

Abstract
Background
The σ54 subunit controls a unique class of promoters in bacteria. Such promoters, without exception, require enhancer binding proteins (EBPs) for transcription initiation. Desulfovibrio vulgaris Hildenborough, a model bacterium for sulfate reduction studies, has a high number of EBPs, more than most sequenced bacteria. The cellular processes regulated by many of these EBPs remain unknown.
Results
To characterize the σ54-dependent regulome of D. vulgaris Hildenborough, we identified EBP binding motifs and regulated genes by a combination of computational and experimental techniques. These predictions were supported by our reconstruction of σ54-dependent promoters by comparative genomics. We reassessed and refined the results of earlier studies on regulation in D. vulgaris Hildenborough and consolidated them with our new findings. It allowed us to reconstruct the σ54 regulome in D. vulgaris Hildenborough. This regulome includes 36 regulons that consist of 201 coding genes and 4 non-coding RNAs, and is involved in nitrogen, carbon and energy metabolism, regulation, transmembrane transport and various extracellular functions. To the best of our knowledge, this is the first report of direct regulation of alanine dehydrogenase, pyruvate metabolism genes and type III secretion system by σ54-dependent regulators.
Conclusions
The σ54-dependent regulome is an important component of transcriptional regulatory network in D. vulgaris Hildenborough and related free-living Deltaproteobacteria. Our study provides a representative collection of σ54-dependent regulons that can be used for regulation prediction in Deltaproteobacteria and other taxa.
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-015-2176-y) contains supplementary material, which is available to authorized users.
Keywords: Transcription factor, Transcriptional regulation, Sigma factor, Desulfovibrio vulgaris, Enhancer binding proteins
Background
Sigma subunits of bacterial RNA polymerase regulate the process of transcriptional initiation through binding to RNA polymerase holoenzyme, recognition of promoter sequences, and promotion of open complex formation. One of the sigma factors, called alternative sigma factor σ54, significantly differs from all other known sigma factor proteins in two respects (reviewed in [1]). First, it recognizes and binds characteristic promoters with GG −24 and TGC −12 elements that are more conserved than the promoters of σ70 family. Second, σ54 strictly requires the ATP hydrolysis activity of an activating transcription factor (TF) for the formation of open promoter complex. TFs necessary for such activation are called prokaryotic enhancer-binding proteins (EBPs) [2]. EBPs specifically bind a conserved upstream activating sequence (UAS) that contains one or several TF binding sites. UASs are located 100 bp or more upstream from the σ54-dependent promoter, and the interaction of σ54-polymerase complex with EBP oligomer requires DNA looping between the UAS and promoter. This looping is often facilitated by bacterial chromatin proteins like integration host factor or histone-like protein HU. Thus, initiation of σ54-dependent transcription involves promoter binding by σ54-containing RNA polymerase, UAS binding by EBP, and activation of σ54-polymerase by ATP hydrolysis with subsequent formation of an open complex.
The analysis of taxonomic diversity of rpoN (the gene encoding σ54) and EBP-encoding genes demonstrated their presence in about 60 % of bacterial genomes [3]. σ54 seems to be an essential protein in some Deltaproteobacteria, since attempts to produce viable rpoN deletion mutants of Myxococcus xanthus [4] and Geobacter sulfurreducens [5] were unsuccessful.
σ54-dependent regulons (called sigmulons) have been extensively studied in several model organisms. It was found that in Escherichia coli, the σ54 sigmulon includes about 30 operons (reviewed in [6]), and 14 of them are involved in nitrogen metabolism. The products of other σ54-dependent operons contribute to formate, propionate and acetolactate metabolism, zinc tolerance, phage shock response and other functions. Most of E. coli EBPs control one or few operons, and only NtrC regulon includes 14 operons. However, in other model organisms EBPs may control a larger number of promoters. For example, NifA in Rhizobium etli regulates 43 operons involved in nitrogen fixation, energy production, transport, secondary metabolism and other diverse functions [7]. In Pseudomonas putida, there are 22 EBP-encoding genes and most of them are located close to σ54-dependent operons [8]. Of 46 σ54-dependent promoters in P. putida, 36 are related to nitrogen and carbon metabolism, flagella and motility. A much larger σ54-dependent sigmulon (108 promoters) was described in Geobacter sulfurreducens [5]. In this bacterium, σ54-dependent genes are involved in nitrogen assimilation, formate and amino acid metabolism, C4-dicarboxylate transport, flagella and pili biogenesis and other functions. Nevertheless, some bacteria have very small σ54 sigmulons. For example, Lactobacillus plantarum has only one EBP that activates a single σ54-dependent promoter upstream of mannose transport operon [9].
