

Isolation of bis(copper) complexes that are kinetically more active than their mononuclear counterparts in the most popular “click chemistry” reaction.
Keywords: Catalysis, Mechanism, carbene
Abstract
The copper-catalyzed 1,3-dipolar cycloaddition of an azide to a terminal alkyne (CuAAC) is one of the most popular chemical transformations, with applications ranging from material to life sciences. However, despite many mechanistic studies, direct observation of key components of the catalytic cycle is still missing. Initially, mononuclear species were thought to be the active catalysts, but later on, dinuclear complexes came to the front. We report the isolation of both a previously postulated π,σ-bis(copper) acetylide and a hitherto never-mentioned bis(metallated) triazole complex. We also demonstrate that although mono- and bis-copper complexes promote the CuAAC reaction, the dinuclear species are involved in the kinetically favored pathway.
The copper-catalyzed azide-alkyne cycloaddition (CuAAC) (1, 2) is the flagship of the “click chemistry” arsenal (3, 4). Because of the straightforward procedures, the high functional group tolerance, and the usually high yields, CuAAC has affected all fields of chemistry from material (5–8) to life sciences (9–13); however, the mechanism of the CuAAC is still under debate (14, 15). The first postulated mechanism involved mono-copper complexes, although more recently, the participation of bis(copper) complexes has been suggested on the basis of kinetic experiments (16) and computational investigations (17–19). In 2013, Fokin and colleagues published a very elegant mechanistic study and stated that “monomeric copper-acetylide complexes 1Cu are not reactive towards organic azides unless an exogenous copper catalyst is added” (20). The marked difference with the previously accepted mechanism is the introduction of the cationic π,σ-bis(copper) acetylide complexes of type 1Cu2 (Fig. 1) as the “catalytically active complex,” instead of 1Cu. However, such a dinuclear species was qualified as a “non-isolable and highly reactive intermediate.” We have recently shown that the strong σ-donating and π-accepting properties of cyclic (alkyl)(amino)carbenes (CAACs) (21–23) allow for the isolation of electron-deficient (24), electron-rich (25), and even paramagnetic species (26). This prompted us to attempt using CAAC ligands to stabilize elusive species involved in the CuAAC catalytic cycle. Here, we report the isolation of a π,σ-bis(copper) complex of type 1Cu2 and of an unprecedented catalytically active 3,5-bis(metallated) triazole 2Cu2. Thanks to the unusual stability of these complexes, we compare the mono- and dinuclear-based mechanisms and bring unambiguous evidence that the latter is indeed kinetically favored.
Fig. 1. Stepwise reproduction of the CuAAC reaction; isolation of mono- and bis(copper) intermediates involved in both catalytic pathways.

(A) Stoichiometric reactions reproducing the different steps of the postulated CuAAC catalytic cycles, which allow for the isolation of a previously postulated π,σ-bis(copper) complex of type 1Cu2 and of the never-mentioned bis(copper) triazole 2Cu2. (B) Kinetic profiles of the stoichiometric reactions of 1Cua and 1Cu2a with benzyl azide, which reveal the critical rate acceleration when the bimetallic complex 1Cu2a is used. (C) Molecular view of complexes 1Cu2a (left, table S1), 2Cu2a (center, table S2), and 1Cu2b (right, table S3) in the solid state (for clarity, H atoms and anions are omitted).
We first prepared the monomeric (27, 28) copper complex 1Cua (figs. S7 and S8) by reacting lithium phenylacetylide with (CAAC)CuOAc (figs. S1 and S2) in tetrahydrofuran (THF) (Fig. 1A). We found that 1Cua cleanly reacted with (CAAC)CuOTf (figs. S3 to S6) in methylene chloride, affording the desired cationic dinuclear complex 1Cu2a (figs. S9 and S10), which was isolated as a white solid in 95% yield. The 1H and 13C nuclear magnetic resonance (NMR) spectra show only one set of signals for both CAAC ligands, even at temperatures as low as −80°C, which is indicative of a very fast exchange between the two “Cu(CAAC)” units. However, in the solid state, a single crystal x-ray diffraction study shows two different types of coordination, π and σ (Fig. 1C, left). The Cu-Cu distance [2.9387(4) Å] is slightly longer than those computationally predicted by Fokin and colleagues for model compounds (2.85 to 2.88 Å) (19). 1Cu2a proved to be air-stable and thermally robust [melting point (mp): 174°C].
