

A metagenomics analysis of Arctic freshwater viral DNA communities uncovers unique genetics and bipolar lineages.
Keywords: Virus, metagenomics, Arctic, environmental microbiology, biodiversity, freshwater lake
Abstract
Viruses constitute the most abundant biological entities and a large reservoir of genetic diversity on Earth. Despite the recent surge in their study, our knowledge on their actual biodiversity and distribution remains sparse. We report the first metagenomic analysis of Arctic freshwater viral DNA communities and a comparative analysis with other freshwater environments. Arctic viromes are dominated by unknown and single-stranded DNA viruses with no close relatives in the database. These unique viral DNA communities mostly relate to each other and present some minor genetic overlap with other environments studied, including an Arctic Ocean virome. Despite common environmental conditions in polar ecosystems, the Arctic and Antarctic DNA viromes differ at the fine-grain genetic level while sharing a similar taxonomic composition. The study uncovers some viral lineages with a bipolar distribution, suggesting a global dispersal capacity for viruses, and seemingly indicates that viruses do not follow the latitudinal diversity gradient known for macroorganisms. Our study sheds light into the global biogeography and connectivity of viral communities.
INTRODUCTION
Viruses constitute the most abundant biological entities and a large reservoir of genetic diversity on Earth (1, 2). They control microbial abundance and community structure (3), and microbial genetic diversity and evolution are shaped by virus-mediated gene transfer and host range (4, 5). In addition, viruses exert a profound effect on food web interactions and affect global geochemical cycles (6–9). However, our current knowledge on viruses in nature is scarce (4, 10).
High-latitude freshwater ecosystems represent few of the last pristine habitats on the planet (11). They are mainly oligotrophic environments, dominated by microorganisms. As predation pressure diminishes with latitude, viral composition might be acting as the primary factor regulating the extreme polar environments (12). Consistent with this, Antarctic lakes display a high rate of visibly phage-infected bacteria (13). High-latitude freshwater habitats constitute a unique ecological model to understand the influence of viruses on natural microbial communities and the overall ecosystem. However, no large-scale analysis has yet assessed the diversity and composition of the Arctic freshwater virome.
An Antarctic freshwater lake was shown to host a diverse viral DNA community, unexpected for such an extreme and high-latitude ecosystem, and to be dominated by viruses belonging to unknown families related to single-stranded DNA (ssDNA) viruses (14). This study raised important questions in the field, and the untested hypothesis of whether the polar regions are biodiversity and evolutionary hot spots was suggested (15). It remains unknown whether the extreme Arctic freshwater environments also host a similar and diverse viral DNA community, and whether polar freshwater and polar oceans share a common diversity and community composition. Arctic and Antarctic freshwater environments share common features: extreme annual cycles of temperature, sunlight, and ice phenology. Nevertheless, these environments are separated by extreme distance and other physical barriers. Hence, a parallel study of these environments would provide valuable information about viral biogeography and connectivity worldwide (16).
We describe the first metagenomic analysis of the Arctic freshwater DNA virome from six large water bodies. Additionally, we provide in-depth sequence data from the Antarctic virome and a comprehensive comparison of the polar viromes and other known freshwater viromes. Our results show that the Arctic DNA virome is composed of unique viral lineages not found in the Antarctic virome and some bipolar lineages. Arctic and Antarctic freshwater viromes are related at the taxonomic level and differ from other studied regions. Viral diversity analysis across viromes indicates that viruses do not follow a latitudinal diversity gradient.
RESULTS
Samples from six water bodies in Spitsbergen (78°N, Svalbard, Norway) were collected in three different years (Fig. 1). DNA viral genomes were extracted from purified virus particles and shotgun-sequenced using Illumina technology, with the exception of the viral DNA from Lv1Pond, which was sequenced using Roche 454 technology. For comparative purposes, the previously reported Antarctic Limnopolar Lake summer viral community DNA was resequenced with Illumina technology.
Fig. 1. Diagram depicting the global location of the freshwater environments studied.

A detailed position of the Arctic lakes in Spitsbergen and photographs of the lakes at the time of sampling are shown. Coordinates (latitude/longitude) of the Arctic lakes: Lv1 (Lake Linnevatnet) (78°03.864′N; 13°46.308′E); Lv1Pond (Borgdammane) (78°04.254′N; 13°47.652′E); IR1 (Lake Tunsjøen) (78°03.375′N; 13°40.313′E); IR2 (78°02.935′N; 13°41973′E); SvL1 (Lake Nordammen) (78°38.279′N; 16°44.025′E); SvL2 (Lake Tenndammen) (78°06.118′N; 15°02.024′E). [Svalbard map was obtained from http://es.wikipedia.org/wiki/Svalbard#mediaviewer/Archivo:Topographic_map_of_Svalbard.svg and published under terms of the GNU Free Documentation License (http://commons.wikimedia.org/wiki/Commons:GNU_Free_Documentation_License_1.2).]
The Arctic virome is dominated by unknown and small ssDNA viruses
On average, blastx searches against known viral genomes could only assign 9.8% of the reads to a previously known taxonomic unit (Fig. 2). The vast majority of these were classified as ssDNA viruses (86%), with an important fraction of Circoviridae (38.1%), followed by dsDNA (double-stranded DNA) viruses (2.8%), mainly composed of Caudovirales (1.8%). Unexpectedly, a fraction of the assigned reads of these DNA viromes were assigned to ssRNA viruses. Comparisons of the taxonomic profiles based on classified reads from the Arctic viromes showed that, overall, they harbored members of the same viral families. However, it is noteworthy that the amount of classified reads was much lower for sample Lv1 than for the other samples (2.5% versus 7.8 to 15.5%) and that the profiles of samples Lv1 and SvL2 separated from the rest mainly because of increased abundances of Circoviridae- and Nanoviridae-classified reads and a lower proportion of Microviridae. The Phi29 polymerase has been shown to preferentially amplify circular ssDNA (17). However, on the basis of the percentage of assigned reads to each of the most abundant viral groups (ssDNA viruses, Siphoviridae, Podoviridae, and Myoviridae) and accounting for the average differences in genome sizes, the relative abundance of ssDNA viruses in the Arctic viromes, assuming the reported 100 × (17) positive amplification bias, was 88.6 ± 8.3%. Therefore, ssDNA viruses were abundant in the Arctic viromes even considering a Phi29 polymerase bias.
Fig. 2. Taxonomic distribution of assigned metagenomic reads from Arctic viromes.