As σ54-dependent transcription requires EBP for initiation, the range of functions regulated by σ54 directly depends on the assortment of EBPs in the bacterium. A search within the MicrobesOnline database demonstrates the presence of EBPs in 1435 bacterial species [10]. Aerobic Deltaproteobacteria with large genomes have the highest total number of EBPs (Fig. 1, Additional file 1). A high number of EBPs in Myxococcus xanthus and related bacteria can be explained not only by their genome size, but also by wide involvement of these regulators in the very complex fruiting body development program [11]. However, the highest relative number of EBPs (with relation to genome size) was observed in anaerobic Deltaproteobacteria, isolated from soil and aquatic habitats. Desulfovibrio vulgaris strains are in the third place on the list after Desulfomonile tiedjei and Desulfomicrobium baculatum.
Fig. 1.
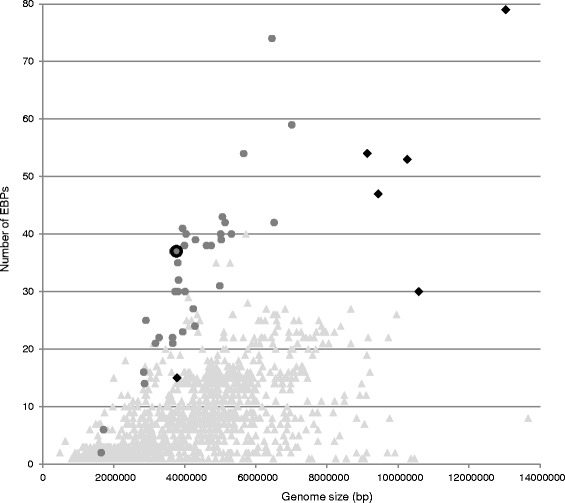
The number of EBPs in the bacterial genomes of different sizes. Grey circles represent anaerobic Deltaproteobacteria, and black diamonds represent aerobic Deltaproteobacteria. D. vulgaris Hildenborough marked by a black ring. Grey triangles represent other bacterial genomes
One of these strains, Desulfovibrio vulgaris Hildenborough, a model bacterium for sulfate reduction studies, has 37 genes encoding putative σ54-associated EBPs [12]. Among these, both target genes and TF binding motifs are known for 14 EBPs [13], and only the target genes for 6 more EBPs [13, 14]. Putative σ54-dependent promoters were predicted computationally upstream of many EBP target operons [13, 14]. A genome-wide analysis of D. vulgaris Hildenborough transcriptome [15] has identified 70 σ54-dependent promoters by a combination of 5′-RNA-seq and computational analysis of promoter motifs.
Despite the existing data on σ54-dependent transcription in D. vulgaris Hildenborough, we still do not have a complete understanding of the role of the σ54-dependent regulome (defined as the compendium of regulatory elements that facilitate σ54-dependent transcription) in this bacterium. For most of σ54-dependent promoters, we do not know EBPs that activate them. We also do not know binding motifs and target genes for multiple EBPs. Thus, the goal of the current study was to comprehensively reconstruct the σ54-dependent regulome in D. vulgaris Hildenborough genome-wide. We inferred several σ54-dependent regulons not known before (including those associated with type III secretion, electron transport, pyruvate and formate metabolism). This allowed us to make important conclusions about the set of functions controlled by σ54-associated regulators in D. vulgaris Hildenborough and related Deltaproteobacteria.
Results and discussion
In order to characterize a particular type of transcriptional regulation in bacteria, one needs to elucidate all the necessary components in this process, which typically are: transcriptional regulator, its binding sites, and a set of regulated genes or operons. In our study we aimed at discovering the compendium of regulatory elements that facilitate σ54-dependent transcription, i.e., σ54-dependent regulome. This regulome consists of all σ54-dependent transcriptional activators (EBPs), EBP binding sites and EBP-regulated operons with σ54-dependent promoters. We started with the computational analysis of all predicted D. vulgaris Hildenborough EBPs to verify the presence of DNA-binding domain and σ54-interacting domain. Next, we reconstructed EBP binding motifs (if unknown) and identified EBP binding sites in the D. vulgaris Hildenborough genome. Finally, we compared EBP regulons with the σ54-dependent sigmulon and identified a set of operons co-regulated by EBPs and σ54 subunit.
