux and TER, but prevented the LPS-induced increase in Caco-2 inulin flux (Fig. 1F) and drop in Caco-2 TER (Fig. 1G). These data suggested that the LPS-induced increase in Caco-2 TJ permeability required FAK activation.
Next, we examined whether FAK activation was sufficient to cause an increase in Caco-2 TJ permeability. For these studies, we examined the effect of known FAK activator intercellular adhesion molecule-1 (ICAM-1, 450 nM). ICAM-1 has been shown to selectively activate FAK in endothelial cells (28). To examine the effect of FAK activation in regulating the increase in TJ permeability, Caco-2 monolayers were treated with ICAM-1 (450 nM). As previously reported, ICAM-1 caused a rapid activation of FAK in Caco-2 monolayers (data not shown). ICAM-1 treatment resulted in a similar proportional increase in Caco-2 inulin flux (Fig. 2A) and drop in TER (Fig. 2B), as the LPS treatment, suggesting that the activation of FAK by itself was sufficient to cause an increase in Caco-2 TJ permeability.
Figure 2.

Effect of FAK activator, ICAM-1, on Caco-2 TJ permeability. ICAM-1 treatment (5d) significantly induced an increase in Caco-2 inulin flux (A) and drop in TER (B), which were similar to LPS treatment. The mean TER for control Caco-2 monolayers was 529 ± 43 Ω • cm2. n=4. *, p<0.0001 vs control.
LPS-induced increase in Caco-2 TJ permeability is mediated by MyD88-dependent pathway
The TLR-4 signal transduction is regulated by two distinct pathways: MyD88-dependent and MyD88-independent pathway (or TRAM/TRIF-dependent pathway). To determine whether these pathways were involved in LPS modulation of Caco-2 TJ permeability, Caco-2 MyD88, TRAM and TRIF were selectively knocked down. The MyD88 siRNA transfection resulted in knock-down of MyD88 (Fig. 3A) and inhibition of LPS-induced increase in Caco-2 inulin flux and drop in TER (Fig. 3B, 3C). In contract, the siRNA-induced silencing of TRAM or TRIF did not affect the LPS-induced increase in Caco-2 inulin flux and drop in TER (Fig. 4). These results suggested that the LPS effect on Caco-2 TJ permeability was mediated by MyD88 dependent pathway but not TRAM/TRIF pathway.
Figure 3.

The involvement of MyD88 and IRAK4 in LPS-induced increase in Caco-2 permeability. (A). siRNA MyD88 transfection resulted in a near-complete depletion of MyD88 expression as assessed by Western blot analysis and relative densitometry. The Western blot analysis was performed 72 h after siRNA MyD88 transfection. siRNA-induced silencing of MyD88 prevented LPS-induced increase in Caco-2 inulin flux (B) and drop in TER (C). The mean TER for control Caco-2 monolayers was 536 ± 20 Ω • cm2. (D). Effect of LPS on phospho-IRAK4 expression assessed by Western blot. LPS (0.3 ng/ml) treatment induced a time-dependent increase in phospho-IRAK4 expression. The densitometry analysis indicates a time-dependent increase in relative levels of phoshpo-IRAK4 after LPS treatment and no significant change in the total IRAK4 levels. (E). Effect of LPS on IRAK4 activity measured by IRAK4 Kinase Enzyme System and ADP-Glo Kinase Assay. (F). Effect of IRAK4 inhibitor (N-(2-Morpholinylethyl)-2-(3-nitrobenzoylamido)-benzimidazole, 200 nM) on Caco-2 IRAK4 activity. IRAK4 inhibitor prevented the LPS-induced increase in Caco-2 IRAK4 activity. (G). Effect of IRAK4 inhibitor on Caco-2 TER. IRAK4 inhibitor prevented the LPS-induced drop in Caco-2 TER. The mean TER for control Caco-2 monolayers was 516 ± 9 Ω • cm2. (H) Effect of siRNA-induced silencing of MyD88 on IRAK4 activity in Caco-2 monolayers. n=4. *, p<0.0001 vs control. **, p<0.0001 vs. LPS treatment.
Figure 4.

Effect of siRNA silencing of TRAM and TRIF on LPS-induced alteration in Caco-2 inulin flux and TER. Caco-2 cells were transfected with TRAM siRNA or TRIF siRNA for 24 h prior to LPS treatment. (A). siRNA TRAM transfection resulted in a near-complete depletion of TRAM expression as assessed by Western blot analysis and relative Densitometry. The Western blot analysis was performed 72 h after siRNA TRAM transfection. The siRNA-induced silencing of TRAM did not prevent LPS-induced increase in Caco-2 inulin flux (B) and drop in TER (C). The mean TER for control Caco-2 monolayers was 551 ± 18 Ω • cm2. (D). siRNA TRIF transfection resulted in a near-complete depletion of TRIF expression as assessed by Western blot analysis and relative densitometry. The Western blot analysis was performed 72 h after siRNA TRIF transfection. The siRNA-induced silencing of TRIF also did not prevent LPS-induced increase in Caco-2 inulin flux (E) and drop in TER (F). The mean TER for control Caco-2 monolayers was 513 ± 19 Ω • cm2. n=4. *, p<0.001 vs control.