Despite its thermodynamic stability, 1Cu2a reacts cleanly with benzyl azide in methylene chloride, affording 2Cu2a (figs. S11 and S12), which was isolated in 87% yield as an air- and moisture-stable, pale yellow solid. 1H and 13C NMR spectra show the presence of one benzyl and one phenyl group, as well as two magnetically nonequivalent CAAC moieties. X-ray diffraction studies of the corresponding tetrafluoroborate salt confirmed the 3,5-dimetallated-1,2,3-triazole structure 2Cu2a (Fig. 1C, center).
Because dinuclear complexes of type 2Cu2a had never been postulated in any CuAAC mechanism, the next question was whether it was involved in the catalytic cycle. Addition of a stoichiometric amount of phenyl acetylene to 2Cu2a led, after 5 hours, to the quantitative release of the 1-benzyl-4-phenyl-1,2,3-triazole 3 accompanied by the regeneration of the bis-copper complex 1Cu2a. As an additional confirmation, the same reaction was repeated using 2Cu2a and a different alkyne (Ph2CHC≡CH), which resulted in the isolation of 3 along with the corresponding π,σ-bis-copper complex 1Cu2b (Fig. 1C, right; figs. S13 and S14) in quantitative yields.
After having shown that the bis-copper complex 1Cu2a could be part of the catalytic cycle, we turned our attention to the corresponding mono-copper complex 1Cua. We found that a stoichiometric amount of benzyl azide slowly reacted with complex 1Cua, leading, after 16 hours, to the C-metallated heterocycle 2Cua (figs. S15 and S16), which was isolated as a pale yellow solid in 61% yield (29). Addition of one equivalent of phenyl acetylene to 2Cua led, after 5 hours, to the quantitative isolation of triazole 3 and regeneration of the mono-copper complex 1Cua.
These results as a whole suggest that both the mono- and bis-copper complexes could lead to the triazole 3, at least under stoichiometric conditions. However, the kinetic profile of the reactions of 1Cua and 1Cu2a with benzyl azide revealed critical rate acceleration when the bimetallic complex is used [kobs (1Cu2a)/kobs (1Cua) > 94] (Fig. 1B and fig. S17) (30). In contrast, the protodemetallation of 2Cu2a and 2Cua affording triazole 3 and regenerating 1Cu2a and 1Cua, respectively, proceeds at a similar rate (fig. S18). It should be noted that in both cases, the protodemetallation arises from the reaction with the terminal alkyne and, thus, does not necessitate an external Brønsted acid. These reactions lead back to the acetylide complexes 1Cu2a and 1Cua, which exclude the copper precatalyst (CAAC)CuOTf from the catalytic cycle.
The experiments discussed above reproduce parts of both postulated catalytic cycles under stoichiometric conditions. Each step proved to be extremely clean and high-yielding, and could be achieved either under strictly dry and anaerobic conditions or using nondried solvent under air. To confirm that the isolated complexes are active catalytic species, phenyl acetylene was reacted with benzyl azide at room temperature in methylene chloride using 10 mol % of 1Cua and 2Cua, and only 5 mol % of 1Cu2a and 2Cu2a. The kinetic profiles of the reactions reveal that the catalytic activity of the dinuclear complexes 1Cu2a and 2Cu2a is drastically higher (94 and 99% yields of 3 after 10 hours) than that of mononuclear complexes 1Cua and 2Cua (2 and 12% yields of 3 after 10 hours) (Fig. 2A and fig. S19). These results are in agreement with those obtained for the stoichiometric reactions. Note that although the reactions promoted by 1Cu2a and 2Cu2a proceed at the same rate, a short initiation period is observed with 1Cu2a, which is concomitant with the formation of 2Cu2a; after the initiation period, the concentration of 2Cu2a remains constant over the course of the catalytic reaction (Fig. 2B). Similar observations can be made from the reactions catalyzed by complexes 1Cua and 2Cua (Fig. 2C), demonstrating that metallated triazoles 2Cua and 2Cu2a are the resting states of their respective catalytic cycles.