The repertoire of σ54-dependent regulators in D. vulgaris Hildenborough
All σ54-dependent EBPs possess a central AAA+ domain responsible for ATP binding and hydrolysis, oligomerization and interaction with σ54 subunit [2]. A common feature of σ54-dependent EBPs is the peptide motif ‘GAFTGA’, a structural element of AAA+ domain that interacts with sigma subunit protein [2]. We checked the presence of this motif in 37 EBPs from D. vulgaris Hildenborough by comparing them with AAA+ domain model from PFAM database (described in Methods). An exact GAFTGA sequence was observed in 25 of 37 EBPs, while in the other 12 EBPs this motif has different substitutions (Table 1). Ten of these 12 EBPs possess eight variations of the motif (GAFSEA, GAFSGA, GAFTDA, GAFTGG, GAFTHA, GAYTDA, GAYTGS, GSFTGA) that would maintain the EBP’s ability to activate transcription [2]. The remaining two EBPs (DVUA0100 and DVU2960) have substitutions in the GAFTGA motif that might interfere with its ability to bind σ54 protein. DVUA0100 possesses three substitutions (GVATGV), but our regulon prediction results (see below) suggest that they do not prevent DVUA0100 from activating σ54-dependent promoters. The second protein, DVU2960, has only one substitution of highly conserved threonine for proline (GAFPGA). Although an earlier work [3] reported a single amino acid deletion in the GAFPGA motif of DVU2960, our comparison with the AAA+ domain model does not confirm the deletion. Nevertheless, the functionality of DVU2960 as a σ54-dependent activator is unclear, because the same substitution of threonine for proline in another σ54-dependent activator, NifA from Bradyrhizobium japonicum, rendered the protein inactive [2].
Table 1.
Enhancer binding proteins of D. vulgaris Hildenborough
| Regulator | Type | GAFTGA motif | Target operons | Function | Regulon reference |
|---|---|---|---|---|---|
| DVU0110 | TCS response regulator | GAFTGA | DVU0132-DVU0133, DVU1152 | Exopolysaccharide and biofilm synthesis,transmembrane transport | Rajeev et al., 2011 [13], this study |
| DVU0118 | TCS response regulator | GAFTGA | DVU0123-DVU0121 | Transmembrane transport | Rajeev et al., 2011 [13] |
| DVU0151 | Single-component regulator | GAFSEA | DVU0150-DVU0146 | Transmembrane transport | This study |
| DVU0539 | TCS response regulator | GAFTGA | DVU0542-DVU0545, DVU0943-DVU0946, DVU2133-DVU2132, DVU3025-DVU3033 | Lactate metabolism, transmembrane transport | Rajeev et al., 2011 [13] |
| DVU0569 | Single-component regulator | GAFTGG | DVU0571 | Amino acid metabolism | This study |
| DVU0619 | TCS response regulator | GAFTGA | DVU0617-DVU0616 | This study | |
| DVU0621 | TCS response regulator | GAFTGA | DVU0624-DVU0625, DVU3025-DVU3033 | Nitrite stress response, lactate metabolism | Rajeev et al., 2011 [13], this study |
| DVU0653 | TCS response regulator | GAFTGA | ncRNA downstream of DVU0653 | Posttranscriptional regulation | Rajeev et al., 2011 [13], this study |
| DVU0679 | TCS response regulator | GAYTGS | ncRNA downstream of DVU0679 | Posttranscriptional regulation | Rajeev et al., 2011 [13] |
| DVU0744 | TCS response regulator | GAFTGA | DVU0682 | Transcriptional regulation | Rajeev et al., 2011 [13], this study |
| DVU0804 | TCS response regulator | GAFTGA | DVU0805-DVU0806 | Posttranscriptional regulation | Rajeev et al., 2011 [13], this study |
| DVU0946 | TCS response regulator | GAFTGA | DVU0542-DVU0545, DVU0943-DVU0946, DVU2133-DVU2132 | Lactate metabolism, transmembrane transport | Rajeev et al., 2011 [13] |
| DVU1063 | TCS response regulator | GAFTGA | DVU0307, DVU0316-DVU0310, DVU0318, DVU0320, DVU0863-DVU0862, DVU1032, DVU1441, DVU1444, DVU1445-DVU1443, DVU1805, DVU1880, DVU2090 | Flagella | Rajeev et al., 2011 [13], this study |
| DVU1156 | TCS response regulator | GAFTGA | DVU1164 | Amide metabolism | Rajeev et al., 2011 [13], this study |
| DVU1419 | TCS response regulator | GAFTGA | DVU0036, DVU1418-DVU1419, ncRNA upstream of DVU3282 | Envelope stress response, posttranscriptional regulation | Rajeev et al., 2011 [13] |