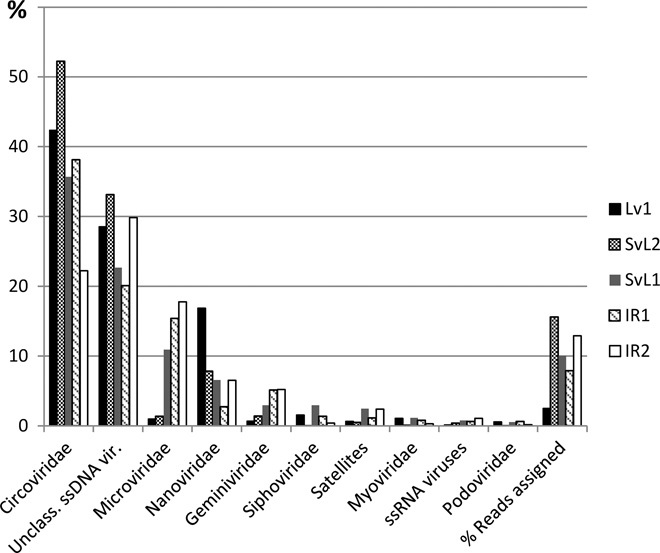
Results represent blastx hits against a viral genome database summarized by MEGAN (Meta Genome Analyzer). Only the 10 most abundant categories are shown.
Most polar contigs do not bear high similarity to known viral genomes
Assembly of polar reads, including those with no similarity in databases, into larger sequences produced an average of 15,541 contigs per sample [5175 at least 1000 base pairs (bp) in length], accounting for a total of over 97 Mbp, including contigs as large as 114,603 bp (Table 1). blastx searches revealed that for all Arctic viromes, contigs with the highest abundance were likely ssDNA viruses (fig. S1A). We cross-aligned the contigs obtained to all known viral genomes. No similarities greater than 65% were found for IR1 (Isfjord Radio 1), IR2, and Lv1. Both SvL1 and SvL2 produced contigs (SvL1-4380 and SvL2-1027) with resemblance to Sclerotinia sclerotiorum hypovirulence–associated DNA virus 1 (507-bp overlap, 79% similarity; and 975-bp overlap, 89% similarity, respectively), a fungus virus related to the Geminiviridae. Also, the resequenced Antarctic virome produced a contig (Ant-0) similar to Bathycoccus sp. RCC1105 virus BpV1 (500 bp, 76%), a phycodnavirus, and another contig (Ant-53) with regions similar to Cafeteria roenbergensis virus BV-PW1 (1439 bp, 87%) and Phaeocystis globosa virus (3024 bp, 93%), members of the Mimiviridae and Phycodnaviridae, respectively. Taxonomic affiliation of the contigs using the METAVIR pipeline confirmed the prevalence of ssDNA viruses and the presence of sequences related to ssRNA viruses within the Arctic viromes. A closer inspection of these sequences revealed that they belonged to small circular contigs composed of replication proteins most similar to Circoviridae-Geminiviridae-Nanoviridae genes (and, in a few cases, Phycodnaviridae) and coat proteins most similar to the ssRNA virus Sclerophthora macrospora virus A.
Table 1. Summary of polar metagenomic assemblies.
Lengths in bp.
|
No. of contigs |
Sum (bp) |
Maximum size |
Average size |
N50 |
No. of contigs ≥1000 |
|
| SvL1 | 22,475 | 23,071,732 | 45,109 | 1,026 | 1,383 | 6,926 |
| IR1 | 13,180 | 14,262,844 | 43,704 | 1,082 | 1,433 | 4,661 |
| IR2 | 25,496 | 28,649,721 | 10,058 | 1,123 | 1,579 | 9,111 |
| SvL2 | 14,637 | 14,096,695 | 6,102 | 963 | 1,300 | 5,259 |
| Lv1 | 12,624 | 12,193,408 | 30,869 | 965 | 1,117 | 3,801 |
| Antarctic | 4,834 | 5,452,144 | 114,603 | 1,127 | 1,528 | 1,294 |
The Arctic freshwater viral communities share genes and genomes
A stringent mapping analysis [minimum 98% similarity, shown to differentiate between closely related phages (18)] of reads to contigs assessing the extent of genetic content overlap between communities (fig. S2) showed very little fine-grain genetic overlap between Arctic and Antarctic viral communities. Lv1 and SvL2 shared the most genetic information. IR1 and IR2 were also genetically very similar to each other, and overall, SvL1 shared the least genetic information with the other Arctic viromes.
Next, we studied to which degree these Arctic freshwater environments might share common species (genomes) and/or highly similar genomic regions, analyzing cross-contig alignments with 98% similarity and 500-bp length thresholds. IR1 and IR2 shared the greatest number of high-similarity genomic regions (table S1), whereas Lv1 and SvL1 shared relatively few of these regions when compared with the rest of the pairwise comparisons. A more detailed analysis detected three groups of highly similar circular contigs (indicative of completion of a genomic element) present in four of the five Arctic communities (fig. S1B). These sets had intra-group similarities >97% and lengths ranging from 1120 to 1710 bp, presenting two to three open reading frames (ORFs). As for the most abundant circular contigs in the Arctic viromes (fig. S1A), blastx searches showed significant similarities for some of these ORFs to known replication proteins of ssDNA viruses, in most cases related to uncultured marine viruses.
The Arctic freshwater virome differs from other environments at fine scale
Analysis of blastx-derived taxonomic profiles showed a strong partitioning of the viromes between polar and nonpolar viromes, mainly driven by the relative abundances of small circular ssDNA viruses and phages (Fig. 3). This clustering includes the Lv1Pond and, significantly, the Antarctic spring virome. On the contrary, Antarctic summer virome segregates from the rest mainly because of increased proportions of Phycodnaviridae-related sequences (table S2). Another feature is that the Arctic Ocean community separates from the Arctic viromes. The Arctic viromes were found to contain mainly genes from the categories of phages, prophages, and transposable elements (not shown), which were also dominant in the temperate Lake Bourget and Antarctic spring viromes. The other prominent feature is the enrichment within all Sahara viromes, Antarctic summer, and Arctic Ocean of reads with similarity to genes coding for functions related to various families of metabolic pathways, including photosynthesis, respiration, and stress response. Nevertheless, we did not proceed with further analysis because the overall percentage of assigned reads was likely too low to provide informative conclusions (average, 1.6 ± 2.9%).
Fig. 3. Ordination of individual freshwater viromes according to their taxonomic profiles.

The figure represents the first two axes of a principal components analysis based on the taxonomic affiliation of assigned reads. (A) Projection of the individual viromes on the model. Viromes from the same environment are represented by equal forms. (B) Projection of the viral groups with a higher contribution to the model. The position of each group represents its association with each virome. The analysis indicates a strong partitioning of the viromes between polar and nonpolar viromes, driven mainly by the relative abundance of small circular ssDNA viruses and phages. U., unclassified; d, distance.