Fig. 2. Kinetic profiles of the CuAAC reaction of phenyl acetylene with benzyl azide using CAAC-supported mono- and bis(copper) catalysts.

(A) The kinetic profiles of the catalytic reaction of phenyl acetylene with benzyl azide demonstrate the superior catalytic activity of the dinuclear complexes 1Cu2a and 2Cu2a over their mononuclear counterparts 1Cua and 2Cua, and show that the (CAAC)CuOTf complex adopts the dinuclear pathway after an initiation period. (B) Evolution of the amount of dimetallated triazole 2Cu2a and free triazole 3 during the early period of the catalytic reaction using 1Cu2a as catalyst, showing that 2Cu2a is the resting state of the catalytic cycle. a.u., arbitrary units. (C) Same as (B), but of 2Cua and 3 using 1Cua as catalyst. (D and E) Et3N, as a proton scavenger, allows for the rapid formation of 1Cu2a from (CAAC)CuOTf and phenyl acetylene (D), and consequently shortens the induction period of the CuAAC reaction described in (A) using (CAAC)CuOTf (E).
The results described above demonstrate that the catalytic reaction operates efficiently once 1Cu2a is formed. When phenyl acetylene was reacted with benzyl azide at room temperature, in the presence of 10 mol % (CAAC)CuOTf, we observed the near quantitative formation of triazole 3, but only after 35 hours. The kinetic profile (Fig. 2A) shows a long initiation period, followed by drastic rate acceleration. Because the formation of the bis(nuclear) acetylide 1Cu2a from 1Cua is a fast reaction (vide supra), we conclude that the formation of 1Cua is the limiting step of CuAAC reaction when (CAAC)CuOTf is the precatalyst. Indeed, when two equivalents of (CAAC)CuOTf were added to phenyl acetylene, in the absence of benzyl azide, no apparent reaction occurred after 8 hours at ambient temperature (Fig. 2D). The trifluoromethanesulfonate anion is too weakly basic to efficiently promote the metallation of the alkyne (31). Addition of one equivalent of triethylamine to the previous solution cleanly afforded 1Cu2a along with Et3NH+OTf− within a few minutes. The effect of the presence of an additional base is also apparent on the kinetic profile of the catalytic reaction using 10 mol % (CAAC)CuOTf and 5 mol % triethylamine (Fig. 2E). As expected, a significant shortening of the initiation period is observed.
In conclusion, the isolation of the mono- and bis-copper acetylide complexes 1Cua and 1Cu2a demonstrates that although both species are active in the catalytic cycle, the dinuclear complex is involved in the kinetically favored pathway (Fig. 3). The stability of 1Cu2a seems to be in contradiction with previous reports (21). The peculiar electronic properties of the CAAC ligand possibly play a role (32), but the use of a weakly coordinating trifluoromethanesulfonate ligand is crucial. Indeed, when (CAAC)CuX complexes bear the more popular chloride or acetate X ligand, no bis(copper) complexes could be observed, although kinetic data demonstrate their involvement. Moreover, when the trifluoromethanesulfonate is used, investigations of the dinuclear pathway permitted the isolation of a never-postulated bis(copper) triazole complex of type 2Cu2, which is the resting state of the catalytic cycle. The alkyne serves as the proton source for the demetallation of 2Cu2, which regenerates the π,σ-bis(copper) acetylide of type 1Cu2, leaving out complexes of type 1Cu from the catalytic cycle. These results led to the broader question of whether bis(metallic) complexes of type 1Cu2 are also key species in other copper-catalyzed organic reactions (33).
Fig. 3. Mechanistic conclusions: Both the mono- and bis-copper pathways are active in the CuAAC reaction, but the latter is kinetically favored.
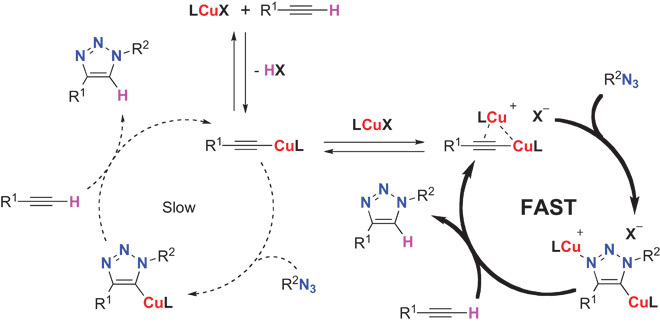
The protodemetallation is performed by the alkyne, which regenerates the metallated acetylide, thereby excluding the σ-copper acetylide from the preferred catalytic cycle.