LPS-induced increase in Caco-2 TJ permeability requires an increase in IRAK4 activity
IRAK4 is a direct substrate of MyD88, and MyD88 activation produces the enzymatic phosphorylation (Thr345) of IRAK4. In the control Caco-2 monolayers, there was very little phosphorylation of IRAK4 (Thr345) (Fig. 3D). LPS (0.3 ng/ml) treatment resulted in a time-dependent increase in Thr345 phosphorylation (Fig. 3D), correlating with the time course of increase in Caco-2 TJ permeability (Fig. 1B). The time course of LPS-induced IRAK4 Thr345 phosphorylation also correlated with the time-course of increase in TLR-4 expression (Supplemental Fig. 1). The LPS effect on IRAK4 activity was also determined by IRAK4 Kinase Enzyme System and ADP-Glo Kinase Assay (using myelin basic protein (MBP) as the substrate). The Kinase Detection Reagent converts ADP to ATP and the newly synthesized ATP is converted to light using the luciferase/luciferin reaction. LPS (0.3 ng/ml) caused a time-dependent increase in IRAK4 activity (Fig. 3E). The time course of LPS-induced increase in IRAK4 activity paralleled the increase in IRAK4 phosphorylation. To validate the involvement of IRAK4, the effect of specific IRAK4 inhibitor, N-(2-Morpholinylethyl)-2-(3-nitrobenzoylamido)-benzimidazole (NMN)(Calbiochem), on LPS-induced increase in Caco-2 TJ permeability was examined. Caco-2 cells were pretreated with NMN for 1 h prior to the LPS treatment. NMN (200 nM) inhibited the LPS-induced increase in IRAK4 activity (Fig. 3F) and drop in Caco-2 TER (Fig. 3G). Next to confirm that IRAK4 activation is indeed regulated by MyD88, Caco-2 MyD88 expression was knocked down. The siRNA-induced knock-down of MyD88 prevented the LPS-induced increase in IRAK4 activity in Caco-2 monolayers (Fig. 3H). MyD88 knock-down also prevented the LPS-induced increase in Caco-2 TJ permeability (Fig. 3B and 3C). Together, these findings suggested that the LPS effect on Caco-2 TJ permeability was mediated by the MyD88 phosphorylation and activation of IRAK4.
Activation of FAK and MyD88 is TLR-4-dependent
Above data suggested that the LPS effect on Caco-2 TJ permeability was dependent on both FAK and MyD88 activation; however the interaction between TLR-4, FAK and MyD88 remains unknown. We hypothesized that FAK plays a regulatory role in the TLR-4 and MyD88 cross-talk. To test this hypothesis, we first assessed binding or co-localization of TLR-4, FAK, and MyD88 by co-immunoprecipitation studies. Following LPS treatment, Caco-2 cell lysates were immunoprecipitated with anti-TLR-4 antibody, and the TLR-4 immunoprecipitate assessed for the presence of FAK and MyD88 via immunoblotting. As shown in Fig. 5, both FAK and MyD88 were present in the TLR-4-immunoprecipitate, confirming the presence of TLR-4/FAK/MyD88 cluster or molecular aggregate. FAK and MyD88 were associated with TLR-4 in both untreated Caco-2 cells and following LPS treatment, and the amount of total FAK and MyD88 in the immunoprecipitated remained the same. However, the level of tyrosine-phosphorylated (Y397) FAK was significantly increased after the LPS treatment.
Figure 5.

Co-immunoprecipitation of TLR-4, FAK and MyD88 using Dynabeads Protein G. After LPS treatment (5 d), Caco-2 monolayers were lysed, and immunoprecipitated with TLR-4 antibody, then phospho-FAK, FAK and MyD88 were detected by Western blot analysis. n=4. Densitometry analysis showed a significant increase in phospho-FAK compared to control and no change in total FAK and MyD88 expression. *, p<0.001 vs control.
To study the molecular interactions between TLR-4, FAK and MyD88, the effect of siRNA-induced knock-down of TLR-4, FAK and MyD88 was examined. The TLR-4 siRNA transfection produced a near-complete knockdown of TLR-4 protein (Fig. 6A). The TLR-4 knockdown resulted in inhibition of the LPS-induced increase in phospho-FAK and phospho-IRAK4 in Caco-2 monolayers (Fig. 6B), suggesting that the LPS-induced activation of FAK and MyD88 were dependent on TLR-4. FAK siRNA transfection also prevented the LPS-induced increase in phospho-IRAK4 in Caco-2 monolayers (Fig. 6C). On the other hand, MyD88 siRNA transfection did not affect the LPS-induced increase in phospho-FAK (Fig. 6D), suggesting that FAK regulates the MyD88 activation but not the reverse. Together, these results show the cross-talk between TLR-4, FAK and MyD88 activation.
Figure 6.

TLR-4 was required for LPS-induced activation of FAK and MyD88. (A). The siRNA TLR-4 transfection resulted in a near-complete depletion of TLR-4 expression as assessed by Western blot analysis and relative Densitometry. The Western blot analysis was performed 72 h after siRNA TLR-4 transfection. (B). The siRNA-induced silencing of TLR-4 prevented LPS-induced increase in phospho-FAK and phospho-IRAK4 expression in Caco-2 monolayers. siTLR-4 transfection was performed 1 d prior to the LPS treatment, and the Western blot analysis was performed after 5 d LPS treatment. Densitometry analysis revealed significant increase in phospho-FAK and phospho-IRAK4 expression after LPS treatment compared to control which were abolished by silencing TLR-4. (C). The siRNA-induced silencing of FAK prevented LPS-induced increase in phospho-IRAK4 expression in Caco-2 monolayers shown by Western Blot and densitometry analysis. siFAK transfection was performed 1 d prior to the LPS treatment, and the Western blot analysis was performed after 5 d LPS treatment. (D) siRNA-induced silencing of MyD88 did not prevent LPS-induced increase in phospho-FAK expression in Caco-2 monolayers as shown by western blot and densitometry analysis. siMyD88 transfection was performed 1 d prior to the LPS treatment, and the Western blot analysis was performed after 5 d LPS treatment. n=4.. *, p<0.001 vs control. **, p<0.001 vs LPS treatment.
LPS effect on junctional localization of TJ proteins
To assess the possible effect of LPS on junctional localization of TJ proteins in Caco-2 cells, we examined the effect of LPS (0.3 ng/ml) on expression of cytoplasmic TJ protein ZO-1 and transmembrane proteins occludin, claudin-1, claudin-3 and claudin-5 using immunostaining. As shown in Figure 1B, LPS caused an increase in TJ permeability by day 4; however, LPS did not have significant effect on junctional localization of occludin, ZO-1 and the claudin-1, 3, and 5 (Supplemental Figure 2). These studies suggested that LPS (0.3 ng/ml) did not have significant effect on junctional localization of TJ proteins studied, and that the LPS-induced increase in tight junction permeability was independent of TJ protein distribution.