Reference-dependent analysis of metagenomic reads returned a small proportion of positive hits. Hence, we also used two reference-independent comparison methods, using all metagenomic reads: crAss, a cross-assembly–based fine-grained analysis, and more coarse-grained cross-tblastx comparisons between viromes. crAss results (Fig. 4) indicated that viromes from the same environment share fine-grained genetic information, with unnoticeable overlap between different environments, including Arctic-Antarctica and Arctic freshwater-seawater. Such clustering by environment is also observable in the cross-tblastx results (fig. S3). However, in this case, some degree of coarse-grain genetic overlap between environments is observable. For instance, Aquaculture and Sahara communities shared some relative overlap (averaging 5.6 ± 1.9%), and the Antarctic showed some overlap with the Arctic (averaging 7.7 ± 4%). Two of the Aquaculture and both temperate lake viromes (especially Bourget) showed some overlap with both the Sahara and Arctic samples, which nevertheless showed little overlap among themselves (0.5 ± 0.64). Finally, the reclaimed water viromes showed little overlap to the other, except for Nursery, which displayed overlap with the Arctic viromes (5.3 ± 3%) except for Lv1 and Lv1Pond. Finally, the Arctic Ocean virome consistently showed very low overlap values (0.3 ± 0.3). We also produced per-environment viromes by pooling all available reads for that particular environment. The results of the cross-tblastx analysis of these pools (Fig. 5) are confirmatory of those obtained with the individual communities with regard to the overall coarse-grain genetic overlap between the different environments. They showed even more clearly how the temperate lake viromes present high relative overlap to all other environments (except Arctic Ocean) and vice versa, likely representing a midpoint between the ssDNA virus–dominated polar communities and the other phage-abundant environments.
Fig. 4. Reference-independent cross-assembly (crAss) comparison of individual freshwater viromes.
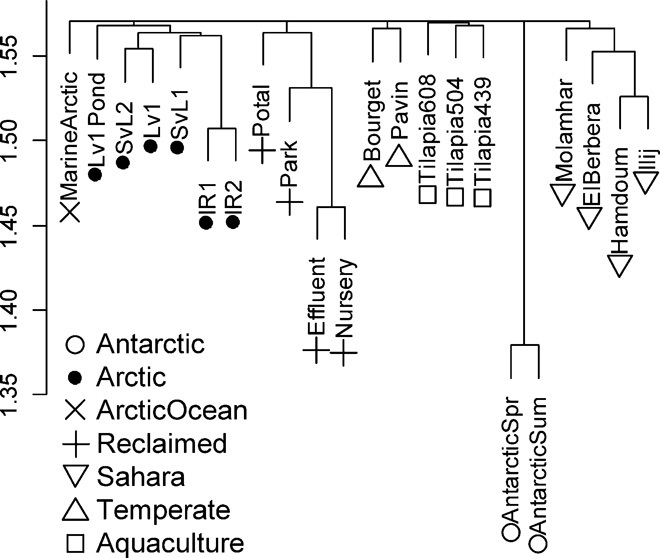
The dendrogram represents a hierarchical clustering (average-linkage method) of Wootters distance values derived from the cross-assembly of freshwater viromes. Viromes from the same environment are represented by equal forms. The analysis evidences very low fine-grain genetic overlap between viromes from different environments.
Fig. 5. Coarse-grain genetic overlap between environments.
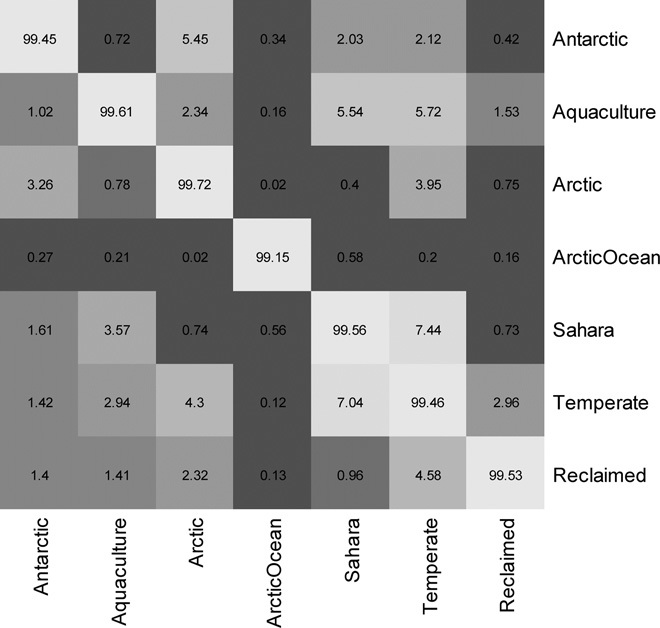
The heatplot represents the results of cross-tblastx searches between per-environment pooled viral metagenomes. Values (% of total reads with hits with E value <0.001) are registered within the boxes and color-coded. Horizontal labels represent viromes acting as query, and vertical labels represent viromes acting as reference.
Some ssDNA viral groups exhibit a bipolar distribution
The per-environment pooled reads were also used for contig reconstruction to look for shared genomes and/or genomic regions between the different environments. Overall, the results (table S3) reveal little contig overlap between the different environments, although there is a noticeable trend of increasing overlap with decreasing similarity threshold. Further analysis revealed that two of the overlapping contig pairs between the Arctic and Antarctica represented circular contigs showing 93.8 and 90.8% similarity along their complete sequence (fig. S1C). The first group includes a viral_rep gene most similar to a likely Circoviridae sequence and another ORF bearing low similarity to a phage integrase, whereas the second group presents two ORFs, one of them with similarity to a Circoviridae-related putative viral capsid protein. Also, one of the overlapping contig pairs between the Arctic and the French temperate lakes represented a 1936-bp sequence with 95.8% similarity. This sequence presented two ORFs with no similarities and a third ORF with similarity to a phage structural protein. Strikingly, all overlapping contigs between the Antarctic and Sahara environments at the highest similarity, and the single overlapping contig between the reclaimed water environment and both Antarctic and Sahara environments, produced blastx results pointing to DNA mobilization or replication-related proteins related to Acinetobacter sequences. The longest among the overlapping contigs between Antarctic and Sahara environments represented a circular 5594-bp sequence (Sahara_97 has a 180-bp deletion and then 99.9% similarity with respect to Antarctic_92). Further analysis showed that this contig is formed by a 2878-bp segment with 83% similarity to Acinetobacter baumannii E7 pRAY-v2 plasmid and a second segment with 99% similarity to a hypothetical Lactamase_B superfamily protein from Acinetobacter sp. Thus, this circular contig likely represents an antibiotic resistance–carrying plasmid. The second group (contigs linking the reclaimed water, Sahara, and Antarctic environments) was formed by circular contigs ranging from 1739 to 2088 bp in length and with an average similarity of 74.6%. These sequences bear great similarity to uncultured bacterial and Acinetobacter sp. plasmids and to Sphinx 1.76, a nuclease-resistant circular DNA (containing a replicase ORF related to Microviridae) found to co-purify with infectivity in various transmissible spongiform encephalopathies (19). A recent study (20) has found experimental evidence that Acinetobacter sp. DS002 plasmid, most similar (67%) to the sibling Sphinx 2.36 circular DNA (also transmissible spongiform encephalopathy–related and carrying the putative Microviridae replicase), is a phage. These Acinetobacter-related contigs may represent bacterial DNA contamination that profited from a Phi29 overamplification, but these sequences were unrelated to genomic regions and corresponded to plasmids or known or suspected Acinetobacter phages. These sequences were detected in three different environments and produced in three laboratories using different sequencing platforms. Moreover, they represent distinct parental sequences, likely indicating a natural community origin rather than a unique contaminant. All three protocols included nuclease treatments, indicating that the sequences could correspond to genetic material protected within virions. The sequences were not detected in our recent Arctic viromes, nor did our Phi29 amplification–negative controls produce noticeable mass. Thus, the origin of these DNA sequences is unclear; they likely represent abundant, varied, and resistant environmental contaminants (plasmids), but some could correspond to true viral sequences of global freshwater viromes.