| DVU1949 | Single-component regulator | GAFTGA | DVU1231-DVU1233, DVU1258, DVU3392*, DVU1952-DVU1949*, DVU0671*, DVU3290-DVU3292*, DVU2343-DVU2340*, DVU1823-DVU1821*, DVU0753-DVU0751* | Nitrogen metabolism and transport | This study |
| DVU2106 | Single-component regulator | GAFTDA | DVU2105-DVU2103, DVU2107-DVU2109 | Cell division | Fievet et al., 2011 |
| DVU2114 | TCS response regulator | GAFTGA | DVU3342-DVU3344, DORF39640-DVU2129 | Pili | Rajeev et al., 2011 [13] |
| DVU2275 | Single-component regulator | GAYTDA | DVU2272-DVU2269 | Pyruvate metabolism and transport | This study |
| DVU2359 | Single-component regulator | GAFTGA | ncRNA upstream of DVU2357 | Posttranscriptional regulation | This study |
| DVU2394 | TCS response regulator | GSFTGA | DVU2405-DVU2399 | Energy metabolism | Rajeev et al., 2011 [13] |
| DVU2827 | Single-component regulator | GAFSGA | DVU2820, DVU2822-DVU2825 | Pyruvate metabolism and transport | This study |
| DVU2894 | Single-component regulator | GAFTHA | DVU0047-DVU0043, DVU0410-DVU0409, DVU2894* | Flagella | This study |
| DVU2934 | TCS response regulator | GAFTGA | DVU2917 | Lipid A biosynthesis | Rajeev et al., 2011 [13] |
| DVU2956 | Single-component regulator | GAFTGA | DVU2957-DVU2964 | Transmembrane transport | This study |
| DVU2960 | Single-component regulator | GAFPGA | |||
| DVU2989 | Single-component regulator | GAFTGA | DVU2988-DVU2986 | Phage shock response | This study |
| DVU3023 | TCS response regulator | GAFTGA | DVU2451, DVU3025-DVU3033, DVU3284* | Lactate metabolism and transport | Rajeev et al., 2011 [13] |
| DVU3142 | Single-component regulator | GAFTGA | DVU3143-DVU3145 | Energy metabolism | This study |
| DVU3220 | TCS response regulator | GSFTGA | DVU1231-DVU1233 | Nitrogen metabolism and transport | Rajeev et al., 2011 [13] |
| DVU3305 | TCS response regulator | GAFSGA | DVU3302-DVU3298, DVU3303-DVU3305* | General stress response, transmembrane transport | Rajeev et al., 2011 [13] |
| DVU3334 | TCS response regulator | GAFTGA | DVU3339.1-DVU3337 | Potassium uptake | Rajeev et al., 2011 [13] |
| DVU3381 | TCS response regulator | GAFTGA | DVU3382-DVU3381, DVU3384 | Envelope stress response | Rajeev et al., 2011 [13] |
| DVUA0024 | TCS response regulator (pseudogene) | GAFTGA | |||
| DVUA0057 | TCS response regulator | GAFTGA | DVU0132-DVU0133, DVU1152, DVUA0030, DVUA0032-DVUA0031, DVUA0089, DVUA0036-DVUA0041 | Exopolysaccharide and biofilm synthesis | Rajeev et al., 2011 [13], this study |
| DVUA0100 | Single-component regulator | GVATGV | DVU4010, DVUA0106-DVUA0099, DVUA203-DVUA0125 | Type III secretion | This study |
| DVUA0143 | Single-component regulator | GAFTGA | DVUA0015-DVUA0007, DVUA0016 | Nitrogen metabolism | This study |
Operons that do not have σ54-dependent promoters are marked with asterisk
Many known EBPs have one or two N-terminal domains that modulate transcriptional activation capability of the regulator [2]. These domains recognize various environmental signals through phosphorylation or ligand binding. In particular, 22 EBPs of D. vulgaris Hildenborough are response regulators of two-component systems (TCS) with a N-terminal response regulator receiver domain (Fig. 2) that mediates phosphotransfer from a sensor histidine kinase. The other 15 EBPs are one-component regulatory systems (OCS) containing both input and output domains in a single protein, and lacking histidine kinase and response regulator domains. The two most common sensing domains of OCSs are GAF (3 proteins) [16] and PAS domains (6 proteins) [17]. One of the response regulator proteins, DVU0774, also has a PAS domain, which is suggestive of signal integration or dual activation. Besides PAS and GAF domains, we observed two OCSs with long conserved N-terminal regions that may encompass unknown sensing domains (DVU0151 and DVUA0100). Finally, 4 out of the 37 EBPs (DVU2894, DVU2956, DVU2989 and DVUA0024) lack N-terminal domains suggesting either an interaction with a partner protein that affects the regulatory activity of the EBP, or participation of these proteins in a regulatory cascade. Such interactions have been described in other species, for example, PspA protein negatively regulating PspF transcription factor [18].