FAK regulates the LPS-induced increase in mouse intestinal permeability in-vivo
In previous studies, intraperitoneal LPS (0.1 mg/kg body weight) injection of mice resulted in a physiologically relevant elevation in serum LPS (0.3–0.6 ng/ml) and an increase in mouse intestinal permeability. The LPS-induced increase in mouse intestinal permeability was also dependent on an increase on mouse enterocyte TLR-4 expression. To examine the role of FAK activation in LPS-induced increase in mouse intestinal permeability in-vivo, we determined the LPS (0.1 mg/Kg body weight) effect on FAK tyrosine (Y397) phosphorylation in mice intestinal tissue by Western blot analysis. As shown in Fig. 7A, there was a time-dependent increase in phospho-FAK following LPS treatment, indicating that LPS causes a time-dependent activation of FAK. Next, mouse enterocyte FAK was selectively KD by in-vivo FAK siRNA transfection. Mouse intestinal epithelial cells were selectively transfected with FAK siRNA in-vivo as recently described by us. In brief, 6 cm segment of small intestinal mucosal surface was exposed to a short pulse (1 hr) of transfecting solution containing FAK siRNA, which leads to the siRNA transfection of surface epithelial cells. The effect of FAK siRNA transfection of intestinal epithelial cells on mouse intestinal permeability was determined following LPS treatment, using Texas Red-labeled dextran (10 KD) as the paracellular marker. Intraperitoneal LPS (0.1 mg/kg body weight) was injected daily for 5 days. FAK siRNA transfection caused a KD of FAK expression in mouse intestinal tissue (Fig. 7B). As shown previously, LPS caused an increase in mouse intestinal permeability. LPS caused a similar increase in mouse intestinal permeability in mice transfected with scrambled or non-target siRNA (Fig. 7C). In-vivo transfection with FAK siRNA prevented the LPS-induced increase in mouse intestinal permeability (Fig. 7C). These results indicated that FAK was required for the LPS-induced increase in mouse intestinal permeability.
Figure 7.

Involvement of FAK and MyD88 in LPS-induced increase in mouse intestinal permeability. (A). Time-course effect of LPS (0.1 mg/kg body weight) on phospho-FAK in mouse intestinal tissue as assessed by Western blot analysis. Densitometry analysis showed significant time-dependent increase in the expression of phospho-FAK. n=3. (B). FAK siRNA silencing knocked down the expression of FAK in mouse intestinal tissue as assessed by Western blot and densitometry analysis. siFAK transfection was performed 1 d prior to the LPS treatment, and the Western blot analysis was performed after 5 d LPS treatment. n=3. (C). Effect of siRNA-induced silencing of FAK on LPS-induced increase in mouse intestinal permeability. n=4. (D). Effect of LPS on intestinal permeability in MyD88−/− mice. n=4. *, p < 0.001 vs WT control; **, p < 0.001 vs WT LPS treatment.
LPS-induced increase in mouse intestinal permeability is through MyD88-dependent pathway
To determine the requirement of MyD88 in LPS-induced increase in mouse intestinal permeability, the LPS effect was also examined in MyD88 deficient mice (C57BL/6 MyD88−/−, Jackson Laboratory). Intraperitoneal LPS (0.1 mg/Kg body weight) did not cause an increase in intestinal permeability in MyD88−/− mice (Fig. 7D), confirming that the LPS-induced increase in mouse intestinal permeability in-vivo was mediated by the MyD88-dependent pathway.
High dose LPS (1mg/Kg body weight) causes a rapid increase in mouse intestinal permeability and intestinal mucosal damage
Previous studies have shown that high doses of LPS (1 or 5 mg/Kg body weight) cause intestinal mucosal damage and intestinal inflammation (29–33). To examine the effect of high dose LPS on mucosal damage and intestinal permeability, 1 mg/Kg body weight LPS was injected by intraperitoneal route. The high dose LPS administration resulted in a peak mouse serum level of 1.3 μg/ml (21). The intestinal permeability was assessed by in-vivo recycling perfusion at 3 and 24 h post LPS treatment. High dose of LPS caused a significant increase in intestinal 10KD dextran flux as early as 3 h and showed a 8–10 fold increase in 10KD dextran flux by 24 h after LPS injection (Fig. 8A).
Figure 8.

Effect of high dose LPS (1 mg/kg body weight) on mouse small intestine. (A). Effect of high dose LPS (1 mg/Kg body weight) on intestinal permeability in mice. High dose LPS (1 mg/Kg body weight) caused a time-dependent increase in intestinal permeability in wild type mice. (B). Histologic changes in small intestine after high dose LPS treatment in wild-type (WT), TLR-4−/− mice and MyD88−/− mice. H&E stain, original magnification, X200. (C). Gross morphologic changes in small intestine after high dose LPS treatment in WT, TLR-4−/− mice and MyD88−/− mice. (D). Effect of high dose LPS on mice body weights. (E). Effect of high dose LPS on disease activity index in mice. Mice were treated with LPS (1 mg/Kg body weight, i.p.) for 24 h. n=4. *, p<0.001 vs WT control.
Next, we investigated the effect of high dose LPS on intestinal mucosal damage. The intraperitoneal injection of LPS (1 mg/Kg body weight) caused a marked increase in neutrophilic and mononuclear infiltration in the lamina propria by 24 h. The villus mucosal surface showed deranged epithelial nuclei, marked sloughing and denudation of the epithelial cells, loss of Goblet cells, decrease in finger-like villus projections and a marked expansion of villus compartment (Fig. 8B). Corresponding to the histologic inflammation, there were also gross morphologic changes consistent with small intestinal inflammation, including intestinal tissue edema, erythema, and increased vasculature (Fig. 8C). High dose LPS also caused a rapid decrease in body weight compared to the untreated mice (Fig. 8D). We also assessed the effect of high dose LPS on clinical disease activity index as previously described (34). The components of clinical disease activity index included ruffled fur, decrease in movement and avoidance behavior, loose stools, and bloody stools. The high dose LPS resulted in significant increase in disease activity index (Fig. 8E). Together, these findings demonstrated that high dose LPS causes a rapid and severe mucosal damage and an accompanying increase in weight loss and intestinal disease activity.