Polar freshwater environments harbor diverse viral communities
The extent of polar microbial diversity remains an outstanding question (15). Thus, we set out to assess possible latitudinal diversity trends within the studied freshwater viromes. Community structure and richness of each viral metagenome was evaluated using the PHACCS tool. The software predicted large differences in both richness and diversity for viromes arising from the same environments (Table 2), and the results obtained for all polar viromes are well within the boundaries defined by the other communities studied, sustaining the idea that polar viral communities are not less diverse than their lower-latitude counterparts.
Table 2. Ecological indices predicted for subsampled viromes.
Results derived from PHACCS and GAAS (Genome relative Abundance and Average Size) analyses. SW, Shannon-Wiener index; Scenario, most likely rank-abundance scenario for the community (Log, logarithmic; Pow, power law).
|
Average genome size (bp) |
Richness | Evenness | SW | Scenario | |
| Bourget | 9,165 | 3,000 | 0.95 | 7.6 | Log |
| Pavin | 21,231 | 315 | 0.85 | 4.9 | Pow |
| Lv1Pond | 2,387 | 289 | 0.97 | 5.5 | Pow |
| Lv1 | 2,893 | 107 | 0.95 | 4.4 | Pow |
| SvL1 | 3,041 | 1,000 | 0.86 | 5.9 | Pow |
| IR1 | 3,785 | 81 | 0.92 | 4.0 | Pow |
| IR2 | 3,368 | 3,806 | 0.81 | 6.68 | Pow |
| SvL2 | 1,570 | 520 | 0.89 | 5.5 | Pow |
| Effluent | 23,747 | 916 | 0.92 | 6.3 | Pow |
| Nursery | 5,314 | 6,099 | 0.91 | 7.9 | Pow |
| Park | 2,939 | 7,200 | 0.93 | 8.3 | Pow |
| Potal | 34,069 | 30 | 0.91 | 3.12 | Pow |
| ArcticOcean | 70,311 | 480 | 0.97 | 6.0 | Log |
| AntarcticSpr | 3,911 | 204 | 0.94 | 5.0 | Pow |
| AntarcticSum | 33,466 | 562 | 0.76 | 4.8 | Pow |
| ElBerbera | 93,666 | 1,297 | 0.74 | 5.3 | Log |
| Hamdoum | 69,976 | 769 | 0.60 | 4.0 | Pow |
| Ilij | 78,752 | 579 | 0.89 | 5.6 | Log |
| Molamhar | 88,180 | 309 | 0.93 | 5.3 | Pow |
| Tilapia608 | 52,161 | 739 | 0.95 | 6.3 | Pow |
| Tilapia504 | 11,011 | 3,804 | 0.96 | 7.9 | Log |
| Tilapia439 | 30,706 | 3,284 | 0.93 | 7.6 | Log |
Then, we focused on the two most abundant viral families detected within the polar viromes and studied the existing family-wise phylogenetic marker genes contained within the contigs generated: the vp1 capsid protein gene for Microviridae and the rep replication protein gene for Circoviridae-Nanoviridae-Geminiviridae. vp1 gene sequences from the polar contigs clustered into 333 groups at 50% identity, and 26 of the representative sequences from these groups were found to cluster within 7 of the 31 groups obtained from previously described vp1 sequences. Also, rep polar sequences grouped into 2648 clusters at the same identity threshold, with 22 of their representative sequences later clustering within 15 of the 111 groups obtained from previously described genes. These results evidence a broad diversity for these viral families contained within the polar viromes (fig. S4).
DISCUSSION
We present in-depth sequence data from Arctic and Antarctic DNA viromes, including viral community deep-sequencing data from six Arctic lakes, combined with a comparative analysis of published freshwater DNA viromes from a range of worldwide geographic locations. The RNA viral community, which has been shown to be highly abundant in other natural aquatic ecosystems (21), has not been characterized here.
As with all metagenomic studies, there were certain limits to studying the composition and structure of viral communities and comparing them with previous reports. A recent study shows that library preparation method and sequencing platform represent a weak source of bias in the study of natural viral communities through next-generation sequencing (22). However, various reports have shown that differing viral community extraction methods produce noticeably different taxonomic profiles from the same virome sample (23, 24). In order to avoid this, our chosen viral community extraction method omitted known strong sources of bias: CsCl gradient–based purification [biased toward tailed phages (25)], use of chloroform (damages the lipid envelope surrounding the viral capsid of some viruses, jeopardizing subsequent genome recovery), and 0.2-μm filtration step [may fail to recover larger viral particles, which are common in aquatic ecosystems (26)]. To obtain sufficient genetic material for sequencing, we relied on Phi29 polymerase amplification, which has been shown to produce a bias toward small circular DNA (17). Nevertheless, all other chosen viral metagenomes compared in this study also relied on Phi29 amplification and used the same enzyme kit. This kit produces a consistent bias with high experimental reproducibility (27), making these data sets comparable without leading the analyses astray.
The Arctic viral metagenomes were similar in their taxonomic composition, mainly dominated by ssDNA and unknown viruses. Finding a large fraction of unaffiliated viral metagenomic reads is a common issue in aquatic environments (28). The abundance of ssDNA viruses was observed even after estimating a 100-fold increase bias of the Phi29 polymerase for circular ssDNA genomes. Most polar contigs obtained had no highly similar (≥65%) genomes in the databases, in agreement with the notion that our knowledge of viral diversity in nature is very sparse (10). We were able to find circular contigs representative of the recently described RNA-DNA hybrid viruses (29), which sustain previous reports indicating that these viruses may be globally distributed (29, 30). Overall, the viral communities retrieved from samples IR1 and IR2 were the most similar of the Arctic viromes. This is not surprising because they originate from the same area and were obtained in the same season (late summer) when both presented no ice cover. The Lake Tenndammen community (SvL2) was most similar to Lake Linnevatnet’s virome (Lv1), despite the fact that they were taken at different seasons and that Lake Linnevatnet presents a more complex food web including fish. The virome from sample SvL1, derived from melted top ice of Lake Nordammen, was found to be the most different among the deeper-sequenced Arctic samples. This seems to indicate that although composed of similar viral taxa, this melted top ice environment is not representative of a large water body environment.