Fig. 2.
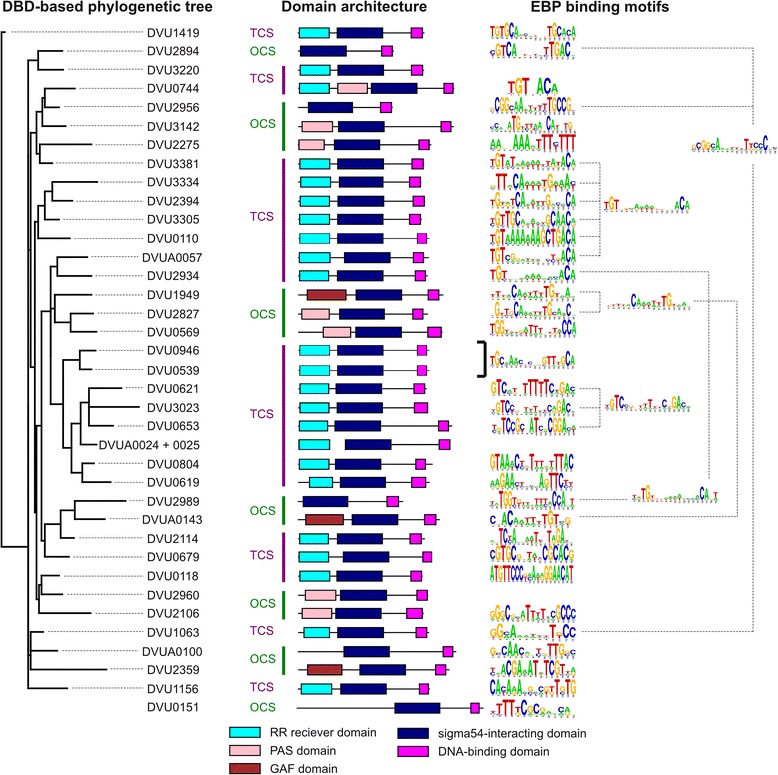
Phylogenetic tree of D. vulgaris Hildenborough EBPs based on DNA-binding domains. DVU0151 protein was omitted from this tree because it has a different DNA-binding domain sequence. Domain architecture and TF binding motifs are shown for each protein, where available. DVU0946 and DVU0539 EBPs share a common binding motif (indicated by bracket). Groups of similar motifs identified by TOMTOM comparison are displayed
The vast majority of known EBPs have a DNA-binding domain that is located at the C-terminus of the protein [2]. In all D. vulgaris Hildenborough EBPs but one, there are annotated Fis-type HTH motifs (Additional file 2), which are typical for σ54-dependent regulators [19]. The only exception is DVU0151 that does not have an annotated C-terminal DNA-binding domain and thus is not included in the phylogenetic tree constructed by DNA-binding domains (Fig. 2).