High dose LPS-induced increase in mouse intestinal permeability and intestinal inflammation is mediated via TLR-4 pathway
To determine the requirement of TLR-4 in high dose LPS-induced increase in mouse intestinal permeability, the effect of high dose LPS (1 mg/Kg body weight) was examined in TLR-4 deficient mice (C57BL/6 TLR-4−/−, Jackson Laboratory). The LPS-induced increase in intestinal permeability was completely prevented in TLR-4−/− mice (Fig. 9A), confirming that the high dose LPS-induced increase in mouse intestinal permeability was also mediated through the TLR-4 signal transduction pathway. High dose LPS did not cause significant decrease in body weight nor increase in clinical disease activity index (Fig. 9B, 9C). Additionally, the histologic and gross morphologic intestinal inflammation were also prevented in TLR-4−/− mice (Fig. 8B, 8C). These data suggested that the LPS-induced increase in intestinal permeability and intestinal inflammation is mediated via TLR-4 signal transduction pathway.
Figure 9.

High dose LPS-induced increase in mouse intestinal permeability and mucosal damage were mediated via TLR-4 pathway. (A). Effect of high dose LPS (1 mg/Kg body weight) on intestinal permeability in TLR-4−/− mice. (B). Effect of high dose LPS on mice body weight change in TLR-4−/− mice. (C). Effect of high dose LPS on disease activity index in TLR-4−/− mice. Mice were treated with LPS (1 mg/Kg body weight, i.p. for 24 h). n=4. *, p < 0.05 vs WT control; **, p < 0.05 vs WT LPS treatment.
High dose LPS induces TLR-4 dependent activation of FAK
In the above studies with Caco-2 monolayers and low dose LPS in-vivo studies, we showed that the LPS-induced increase in intestinal TJ permeability was mediated by TLR-4-dependent activation of FAK and MyD88. In the following studies, the effect of high dose LPS on FAK tyrosine (Y397) phosphorylation in mice intestinal tissue was determined by western blot analysis. As shown in Fig. 10A, there was a time-dependent increase in phospho-FAK following LPS treatment as early as 1 h post LPS treatment, indicating that LPS causes a time-dependent activation of FAK. Similarly, we assessed the expression of FAK tyrosine (Y397) phosphorylation in TLR-4−/− mice treated with high dose LPS. There was no increase in the expression of phosphorylated FAK in TLR-4−/− mice (Fig. 10B), confirming that FAK is also phosphorylated in high dose LPS-treated mice via TLR-4 pathway. The total expression of FAK in LPS-treated WT and TLR-4−/− mice was unchanged compared to the control mice (Fig. 10B). These results suggested that phosphorylation of FAK preceded the LPS-induced increase in mouse intestinal permeability, and was dependent on TLR-4 activation.
Figure 10.
FAK regulates the high dose LPS-induced increase mucosal damage. (A). Time-course effect of high dose LPS (1 mg/kg body weight) on phospho-FAK in mouse intestinal tissue as assessed by Western blot and densitometry analysis. (B) Effect of high dose LPS on phospho-FAK in TLR-4−/− mice as assessed by Western blot and densitometry analysis. n=4. *, p < 0.05 vs WT control; **, p < 0.05 vs WT LPS treatment.
High dose LPS-induced increase in mouse intestinal permeability and intestinal inflammation is mediated via TLR-4-MyD88-IRAK4 axis
Above studies suggested that the LPS-induced activation of MyD88 was dependent on TLR-4/FAK activation. To assess the role of MyD88 in high dose LPS-induced increase in mouse intestinal permeability and intestinal inflammation, the effect of high dose LPS was examined in MyD88 deficient mice (C57BL/6 MyD88−/−, Jackson Laboratory). As shown in our in-vitro studies, LPS caused a time-dependent increase in IRAK4 phosphorylation (Fig. 3D). High dose LPS (1 mg/Kg body weight) caused a significant increase in small intestine IRAK4 phosphorylation in WT mice; however, IRAK4 phosphorylation was completely inhibited in MyD88−/− mice (Figs. 11A), confirming that MyD88 is required for IRAK4 phosphorylation. High dose LPS also did not cause an increase in intestinal permeability or intestinal inflammation in MyD88−/− mice (Fig. 11). High dose LPS did not cause histologic or gross morphologic intestinal inflammation in MyD88−/− mice (Fig. 8B, 8C). The LPS-induced decrease in body weight and increase in disease activity index were also prevented in MyD88−/− mice (Fig. 11C, 11D). Lastly, LPS-induced increase in IRAK4 phosphorylation was prevented in TLR-4−/− mice (Fig. 11E). Together, these findings suggested that the high dose LPS effect on mouse intestinal permeability and intestinal inflammation was regulated by TLR-4 dependent activation of FAK/MyD88/IRAK4 axis.
Figure 11.

High dose LPS-induced increase in mouse intestinal permeability and mucosal damage were mediated via MyD88-dependent pathway. (A). Effect of high dose LPS on phospho-IRAK4 in MyD88−/− mice as assessed by Western blot and densitometry analysis. Mice were treated with LPS (1 mg/Kg body weight, i.p. for 24 h). (B). Effect of high dose LPS (1 mg/Kg body weight) on intestinal permeability in MyD88−/− mice. (C). Effect of high dose LPS on mice body weight change in MyD88−/− mice. (D). Effect of high dose LPS on disease activity index in MyD88−/− mice. (E). Effect of high dose LPS on phospho-IRAK4 in TLR-4−/− mice as assessed by Western blot and densitometry analysis. n=4. *, p < 0.05 vs WT control; **, p < 0.05 vs WT LPS treatment.