The taxonomic composition of Arctic communities separated them from viromes from freshwater samples in other regions of the world studied but was similar to that of the Antarctic spring. Arctic viral communities were found to share genes and large genomic regions. We detected three frequent genomes, likely corresponding to highly stable and abundant viral species represented in four of five Arctic viral communities. The fact that we were not able to detect more frequent species from these similar and neighboring environments, despite the high coverage attained, indicates that viral communities from these environments are highly dynamic in their rank abundance structure, which agrees with theoretical predictions made for marine phage communities (31).
Overall, viromes from the same environment were most similar to each other but showed some degree of coarse-grain genetic overlap with other environments, a trend consistently reported for both viral and bacterial communities (32, 33). The Arctic Ocean virome clearly separated not only from the Arctic freshwater samples but also from the other freshwater viromes, which is consistent with the saline/nonsaline split being a most important driver of microbial community structure (34). The latitudinal diversity gradient (that is, species richness decreasing toward the poles) is one of the most prominent patterns in ecology (35). Our results based on both PHACCS and the analysis of the diversity contained within the phylogenetic marker genes of predominant viruses in polar viromes are not supportive of this latitudinal diversity gradient existing in freshwater viromes but rather in agreement with reports indicating that such a trend is not observable in other microbial communities (16, 36).
Although similar in their taxonomic distribution of metagenomic reads, Arctic and Antarctic viromes differed at the fine-grain genetic level, indicating that they are dominated by different viral species. Yet, we were able to find circular contigs in both environments showing sequence similarities greater than 90%. The fact that lineages of highly similar ssDNA viruses thrive in both the Arctic and Antarctica not only indicates the presence of similar environmental filters but also is in conflict with simple allopatric speciation and seemingly indicates an important global dispersal capacity for some viruses.
The metagenomic analysis of DNA viruses in freshwater bodies in the Arctic addresses many questions in polar microbiology (15). First, it shows that viral communities in the Arctic and Antarctic freshwater ecosystems share taxonomic composition of viruses, dominated by unknown and small ssDNA viruses, but show very low fine-grain genetic overlap. Second, it defines an Arctic freshwater viral community very different from that of the Arctic Ocean. Third, it identifies some viral lineages with bipolar distribution, suggesting the capacity of some viruses to disperse at a global scale. Fourth, it shows that viral species richness does not decrease in the Arctic, indicating that viruses may not follow a latitudinal diversity gradient. Last, our comparative analysis sheds light into the global biogeography and connectivity of viral communities and highlights not only the uniqueness of the polar environments but also the differences between Arctic and Antarctic microbial ecosystems, despite their exposure to similar environmental conditions.
MATERIALS AND METHODS
Sampling sites
Samples were taken from several Arctic freshwater bodies in Spitsbergen (Svalbard, Norway) (Fig. 1). Lakes are defined as water bodies whose bottom waters remain unfrozen all year round, whereas ponds may freeze entirely over winter. Lake Linnevatnet (Lv1) presents a developed food web including fish. Sample Lv1Pond was obtained from the nearby pond Borgdammane (summer 2010). IR1 (Lake Tunsjøen) and IR2 (pond) are from the vicinity of Kapp Linné (late summer 2011). Lake Nordammen (SvL1) was completely frozen (samples represent a combination of melted top ice from three different sites), whereas Lake Tenndammen (SvL2) represents a shallow lake with frozen surface at the time of sampling (spring 2012). Lv1 and Lv1Pond water samples were taken from different depths and mixed to give a representation of the water column.
Virome extraction
Viral metagenomic DNA was obtained as described (14) with minor modifications. Ninety liters were filtered through a 30-μm nylon mesh and by 0.45-μm tangential flow filtration (TFF) using a Centramate holder (Pall) to remove bacteria and smaller eukaryotes. Viral fractions were concentrated 100 times by 70-kD TFF. Viral stocks were preserved at −20°C/−80°C before DNA extraction.
DNA extraction and sequencing
Frozen stocks were thawed at 4°C and passed through a 25% sucrose cushion by centrifugation for 16 hours at 60,000g and 4°C. The pellets were resuspended in 10 mM tris (pH 8) and 1 mM EDTA and filtered using a 0.45-μm filter. Viral concentrates were then treated with deoxyribonuclease I (DNase I) (500 U ml−1), nuclease S7 (420 U ml−1), ribonuclease A (RNAse A) (100 μg ml−1), and RNAse H (2 U per reaction) for 30 min at room temperature to remove free nucleic acids. Nuclease reactions were stopped with 12 mM EDTA/2 mM EGTA, and then viral capsids and envelopes were disrupted with SDS (0.5%) and proteinase K (200 μg ml−1) treatment. Viral DNA was extracted with phenol-chloroform, and ethanol was precipitated. The resulting DNA was randomly amplified using Phi29 polymerase and modified random hexamers (GenomiPhi HY, GE Healthcare) for 2.5 hours, according to the manufacturer’s instructions. Finally, the nucleic acids were shotgun-sequenced using an Illumina HiSeq apparatus (Parque Científico de Madrid), resulting in 5.4 million to 7.4 million 2 × 100–bp reads per sample. The extraction protocol used successfully limited possible bacterial contamination, as evidenced by transmission electron microscopy and BLAST searches against an all-inclusive 16S ribosomal RNA gene database. For comparative purposes, metagenomic DNA from the Limnopolar Lake summer sample previously reported (14) was also resequenced with the same device obtaining more than 1.8 million 2 × 75–bp reads. Additionally, DNA derived in the same fashion from Lv1Pond was sequenced with a Roche 454 FLX device obtaining more than 228 thousand reads circa 250 bp in length. Because of its relatively small amount of information, this last data set was only used in the comparisons to other published freshwater viromes.
