

Abstract
Attention deficits in Alzheimer's disease can exacerbate its other cognitive symptoms, yet relevant disruptions of key prefrontal circuitry are not well understood. Here, in the TgCRND8 mouse model of this neurological disorder, we demonstrate and characterize a disruption of cholinergic excitation in the major corticothalamic layer of the prefrontal cortex, in which modulation by acetylcholine is essential for optimal attentional function. Using electrophysiology with concurrent multiphoton imaging, we show that layer 6 pyramidal cells are unable to sustain cholinergic excitation to the same extent as their nontransgenic littermate controls, as a result of the excessive activation of calcium-activated hyperpolarizing conductances. We report that cholinergic excitation can be improved in TgCRND8 cortex by pharmacological blockade of SK channels, suggesting a novel target for the treatment of cognitive dysfunction in Alzheimer's disease.
SIGNIFICANCE STATEMENT Alzheimer's disease is accompanied by attention deficits that exacerbate its other cognitive symptoms. In brain slices of a mouse model of this neurological disorder, we demonstrate, characterize, and rescue impaired cholinergic excitation of neurons essential for optimal attentional performance. In particular, we show that the excessive activation of a calcium-activated potassium conductance disrupts the acetylcholine excitation of prefrontal layer 6 pyramidal neurons and that its blockade normalizes responses. These findings point to a novel potential target for the treatment of cognitive dysfunction in Alzheimer's disease.
Keywords: acetylcholine receptors, afterhyperpolarization potential (AHP), Alzheimer's model, corticothalamic, prefrontal cortex, SK channels
Introduction
Alzheimer's disease (AD) is a neurodegenerative disorder that constitutes the main cause of dementia. It is associated with profound cognitive impairments that include severe memory loss and early attention deficits (Grady et al., 1988; Perry and Hodges, 1999; Baddeley et al., 2001; Romberg et al., 2013a). Impaired attention negatively affects the day-to-day life of AD patients, contributing to distraction, confusion, and poor executive control (Perry and Hodges, 1999). It is likely that disruptions in attention further exacerbate the memory deficits that constitute a telltale sign of the disease: if you can't attend, you can't encode (Romberg et al., 2013a,b). However, little is understood of the cellular mechanisms that underlie the non-mnemonic deficits of executive function in AD and their contribution to the disease.
To date, acetylcholinesterase inhibitors constitute the standard of care for AD (Citron, 2010), although loss of cholinergic cells is an end-stage manifestation of the disease that is unlikely to account for cognitive deficits that may arise much earlier (Davis et al., 1999; Terry and Buccafusco, 2003). We now know that the cholinergic modulation of the prefrontal cortex (PFC) is essential for optimal attentional performance (Dalley et al., 2004; Parikh et al., 2007; Bailey et al., 2010; Guillem et al., 2011; Proulx et al., 2014a). Acetylcholine (ACh) is released in the PFC during attention tasks (Parikh et al., 2007), in which it exerts robust direct excitation of the deep cortical layers (Kassam et al., 2008; Poorthuis et al., 2013; Proulx et al., 2014b). The powerful and prolonged activation of layer 6 pyramidal neurons by ACh is consistent with the sustained firing necessary for attention and other executive functions (Funahashi et al., 1989; Hasselmo and Sarter, 2011; Ikkai and Curtis, 2011; Zhou et al., 2011) and depends on a complex interplay of nicotinic- and muscarinic-mediated signaling (Bailey et al., 2010; Tian et al., 2011). Manifestations of AD in people and animal models have long been linked to aberrations in cholinergic enzymatic activity (Davies and Maloney, 1976; Perry et al., 1978; Henke and Lang, 1983; Zubenko et al., 1989; Davis et al., 1999; Selkoe, 2001; Auld et al., 2002; DeKosky et al., 2002), nicotinic receptor binding (Nordberg et al., 1988; Whitehouse et al., 1988; Marutle et al., 1999; Nordberg, 2001; O'Brien et al., 2007; Kendziorra et al., 2011), and cholinergic signaling pathways (Kelly et al., 1996; Jope et al., 1997; Jope, 1999; Liu et al., 2001; Pettit et al., 2001; Auld et al., 2002). How do these broad changes affect the activation and sustained firing of prefrontal layer 6 attention circuitry?
To address this question, we examined cholinergic signaling in prefrontal attention circuitry of a well characterized animal model of AD. The TgCRND8 mouse model is used widely in Alzheimer's research and encodes both the Swedish and Indiana mutations of amyloid precursor protein (APP) found in inherited familial AD (Chishti et al., 2001). Here, we report significant impairments in the cholinergic excitation of layer 6 prefrontal neurons of TgCRND8 mice by 3–4 months of age and show that these deficits can be rescued pharmacologically.
Materials and Methods
Animals.
All experiments were approved by the University of Toronto Animal Care Committee. We used adult male and female TgCRND8 mice on a mixed C57BL/6–C3H background, as reported previously (Chishti et al., 2001), along with their nontransgenic littermate siblings [wild type (WT)] as controls (mean ± SEM age: WT, 109 ± 2 d, n = 20 animals; TgCRND8, 109 ± 2 d, n = 26 animals).
Brain slice preparation and recording conditions.
Each brain was cooled as rapidly as possible with 4°C oxygenated sucrose artificial CSF (ACSF; 254 mm sucrose substituted for NaCl). Coronal slices (400 μm thick) of the PFC were cut on a Dosaka Pro-7 Linear Slicer (SciMedia) and were transferred to 30°C oxygenated ACSF (in mm: 128 NaCl, 10 d-glucose, 26 NaHCO3, 2 CaCl2, 2 MgSO4, 3 KCl, and 1.25 NaH2PO4, pH 7.4) in a prechamber (Automate Scientific) and allowed to recover for at least 1.5 h before the beginning of an experiment. Slices were placed in a chamber on the stage of an upright microscope for whole-cell recordings. ACSF was bubbled with 95% oxygen and 5% carbon dioxide and flowed over the slice at 30°C with a rate of 3–4 ml/min.
Electrophysiological recordings.
Layer 6 pyramidal neurons were patched under visual control using infrared differential interference contrast microscopy in the cingulate and prelimbic regions of the PFC. Intracellular patch solution contained the following (in mm): 120 K-gluconate, 5 KCl, 2 MgCl2, 4 K-ATP, 0.4 Na2-GTP, 10 Na2-phosphocreatine, and 10 HEPES buffer, adjusted to pH 7.33 with KOH. Data were acquired and low-pass filtered at 20 kHz with an Axopatch 200b amplifier (Molecular Devices) and Digidata1440 digitizer/pClamp10.2 software (Molecular Devices). Intrinsic cellular properties were assessed in current-clamp mode and did not differ significantly between genotypes (Table 1; mean ± SEM; sample size for this analysis, n = 49 WT neurons; n = 58 TgCRND8 neurons).
Table 1.
Electrophysiological properties of PFC layer 6 pyramidal neurons of TgCRND8 mice and littermate WT controls
| WT | TgCRND8 | P | |
|---|---|---|---|
| Capacitance (pF) | 72 ± 2 | 71 ± 2 | 0.5 |
| Input resistance (MΩ) | 313 ± 18 | 285 ± 16 | 0.2 |
| Membrane potential (mV) | −83 ± 1 | −85 ± 1 | 0.3 |
| Spike threshold (mV) | −52 ± 0.4 | −51 ± 0.5 | 0.4 |
| Spike amplitude (mV) | 79 ± 1 | 80 ± 1 | 0.5 |
All drugs were bath applied. ACh chloride and atropine were obtained from Sigma, apamin from Alomone Labs, dihydro-β-erythroidine hydrobromide (DHβE) and AP-5 from R&D Systems, and cadmium chloride from Emerald BioSystems.
Calcium imaging.
The calcium dye Oregon Green BAPTA-1 (OGB-1; 100 μm) was included in the pipette in a subset of cells, along with Alexa Fluor-594 hydrazide (20 μm), to aid in the visualization of the neuron. Multiphoton imaging was performed using a titanium:sapphire laser (Newport) tuned to wavelength 800 nm and an Olympus Fluoview FV1000 microscope with a 60× water-immersion 0.90 numerical aperture objective. The emitted fluorescence was separated into green (OGB-1 signal) and red (Alexa Fluor-594 signal) channels with a dichroic mirror at 570 nm and filtered (green, barrier filter 495–540; red, barrier filter 570–620) before detection. Images were acquired at a rate of ∼10 frames/s and analyzed with Fluoview software. A pan-somatic area of interest was selected for analysis, and green fluorescence increases were calculated relative to baseline fluorescence (dF/F). The green/red fluorescence (G/R) ratio was calculated by dividing the green signal by that of the red channel for the purpose of evaluating potential differences in basal calcium levels between genotypes.
Statistical analyses.