MATERIALS AND METHODS
General
1H and 13C NMR spectra were recorded on Bruker Avance 300, Jeol ECA 500, and Varian Inova 500 spectrometers. NMR multiplicities are abbreviated as follows: s = singlet, d = doublet, t = triplet, q = quartet, sept = septet, m = multiplet, br = broad signal. Chemical shifts are given in parts per million (ppm) and are referenced to SiMe4 (1H, 13C) and CFCl3 (19F). All spectra were obtained at 25°C in the solvent indicated. Coupling constants J are given in hertz (Hz). Mass spectra were performed at the University of California San Diego Mass Spectrometry Laboratory. Melting points were measured with an electrothermal MEL-TEMP apparatus. Single crystal x-ray diffraction data were collected on a Bruker Apex II CCD (charge-coupled device) detector using Mo-Kα radiation (λ = 0.71073 Å). Crystals were selected under oil, mounted on nylon loops, and then immediately placed in a cold stream of nitrogen. Structures were solved and refined using Olex2 and SHELXTL. The CAAC and 3,3-diphenyl-1-propyne were prepared following literature procedures, whereas all other starting materials were purchased from commercial sources.
Synthesis of (CAAC)CuCCPh 1Cua
n-BuLi (2.4 mmol, 2.5 M in hexane) was slowly added to a solution of phenyl acetylene (234 mg, 2.3 mmol) in THF (15 ml) at −78°C. After 30 min, a solution of (CAAC)CuOAc (1.0 g, 2.3 mmol) in THF (10 ml) was added, and the mixture was stirred at ambient temperature for 1 hour. Volatiles were removed under vacuum, and the remaining solid was extracted with benzene (3 × 10 ml). After removal of benzene under vacuum, the solid was washed with hexane (20 ml). 1Cua was obtained as a light yellow solid. Yield 88% (970 mg). mp 152°C [decomposition (dec.)]. 1H NMR (CD2Cl2, 500 MHz): δ = 7.48 (t, J = 7.8 Hz, 1 H, p-H), 7.34 (d, J = 7.8 Hz, 2 H, m-H), 7.23 (br d, J = 7.1 Hz, 2 H, o-HC6H5), 7.14 (br t, J = 7.1 Hz, 2 H, m-HC6H5), 7.08 (br t, J = 7.1 Hz, 1 H, p-HC6H5), 2.88 (sept, J = 6.9 Hz, 2 H, CHMe2), 1.98 (s, 2 H, CH2), 1.96 to 1.87 (m, 2 H, CH2), 1.86 to 1.74 (m, 2 H, CH2), 1.39 to 1.30 (m, 18 H, CH3, CHCH3), 1.11 (t, J = 7.5 Hz, 6 H, CH3); 13C NMR (CD2Cl2, 125 MHz): δ = 253.5 (Ccarbene), 145.6 (Cq), 135.1 (Cq), 131.8 (CHC6H5), 129.9 (CHAr), 128.1 (CHC6H5), 127.7 (PhCCCu), 125.7 (CHAr), 125.0 (CHC6H5), 121.9 (Cq-C6H5), 106.3 (PhCCCu), 81.3 (Cq), 63.3 (Cq), 42.8 (CH2), 31.4 (CH2), 29.5, 29.4, 27.3, 22.4, 9.9; high-resolution mass spectrometry (HRMS) [electrospray ionization–time-of-flight mass spectrometry (ESI-TOFMS)]: mass/charge ratio (m/z) calculated for C30H40CuNNa+ 500.2349, found 500.2355.