Discussion
Defective intestinal epithelial TJ barrier is an important pathogenic factor contributing to the development of intestinal inflammation and systemic inflammatory responses by allowing increased intestinal permeation and systemic circulation of gut-derived bacterial antigens (1, 2). Plasma LPS levels are markedly elevated in IBD and NEC and play an important role in the inflammatory process by activating the immune response. Previous studies in various experimental model systems have shown that LPS challenge with high pharmacologic doses (50 μg/ml) impairs the integrity of the intestinal barrier and leads to rapid death in cell and animal models (19, 35). Previous study from our laboratory suggested that LPS, at low, clinically relevant concentrations (0.1–1 ng/ml), causes a selective increase in intestinal TJ permeability by inducing enterocyte membrane expression and localization of TLR-4 without causing cell damage or cell death (21). The increase in TLR-4 expression also caused an increase in expression and membrane recruitment of CD14 and TLR-4 signal transduction pathway dependent increase in intestinal TJ permeability (21). Herein, we extend on our previous findings to delineate the adaptor proteins that mediate the LPS modulation of intestinal TJ barrier. Additionally, we extend our studies to demonstrate the clinical relevance of adaptor protein activation in mediating high dose LPS-induced intestinal inflammation and mucosal damage. Our results suggest that the LPS-induced increase in intestinal TJ permeability and intestinal inflammation is regulated by TLR-4 receptor complex activation of adaptor protein FAK and FAK activation of MyD88/IRAK4 but not TRAM/TRIF pathway
The LPS stimulation of mammalian cells occurs through series of interactions with several membrane associated proteins including the LPS binding protein (LBP), CD14, MD2 and TLR-4. LBP is a soluble shuttle protein which directly binds to LPS and facilitates the association between LPS and CD14 (36). CD14 facilitates the transfer of LPS to the TLR-4/MD2 receptor complex and modulates LPS recognition (37). MD2 is a soluble protein that non-covalently associates with TLR-4 but can directly form a complex with LPS in the absence TLR-4 (38). Upon LPS recognition, TLR-4 undergoes oligomerization and recruits its downstream adaptor proteins through interactions with the TIR (Toll-interleukin-1 receptor) domains. There are five TIR domain-containing adaptor proteins: MyD88, TIRAP, TRIF, TRAM, and SARM (sterile α and HEAT-Armadillo motifs-containing protein) (39). TLR-4 signaling has been divided into MyD88-dependent and MyD88-independent or TRIF-dependent pathways. MyD88-dependent pathway includes adaptor proteins TIRAP and MyD88, and MyD88-independent pathway includes adaptor proteins TRAM and TRIF (22). In MyD88-dependent pathway, TIRAP functions as a bridging adaptor for MyD88, and recruits MyD88 to the cytoplasmic domain of TLR-4. TIRAP contains a putative TRAF6-binding motif within its TIR domain and a C-terminal phosphatidylinositol 4,5-bisphosphate (PIP2)-binding domain distinct from its TIR domain which serves to localize it to the plasma membrane. The tyrosine phosphorylation (Tyr-86, Tyr-106, and Tyr-187) causes a conformational change in the TIR domain of TIRAP, and regulates its interaction with other proteins. After recruitment, MyD88 directly interacts with IRAK4 and IRAK1 via homophilic death domain (DD) interactions (22). In TRIF-dependent pathway, TRAM is utilized solely by TLR-4. It functions as a bridging adaptor for the MyD88-independent signaling pathway, serving to recruit TRIF to TLR-4 (22). In our studies, the targeted KD of MyD88 prevented the LPS-induced increase in Caco-2 TJ permeability, indicating the requirement of MyD88 signal transduction pathway in mediating the LPS-induced increase in Caco-2 TJ permeability. The possibility of TRAM and TRIF involvement was also considered, but KD of TRAM or TRIF did not affect the LPS-induced increase in intestinal TJ permeability, suggesting that the LPS effect was regulated specifically by the MyD88-dependent pathway.
FAK, a nonreceptor protein tyrosine kinase involved in signaling downstream of integrins, was recently shown to trigger inflammatory responses. FAK expression and signaling have been shown to play significant role in the regulation of cell adhesion, apoptosis, migration, and proliferation in variety of cell types, both under basal conditions and during inflammatory stress (40); however the role of FAK on TJ barrier regulation remains unknown. FAK is an adaptor protein that appears to be involved in TLR-4 signal transduction process to initiate proinflammatory response. It has been shown that TLR-4 signaling leads to an increase in serine phosphorylation of intestinal FAK in NEC mice model (41) and FAK appeared to be involved in NF-κB phosphorylation and transcriptional activity in endothelial cells (42). How FAK is involved in the TLR-4 signaling pathway and TJ barrier regulation remains unclear. Herein, we show for the first time that FAK plays a central role in the TLR-4 signal transduction process and in TJ barrier regulation. We also show for the first time that FAK is a direct regulator of MyD88 and its downstream effects. Our co-immunoprecipitation experiments showed that FAK and MyD88 were associated with enterocyte TLR-4. The regulatory role of FAK in LPS modulation of Caco-2 TJ barrier was confirmed by FAK inhibition studies, which showed that FAK inhibition or silencing prevented the LPS-induced MyD88 activation and increase in TJ permeability. Together, these studies suggested that LPS binding to TLR-4 caused the activation of FAK; which, in turn, caused the activation of MyD88 and IRAK4. Thus, FAK appears to play a crucial role in mediating the TLR-4 receptor activation of MyD88 pathway and increase in TJ permeability.
In addition to the TIR domain, MyD88 also contains the death domain (DD), which can recruit other DD containing molecules through homotypic interactions (22). Upon LPS stimulation, MyD88 recruits and activates the DD-containing kinase, IL-1 receptor-associated kinase 4 (IRAK4) (22). IRAK4 belongs to the IRAK family, and contains both the DD and the kinase domain. Previous studies have suggested that IRAK4 kinase activity is important for transmitting TLR signals, including the induction of proinflammatory cytokines. IRAK4 knockout macrophages show severely impaired production of proinflammatory cytokines upon LPS stimulation. IRAK4 KO mice are also resistant to LPS-induced septic shock (43). LC–MS/MS analysis has identified phosphorylations at Thr342, Thr345, and Ser346 of IRAK4, which reside within the activation loop. Among three single mutations, T345A showed the most significant decrease in activity, ~70%, whereas T342A and S346A showed moderate reductions, 57% and 50%, respectively. These results indicated that phosphorylation at each of the three sites contributes to IRAK4 kinase activity with Thr345 being the dominant site (44). Our data showed that LPS treatment induces a time-dependent increase in IRAK4 phosphorylation at Thr345 and inhibition of IRAK4 phosphorylation and activity prevented the LPS-induced increase in Caco-2 TJ permeability. The LPS-induced activation of intestinal epithelial IRAK4 required up-stream activation of TLR-4, FAK and MyD88. Thus, our data suggested that IRAK4 acts as a downstream target of TLR-4-FAK-MyD88 signal transduction axis and that IRAK4 phosphorylation signals the TJ barrier opening.