Results are expressed as mean ± SEM, and all statistical comparisons were made at a significance level of 0.05 (Prism versions 5.0d/6.0; GraphPad Software). Average traces of postburst potentials and fluorescent increases were generated with Axograph X. For the purpose of quantifying ACh-elicited firing, input–output curves and postburst afterhyperpolarization potentials (AHPs), action potentials were detected automatically with a derivative threshold of 20 mV/ms. To assess the ability of cells to sustain near-maximal firing in response to ACh, the cumulative probability of the instantaneous firing frequency of individual action potentials was normalized to the maximum instantaneous firing frequency within each ACh-elicited train of action potentials. By normalizing to maximum instantaneous frequency within the cell, neurons with a greater number of spikes will not unduly skew the cumulative probability distribution. This analysis further provides insight about the extent to which each spike train can sustain near-maximal firing. Genotype and treatment differences of cumulative probabilities were assessed with Kolmogorov–Smirnov (K–S) tests. The peak and area under the curve (AUC) of postburst potentials were measured from the peak of the last action potential in each burst up to 1 s post-burst.
Results
TgCRND8 mice cannot sustain peak excitation by ACh in prefrontal layer 6
Because there is a panoply of cholinergic-related deficits in AD, we investigated the effects of known concentrations of bath-applied ACh on brain slices from littermate WT and TgCRND8 mice at an age when attention deficits may be developing. In layer 6 pyramidal neurons of the PFC, ACh elicited cellular depolarization in a concentration-dependent manner in both WT and TgCRND8 mice (Fig. 1). Spiking was elicited in fewer cells in TgCRND8 than WT mice (Fig. 1A; three-way contingency test, Gdf = 72 = 70.72, p < 0.0001). Furthermore, in cells in which ACh (1 mm) elicited suprathreshold depolarizations, ACh-elicited firing patterns were revealed to be distinct. The distribution of instantaneous frequency measurements for individual action potentials were significantly different between groups (Fig. 1B; p < 0.004, Mann–Whitney test), and individual TgCRND8 neurons failed to sustain near-maximal firing frequencies to the same extent as WT cells (Fig. 1C): the cumulative probability of action potential frequency—normalized to maximal instantaneous frequency within each cell—was reduced dramatically in TgCRND8 neurons (K–S test, p < 0.00001). However, the maximal instantaneous firing frequency (WT, 9 ± 1 Hz, n = 17; TgCRND8, 11 ± 3 Hz, n = 15; t test, p = 0.4) and duration of spiking (WT, 47 ± 11 s, n = 17; TgCRND8, 42 ± 9 s, n = 15; t test, p = 0.7) achieved were similar between genotypes. The depolarization elicited by ACh was not significantly different between genotypes as revealed by two-way repeated-measures ANOVA (Fig. 1D; effect of genotype, F(1,40) = 0.29, p = 0.6). Additional probes of the electrophysiological effects of ACh in layer 6 pyramidal cells in voltage clamp found that, near resting membrane potential (Vhold = −75 mV), ACh currents were modestly but significantly smaller in TgCRND8 mice (WT, −121 ± 8 pA, n = 23; TgCRND8, −92 ± 9 pA, n = 28; p = 0.02). However, pharmacological dissection of these current revealed similar nicotinic (WTatropine, −83 ± 12 pA, n = 7; TgCRND8atropine, −86 ± 12 pA, n = 9; p = 0.9) and muscarinic (WTDHBE, −33 ± 7 pA, n = 8; TgCRND8DHBE, −22 ± 6 pA, n = 7; p = 0.3) contributions, suggesting that the ACh current may be decreased in the TgCRND8 mice through an interaction of nicotinic and muscarinic signaling. Therefore, to compensate for any potential differences in the cholinergic drive toward threshold between the genotypes, we applied current to the cells to elicit baseline firing at 1–3 Hz. Under these conditions, TgCRND8 neurons also failed to achieve equivalent levels of peak firing (maximal instantaneous frequency: WT, 21 ± 2 Hz, n = 10 cells; TgCRND8, 15 ± 2 Hz, n = 13 cells; p < 0.05; cumulative probability of action potential instantaneous frequencies, K–S test, p < 0.00001; data not shown). Altogether, these data suggest that the deficit in layer 6 neurons at this early stage in TgCRND8 mice appears to be selectively limited to a marked impairment in the ability to maintain peak excitation to ACh.
Figure 1.
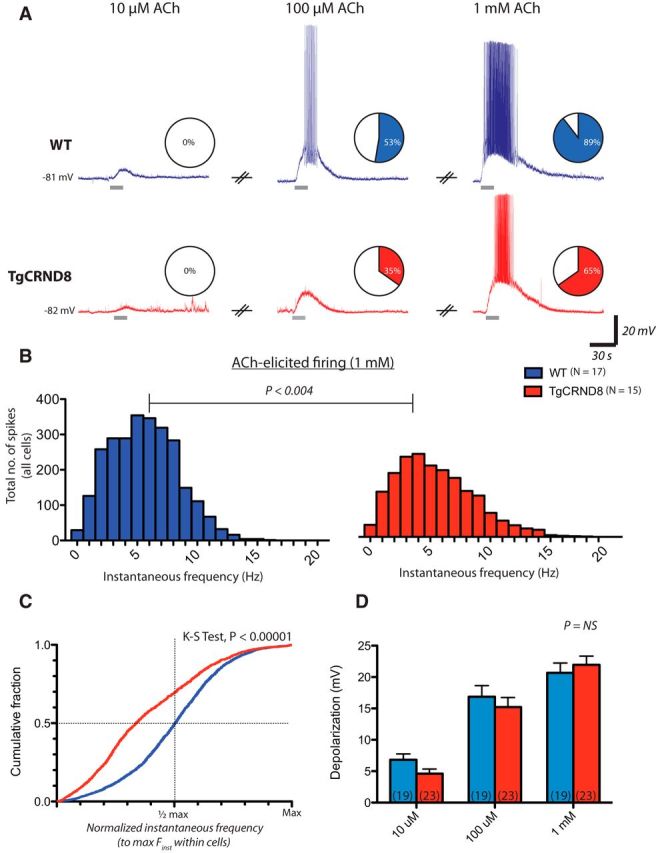
Impaired excitation in response to ACh in layer 6 PFC of TgCRND8 mice. Bath application of ACh elicits cellular depolarization from resting membrane potential in a concentration-dependent manner in both WT and TgCRND8 neurons. ACh was applied at 10 μm, 100 μm, and 1 mm in WT (n = 19 matched pairs) and TgCRND8 (n = 23 matched pairs). A, Representative example of responses in a WT cell (top, blue traces) and a TgCRND8 neuron (bottom, red traces). Pie charts depict the proportion of cells in which ACh elicited depolarizations that led to spiking. ACh elicited spiking in 0 of 19 cells in WT and 0 of 23 cells in TgCRND8 when applied at 10 μm, in 10 of 19 (53%) cells in WT and 8 of 23 (35%) cells in TgCRND8 at 100 μm, and in 17 of 19 (89%) cells in WT and 15 of 23 (65%) cells in TgCRND8 mice when applied at 1 mm (three-way contingency test, G2df = 7 = 70.72, p < 0.0001). B, Frequency histograms of instantaneous firing frequencies elicited by ACh (1 mm) in all cells (p < 0.004, Mann–Whitney test). C, The cumulative probability of action potential firing frequency elicited by 1 mm ACh [normalized to maximal instantaneous frequency (Finst) within each cell] reveals that TgCRND8 neurons fail to sustain near maximal firing frequencies to the same extent as WT cells (K–S test, p < 0.00001). Note that the distribution is left-skewed in TgCRND8 neurons, with a greater number of observations falling below half-maximal frequency values. D, Mean depolarization was 7 ± 1 mV in WT and 5 ± 1 mV in TgCRND8 at 10 μm, 17 ± 2 mV in WT and 15 ± 2 mV in TgCRND8 at 100 μm, and 21 ± 2 mV in WT and 22 ± 1 mV in TgCRND8 at 1 mm ACh. The depolarization achieved was not significantly different between genotypes as revealed by two-way repeated-measures ANOVA (effect of genotype, F(1,40) = 0.29, p = 0.6).