Synthesis of bis(copper) acetylide complexes 1Cu2a
(CAAC)CuOTf (530 mg, 1 mmol) was added to a solution of 1Cua (480 mg, 1 mmol) in methylene chloride (20 ml). The reaction was stirred for 5 min at ambient temperature. After removing the solvent under vacuum, 1Cu2a was obtained as a white solid. Yield 95% (952 mg). mp 174°C (dec.). 1H NMR (CD2Cl2, 500 MHz): δ = 7.44 (t, J = 7.8 Hz, 2 H, p-H), 7.26 (d, J = 7.8 Hz, 4 H, m-H), 7.22 (t, J = 7.6 Hz, 1 H, p-H), 7.04 (t, J = 7.6 Hz, 2 H, m-H), 6.41 (d, J = 7.6 Hz, 2 H, o-H), 2.82 (sept, J = 6.9 Hz, 4 H, CHMe2), 2.05 (s, 4 H, CH2), 1.96 to 1.83 (m, 4 H, CH2), 1.80 to 1.68 (m, 4 H, CH2), 1.38 (s, 12H, CH3), 1.29 (d, J = 6.9 Hz, 12 H, CHCH3), 1.18 to 1.02 (m, 24 H); 13C NMR (CD2Cl2, 125 MHz): δ = 249.4 (Ccarbene), 145.3 (Cq-Aro), 134.7 (Cq-Aro), 132.0 (CH-Aro), 130.4 (CH-Aro), 129.4 (CH-Aro), 128.7 (CH-Aro), 125.4 (CH-Aro), 122.2 (PhCCCu2), 118.9 (Cq-Aro), 110.8 (PhCCCu2), 82.5 (Cq), 63.1 (Cq), 42.1 (CH2), 31.5 (CH2), 29.4, 27.2, 22.4, 9.9; HRMS (ESI-TOFMS): m/z calculated for C52H75Cu2N2+ 853.4517, found 853.4507.
Synthesis of bis(copper) triazole complexes 2Cu2a
Benzyl azide (40.0 mg, 0.30 mmol) was added to a solution of 1Cu2a (250 mg, 0.25 mmol) in methylene chloride (0.5 ml). After 1 hour, diethyl ether (15 ml) was added to induce the precipitation of the product. After filtration, 2Cu2a was isolated as a pale yellow solid. Yield 87% (246 mg). mp 175°C (dec.). 1H NMR (CD2Cl2, 500 MHz): δ = 7.48 to 7.38 (m, 2 H, p-H), 7.34 to 7.14 (m, 12 H), 6.82 (br, 2 H), 5.11 (s, 2 H, CH2Ph), 2.77 (sept, J = 6.9 Hz, 4 H, CHMe2), 2.01 (s, 2 H, CH2), 1.96 (s, 2 H, CH2), 1.85 to 1.73 (m, 2 H, CH2), 1.73 to 1.61 (m, 4 H, CH2), 1.61 to 1.51 (m, 2 H, CH2), 1.34 (br, 12 H, CH3), 1.29 to 1.20 (m, 12 H, CHCH3), 1.04 to 0.86 (m, 24 H); 13C NMR (CD2Cl2, 125 MHz): δ = 252.7 (Ccarbene), 249.3 (Ccarbene), 156.7 (Ctriazolide), 155.1 (Ctriazolide), 145.4 (Cq), 145.3 (Cq), 136.7 (CqAr), 135.2 (Cq), 134.8 (Cq), 133.3 (CqAr), 130.3 (CHAr), 130.2 (CHAr), 129.1 (CHAr), 128.8 (CHAr), 128.2 (CHAr), 128.1 (Cq-Ar), 128.0 (CHAr), 127.1 (CHAr), 125.2 (CHAr), 125.1 (CHAr), 82.3 (Cq), 81.9 (Cq), 63.3 (Cq), 63.0 (Cq), 57.1 (CH2Ph), 42.4 (CH2), 31.3 (CH2), 29.4, 27.0, 22.3, 9.7. HRMS (ESI-TOFMS): m/z calculated for C59H82Cu2N5+ 986.5157, found 986.5161.
Protodemetallation of 2Cu2a leading to the regeneration of 1Cu2a
Phenyl acetylene (15 mg, 0.15 mmol) was added to the solution of 2Cu2a (136 mg, 0.12 mmol) in methylene chloride (5 ml). After stirring for 16 hours at room temperature, the solvent was removed under vacuum. The remaining solid was washed with diethyl ether (3 × 10 ml) to remove triazole 3, and after drying, 1Cu2a was obtained as a yellow solid. Yield 91% (108 mg). NMR data are identical to those of a sample prepared as described above.