In this study, we also investigated the in-vivo relevance of FAK and MyD88 in the regulation of mouse intestinal permeability. The in-vivo studies indicated that LPS at physiologic serum concentrations (0.1–1 ng/ml) causes the activation of FAK in mouse enterocytes; and targeted KD of mouse enterocyte FAK by in-vivo siRNA transfection prevented the LPS-induced increase in mouse intestinal permeability. The LPS-induced increase in intestinal permeability was also inhibited in MyD88−/− mice, confirming the requirement of MyD88 in LPS-induced increase in mouse intestinal permeability.
We also studied the effects of high dose LPS (1 mg/Kg body weight, i.p.) on small intestinal inflammation and the mechanistic involvement of the TLR-4/FAK/MyD88 axis in the pathogenesis of increase in intestinal permeability and intestinal inflammation. High dose LPS (1 mg/Kg body weight) administration caused an immediate increase in small intestinal permeability (within 3 h); which was followed by a rapid development of intestinal mucosal damage and inflammation (by 24 h), weight loss, and increase in clinical disease activity (Fig. 8). Our data also indicated that the high dose LPS-induced small intestinal inflammation was preceded by FAK and IRAK4 phosphorylation and increase in intestinal permeability.
Additionally, our results indicated that, in TLR-4−/− and MyD88−/− mice, high dose LPS-induced increase in FAK and IRAK4 phosphorylation are inhibited and that the increase in intestinal permeability is prevented. Furthermore, the high dose LPS-induced development of intestinal inflammation and increase in clinical disease activity were completely prevented in TLR-4−/− and MyD88−/− mice. Thus, our findings demonstrated that the TLR-4/FAK/MyD88 axis also plays a crucial role in mediating high dose LPS-induced intestinal mucosal damage. Thus, our results suggested that TLR-4/FAK/MyD88 signal transduction axis plays an important role in regulating both LPS-induced increase in intestinal permeability and intestinal inflammation.
In conclusion, our results provide important novel insight into the intracellular processes and signal transduction pathway that mediate LPS-induced increase in intestinal TJ permeability and intestinal inflammation. Our studies show that LPS, at physiologically relevant concentrations causes an activation of the TLR-4 signal transduction cascade leading to the phosphorylation and activation of the intestinal epithelial cell FAK; the activated enterocyte FAK regulates the activation of MyD88 and IRAK4, which transmits the signal that culminates in the opening of the intestinal TJ barrier. Additionally, our studies also show that high, pharmacologic doses of LPS cause a rapid increase in intestinal permeability and intestinal mucosal damage; and that the LPS-induced mucosal damage is also regulated in part by TLR-4/FAK/MyD88 signal transduction axis. Our studies provide proof-of-concept data showing that TLR-4, FAK and MyD88 can be targeted in-vivo to preserve the intestinal TJ barrier function and to prevent intestinal mucosal damage.
Supplementary Material
Acknowledgments
Grant support: This research project was supported by a Veterans Affairs (VA) Merit Review grant from the VA Research Service and National Institute of Diabetes and Digestive and Kidney Diseases Grant RO 1-DK-64165–01 (to T. Y. Ma).
Abbreviations
- CD14
cluster of differentiation 14
- Cont
control
- EDTA
ethylenediaminetetraacetic acid
- FAK
focal adhesion kinase
- IBD
inflammatory bowel disease
- IRAK4
interleukin-1 receptor associated kinase 4
- LPS
lipopolysaccharides
- MD2
myeloid differentiation protein 2
- MyD88
myeloid differentiation primary response gene 88
- NEC
necrotizing enterocolitis
- PBS
phosphate buffered saline
- Scr
scramble
- SDS-PAGE
sodium dodecyl sulfate polyacrylamide gel electrophoresis
- TER
transepithelial electrical resistance
- TJ
tight junction
- TLR
toll-like receptor
- TRAM
TRIF-related adapter molecule
- TRIF
TIR domain-containing adaptor inducing IFN-β
- WT
wild type
Footnotes
Author contributions: Shuhong Guo designed and conducted experiments and wrote the manuscript. Meghali Nighot conducted animal experiments. Rana Al-Sadi conducted western blot, siRNA transfection and Caco-2 inulin flux studies. Tarik Alhmoud conducted western blot. Prashant Nighot conducted animal experiments. Thomas Y. Ma evaluated the results, supervised this study and wrote the manuscript.
Disclosures: This is no conflict of interest for all authors.