Probing mechanisms of altered excitability in TgCRND8 prefrontal layer 6 neurons
Given the observed differences in sustaining ACh-elicited peak firing, we investigated whether there were differences in intrinsic excitability in prefrontal layer 6 pyramidal neurons. We generated input–output curves by applying depolarizing current steps of 500 ms duration in 50 pA increments. A clear difference between genotypes was evident in the layer 6 neurons at stronger depolarizations, with fewer action potentials elicited in TgCRND8 mice (Fig. 2; effect of genotype, two-way ANOVA, F(1,253) = 8.12, p = 0.005). However, at rheobase, there were no significant differences in action potential amplitude (WT, 79 ± 3 mV, n = 13; TgCRND8, 77 ± 2, n = 12; p = 0.5), rise time (WT, 282 ± 12 μs, n = 13; TgCRND8, 289 ± 11 μs, n = 12; p = 0.7), half-width (WT, 1 ± 0.03 ms, n = 13; TgCRND8, 1 ± 0.03 ms, n = 12; p = 0.4), or current applied (WT, 66 ± 10 pA, n = 13; TgCRND8, 84 ± 11 pA, n = 12; p = 0.2) in the same subset of cells.
Figure 2.

Reduced intrinsic excitability in layer 6 prefrontal pyramidal neurons of TgCRND8 mice. Input–output curves were generated by applying depolarizing current steps of 500 ms duration in 50 pA increments from resting membrane potential. A, Representative example of a train of action potentials elicited by a 250 pA current step in WT (top, blue trace) and TgCRND8 (bottom, red trace) neurons. Bottom, A schematic representation of the current injection. B, Input–output curves in WT cells (blue, n = 13) and TgCRND8 cells (red, n = 12 cells). Data points represent mean ± SEM. Effect of genotype is significant as revealed by two-way ANOVA (F(1,253) = 8.12, p = 0.005).
The neuronal ability to sustain repetitive spike firing is dependent on the electrophysiological phenomenon known as the AHP (Kandel and Spencer, 1961; Sah, 1996; Disterhoft and Oh, 2006; Bean, 2007), which constitutes the undershoot of the action potential and results from hyperpolarizing conductances activated during spiking. Accordingly, we examined systematically whether there were genotype differences in the AHP that occurs in response to the identical numbers of action potentials in TgCRND8 cells and WT controls. Action potentials were elicited by applying brief depolarizing current pulses (2 nA, 2 ms) at 50 Hz from a membrane potential of −70 mV (Fig. 3). Both the peak AHP and postburst area were significantly greater in TgCRND8 mice as revealed by two-way ANOVA (Fig. 3B,C; effect of genotype, peak AHP, F(1,259) = 8.35, p = 0.004; postburst area, F(1,222) = 18.53, p < 0.0001). A more detailed analysis further revealed that the greatest differences were observed between 100 and 500 ms after burst (data not shown), suggesting differences in what is considered the medium AHP (Sah, 1996; Faber and Sah, 2003).
Figure 3.

The AHP is enhanced in layer 6 pyramidal neurons of TgCRND8 mice. Action potentials were elicited by applying brief depolarizing current pulses (2 nA, 2 ms) at 50 Hz from a membrane potential of −70 mV. A depicts the overlay of the average responses in WT (blue, n = 16 cells) and TgCRND8 (red, n = 23 cells). A schematic of the spike burst protocol is shown below. B, Peak AHP in WT and TgCRND8 (two-way ANOVA, effect of genotype, F(1,259) = 8.35, p = 0.004). C, Mean postburst AUC in WT and TgCRND8 (two-way ANOVA, effect of genotype, F(1,222) = 18.53, p < 0.0001). Data represented in B and C are mean ± SEM.
Because several components of the AHP exhibit calcium dependence (Sah, 1996; Faber and Sah, 2003), we sought to determine whether enhanced postburst AHPs were correlated with increased calcium responses in layer 6 TgCRND8 neurons. Accordingly, this subset of our AHP experiments were performed with concurrent multiphoton imaging of the intracellular calcium indicator OGB-1 (100 μm) in the recorded layer 6 pyramidal neurons. The spike burst protocols elicited changes in intracellular calcium levels, as measured by changes in fluorescence with respect to baseline levels (dF/F). These changes depended on the number of spikes in both genotypes (effect of number of spikes, peak dF/F, F(5,222) = 93.06, p < 0.0001; dF/F area, F(5,122) = 80.48, p < 0.0001) but were significantly greater in layer 6 TgCRND8 neurons (Fig. 4; peak dF/F, effect of genotype, F(1,222) = 11.82, p = 0.0007; dF/F area, effect of genotype, F(1,122) = 12.61, p = 0.0005). Baseline fluorescence was not different between genotypes, as assessed by the G/R ratio (WT, 233 ± 13 units, n = 16; TgCRND8, 230 ± 15 units, n = 23; p = 0.9). Moreover, the linear regression of mean peak AHP against mean peak dF/F in WT and TgCRND8 neurons were similar between genotypes, suggesting that AHPs are enhanced in TgCRND8 mice as a result of enhanced calcium increases (slopes of the linear regressions, p = 0.2).
Figure 4.

Spiking elicits greater Ca2+ responses in TgCRND8 neurons. A, In the same subset of cells as Figure 3, changes in calcium with respect to baseline levels (dF/F) elicited by the spike burst protocol detected by the calcium indicator OGB-1 (100 μm) with two-photon microscopy. An overlay of the average responses in WT (blue, n = 16) and TgCRND8 (red, n = 23) is shown, and a schematic of the spike burst protocol is depicted below. B, Quantification of the peak dF/F (mean ± SEM) in WT and TgCRND8 neurons (two-way ANOVA, effect of genotype, F(1,222) = 11.82, p = 0.0007). C, AUC of dF/F in WT and TgCRND8 (two-way ANOVA, effect of genotype, F(1,122) = 12.61, p = 0.0005).
One candidate channel for a calcium-dependent medium AHP is the small-conductance calcium-activated potassium channel known as SK (Sah, 1996; Faber and Sah, 2003). We found that, in WT layer 6 PFC pyramidal neurons, the postburst AHP was reduced by the SK-specific toxin apamin, particularly between 50 and 100 ms after the burst (Fig. 5). In TgCRND8 layer 6 neurons, the postburst AHP was also reduced by apamin (TgCRND8control vs TgCRND8apamin, peak AHP, F(1,258) = 39.14, p < 0.0001; AUC, F(1,258) = 33.59, p < 0.0001). Moreover, between 100 and 500 ms—at which the genotype difference was most pronounced—differences were abolished in the presence of apamin (Fig. 6; WTapamin vs TgCRND8apamin, peak AHP100–500 ms, F(1,234) = 0.04, p = 0.84; AUC100–500 ms, F(1,234) = 0.40, p = 0.53), suggesting that the enhanced AHPs in TgCRND8 mice result from a greater SK contribution. Interestingly, the apamin experiments did reveal a residual, apamin-resistant early AHP in TgCRND8 neurons (one to four spikes). Overall, however, the SK inhibitor apamin restored AHP area to WT control levels in TgCRND8 neurons (WTcontrol vs TgCRND8apamin, two-way ANOVA, F(1,216) = 1.03, p = 0.3) and also abolished genotype differences in intrinsic excitability as assessed by input–output curves (WTcontrol vs TgCRND8apamin, two-way ANOVA, F(1,168) = 0.01, p = 0.9).
Figure 5.

The postburst AHP is apamin sensitive in WT. A, An example of postburst AHPs elicited under control condition in WT and an example of postburst AHPs elicited in the presence of the SK antagonist apamin (200 nm) in another layer 6 PFC neuron from WT (bottom). B, C, In control versus apamin conditions, measurements of the peak AHP (two-way ANOVA, treatment effect, F(1,198) = 39.62, p < 0.0001) and postburst area (two-way ANOVA, treatment effect, F(1,198) = 10.70, p = 0.001).
Figure 6.

Enhanced AHPs in TgCRND8 neurons result from a greater SK contribution. A, Overlay of the postburst AHPs elicited by the spike burst protocol in the presence of apamin (200 nm) in WT (blue, n = 19 cells) and TgCRND8 (red, n = 22 cells) mice. B, C, Measurements of the peak AHP (two-way ANOVA, effect of genotype, F(1,234) = 4.90, p = 0.03) and postburst area (two-way ANOVA, effect of genotype, F(1,234) = 5.94, p = 0.02). Apamin was most effective at normalizing the post-burst AHP in TgCRND8 in which the genotype difference was most pronounced (WTapamin vs TgCRND8apamin, peak AHP100–500 ms, F(1,234) = 0.04, p = 0.84; AUC100–500 ms, F(1,234) = 0.40, p = 0.53) but reveals a residual, apamin-resistant early AHP in TgCRND8 (1–4 spikes, arrowheads).