Supplementary Material
Acknowledgments
We are grateful to the U.S. Department of Energy (DE-FG02-13ER16370) for financial support of this work. Metrical data for the solid-state structures of 1Cu2b, 1Cu2a, and 2Cu2a are available free of charge from the Cambridge Crystallographic Data Centre under reference numbers CCDC-1042482, CCDC-1042483, and CCDC-1042484, respectively. Competing interests: The authors declare that they have no competing interests.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/1/5/e1500304/DC1
Supplementary text
Fig. S1. 1H NMR of (CAAC)CuOAc in CD2Cl2.
Fig. S2. 13C NMR of (CAAC)CuOAc in CD2Cl2.
Fig. S3. 1H NMR of (CAAC)CuCl in CD2Cl2.
Fig. S4. 13C NMR of (CAAC)CuCl in CD2Cl2.
Fig. S5. 1H NMR of (CAAC)CuOTf in CDCl3.
Fig. S6. 13C NMR of (CAAC)CuOTf in CDCl3.
Fig. S7. 1H NMR of 1Cua in CD2Cl2.
Fig. S8. 13C NMR of 1Cua in CD2Cl2.
Fig. S9. 1H NMR of 1Cu2a in CD2Cl2.
Fig. S10. 13C NMR of 1Cu2a in CD2Cl2.
Fig. S11. 1H NMR of 2Cu2a in CD2Cl2.
Fig. S12. 3C NMR of 2Cu2a in CD2Cl2.
Fig. S13. 1H NMR of 1Cu2b in CD2Cl2.
Fig. S14. 3C NMR of 1Cu2b in CD2Cl2.
Fig. S15. 1H NMR of 2Cua in CD2Cl2.
Fig. S16. 13C NMR of 2Cua in CD2Cl2.
Fig. S17. Evolution of the annulation reaction monitored by 1H NMR.
Fig. S18. Evolution of the protodemetallation reaction monitored by 1H NMR.
Fig. S19. Evolution of the catalytic reaction between benzyl azide and phenyl acetylene monitored by 1H NMR.
Table S1. Crystal data and structure refinement for 1Cu2a.
Table S2. Crystal data and structure refinement for 2Cu2a (X = BF4).
Table S3. Crystal data and structure refinement for 1Cu2b (X = BF4).
REFERENCES AND NOTES
- 1.Rostovtsev V. V., Green L. G., Fokin V. V., Sharpless K. B., A stepwise Huisgen cycloaddition process: Copper(I)-catalyzed regioselective “ligation” of azides and terminal alkynes. Angew. Chem. Int. Ed. Engl. 41, 2596–2599 (2002). [DOI] [PubMed] [Google Scholar]
- 2.Tornøe C. W., Christensen C., Meldal M., Peptidotriazoles on solid phase: [1,2,3]-Triazoles by regiospecific copper(I)-catalyzed 1,3-dipolar cycloadditions of terminal alkynes to azides. J. Org. Chem. 67, 3057–3064 (2002). [DOI] [PubMed] [Google Scholar]
- 3.Kolb H. C., Finn M. G., Sharpless K. B., Click chemistry: Diverse chemical function from a few good reactions. Angew. Chem. Int. Ed. Engl. 40, 2004–2021 (2001). [DOI] [PubMed] [Google Scholar]
- 4.Finn M. G., Fokin V. V., Click chemistry: Function follows form. Chem. Soc. Rev. 39, 1231–1232 (2010). [DOI] [PubMed] [Google Scholar]
- 5.Evans R. A., The rise of azide–alkyne 1,3-dipolar ‘click’ cycloaddition and its application to polymer science and surface modification. Aust. J. Chem. 60, 384–395 (2007). [Google Scholar]
- 6.Golas P. L., Matyjaszewski K., Marrying click chemistry with polymerization: Expanding the scope of polymeric materials. Chem. Soc. Rev. 39, 1338–1354 (2010). [DOI] [PubMed] [Google Scholar]
- 7.Hua Y., Flood A. H., Click chemistry generates privileged CH hydrogen-bonding triazoles: The latest addition to anion supramolecular chemistry. Chem. Soc. Rev. 39, 1262–1271 (2010). [DOI] [PubMed] [Google Scholar]
- 8.Liang L., Astruc D., The copper(I)-catalyzed alkyne-azide cycloaddition (CuAAC) “click” reaction and its applications. An overview. Coord. Chem. Rev. 255, 2933–2945 (2011). [Google Scholar]
- 9.He X. P., Xie J., Tang Y., Li J., Chen G. R., CuAAC click chemistry accelerates the discovery of novel chemical scaffolds as promising protein tyrosine phosphatases inhibitors. Curr. Med. Chem. 19, 2399–2405 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nájera C., Sansano J. M., 1,3-Dipolar cycloadditions: Applications to the synthesis of antiviral agents. Org. Biomol. Chem. 7, 4567–4581 (2009). [DOI] [PubMed] [Google Scholar]