References
- 1.Turner JR. Intestinal mucosal barrier function in health and disease. Nat Rev Immunol. 2009;9:799–809. doi: 10.1038/nri2653. [DOI] [PubMed] [Google Scholar]
- 2.Ma TY, Anderson JM. Physiology of the Gastrointestinal Tract. Elsevier Academic Press; Burlington, MA: 2006. Tight junctions and the intestinal barrier. [Google Scholar]
- 3.Berman L, Moss RL. Necrotizing enterocolitis: an update. Semin Fetal Neonatal Med. 2011;16:145–150. doi: 10.1016/j.siny.2011.02.002. [DOI] [PubMed] [Google Scholar]
- 4.Clark JA, Doelle SM, Halpern MD, Saunders TA, Holubec H, Dvorak K, Boitano SA, Dvorak B. Intestinal barrier failure during experimental necrotizing enterocolitis: protective effect of EGF treatment. Am J Physiol Gastrointest Liver Physiol. 2006;291:G938–949. doi: 10.1152/ajpgi.00090.2006. [DOI] [PubMed] [Google Scholar]
- 5.Oriishi T, Sata M, Toyonaga A, Sasaki E, Tanikawa K. Evaluation of intestinal permeability in patients with inflammatory bowel disease using lactulose and measuring antibodies to lipid A. Gut. 1995;36:891–896. doi: 10.1136/gut.36.6.891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Munkholm P, Langholz E, Hollander D, Thornberg K, Orholm M, Katz KD, Binder V. Intestinal permeability in patients with Crohn’s disease and ulcerative colitis and their first degree relatives. Gut. 1994;35:68–72. doi: 10.1136/gut.35.1.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Arrieta MC, Madsen K, Doyle J, Meddings J. Reducing small intestinal permeability attenuates colitis in the IL10 gene-deficient mouse. Gut. 2009;58:41–48. doi: 10.1136/gut.2008.150888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mennigen R, Nolte K, Rijcken E, Utech M, Loeffler B, Senninger N, Bruewer M. Probiotic mixture VSL#3 protects the epithelial barrier by maintaining tight junction protein expression and preventing apoptosis in a murine model of colitis. Am J Physiol Gastrointest Liver Physiol. 2009;296:G1140–1149. doi: 10.1152/ajpgi.90534.2008. [DOI] [PubMed] [Google Scholar]
- 9.Benoit R, Rowe S, Watkins SC, Boyle P, Garrett M, Alber S, Wiener J, Rowe MI, Ford HR. Pure endotoxin does not pass across the intestinal epithelium in vitro. Shock. 1998;10:43–48. doi: 10.1097/00024382-199807000-00008. [DOI] [PubMed] [Google Scholar]
- 10.Ge Y, Ezzell RM, Warren HS. Localization of endotoxin in the rat intestinal epithelium. J Infect Dis. 2000;182:873–881. doi: 10.1086/315784. [DOI] [PubMed] [Google Scholar]
- 11.Hurley JC. Endotoxemia: methods of detection and clinical correlates. Clin Microbiol Rev. 1995;8:268–292. doi: 10.1128/cmr.8.2.268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Andreasen AS, Krabbe KS, Krogh-Madsen R, Taudorf S, Pedersen BK, Moller K. Human endotoxemia as a model of systemic inflammation. Curr Med Chem. 2008;15:1697–1705. doi: 10.2174/092986708784872393. [DOI] [PubMed] [Google Scholar]
- 13.Marshall JC, Walker PM, Foster DM, Harris D, Ribeiro M, Paice J, Romaschin AD, Derzko AN. Measurement of endotoxin activity in critically ill patients using whole blood neutrophil dependent chemiluminescence. Crit Care. 2002;6:342–348. doi: 10.1186/cc1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wellmann W, Fink PC, Benner F, Schmidt FW. Endotoxaemia in active Crohn’s disease. Treatment with whole gut irrigation and 5-aminosalicylic acid. Gut. 1986;27:814–820. doi: 10.1136/gut.27.7.814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sharma R, Tepas JJ, 3rd, Hudak ML, Mollitt DL, Wludyka PS, Teng RJ, Premachandra BR. Neonatal gut barrier and multiple organ failure: role of endotoxin and proinflammatory cytokines in sepsis and necrotizing enterocolitis. J Pediatr Surg. 2007;42:454–461. doi: 10.1016/j.jpedsurg.2006.10.038. [DOI] [PubMed] [Google Scholar]
- 16.Leal RF, Milanski M, de Ayrizono ML, Coope A, Rodrigues VS, Portovedo M, Oliveira LM, Fagundes JJ, Coy CS, Velloso LA. Toll-like receptor 4, F4/80 and pro-inflammatory cytokines in intestinal and mesenteric fat tissue of Crohn’s disease. Int J Clin Exp Med. 2013;6:98–104. [PMC free article] [PubMed] [Google Scholar]
- 17.Le Mandat Schultz A, Bonnard A, Barreau F, Aigrain Y, Pierre-Louis C, Berrebi D, Peuchmaur M. Expression of TLR-2, TLR-4, NOD2 and pNF-kappaB in a neonatal rat model of necrotizing enterocolitis. PLoS One. 2007;2:e1102. doi: 10.1371/journal.pone.0001102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.De Jager PL, Franchimont D, Waliszewska A, Bitton A, Cohen A, Langelier D, Belaiche J, Vermeire S, Farwell L, Goris A, Libioulle C, Jani N, Dassopoulos T, Bromfield GP, Dubois B, Cho JH, Brant SR, Duerr RH, Yang H, Rotter JI, Silverberg MS, Steinhart AH, Daly MJ, Podolsky DK, Louis E, Hafler DA, Rioux JD. The role of the Toll receptor pathway in susceptibility to inflammatory bowel diseases. Genes Immun. 2007;8:387–397. doi: 10.1038/sj.gene.6364398. [DOI] [PubMed] [Google Scholar]
- 19.Yu LC, Flynn AN, Turner JR, Buret AG. SGLT-1-mediated glucose uptake protects intestinal epithelial cells against LPS-induced apoptosis and barrier defects: a novel cellular rescue mechanism? FASEB J. 2005;19:1822–1835. doi: 10.1096/fj.05-4226com. [DOI] [PubMed] [Google Scholar]
- 20.Hirotani Y, Ikeda K, Kato R, Myotoku M, Umeda T, Ijiri Y, Tanaka K. Protective effects of lactoferrin against intestinal mucosal damage induced by lipopolysaccharide in human intestinal Caco-2 cells. Yakugaku Zasshi. 2008;128:1363–1368. doi: 10.1248/yakushi.128.1363. [DOI] [PubMed] [Google Scholar]
- 21.Guo S, Al-Sadi R, Said HM, Ma TY. Lipopolysaccharide causes an increase in intestinal tight junction permeability in vitro and in vivo by inducing enterocyte membrane expression and localization of TLR-4 and CD14. Am J Pathol. 2013;182:375–387. doi: 10.1016/j.ajpath.2012.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lu YC, Yeh WC, Ohashi PS. LPS/TLR4 signal transduction pathway. Cytokine. 2008;42:145–151. doi: 10.1016/j.cyto.2008.01.006. [DOI] [PubMed] [Google Scholar]