Enhanced suprathreshold calcium flux underlies aberrations in the cholinergic excitation of layer 6 pyramidal neurons of TgCRND8 animals
Cholinergic stimulation of layer 6 pyramidal neurons has the potential to raise intracellular calcium levels substantially. Not only does ACh bring the majority of these neurons to spiking—thereby permitting the activation of voltage-sensitive channels that flux calcium (i.e., NMDA receptors, voltage-gated calcium channels)—but the ACh receptors themselves are poised to contribute to intracellular calcium increases. Indeed, in layer 6 PFC, ACh stimulates an unusual subtype of nicotinic receptor (Bailey et al., 2010) known to have high calcium flux (Tapia et al., 2007) and stimulates excitatory muscarinic receptors (Tian et al., 2011), which can also release calcium from intracellular stores in prefrontal neurons (Gulledge and Stuart, 2005; Gulledge et al., 2009; Proulx et al., 2014b). Thus, given the observation that cholinergic firing is impaired in TgCRND8 neurons and that calcium-dependent AHPs are enhanced in these same cells, we next investigated whether ACh-elicited spiking is accompanied by greater calcium responses in the TgCRND8 neurons. ACh (1 mm, 15 s) elicited spiking in eight of nine cells in WT and six of seven cells in TgCRND8 mice with calcium increases that were significantly greater in the TgCRND8, as detected by the calcium-sensitive dye OGB-1 (100 μm; Fig. 7; peak dF/F: WT, 50 ± 9%, n = 8; TgCRND8, 78 ± 7%, n = 6, p = 0.04; area of dF/F: WT, 1090 ± 240% × s, n = 8, TgCRND8, 1950 ± 500% × s, n = 6, p = 0.1). Furthermore, in this independent replication, we also observed that TgCRND8 cells failed to sustain firing frequencies to the same extent as WT neurons (cumulative probabilities of normalized instantaneous frequency, K–S test, p < 0.00001). Thus, impairments in cholinergic firing in the TgCRND8 neurons were accompanied by increased calcium responses.
Figure 7.
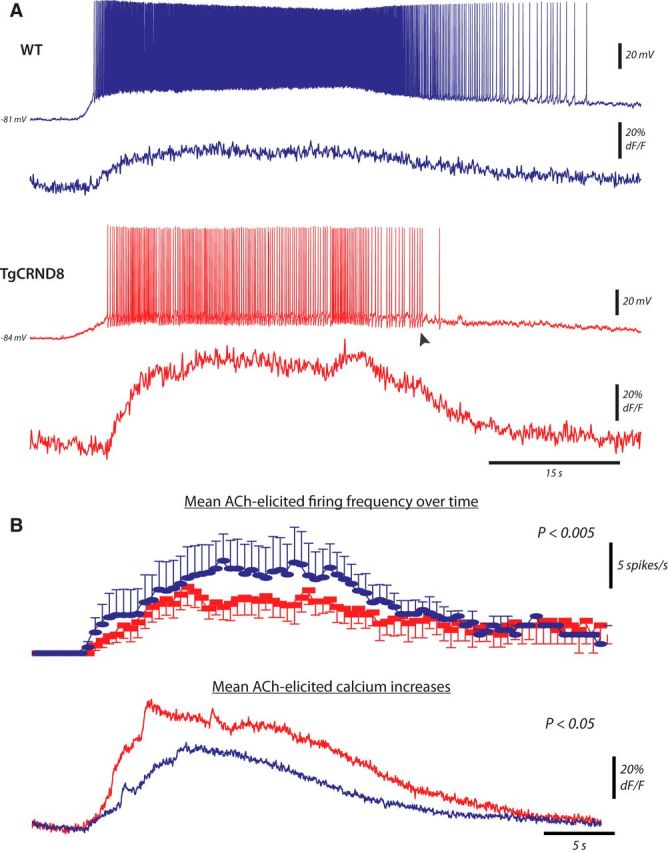
Greater Ca2+ responses accompany ACh-elicited firing in the TgCRND8. ACh (1 mm, 15 s) elicited spiking in eight of nine cells in WT and six of seven in TgCRND8, with calcium increases that were significantly greater in the TgCRND8 cells as detected by the calcium-sensitive dye OGB-1 (100 μm). A, Example of whole-cell membrane potential (top) and calcium increases (bottom) simultaneously recorded in response to bath application of ACh in WT (top panel) and TgCRND8 (bottom panel). Note the large AHPs in the TgCRND8 trace (arrowhead). B, Average number of spikes per seconds (top) and calcium responses (bottom) elicited by ACh in the WT (blue, n = 8) and TgCRND8 (red, n = 6 cells) cells.
Because multiple sources of calcium may contribute to these cholinergic calcium responses in layer 6 neurons of PFC, we examined genotype differences in both subthreshold and suprathreshold sources of calcium ion flux. In voltage clamp, to prevent spiking and the opening of voltage-gated channels, calcium responses elicited by ACh were not different between genotypes when the membrane potential was clamped at −75 mV (peak dF/F: WT, 46 ± 6%, n = 16; TgCRND8, 48 ± 7%, n = 18; p = 0.8; area of dF/F: WT, 638 ± 110% * s, n = 16; TgCRND8, 606 ± 65% * s, n = 18, p = 0.8). These data suggest that cholinergic stimulation greatly enhances cellular calcium increases in the TgCRND8 mice only when coincident with trains of action potentials. To probe these genotype differences in suprathreshold calcium influx, we next examined ACh calcium responses in current clamp in the presence of the NMDA receptor channel antagonist AP-5 (50 μm) or in the presence of the voltage-gated calcium channel blocker CdCl2 (100 μm). We found that ACh-elicited calcium responses were significantly reduced with respect to ACSF control in the TgCRND8 neurons but not in WT cells, in either AP-5 (peak dF/F: WTAP-5, 68 ± 9, n = 7, p = 0.2; TgCRND8AP-5, 38 ± 7%, n = 7, p = 0.002) or CdCl2 (peak dF/F: WTCdCl2, 32 ± 9%, n = 5, p = 0.2; TgCdCl2, 15 ± 4%, n = 7, p < 0.0001). In addition, each manipulation could improve the ability of TgCRND8 neurons to sustain cholinergic firing (K–S tests, TgCRND8control vs TgCRND8AP5, p < 0.0005; TgCRND8control vs TgCRND8CdCl2, p < 0.00001), consistent with the interpretation that the dysregulation of a calcium-sensitive conductance underlies impairments in the cholinergic excitation of TgCRND8 neurons.
Inhibition of SK channels improves cholinergic excitability of TgCRND8 neurons
Because the layer 6 TgCRND8 neurons show reduced ability to maintain peak firing, elevated calcium levels in response to ACh, and enhanced apamin-sensitive AHPs, we sought to determine whether the inhibition of SK channels could help improve cholinergic excitability in layer 6 PFC of TgCRND8 mice. In the presence of apamin, an SK-antagonist, 1 mm ACh elicited spiking in 8 of 12 (67%) cells, and, under these conditions, higher-frequency spiking could now be sustained to a significantly greater extent, leading to a rightward shift of the cumulative probability distribution of ACh-elicited firing frequency in TgCRND8 neurons (Fig. 8B; K–S test, p < 0.00001). These data support strongly the hypothesis that impairments in cholinergic excitability arise from enhanced activity of calcium-activated SK channels in layer 6 of the TgCRND8 mice.
Figure 8.
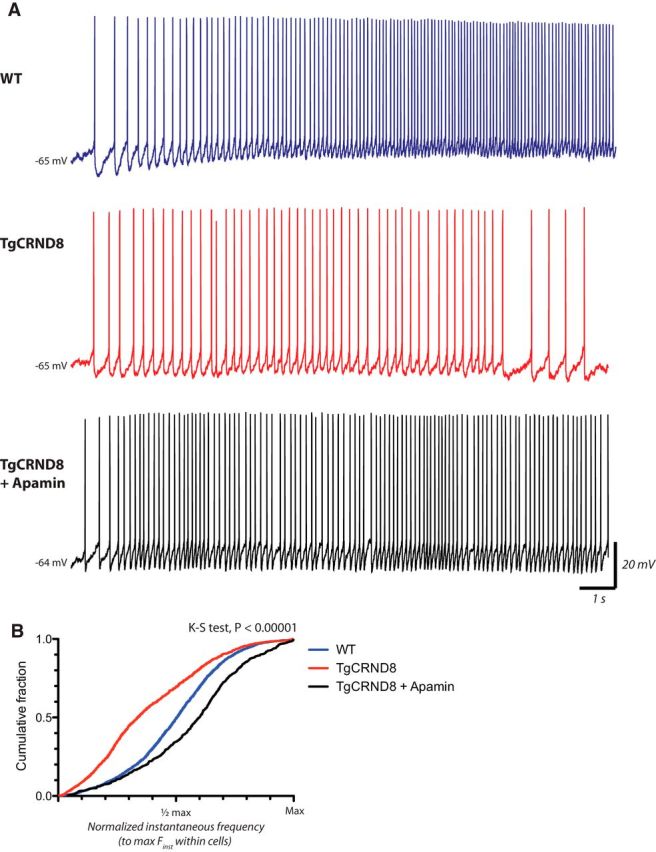
Inhibition of SK improves cholinergic excitability in TgCRND8 mice. A, Representative example of cell firing elicited by bath application of 1 mm ACh (15 s) in WT (top, blue trace), TgCRND8 (middle, red trace), and a TgCRND8 cell in the presence apamin (bottom, black trace). Traces represent the first 15 s of ACh-elicited firing. B, Apamin induces a rightward shift of the cumulative probability distribution of ACh-elicited firing frequency in TgCRND8 (K–S test, p < 0.00001; TgCRND8 control, n = 15 cells, TgCRND8 + Apamin, n = 8 cells).