Metagenomic read analysis
A series of filtering and trimming steps were undertaken to remove low-quality reads and bases using prinseq-lite software (37). Taxonomic assignment of reads was carried out by performing blastx (E score <10−3) searches using as reference set the most recent release of viral genomes from the National Center for Biotechnology Information (NCBI) containing 4788 sequences, and then summarizing the results with MEGAN 4 (38). To compare the viromes obtained in this study with other published freshwater viromes, we downloaded their sequences from the relevant sources. These included data sets from an aquaculture facility (Tilapia504, Tilapia608, and Tilapia439; SEED accessions 4440412.3, 4440439.3, and 4440424.3) (32), samples from an Antarctic lake [Antarctic summer and Antarctic spring; SRA (Sequence Read Archive) accession SRA008157] (14), viromes from Sahara desert perennial ponds [ElBerbera, Hamdoum, Ilij, and Molamhar; MG-RAST (Metagenomic Rapid Annotations using Subsystems Technology) ID 4446033.3, 4445715.3, 4445716.3, and 4445718.3] (39), data sets from a reclaimed-waters study (Potal, Effluent, Park, and Nursery; SRA accession SRA008294) (40), samples from French temperate lakes (Pavin and Bourget; SRA accession ERP000339) (41), and one virome from the Arctic Ocean (ArcticOcean; MG-RAST ID 4441621.3) (42). All data sets were subsampled to a common depth (the minimum number of sequences in a virome; 39,351) and then trimmed to 100 bp to homogenize sequence length, thus effectively normalizing sampling effort. Taxonomic and functional assignments were carried out as above but in the latter case using NCBI’s nonredundant database and then using the SEED (43) classification in MEGAN. A cross-tblastx (E score <10−3) between these data sets was undertaken to study the degree of putative coarse-grain genetic overlap between these environments. Then, the percentage of reads giving above-threshold results were graphically represented as a heat map using the gplots R package. To further study the relationships between the different freshwater viral communities, all reads per environment were pooled. The genetic overlap was studied as above using a cross-tblastx (using subsamples of 252,867 reads, 70 bp in length). To further study the relationships between the subsampled individual viromes, we followed the recently described reference-independent strategy crAss (44). Briefly, all sequences were combined in a single pool and then assembled using idba_ud (see below) generating cross-contigs. Then, reads from each environment were mapped onto these cross-contigs using Bowtie 2 (--score-min L,0,−0.2) (45). The degree of similarity between these data sets was assessed using the Wootters distance formula on cross-contig mapped-read counts as previously suggested (44) and represented as a cladogram with R. Then, several ecological indices were calculated for the subsampled individual viromes using the PHACCS software (46). To this end, the average genome length was assessed using GAAS (47), and their contig spectra were calculated with Circonspect (48) using the Minimo assembler (49).
Metagenomic assembly and analysis
Arctic and resequenced Antarctic reads were assembled into contigs using idba_ud (50). Assignment of reads to contigs was also done with Bowtie 2. Cross-contig alignments and alignments of contigs to reference genomes were carried out using NUCmer (51) (500-bp minimum overlap, similarity thresholds ranging from 65 to 98%). On the other hand, to assess the viral genomic connectivity between the environments studied, their per-environment pooled reads were assembled into contigs as above and cross-contig comparisons (again using NUCmer) were carried out.
Analysis of vp1 and rep phylogenetic marker genes
The METAVIR online pipeline (52) was used to look for phylogenetic marker genes in the polar contigs obtained via a hidden Markov model profile of PFAM-derived sequences. The amino acid sequences of marker genes from majority groups (rep gene for Circoviridae-Nanoviridae-Geminiviridae and vp1 gene for Microviridae) were then retrieved, along with the reference genes contained within the resource. Additional genes were obtained from NCBI, genes derived from the contigs obtained in the study of the temperate lakes (41) (where these viral groups were also shown to dominate), and genes from putative genomes recently described in the literature for both Circoviridae-Nanoviridae-Geminiviridae (30) and Microviridae (53). Each gene type data set was divided into two groups, one carrying all amino acid sequences derived from the new polar contigs and another containing all previously described sequences. Then, each group was clustered at 50% identity using CD-HIT (54) (-aS 0.9). Similar gene clusters among data sets were detected using CD-HIT-2D to compare their representative sequences (-c 0.5 -aS 0.7 –s2 0.5). Additionally, the amino acid sequences from each gene type were aligned with muscle package (55), their distances were obtained using the phangorn package (56) in R, under the Whelan and Goldman substitution model (57), and neighbor-joining (NJ) trees were constructed using the njs function. The NJ trees were visualized and edited using FigTree version 1.4 (http://tree.bio.ed.ac.uk/software/figtree).
Supplementary Material
Acknowledgments
We thank A. Quesada and M. L. Avila for helpful comments on the manuscript. Sample collection of Arctic lakes was done as part of the University Centre in Svalbard Master Course AB-327 on Arctic Microbiology (2010–2012), and we thank M. L. Avila, L. Little, and the master students for their help. Funding: This work was funded by the Spanish Ministry of Economy and Competitiveness grant CTM2011-15091-E/ANT. D.A.d.C. was supported by the Marie Curie International Incoming Fellowship grant PIIF-GA-2012-328287, and A.L.-B. was supported by a Ramón y Cajal Fellowship (RYC-2010-06300). Author contributions: D.A.P. and A.A. conceived the project and obtained and concentrated all virome samples. D.A.d.C. and A.L.-B. extracted the metagenomic DNA and prepared the libraries. D.A.d.C. analyzed the sequence data. D.A.d.C., A.L.-B., and A.A. wrote the initial manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: Metagenomic data have been deposited at SRA under accession PRJEB5265, and polar contigs have been deposited at METAVIR under IDs 3295 to 3300.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/1/5/e1400127/DC1
Fig. S1. Significant circular ssDNA-related contigs.
Fig. S2. Fine-grain genetic overlap between polar freshwater viromes.
Fig. S3. Coarse-grain genetic overlap between viromes.
Fig. S4. NJ trees depicting the relationships between phylogenetic marker genes.
Table S1. Cross-contig analysis of Arctic viromes.
Table S2. Taxonomic distribution (%) of assigned metagenomic reads from subsampled freshwater viromes.
Table S3. Cross-contig analysis of pooled freshwater environments.