- 23.Xu B, Song G, Ju Y. Effect of focal adhesion kinase on the regulation of realignment and tenogenic differentiation of human mesenchymal stem cells by mechanical stretch. Connect Tissue Res. 2011;52:373–379. doi: 10.3109/03008207.2010.541961. [DOI] [PubMed] [Google Scholar]
- 24.Zeisel MB, V, Druet A, Sibilia J, Klein JP, Quesniaux V, Wachsmann D. Cross talk between MyD88 and focal adhesion kinase pathways. J Immunol. 2005;174:7393–7397. doi: 10.4049/jimmunol.174.11.7393. [DOI] [PubMed] [Google Scholar]
- 25.Ye D, Guo S, Al-Sadi R, Ma TY. MicroRNA Regulation of Intestinal Epithelial Tight Junction Permeability. Gastroenterology. 2011 doi: 10.1053/j.gastro.2011.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Al-Sadi R, Khatib K, Guo S, Ye D, Youssef M, Ma T. Occludin regulates macromolecule flux across the intestinal epithelial tight junction barrier. Am J Physiol Gastrointest Liver Physiol. 2011;300:G1054–1064. doi: 10.1152/ajpgi.00055.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen HC, Appeddu PA, Isoda H, Guan JL. Phosphorylation of tyrosine 397 in focal adhesion kinase is required for binding phosphatidylinositol 3-kinase. J Biol Chem. 1996;271:26329–26334. doi: 10.1074/jbc.271.42.26329. [DOI] [PubMed] [Google Scholar]
- 28.Etienne S, Adamson P, Greenwood J, Strosberg AD, Cazaubon S, Couraud PO. ICAM-1 signaling pathways associated with Rho activation in microvascular brain endothelial cells. J Immunol. 1998;161:5755–5761. [PubMed] [Google Scholar]
- 29.Liu C, Li A, Weng YB, Duan ML, Wang BE, Zhang SW. Changes in intestinal mucosal immune barrier in rats with endotoxemia. World J Gastroenterol. 2009;15:5843–5850. doi: 10.3748/wjg.15.5843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Huber NL, Bailey SR, Schuster RM, Ogle CK, Lentsch AB, Pritts TA. Remote thermal injury increases LPS-induced intestinal IL-6 production. J Surg Res. 2010;160:190–195. doi: 10.1016/j.jss.2009.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tomita M, Ohkubo R, Hayashi M. Lipopolysaccharide transport system across colonic epithelial cells in normal and infective rat. Drug Metab Pharmacokinet. 2004;19:33–40. doi: 10.2133/dmpk.19.33. [DOI] [PubMed] [Google Scholar]
- 32.Buchholz BM, Chanthaphavong RS, Bauer AJ. Nonhemopoietic cell TLR4 signaling is critical in causing early lipopolysaccharide-induced ileus. J Immunol. 2009;183:6744–6753. doi: 10.4049/jimmunol.0901620. [DOI] [PubMed] [Google Scholar]
- 33.Aoshiba K, Onizawa S, Tsuji T, Nagai A. Therapeutic effects of erythropoietin in murine models of endotoxin shock. Crit Care Med. 2009;37:889–898. doi: 10.1097/CCM.0b013e31819b8371. [DOI] [PubMed] [Google Scholar]
- 34.Wirtz S, Neufert C, Weigmann B, Neurath MF. Chemically induced mouse models of intestinal inflammation. Nat Protoc. 2007;2:541–546. doi: 10.1038/nprot.2007.41. [DOI] [PubMed] [Google Scholar]
- 35.Sukhotnik I, Mogilner J, Krausz MM, Lurie M, Hirsh M, Coran AG, Shiloni E. Oral arginine reduces gut mucosal injury caused by lipopolysaccharide endotoxemia in rat. J Surg Res. 2004;122:256–262. doi: 10.1016/j.jss.2004.07.004. [DOI] [PubMed] [Google Scholar]
- 36.Wright SD, Tobias PS, Ulevitch RJ, Ramos RA. Lipopolysaccharide (LPS) binding protein opsonizes LPS-bearing particles for recognition by a novel receptor on macrophages. J Exp Med. 1989;170:1231–1241. doi: 10.1084/jem.170.4.1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wright SD, Ramos RA, Tobias PS, Ulevitch RJ, Mathison JC. CD14, a receptor for complexes of lipopolysaccharide (LPS) and LPS binding protein. Science. 1990;249:1431–1433. doi: 10.1126/science.1698311. [DOI] [PubMed] [Google Scholar]
- 38.Shimazu R, Akashi S, Ogata H, Nagai Y, Fukudome K, Miyake K, Kimoto M. MD-2, a molecule that confers lipopolysaccharide responsiveness on Toll-like receptor 4. J Exp Med. 1999;189:1777–1782. doi: 10.1084/jem.189.11.1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.O’Neill LA, Bowie AG. The family of five: TIR-domain-containing adaptors in Toll-like receptor signalling. Nat Rev Immunol. 2007;7:353–364. doi: 10.1038/nri2079. [DOI] [PubMed] [Google Scholar]
- 40.Parsons JT. Focal adhesion kinase: the first ten years. J Cell Sci. 2003;116:1409–1416. doi: 10.1242/jcs.00373. [DOI] [PubMed] [Google Scholar]
- 41.Leaphart CL, Cavallo J, Gribar SC, Cetin S, Li J, Branca MF, Dubowski TD, Sodhi CP, Hackam DJ. A critical role for TLR4 in the pathogenesis of necrotizing enterocolitis by modulating intestinal injury and repair. J Immunol. 2007;179:4808–4820. doi: 10.4049/jimmunol.179.7.4808. [DOI] [PubMed] [Google Scholar]
- 42.Petzold T, Orr AW, Hahn C, Jhaveri KA, Parsons JT, Schwartz MA. Focal adhesion kinase modulates activation of NF-kappaB by flow in endothelial cells. Am J Physiol Cell Physiol. 2009;297:C814–822. doi: 10.1152/ajpcell.00226.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Suzuki N, Suzuki S, Duncan GS, Millar DG, Wada T, Mirtsos C, Takada H, Wakeham A, Itie A, Li S, Penninger JM, Wesche H, Ohashi PS, Mak TW, Yeh WC. Severe impairment of interleukin-1 and Toll-like receptor signalling in mice lacking IRAK-4. Nature. 2002;416:750–756. doi: 10.1038/nature736. [DOI] [PubMed] [Google Scholar]
- 44.Cheng H, Addona T, Keshishian H, Dahlstrand E, Lu C, Dorsch M, Li Z, Wang A, Ocain TD, Li P, Parsons TF, Jaffee B, Xu Y. Regulation of IRAK-4 kinase activity via autophosphorylation within its activation loop. Biochem Biophys Res Commun. 2007;352:609–616. doi: 10.1016/j.bbrc.2006.11.068. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.