Together, our findings demonstrate early disruption of cholinergic excitation of layer 6 PFC neurons in an animal model of AD, identify mechanisms underlying this disruption, and use this information to restore the maintenance of peak cholinergic excitability in a critical component of attention circuitry.
Discussion
We have found that layer 6 pyramidal neurons of the PFC are susceptible to disruptions of cellular excitability in the TgCRND8 brain. In particular, we show that these cells are unable to sustain cholinergic excitation to the same extent as nontransgenic littermate mice as a result of the excessive activation of calcium-activated hyperpolarizing conductances. Cholinergic excitation can be improved in layer 6 pyramidal cells of TgCRND8 mice by pharmacological blockade of SK channels, suggesting a novel target for the treatment of attentional impairments in AD.
The TgCRND8 mouse model is a widely used model of disease that recapitulates several key neuropathological and cognitive features of AD (Chishti et al., 2001; Webster et al., 2014), including attention deficits (Romberg et al., 2013b). Because TgCRND8 mice harbor APP mutations without disruptions to other genetic loci of AD vulnerability, namely the tau and presenilin genes (Selkoe, 2001; Goedert and Spillantini, 2006), these previously documented attentional impairments appear related to the overexpression of APP, aberrant APP processing, and/or the production of β-amyloid (Aβ) pathology. The disruption of the cholinergic excitation of PFC layer 6 neurons described here is a plausible proximate cause for the attention deficits observed in TgCRND8 mice. These animals display impairments on the five-choice serial reaction time task (for review, see Bari et al., 2008; Romberg et al., 2013b), an attentional task that requires cholinergic modulation of the deep layer PFC neurons for optimal performance (Bailey et al., 2010; Guillem et al., 2011). The PFC constitutes an important hub for higher cognitive function (Duncan and Owen, 2000; Buckner et al., 2009; Cole et al., 2013; Sporns, 2014), and its neuromodulation exerts powerful influences on the brain as a whole. The ACh released in the PFC during attention tasks (Parikh et al., 2007) robustly and directly excites the deep cortical layers (Kassam et al., 2008; Poorthuis et al., 2013; Proulx et al., 2014a) in a manner consistent with the sustained firing necessary for attention and other executive functions (Funahashi, 2001; Hasselmo and Sarter, 2011; Ikkai and Curtis, 2011; Zhou et al., 2011). The cholinergic modulation of PFC layer 6 would be especially important in this regard given its dense corticothalamic feedback projections and local influence over the activity of the cortical column (Thomson, 2010; Olsen et al., 2012; Kim et al., 2014; Proulx et al., 2014a). It will be most interesting in future work to determine whether and how the targeted modulation of prefrontal layer 6 pyramidal cell excitability by SK channels can impinge on cognitive performance.
The impairments in cholinergic excitability we document would be exacerbated by previously described cholinergic changes that accompany the progression of AD. Such deficits include the neurodegeneration of the cholinergic cells of the basal forebrain (Davies and Maloney, 1976; Whitehouse et al., 1982), as well as a decrease in the enzymatic activity related to the synthesis and breakdown of ACh (Davies and Maloney, 1976; Perry et al., 1978; Henke and Lang, 1983; Zubenko et al., 1989; Davis et al., 1999; Selkoe, 2001; Auld et al., 2002; DeKosky et al., 2002). In the current study, however, bath application of ACh would circumvent potential differences in endogenous ACh levels between the genotypes, indicating that significant differences in cholinergic responses are observed in the TgCRND8 neurons independently of the disruptions in ACh levels that accompany disease progression. Significant reductions in nicotinic receptor binding have also been documented in the AD brain, and this holds particularly true for the α4β2* nicotinic receptor subtype (Nordberg et al., 1988; Whitehouse et al., 1988; Marutle et al., 1999; Nordberg, 2001; O'Brien et al., 2007; Kendziorra et al., 2011) that is expressed highly in layer 6 PFC (Wada et al., 1989, 1990; Kassam et al., 2008; Alves et al., 2010). There is also some evidence of disrupted functional coupling of excitatory muscarinic receptors to downstream cellular effectors (Jope et al., 1997; Jope, 1999). Furthermore, Aβ peptides can impair both muscarinic (Kelly et al., 1996) and nicotinic (Liu et al., 2001; Pettit et al., 2001) receptor function and indeed interfere with the cholinergic system as a whole (for review, see Auld et al., 2002). It is striking that we found only modest reductions in TgCRND8 ACh currents near resting membrane potential and neither differences in muscarinic or nicotinic currents between the genotypes nor in the level of depolarization achieved. Instead, the greatest difference rested in the degree to which layer 6 cells could sustain firing in response to ACh. The extent to which nicotinic and muscarinic receptors contribute to this threshold effect will be critical to elucidate in future work, particularly in view of the accompanying suprathreshold calcium differences that are reported here.
Cholinergic signaling can alter intracellular calcium levels in a multitude of ways. The subtype of nicotinic receptors that play a prominent role in the excitation of PFC layer 6 cells (Kassam et al., 2008; Tian et al., 2011; Poorthuis et al., 2013) are characterized by unusual calcium permeability (Fucile, 2004; Tapia et al., 2007). Although the M1 muscarinic receptors contribute only modestly to the depolarization of PFC layer 6 cells (Tian et al., 2014), they can trigger the release of calcium from intracellular stores (Berridge and Irvine, 1984; Neher et al., 1988; Gulledge and Stuart, 2005; Proulx et al., 2014b). Disruptions in intracellular calcium have been documented in several models of AD (Stutzmann et al., 2004; Stutzmann, 2006; Wykes et al., 2012; Chakroborty and Stutzmann, 2014). Potential contributors suggested to be perturbed in models of AD include calcium-permeable nicotinic receptors (Liu et al., 2001; Pettit et al., 2001), excitatory muscarinic receptors (Kelly et al., 1996), voltage-gated calcium channels (Wang and Mattson, 2014), calcium-induced calcium release (Stutzmann et al., 2004; Stutzmann, 2006), and extra-sensitive NMDA receptors (Shah and Haylett, 2002; Goussakov et al., 2010; Romberg et al., 2012; Wang and Mattson, 2014). Here we find that layer 6 neurons in the TgCRND8 mice do not have aberrant calcium levels at baseline and show only moderate elevation in response to strong depolarizing stimuli and, indeed, to cholinergic stimulation below threshold. Instead, the combination of cholinergic stimuli and depolarization is required to bring about strongly elevated intracellular calcium levels. Our data indicate that aberrations in voltage-gated calcium channel and NMDA receptor function may contribute to this suprathreshold effect. It is interesting to note that calcium flux through both voltage-gated calcium channels and NMDA receptors can couple to SK channels (Marrion and Tavalin 1998; Wolfart and Roeper, 2002; Ngo-Anh et al., 2005; Giessel and Sabatini, 2010), which could account for the impairments in cholinergic firing of layer 6 TgCRND8 neurons that are documented in the current study. However, much additional work will be necessary to determine the cellular mechanisms underlying suprathreshold calcium responses in the TgCRND8 mice and its subsequent dysregulation of cholinergic firing and whether such an interaction is specific to the cholinergic system remains an open question. Nonetheless, it is clear that the cholinergic stimulation of layer 6 pyramidal cells of the PFC can be regarded as a highly relevant physiological stimulus essential to optimal attentional performance (Bailey et al., 2010; Guillem et al., 2011; Proulx et al., 2014a).
In the major corticothalamic layer, in which modulation by ACh is essential for optimal attentional function, we have found that there is impaired cholinergic excitability in a mouse model of AD. Furthermore, we have found that blockade of the SK class of calcium-activated potassium ion channels significantly restores persistent cholinergic excitability in PFC of TgCRND8. Consistent with this finding, local PFC administration of an SK blocker has been shown to improve executive function (Brennan et al., 2008). Of note, the SK-mediated AHP is modifiable by experience (Disterhoft and Oh, 2006), a modulatory control that appears to be essential for learning. However, the dynamic control of its magnitude, and even its calcium sensitivity, is complex (Bildl et al., 2004; Kramár et al., 2004; Ren et al., 2006; Allen et al., 2007; Giessel and Sabatini, 2010; El-Hassar et al., 2014). Regulation of the SK-mediated AHP by BDNF points to a potential locus of disruption in AD and the TgCRND8 model that would be correlated with early cognitive decline (Peng et al., 2005; Francis et al., 2012). Understanding the regulation of persistent cholinergic excitation in PFC attention circuits is a key step in understanding normal control of executive function and its vulnerability to disruption in AD.