REFERENCES AND NOTES
- 1.Suttle C. A., Viruses in the sea. Nature 437, 356–361 (2005). [DOI] [PubMed] [Google Scholar]
- 2.Ignacio-Espinoza J. C., Solonenko S. A., Sullivan M. B., The global virome: Not as big as we thought? Curr. Opin. Virol. 3, 566–571 (2013). [DOI] [PubMed] [Google Scholar]
- 3.Fuhrman J. A., Marine viruses and their biogeochemical and ecological effects. Nature 399, 541–548 (1999). [DOI] [PubMed] [Google Scholar]
- 4.Mann N. H., Cook A., Millard A., Bailey S., Clokie M., Marine ecosystems: Bacterial photosynthesis genes in a virus. Nature 424, 741 (2003). [DOI] [PubMed] [Google Scholar]
- 5.Williamson S. J., Rusch D. B., Yooseph S., Halpern A. L., Heidelberg K. B., Glass J. I., Andrews-Pfannkoch C., Fadrosh D., Miller C. S., Sutton G., Frazier M., Venter J. C., The Sorcerer II Global Ocean Sampling Expedition: Metagenomic characterization of viruses within aquatic microbial samples. PLOS One 3, e1456 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Evans C., Pearce I., Brussaard C. P. D., Viral-mediated lysis of microbes and carbon release in the sub-Antarctic and Polar Frontal zones of the Australian Southern Ocean. Environ. Microbiol. 11, 2924–2934 (2009). [DOI] [PubMed] [Google Scholar]
- 7.Lindell D., Jaffe J. D., Johnson Z. I., Church G. M., Chisholm S. W., Photosynthesis genes in marine viruses yield proteins during host infection. Nature 438, 86–89 (2005). [DOI] [PubMed] [Google Scholar]
- 8.Hurwitz B. L., Hallam S. J., Sullivan M. B., Metabolic reprogramming by viruses in the sunlit and dark ocean. Genome Biol. 14, R123 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Béjà O., Spudich E. N., Spudich J. L., Leclerc M., DeLong E. F., Proteorhodopsin phototrophy in the ocean. Nature 411, 786–789 (2001). [DOI] [PubMed] [Google Scholar]
- 10.Suttle C. A., Marine viruses—Major players in the global ecosystem. Nat. Rev. Microbiol. 5, 801–812 (2007). [DOI] [PubMed] [Google Scholar]
- 11.Convey P., Stevens M. I., Antarctic biodiversity. Science 317, 1877–1878 (2007). [DOI] [PubMed] [Google Scholar]
- 12.Laybourn-Parry J., No place too cold. Science 324, 1521–1522 (2009). [DOI] [PubMed] [Google Scholar]
- 13.Säwström C., Lisle J., Anesio A., Priscu J., Laybourn-Parry J., Bacteriophage in polar inland waters. Extremophiles 12, 167–175 (2008). [DOI] [PubMed] [Google Scholar]
- 14.López-Bueno A., Tamames J., Velázquez D., Moya A., Quesada A., Alcamí A., High diversity of the viral community from an Antarctic lake. Science 326, 858–861 (2009). [DOI] [PubMed] [Google Scholar]
- 15.Anesio A. M., Bellas C. M., Are low temperature habitats hot spots of microbial evolution driven by viruses? Trends Microbiol. 19, 52–57 (2011). [DOI] [PubMed] [Google Scholar]
- 16.Ghiglione J. F., Galand P. E., Pommier T., Pedrós-Alió C., Maas E. W., Bakker K., Bertilson S., Kirchmanj D. L., Lovejoy C., Yager P. L., Murray A. E., Pole-to-pole biogeography of surface and deep marine bacterial communities. Proc. Natl. Acad. Sci. U.S.A. 109, 17633–17638 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kim K. H., Bae J. W., Amplification methods bias metagenomic libraries of uncultured single-stranded and double-stranded DNA viruses. Appl. Environ. Microbiol. 77, 7663–7668 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Breitbart M., Salamon P., Andresen B., Mahaffy J. M., Segall A. M., Mead D., Azam F., Rohwer F., Genomic analysis of uncultured marine viral communities. Proc. Natl. Acad. Sci. U.S.A. 99, 14250–14255 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Manuelidis L., Nuclease resistant circular DNAs copurify with infectivity in scrapie and CJD. J. Neurovirol. 17, 131–145 (2011). [DOI] [PubMed] [Google Scholar]
- 20.Longkumer T., Kamireddy S., Muthyala V. R., Akbarpasha S., Pitchika G. K., Kodetham G., Ayaluru M., Siddavattam D., Acinetobacter phage genome is similar to Sphinx 2.36, the circular DNA copurified with TSE infected particles. Sci. Rep. 3, 2240 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Steward G. F., Culley A. I., Mueller J. A., Wood-Charlson E. M., Belcaid M., Poisson G., Are we missing half of the viruses in the ocean? ISME J. 7, 672–679 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Solonenko S., Ignacio-Espinoza J., Alberti A., Cruaud C., Hallam S., Konstantinidis K., Tyson G., Wincker P., Sullivan M., Sequencing platform and library preparation choices impact viral metagenomes. BMC Genomics 14, 320 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hurwitz B. L., Deng L., Poulos B. T., Sullivan M. B., Evaluation of methods to concentrate and purify ocean virus communities through comparative, replicated metagenomics. Environ. Microbiol. 15, 1428–1440 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Willner D., Furlan M., Schmieder R., Grasis J. A., Pride D. T., Relman D. A., Angly F. E., McDole T., Mariella R. P. Jr, Rohwer F., Haynes M., Metagenomic detection of phage-encoded platelet-binding factors in the human oral cavity. Proc. Natl. Acad. Sci. U.S.A. 108, 4547–4553 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Thurber R. V., Haynes M., Breitbart M., Wegley L., Rohwer F., Laboratory procedures to generate viral metagenomes. Nat. Protoc. 4, 470–483 (2009). [DOI] [PubMed] [Google Scholar]
- 26.Claverie J. M., Giant viruses in the oceans: The 4th Algal Virus Workshop. Virol. J. 2, 52 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yilmaz S., Allgaier M., Hugenholtz P., Multiple displacement amplification compromises quantitative analysis of metagenomes. Nat. Methods 7, 943–944 (2010). [DOI] [PubMed] [Google Scholar]
- 28.Hurwitz B. L., Sullivan M. B., The Pacific Ocean virome (POV): A marine viral metagenomic dataset and associated protein clusters for quantitative viral ecology. PLOS One 8, e57355 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Diemer G., Stedman K., A novel virus genome discovered in an extreme environment suggests recombination between unrelated groups of RNA and DNA viruses. Biol. Direct 7, 13 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Labonté J. M., Suttle C. A., Previously unknown and highly divergent ssDNA viruses populate the oceans. ISME J. 7, 2169–2177 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hoffmann K. H., Rodriguez-Brito B., Breitbart M., Bangor D., Angly F., Felts B., Nulton J., Rohwer F., Salamon P., Power law rank–abundance models for marine phage communities. FEMS Microbiol. Lett. 273, 224–228 (2007). [DOI] [PubMed] [Google Scholar]
- 32.Dinsdale E. A., Edwards R. A., Hall D., Angly F., Breitbart M., Brulc J. M., Furlan M., Desnues C., Haynes M., Li L., McDaniel L., Moran M. A., Nelson K. E., Nilsson C., Olson R., Paul J., Brito B. R., Ruan Y., Swan B. K., Stevens R., Valentine D. L., Thurber R. V., Wegley L., White B. A., Rohwer F., Functional metagenomic profiling of nine biomes. Nature 452, 629–632 (2008). [DOI] [PubMed] [Google Scholar]