- 11.El-Sagheer A. H., Brown T., Click chemistry with DNA. Chem. Soc. Rev. 39, 1388–1405 (2010). [DOI] [PubMed] [Google Scholar]
- 12.Holub J. M., Kirshenbaum K., Tricks with clicks: Modification of peptidomimetic oligomers via copper-catalyzed azide-alkyne [3 + 2] cycloaddition. Chem. Soc. Rev. 39, 1325–1337 (2010). [DOI] [PubMed] [Google Scholar]
- 13.Thirumurugan P., Matosiuk D., Jozwiak K., Click chemistry for drug development and diverse chemical-biology applications. Chem. Rev. 113, 4905–4979 (2013). [DOI] [PubMed] [Google Scholar]
- 14.Meldal M., Tornøe C. W., Cu-catalyzed azide-alkyne cycloaddition. Chem. Rev. 108, 2952–3015 (2008). [DOI] [PubMed] [Google Scholar]
- 15.Berg R., Straub B. F., Advancements in the mechanistic understanding of the copper-catalyzed azide–alkyne cycloaddition. Beilstein J. Org. Chem. 9, 2715–2750 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rodionov V. O., Fokin V. V., Finn M. G., Mechanism of the ligand-free CuI-catalyzed azide–alkyne cycloaddition reaction. Angew. Chem. Int. Ed. Engl. 44, 2210–2215 (2005). [DOI] [PubMed] [Google Scholar]
- 17.Himo F., Lovell T., Hilgraf R., Rostovtsev V. V., Noodleman L., Sharpless K. B., Fokin V. V., Copper(I)-catalyzed synthesis of azoles. DFT study predicts unprecedented reactivity and intermediates. J. Am. Chem. Soc. 127, 210–216 (2005). [DOI] [PubMed] [Google Scholar]
- 18.Ahlquist M., Fokin V. V., Enhanced reactivity of dinuclear copper(I) acetylides in dipolar cycloadditions. Organometallics 26, 4389–4391 (2007). [Google Scholar]
- 19.Straub B. F., μ-Acetylide and μ-alkenylidene ligands in “click” triazole syntheses. Chem. Commun. 2007, 3868–3870 (2007). [DOI] [PubMed] [Google Scholar]
- 20.Worrell B. T., Malik J. A., Fokin V. V., Direct evidence of a dinuclear copper intermediate in Cu(I)-catalyzed azide-alkyne cycloadditions. Science 340, 457–460 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lavallo V., Canac Y., Präsang C., Donnadieu B., Bertrand G., Stable cyclic (alkyl)(amino)carbenes as rigid or flexible, bulky, electron-rich ligands for transition-metal catalysts: A quaternary carbon atom makes the difference. Angew. Chem. Int. Ed. Engl. 44, 5705–5709 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Melaimi M., Soleilhavoup M., Bertrand G., Stable cyclic carbenes and related species beyond diaminocarbenes. Angew. Chem. Int. Ed. Engl. 49, 8810–8849 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Soleilhavoup M., Bertrand G., Cyclic (alkyl)(amino)carbenes (CAACs): Stable carbenes on the rise. Acc. Chem. Res. 48, 256–266 (2015). [DOI] [PubMed] [Google Scholar]
- 24.Ung G., Rittle J., Soleilhavoup M., Bertrand G., Peters J. C., Two-coordinate Fe⁰ and Co⁰ complexes supported by cyclic (alkyl)(amino)carbenes. Angew. Chem. Int. Ed. Engl. 53, 8427–8431 (2014). [DOI] [PubMed] [Google Scholar]
- 25.Weinberger D. S., Melaimi M., Moore C. E., Rheingold A. L., Frenking G., Jerabek P., Bertrand G., Isolation of neutral mono- and dinuclear gold complexes of cyclic (alkyl)(amino)carbenes. Angew. Chem. Int. Ed. Engl. 52, 8964–8967 (2013). [DOI] [PubMed] [Google Scholar]
- 26.Martin C. D., Soleilhavoup M., Bertrand G., Carbene-stabilized main group radicals and radical ions. Chem. Sci. 4, 3020–3030 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Most copper acetylides are polymeric in nature (29) with the exception of a (NHC)CuC ≡ CPh complex (16).