Footnotes
This work was funded by Canadian Institutes of Health Research Grants MOP-89825 (E.K.L.), TAD-117950 (P.F.), and MOP-102467 (J.M.), the Canada Research Chairs Program (E.K.L.), an Ontario Early Researcher Award (E.K.L.), the Canadian Foundation for Innovation (E.K.L.), the Alzheimer's Society of Ontario (P.F.), and the Cryptic Rite Charitable Foundation (J.M.). We thank Mary Hill and Rosemary Ahrens for expert technical assistance.
References
- Allen D, Fakler B, Maylie J, Adelman JP. Organization and regulation of small conductance Ca2+-activated K+ channel multiprotein complexes. J Neurosci. 2007;27:2369–2376. doi: 10.1523/JNEUROSCI.3565-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alves NC, Bailey CDC, Nashmi R, Lambe EK. Developmental sex differences in nicotinic currents of prefrontal layer VI neurons in mice and rats. PLoS One. 2010;5:e9261. doi: 10.1371/journal.pone.0009261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auld DS, Kornecook TJ, Bastianetto S, Quirion R. Alzheimer's disease and the basal forebrain cholinergic system: relations to beta-amyloid peptides, cognition, and treatment strategies. Prog Neurobiol. 2002;68:209–245. doi: 10.1016/S0301-0082(02)00079-5. [DOI] [PubMed] [Google Scholar]
- Baddeley AD, Baddeley HA, Bucks RS, Wilcock GK. Attentional control in Alzheimer's disease. Brain. 2001;124:1492–1508. doi: 10.1093/brain/124.8.1492. [DOI] [PubMed] [Google Scholar]
- Bailey CD, De Biasi M, Fletcher PJ, Lambe EK. The nicotinic acetylcholine receptor alpha5 subunit plays a key role in attention circuitry and accuracy. J Neurosci. 2010;30:9241–9252. doi: 10.1523/JNEUROSCI.2258-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bari A, Dalley JW, Robbins TW. The application of the 5-choice serial reaction time task for the assessment of visual attentional processes and impulse control in rats. Nat Protoc. 2008;3:759–767. doi: 10.1038/nprot.2008.41. [DOI] [PubMed] [Google Scholar]
- Bean BP. The action potential in mammalian central neurons. Nat Rev Neurosci. 2007;8:451–465. doi: 10.1038/nrn2148. [DOI] [PubMed] [Google Scholar]
- Berridge MJ, Irvine RF. Inositol trisphosphate, a novel second messenger in cellular signal transduction. Nature. 1984;312:315–321. doi: 10.1038/312315a0. [DOI] [PubMed] [Google Scholar]
- Bildl W, Strassmaier T, Thurm H, Andersen J, Eble S, Oliver D, Knipper M, Mann M, Schulte U, Adelman JP, Fakler B. Protein kinase CK2 is coassembled with small conductance Ca(2+)-activated K+ channels and regulates channel gating. Neuron. 2004;43:847–858. doi: 10.1016/j.neuron.2004.08.033. [DOI] [PubMed] [Google Scholar]
- Brennan AR, Dolinsky B, Vu MA, Stanley M, Yeckel MF, Arnsten AF. Blockade of IP3-mediated SK channel signaling in the rat medial prefrontal cortex improves spatial working memory. Learn Mem. 2008;15:93–96. doi: 10.1101/lm.767408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckner RL, Sepulcre J, Talukdar T, Krienen FM, Liu H, Hedden T, Andrews-Hanna JR, Sperling RA, Johnson KA. Cortical hubs revealed by intrinsic functional connectivity: mapping, assessment of stability, and relation to Alzheimer's disease. J Neurosci. 2009;29:1860–1873. doi: 10.1523/JNEUROSCI.5062-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakroborty S, Stutzmann GE. Calcium channelopathies and Alzheimer's disease: insight into therapeutic success and failures. Eur J Pharmacol. 2014;39:83–95. doi: 10.1016/j.ejphar.2013.11.012. [DOI] [PubMed] [Google Scholar]
- Chishti MA, Yang DS, Janus C, Phinney AL, Horne P, Pearson J, Strome R, Zuker N, Loukides J, French J, Turner S, Lozza G, Grilli M, Kunicki S, Morissette C, Paquette J, Gervais F, Bergeron C, Fraser PE, Carlson GA, George-Hyslop PS, Westaway D. Early-onset amyloid deposition and cognitive deficits in transgenic mice expressing a double mutant form of amyloid precursor protein 695. J Biol Chem. 2001;276:21562–21570. doi: 10.1074/jbc.M100710200. [DOI] [PubMed] [Google Scholar]
- Citron M. Alzheimer's disease: strategies for disease modification. Nat Rev Drug Discov. 2010;9:387–398. doi: 10.1038/nrd2896. [DOI] [PubMed] [Google Scholar]
- Cole MW, Reynolds JR, Power JD, Repovs G, Anticevic A, Braver TS. Multi-task connectivity reveals flexible hubs for adaptive task control. Nat Neurosci. 2013;16:1348–1355. doi: 10.1038/nn.3470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalley JW, Theobald DE, Bouger P, Chudasama Y, Cardinal RN, Robbins TW. Cortical cholinergic function and deficits in visual attentional performance in rats following 192 IgG-saporin-induced lesions of the medial prefrontal cortex. Cereb Cortex. 2004;14:922–932. doi: 10.1093/cercor/bhh052. [DOI] [PubMed] [Google Scholar]
- Davies P, Maloney AJ. Selective loss of central cholinergic neurons in Alzheimer's disease. Lancet. 1976;2:1403. doi: 10.1016/S0140-6736(76)91936-X. [DOI] [PubMed] [Google Scholar]
- Davis KL, Mohs RC, Marin D, Purohit DP, Perl DP, Lantz M, Austin G, Haroutunian V. Cholinergic markers in elderly patients with early signs of Alzheimer disease. JAMA. 1999;281:1401–1406. doi: 10.1001/jama.281.15.1401. [DOI] [PubMed] [Google Scholar]
- DeKosky ST, Ikonomovic MD, Styren SD, Beckett L, Wisniewski S, Bennett DA, Cochran EJ, Kordower JH, Mufson EJ. Upregulation of choline acetyltransferase activity in hippocampus and frontal cortex of elderly subjects with mild cognitive impairment. Ann Neurol. 2002;51:145–155. doi: 10.1002/ana.10069. [DOI] [PubMed] [Google Scholar]
- Disterhoft JF, Oh MM. Learning, aging and intrinsic neuronal plasticity. Trends Neurosci. 2006;29:587–599. doi: 10.1016/j.tins.2006.08.005. [DOI] [PubMed] [Google Scholar]
- Duncan J, Owen AM. Common regions of the human frontal lobe recruited by diverse cognitive demands. Trends Neurosci. 2000;23:475–483. doi: 10.1016/S0166-2236(00)01633-7. [DOI] [PubMed] [Google Scholar]
- El-Hassar L, Simen AA, Duque A, Patel KD, Kaczmarek LK, Arnsten AF, Yeckel MF. Disrupted in schizophrenia 1 modulates medial prefrontal cortex pyramidal neuron activity through cAMP regulation of transient receptor potential C and small-conductance K+ channels. Biol Psychiatry. 2014;76:476–485. doi: 10.1016/j.biopsych.2013.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faber ES, Sah P. Calcium-activated potassium channels: multiple contributions to neuronal function. Neuroscientist. 2003;9:181–194. doi: 10.1177/1073858403009003011. [DOI] [PubMed] [Google Scholar]
- Francis BM, Kim J, Barakat ME, Fraenkl S, Yücel YH, Peng S, Michalski B, Fahnestock M, McLaurin J, Mount HT. Object recognition memory and BDNF expression are reduced in young TgCRND8 mice. Neurobiol Aging. 2012;33:555–563. doi: 10.1016/j.neurobiolaging.2010.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fucile S. Ca2+ permeability of nicotinic acetylcholine receptors. Cell Calcium. 2004;35:1–8. doi: 10.1016/j.ceca.2003.08.006. [DOI] [PubMed] [Google Scholar]
- Funahashi S. Neuronal mechanisms of executive control by the prefrontal cortex. Neurosci Res. 2001;39:147–165. doi: 10.1016/S0168-0102(00)00224-8. [DOI] [PubMed] [Google Scholar]
- Funahashi S, Bruce CJ, Goldman-Rakic PS. Mnemonic coding of visual space in the monkey's dorsolateral prefrontal cortex. J Neurophysiol. 1989;61:331–349. doi: 10.1152/jn.1989.61.2.331. [DOI] [PubMed] [Google Scholar]
- Giessel AJ, Sabatini BL. M1 Muscarinic receptors boost synaptic potentials and calcium influx in dendritic spines by inhibiting postsynaptic SK channels. Neuron. 2010;68:936–947. doi: 10.1016/j.neuron.2010.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goedert M, Spillantini MG. A century of Alzheimer's disease. Science. 2006;314:777–781. doi: 10.1126/science.1132814. [DOI] [PubMed] [Google Scholar]