- 33.Rodriguez-Brito B., Li L., Wegley L., Furlan M., Angly F., Breitbart M., Buchanan J., Desnues C., Dinsdale E., Edwards R., Felts B., Haynes M., Liu H., Lipson D., Mahaffy J., Martin-Cuadrado A. B., Mira A., Nulton J., Pasić L., Rayhawk S., Rodriguez-Mueller J., Rodriguez-Valera F., Salamon P., Srinagesh S., Thingstad T. F., Tran T., Thurber R. V., Willner D., Youle M., Rohwer F., Viral and microbial community dynamics in four aquatic environments. ISME J. 4, 739–751 (2010). [DOI] [PubMed] [Google Scholar]
- 34.Lozupone C. A., Knight R., Global patterns in bacterial diversity. Proc. Natl. Acad. Sci. U.S.A. 104, 11436–11440 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hawkins B. A., Ecology’s oldest pattern? Endeavour 25, 133–134 (2001). [DOI] [PubMed] [Google Scholar]
- 36.Chu H., Fierer N., Lauber C. L., Caporaso J. G., Knight R., Grogan P., Soil bacterial diversity in the Arctic is not fundamentally different from that found in other biomes. Environ. Microbiol. 12, 2998–3006 (2010). [DOI] [PubMed] [Google Scholar]
- 37.Schmieder R., Edwards R., Quality control and preprocessing of metagenomic datasets. Bioinformatics 27, 863–864 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Huson D. H., Auch A. F., Qi J., Schuster S. C., MEGAN analysis of metagenomic data. Genome Res. 17, 377–386 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fancello L., Trape S., Robert C., Boyer M., Popgeorgiev N., Raoult D., Desnues C., Viruses in the desert: A metagenomic survey of viral communities in four perennial ponds of the Mauritanian Sahara. ISME J. 7, 359–369 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rosario K., Nilsson C., Lim Y. W., Ruan Y., Breitbart M., Metagenomic analysis of viruses in reclaimed water. Environ. Microbiol. 11, 2806–2820 (2009). [DOI] [PubMed] [Google Scholar]
- 41.Roux S., Enault F., Robin A., Ravet V., Personnic S., Theil S., Colombet J., Sime-Ngando T., Debroas D., Assessing the diversity and specificity of two freshwater viral communities through metagenomics. PLOS One 7, e33641 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Angly F. E., Felts B., Breitbart M., Salamon P., Edwards R. A., Carlson C., Chan A. M., Haynes M., Kelley S., Liu H., Mahaffy J. M., Mueller J. E., Nulton J., Olson R., Parsons R., Rayhawk S., Suttle C. A., Rohwer F., The marine viromes of four oceanic regions. PLOS Biol. 4, e368 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Overbeek R., Begley T., Butler R. M., Choudhuri J. V., Chuang H.-Y., Cohoon M., de Crécy-Lagard V., Diaz N., Disz T., Edwards R., Fonstein M., Frank E. D., Gerdes S., Glass E. M., Goesmann A., Hanson A., Iwata-Reuyl D., Jensen R., Jamshidi N., Krause L., Kubal M., Larsen N., Linke B., McHardy A. C., Meyer F., Neuweger H., Olsen G., Olson R., Osterman A., Portnoy V., Pusch G. D., Rodionov D. A., Rückert C., Steiner J., Stevens R., Thiele I., Vassieva O., Ye Y., Zagnitko O., Vonstein V., The subsystems approach to genome annotation and its use in the project to annotate 1000 genomes. Nucleic Acids Res. 33, 5691–5702 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dutilh B. E., Schmieder R., Nulton J., Felts B., Salamon P., Edwards R. A., Mokili J. L., Reference-independent comparative metagenomics using cross-assembly: crAss. Bioinformatics 28, 3225–3231 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Langmead B., Salzberg S. L., Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Angly F., Rodriguez-Brito B., Bangor D., McNairnie P., Breitbart M., Salamon P., Felts B., Nulton J., Mahaffy J., Rohwer F., PHACCS, an online tool for estimating the structure and diversity of uncultured viral communities using metagenomic information. BMC Bioinformatics 6, 41 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Angly F. E., Willner D., Prieto-Davó A., Edwards R. A., Schmieder R., Vega-Thurber R., Antonopoulos D. A., Barott K., Cottrell M. T., Desnues C., Dinsdale E. A., Furlan M., Haynes M., Henn M. R., Hu Y., Kirchman D. L., McDole T., McPherson J. D., Meyer F., Miller R. M., Mundt E., Naviaux R. K., Rodriguez-Mueller B., Stevens R., Wegley L., Zhang L., Zhu B., Rohwer F., The GAAS metagenomic tool and its estimations of viral and microbial average genome size in four major biomes. PLOS Comput. Biol. 5, e1000593 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Allen H., Bunge J., Foster J., Bayles D., Stanton T., Estimation of viral richness from shotgun metagenomes using a frequency count approach. Microbiome 1, 5 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Treangen T. J., Sommer D. D., Angly F. E., Koren S., Pop M., Next generation sequence assembly with AMOS. Curr. Protoc. Bioinformatics Chapter 11, Unit 11.8 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Peng Y., Leung H. C. M., Yiu S. M., Chin F. Y. L., IDBA-UD: A de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics 28, 1420–1428 (2012). [DOI] [PubMed] [Google Scholar]
- 51.Kurtz S., Phillippy A., Delcher A. L., Smoot M., Shumway M., Antonescu C., Salzberg S. L., Versatile and open software for comparing large genomes. Genome Biol. 5, R12 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Roux S., Faubladier M., Mahul A., Paulhe N., Bernard A., Debroas D., Enault F., Metavir: A web server dedicated to virome analysis. Bioinformatics 27, 3074–3075 (2011). [DOI] [PubMed] [Google Scholar]
- 53.Roux S., Krupovic M., Poulet A., Debroas D., Enault F., Evolution and diversity of the Microviridae viral family through a collection of 81 new complete genomes assembled from virome reads. PLOS One 7, e40418 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Huang Y., Niu B., Gao Y., Fu L., Li W., CD-HIT Suite: A web server for clustering and comparing biological sequences. Bioinformatics 26, 680–682 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Edgar R. C., MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schliep K. P., phangorn: Phylogenetic analysis in R. Bioinformatics 27, 592–593 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Whelan S., Goldman N., A general empirical model of protein evolution derived from multiple protein families using a maximum-likelihood approach. Mol. Biol. Evol. 18, 691–699 (2001). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/1/5/e1400127/DC1
Fig. S1. Significant circular ssDNA-related contigs.
Fig. S2. Fine-grain genetic overlap between polar freshwater viromes.
Fig. S3. Coarse-grain genetic overlap between viromes.
Fig. S4. NJ trees depicting the relationships between phylogenetic marker genes.
Table S1. Cross-contig analysis of Arctic viromes.
Table S2. Taxonomic distribution (%) of assigned metagenomic reads from subsampled freshwater viromes.
Table S3. Cross-contig analysis of pooled freshwater environments.