- 28.Garbuzova I. A., Gol’ding I. R., Schegolikhin A. N., Peculiar intensity distribution in the Raman spectra of copper organoacetylides. J. Raman Spectrosc. 26, 391–395 (1995). [Google Scholar]
- 29.Nolte C., Mayer P., Straub B. F., Isolation of a copper(I) triazolide: A “click” intermediate. Angew. Chem. Int. Ed. Engl. 46, 2101–2103 (2007). [DOI] [PubMed] [Google Scholar]
- 30.One of the reviewers suggested that traces of copper(I) dirt could be responsible for the monocopper cycloaddition, a hypothesis that cannot be totally ruled out. Consequently, the rate acceleration can only be greater than 94.
- 31.Bai R., Zhang G., Yi H., Huang Z., Qi X., Liu C., Miller J. T., Kropf A. J., Bunel E. E., Lan Y., Lei A., Cu(II)-Cu(I) synergistic cooperation to lead the alkyne C-H activation. J. Am. Chem. Soc. 136, 16760–16763 (2014). [DOI] [PubMed] [Google Scholar]
- 32.Back O., Henry-Ellinger M., Martin C. D., Martin D., Bertrand G., 31P NMR chemical shifts of carbene–phosphinidene adducts as an indicator of the π-accepting properties of carbenes. Angew. Chem. Int. Ed. Engl. 52, 2939–2943 (2013). [DOI] [PubMed] [Google Scholar]
- 33.Allen S. E., Walvoord R. R., Padilla-Salinas R., Kozlowski M. C., Aerobic copper-catalyzed organic reactions. Chem. Rev. 113, 6234–6458 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/1/5/e1500304/DC1
Supplementary text
Fig. S1. 1H NMR of (CAAC)CuOAc in CD2Cl2.
Fig. S2. 13C NMR of (CAAC)CuOAc in CD2Cl2.
Fig. S3. 1H NMR of (CAAC)CuCl in CD2Cl2.
Fig. S4. 13C NMR of (CAAC)CuCl in CD2Cl2.
Fig. S5. 1H NMR of (CAAC)CuOTf in CDCl3.
Fig. S6. 13C NMR of (CAAC)CuOTf in CDCl3.
Fig. S7. 1H NMR of 1Cua in CD2Cl2.
Fig. S8. 13C NMR of 1Cua in CD2Cl2.
Fig. S9. 1H NMR of 1Cu2a in CD2Cl2.
Fig. S10. 13C NMR of 1Cu2a in CD2Cl2.
Fig. S11. 1H NMR of 2Cu2a in CD2Cl2.
Fig. S12. 3C NMR of 2Cu2a in CD2Cl2.
Fig. S13. 1H NMR of 1Cu2b in CD2Cl2.
Fig. S14. 3C NMR of 1Cu2b in CD2Cl2.
Fig. S15. 1H NMR of 2Cua in CD2Cl2.
Fig. S16. 13C NMR of 2Cua in CD2Cl2.
Fig. S17. Evolution of the annulation reaction monitored by 1H NMR.
Fig. S18. Evolution of the protodemetallation reaction monitored by 1H NMR.
Fig. S19. Evolution of the catalytic reaction between benzyl azide and phenyl acetylene monitored by 1H NMR.
Table S1. Crystal data and structure refinement for 1Cu2a.
Table S2. Crystal data and structure refinement for 2Cu2a (X = BF4).
Table S3. Crystal data and structure refinement for 1Cu2b (X = BF4).