- Goussakov I, Miller MB, Stutzmann GE. NMDA-Mediated Ca2+ influx drives aberrant ryanodine receptor activation in dendrites of young Alzheimer's disease mice. J Neurosci. 2010;30:12128–12137. doi: 10.1523/JNEUROSCI.2474-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grady CL, Haxby JV, Horwitz B, Sundaram M, Berg G, Schapiro M, Friedland RP, Rapoport SI. Longitudinal study of the early neuropsychological and cerebral metabolic changes in dementia of the Alzheimer type. J Clin Exp Neuropsychol. 1988;10:576–596. doi: 10.1080/01688638808402796. [DOI] [PubMed] [Google Scholar]
- Guillem K, Bloem B, Poorthuis RB, Loos M, Smit AB, Maskos U, Spijker S, Mansvelder HD. Nicotinic acetylcholine receptor beta2 subunits in the medial prefrontal cortex control attention. Science. 2011;333:888–891. doi: 10.1126/science.1207079. [DOI] [PubMed] [Google Scholar]
- Gulledge AT, Stuart GJ. Cholinergic inhibition of neocortical pyramidal neurons. J Neurosci. 2005;25:10308–10320. doi: 10.1523/JNEUROSCI.2697-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulledge AT, Bucci DJ, Zhang SS, Matsui M, Yeh HH. M1 Receptors mediate cholinergic modulation of excitability in neocortical pyramidal neurons. J Neurosci. 2009;29:9888–9902. doi: 10.1523/JNEUROSCI.1366-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasselmo ME, Sarter M. Modes and models of forebrain cholinergic neuromodulation of cognition. Neuropsychopharmacology. 2011;36:52–73. doi: 10.1038/npp.2010.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henke H, Lang W. Cholinergic enzymes in neocortex, hippocampus and basal forebrain of non-neurological and senile dementia of Alzheimer-type patients. Brain Res. 1983;267:281–291. doi: 10.1016/0006-8993(83)90880-6. [DOI] [PubMed] [Google Scholar]
- Ikkai A, Curtis CE. Common neural mechanisms supporting spatial working memory, attention and motor intention. Neuropsychologia. 2011;49:1428–1434. doi: 10.1016/j.neuropsychologia.2010.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jope RS. Cholinergic muscarinic receptor signaling by the phosphoinositide signal transduction system in Alzheimer's disease. J Alzheimers Dis. 1999;1:231–247. doi: 10.3233/jad-1999-14-505. [DOI] [PubMed] [Google Scholar]
- Jope RS, Song L, Powers RE. Cholinergic activation of phosphoinositide signaling is impaired in Alzheimer's disease brain. Neurobiol Aging. 1997;18:111–120. doi: 10.1016/S0197-4580(96)00205-9. [DOI] [PubMed] [Google Scholar]
- Kandel ER, Spencer WA. Electrophysiology of hippocampal neurons. II. After-potentials and repetitive firing. J Neurophysiol. 1961;24:243–259. doi: 10.1152/jn.1961.24.3.243. [DOI] [PubMed] [Google Scholar]
- Kassam SM, Herman PM, Goodfellow NM, Alves NC, Lambe EK. Developmental excitation of corticothalamic neurons by nicotinic acetylcholine receptors. J Neurosci. 2008;28:8756–8764. doi: 10.1523/JNEUROSCI.2645-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly JF, Furukawa K, Barger SW, Rengen MR, Mark RJ, Blanc EM, Roth GS, Mattson MP. Amyloid beta-peptide disrupts carbachol-induced muscarinic cholinergic signal transduction in cortical neurons. Proc Natl Acad Sci U S A. 1996;93:6753–6758. doi: 10.1073/pnas.93.13.6753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kendziorra K, Wolf H, Meyer PM, Barthel H, Hesse S, Becker GA, Luthardt J, Schildan A, Patt M, Sorger D, Seese A, Gertz HJ, Sabri O. Decreased cerebral α4β2∗ nicotinic acetylcholine receptor availability in patients with mild cognitive impairment and Alzheimer's disease assessed with positron emission tomography. Eur J Nucl Med Mol Imaging. 2011;38:515–525. doi: 10.1007/s00259-010-1644-5. [DOI] [PubMed] [Google Scholar]
- Kim J, Matney CJ, Blankenship A, Hestrin S, Brown SP. Layer 6 corticothalamic neurons activate a cortical output layer, layer 5a. J Neurosci. 2014;34:9656–9664. doi: 10.1523/JNEUROSCI.1325-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramár EA, Lin B, Lin CY, Arai AC, Gall CM, Lynch G. A novel mechanism for the facilitation of theta-induced long-term potentiation by brain-derived neurotrophic factor. J Neurosci. 2004;24:5151–5161. doi: 10.1523/JNEUROSCI.0800-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Kawai H, Berg DK. beta-Amyloid peptide blocks the response of alpha 7-containing nicotinic receptors on hippocampal neurons. Proc Natl Acad Sci U S A. 2001;98:4734–4739. doi: 10.1073/pnas.081553598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marrion NV, Tavalin SJ. Selective activation of Ca2+-activated K+ channels by co-localized Ca2+ channels in hippocampal neurons. Nature. 1998;395:900–905. doi: 10.1038/27674. [DOI] [PubMed] [Google Scholar]
- Marutle A, Warpman U, Bogdanovic N, Lannfelt L, Nordberg A. Neuronal nicotinic receptor deficits in Alzheimer patients with the Swedish amyloid precursor protein 670/671 mutation. J Neurochem. 1999;72:1161–1169. doi: 10.1046/j.1471-4159.2000.0721161.x. [DOI] [PubMed] [Google Scholar]
- Neher E, Marty A, Fukuda K, Kubo T, Numa S. Intracellular calcium release mediated by two muscarinic receptor subtypes. FEBS Lett. 1988;240:88–94. doi: 10.1016/0014-5793(88)80345-4. [DOI] [PubMed] [Google Scholar]
- Ngo-Anh TJ, Bloodgood BL, Lin M, Sabatini BL, Maylie J, Adelman JP. SK channels and NMDA receptors form a Ca2+-mediated feedback loop in dendritic spines. Nat Neurosci. 2005;8:642–649. doi: 10.1038/nn1449. [DOI] [PubMed] [Google Scholar]
- Nordberg A. Nicotinic receptor abnormalities of Alzheimer's disease: therapeutic implications. Biol Psychiatry. 2001;49:200–210. doi: 10.1016/S0006-3223(00)01125-2. [DOI] [PubMed] [Google Scholar]
- Nordberg A, Adem A, Hardy J, Winblad B. Change in nicotinic receptor subtypes in temporal cortex of Alzheimer brains. Neurosci Lett. 1988;86:317–321. doi: 10.1016/0304-3940(88)90503-4. [DOI] [PubMed] [Google Scholar]
- O'Brien JT, Colloby SJ, Pakrasi S, Perry EK, Pimlott SL, Wyper DJ, McKeith IG, Williams ED. Alpha4beta2 nicotinic receptor status in Alzheimer's disease using 123I–5IA-85380 single-photon-emission computed tomography. J Neurol Neurosurg Psychiatry. 2007;78:356–362. doi: 10.1136/jnnp.2006.108209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen SR, Bortone DS, Adesnik H, Scanziani M. Gain control by layer six in corticalcircuits of vision. Nature. 2012;483:47–52. doi: 10.1038/nature10835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parikh V, Kozak R, Martinez V, Sarter M. Prefrontal acetylcholine release controls cue detection on multiple timescales. Neuron. 2007;56:141–154. doi: 10.1016/j.neuron.2007.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng S, Wuu J, Mufson EJ, Fahnestock M. Precursor form of brain-derived neurotrophic factor and mature brain-derived neurotrophic factor are decreased in the pre-clinical stages of Alzheimer's disease. J Neurochem. 2005;93:1412–1421. doi: 10.1111/j.1471-4159.2005.03135.x. [DOI] [PubMed] [Google Scholar]
- Perry EK, Tomlinson BE, Blessed G, Bergmann K, Gibson PH, Perry RH. Correlation of cholinergic abnormalities with senile plaques and mental test scores in senile dementia. Br Med J. 1978;2:1457–1459. doi: 10.1136/bmj.2.6150.1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry RJ, Hodges JR. Attention and executive deficits in Alzheimer's disease. A critical review. Brain. 1999;122:383–404. doi: 10.1093/brain/122.3.383. [DOI] [PubMed] [Google Scholar]
- Pettit DL, Shao Z, Yakel JL. beta-Amyloid(1–42) peptide directly modulates nicotinic receptors in the rat hippocampal slice. J Neurosci. 2001;21:RC120. doi: 10.1523/JNEUROSCI.21-01-j0003.2001. (1–5) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poorthuis RB, Bloem B, Schak B, Wester J, de Kock CP, Mansvelder HD. Layer-specific modulation of the prefrontal cortex by nicotinic acetylcholine receptors. Cereb Cortex. 2013;23:148–161. doi: 10.1093/cercor/bhr390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proulx E, Piva M, Tian MK, Bailey CD, Lambe EK. Nicotinic acetylcholine receptors in attention circuitry: the role of layer VI neurons of prefrontal cortex. Cell Mol Life Sci. 2014a;71:1225–1244. doi: 10.1007/s00018-013-1481-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proulx É, Suri D, Heximer SP, Vaidya VA, Lambe EK. Early stress prevents the potentiation of muscarinic excitation by calcium release in adult prefrontal cortex. Biol Psychiatry. 2014b;76:315–323. doi: 10.1016/j.biopsych.2013.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren Y, Barnwell LF, Alexander JC, Lubin FD, Adelman JP, Pfaffinger PJ, Schrader LA, Anderson AE. Regulation of surface localization of the small conductance Ca2+-activated potassium channel, Sk2, through direct phosphorylation by cAMP-dependent protein kinase. J Biol Chem. 2006;281:11769–11779. doi: 10.1074/jbc.M513125200. [DOI] [PubMed] [Google Scholar]
- Romberg C, McTighe SM, Heath CJ, Whitcomb DJ, Cho K, Bussey TJ, Saksida LM. False recognition in a mouse model of Alzheimer's disease: rescue with sensory restriction and memantine. Brain. 2012;135:2103–2114. doi: 10.1093/brain/aws074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romberg C, Bussey TJ, Saksida LM. Paying more attention to attention: towards more comprehensive cognitive translation using mouse models of Alzheimer's disease. Brain Res Bull. 2013a;92:49–55. doi: 10.1016/j.brainresbull.2012.02.007. [DOI] [PubMed] [Google Scholar]
- Romberg C, Horner AE, Bussey TJ, Saksida LM. A touch screen-automated cognitive test battery reveals impaired attention, memory abnormalities, and increased response inhibition in the TgCRND8 mouse model of Alzheimer's disease. Neurobiol Aging. 2013b;34:731–744. doi: 10.1016/j.neurobiolaging.2012.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sah P. Ca(2+)-activated K+ currents in neurones: types, physiological roles and modulation. Trends Neurosci. 1996;19:150–154. doi: 10.1016/S0166-2236(96)80026-9. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Alzheimer's disease: genes, proteins, and therapy. Physiol Rev. 2001;81:741–766. doi: 10.1152/physrev.2001.81.2.741. [DOI] [PubMed] [Google Scholar]
- Shah MM, Haylett DG. K+ currents generated by NMDA receptor activation in rat hippocampal pyramidal neurons. J Neurophysiol. 2002;87:2983–2989. doi: 10.1152/jn.2002.87.6.2983. [DOI] [PubMed] [Google Scholar]
- Sporns O. Contributions and challenges for network models in cognitive neuroscience. Nat Neurosci. 2014;17:652–660. doi: 10.1038/nn.3690. [DOI] [PubMed] [Google Scholar]
- Stutzmann GE, Caccamo A, LaFerla FM, Parker I. Dysregulated IP3 signaling in cortical neurons of knock-in mice expressing an Alzheimer's-linked mutation in presenilin1 results in exaggerated Ca2+ signals and altered membrane excitability. J Neurosci. 2004;24:508–513. doi: 10.1523/JNEUROSCI.4386-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stutzmann GE. Enhanced ryanodine receptor recruitment contributes to Ca2+ disruptions in young, adult, and aged Alzheimer's disease mice. J Neurosci. 2006;26:5180–5189. doi: 10.1523/JNEUROSCI.0739-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tapia L, Kuryatov A, Lindstrom J. Ca2+ permeability of the (alpha4)3(beta2)2 stoichiometry greatly exceeds that of (alpha4)2(beta2)3 human acetylcholine receptors. Mol Pharmacol. 2007;71:769–776. doi: 10.1124/mol.106.030445. [DOI] [PubMed] [Google Scholar]
- Terry AV, Jr, Buccafusco JJ. The cholinergic hypothesis of age and Alzheimer's disease-related cognitive deficits: Recent challenges and their implications for novel drug development. J Pharmacol Exp Ther. 2003;306:821–827. doi: 10.1124/jpet.102.041616. [DOI] [PubMed] [Google Scholar]
- Thomson AM. Neocortical layer 6, a review. Front Neuroanat. 2010;4:13. doi: 10.3389/fnana.2010.00013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian MK, Bailey CD, De Biasi M, Picciotto MR, Lambe EK. Plasticity of prefrontal attention circuitry: upregulated muscarinic excitability in response to decreased nicotinic signaling following deletion of alpha5 or beta2 subunits. J Neurosci. 2011;31:16458–16463. doi: 10.1523/JNEUROSCI.3600-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian MK, Bailey CD, Lambe EK. Cholinergic excitation in mouse primary vs. associative cortex: region-specific magnitude and receptor balance. Eur J Neurosci. 2014;40:2608–2618. doi: 10.1111/ejn.12622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wada E, Wada K, Boulter J, Deneris E, Heinemann S, Patrick J, Swanson LW. Distribution of alpha 2, alpha 3, alpha 4, and beta 2 neuronal nicotinic receptor subunit mRNAs in the central nervous system: a hybridization histochemical study in the rat. J Comp Neurol. 1989;284:314–335. doi: 10.1002/cne.902840212. [DOI] [PubMed] [Google Scholar]
- Wada E, McKinnon D, Heinemann S, Patrick J, Swanson LW. The distribution of mRNA encoded by a new member of the neuronal nicotinic acetylcholine receptor gene family (alpha 5) in the rat central nervous system. Brain Res. 1990;526:45–53. doi: 10.1016/0006-8993(90)90248-A. [DOI] [PubMed] [Google Scholar]
- Wang Y, Mattson MP. L-type Ca2+ currents at CA1 synapses, but not CA3 or dentate granule neuron synapses, are increased in 3xTgAD mice in an age-dependent manner. Neurobiol Aging. 2014;35:88–95. doi: 10.1016/j.neurobiolaging.2013.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster SJ, Bachstetter AD, Nelson PT, Schmitt FA, Van Eldik LJ. Using mice to model Alzheimer's dementia: an overview of the clinical disease and the preclinical behavioral changes in 10 mouse models. Front Genet. 2014;5:88. doi: 10.3389/fgene.2014.00088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitehouse PJ, Price DL, Struble RG, Clark AW, Coyle JT, Delon MR. Alzheimer's disease and senile dementia: loss of neurons in the basal forebrain. Science. 1982;215:1237–1239. doi: 10.1126/science.7058341. [DOI] [PubMed] [Google Scholar]
- Whitehouse PJ, Martino AM, Wagster MV, Price DL, Mayeux R, Atack JR, Kellar KJ. Reductions in [3H]nicotinic acetylcholine binding in Alzheimer's disease and Parkinson's disease: an autoradiographic study. Neurology. 1988;38:720–723. doi: 10.1212/WNL.38.5.720. [DOI] [PubMed] [Google Scholar]
- Wolfart J, Roeper J. Selective coupling of T-type calcium channels to SK potassium channels prevents intrinsic bursting in dopaminergic midbrain neurons. J Neurosci. 2002;22:3404–3413. doi: 10.1523/JNEUROSCI.22-09-03404.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wykes R, Kalmbach A, Eliava M, Waters J. Changes in the physiology of CA1 hippocampal pyramidal neurons in preplaque CRND8 mice. Neurobiol Aging. 2012;33:1609–1623. doi: 10.1016/j.neurobiolaging.2011.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Qi XL, Douglas K, Palaninathan K, Kang HS, Buccafusco JJ, Blake DT, Constantinidis C. Cholinergic modulation of working memory activity in primate prefrontal cortex. J Neurophysiol. 2011;106:2180–2188. doi: 10.1152/jn.00148.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zubenko GS, Moossy J, Martinez AJ, Rao GR, Kopp U, Hanin I. A brain regional analysis of morphologic and cholinergic abnormalities in Alzheimer's disease. Arch Neurol. 1989;46:634–638. doi: 10.1001/archneur.1989.00520420054022. [DOI] [PubMed] [Google Scholar]