Our examination of DVU0151 and its orthologs from other Desulfovibrionales revealed that some of them (DvMF_2340, Dde_0697 and Dbac_2039) have matches to the winged helix DNA-binding domain model (SSF46785) from the SUPERFAMILY database [20], thus it is likely that DVU0151 orthologs have DNA-binding activity. Multiple sequence alignment of these proteins demonstrates the presence of strong sequence conservation at the location of the SSF46785 domain across all proteins, including DVU0151 (Additional file 3). This similarity further suggests that DVU0151 is a DNA-binding regulatory protein indeed. Thus, we predict that all 37 σ54-dependent regulators from D. vulgaris Hildenborough bind DNA in a sequence-specific manner, similar to a vast majority of other bacterial EBPs [2].
Reconstructed TF binding motifs for σ54-dependent regulators in D. vulgaris Hildenborough
In order to find the genes and operons that compose the regulon of each EBP, we reconstructed TF binding motifs for all these regulators and used those motifs in comparative genomics analysis. As the first step in motif reconstruction, we identified orthologs of all 37 D. vulgaris Hildenborough EBPs in nine species of Desulfovibrionales, as described in Methods. The number of EBPs in these genomes varies significantly, from 6 in Lawsonia intracellularis to 41 in Desulfomicrobium baculatum (Fig. 3). As expected, D. vulgaris Miyazaki, the closest relative of D. vulgaris Hildenborough, has 33 EBPs conserved between these two species, the highest number among the studied genomes. A little more distant L. intracellularis, D. desulfuricans and D. piger (see phylogenetic tree on Fig. 3) have strikingly lower numbers of EBP orthologs (5–9), due to a lower number of EBPs in these genomes in general. The number of EBP orthologs in the more distant Desulfovibrionales species gradually decrease from 24 in Desulfovibrio alaskensis G20 to 15 in Desulfohalobium retbaensae, with increase of phylogenetic distance from D. vulgaris Hildenborough (Fig. 3). There is no significant difference between conservation of TCS response regulators and that of one-component EBPs.
Fig. 3.
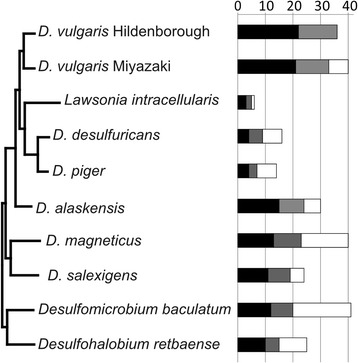
Phylogeny of Desulfovibrionales and conservation of D. vulgaris Hildenborough EBPs in closely related genomes. One-component EBPs from D. vulgaris Hildenborough and their orthologs are marked grey, σ54-dependent response regulators of TCSs from D. vulgaris Hildenborough and their orthologs are marked black, other EBPs (that lack orthologs in D. vulgaris Hildenborough) are marked white. Phylogenetic tree of Desulfovibrionales was extracted from the MicrobesOnline species tree [10], EBP orthologs were calculated as described in Methods section
It is interesting that DVUA0024 protein lacks any N-terminal signal domain but all its orthologs have response regulator receiver domain. We determined that DVUA0025 protein, encoded by a gene immediately upstream of DVUA0024, is an ortholog of this domain. In addition, DVUA0024 protein lacks eight N-terminal amino acids in the AAA+ domain interacting with σ54 subunit. By comparing the nucleotide sequences of the closely related D. vulgaris strains Hildenborough, DP4 and RCH1, we identified a single-nucleotide deletion in the genome of Hildenborough strain (data not shown). This deletion leads to the premature termination of DVUA0025 protein synthesis within the σ54-interacting domain. As D. vulgaris Hildenborough is unable to synthesize the complete functional protein, we excluded DVUA0024 from subsequent regulon analysis. Nevertheless, it is possible that this protein retains some transcriptional activity.
Our analysis of EBP conservation in Desulfovibrionales demonstrated a good potential for comparative reconstruction of regulons. Previous studies of EBPs in D. vulgaris Hildenborough [13, 14] identified binding sites for 14 EBPs. In this work, we reconstructed TF binding motifs for 19 other EBPs (Fig. 2, see Methods for details). Briefly, we used comparative reconstruction of σ54-dependent promoters in combination with regulator conservation results and existing DAP-chip data for several regulators [13]. In many cases, we found EBP-encoding genes to be co-localized with the regulated σ54-dependent promoters, and this co-localization was conserved within the Desulfovibrionales order. In most cases, such co-localization allowed the identification of a single regulatory gene. Only in one case, two regulatory genes, DVU2956 and DVU2960, are co-localized with the same σ54-dependent promoter in several genomes. Using purified proteins, we tested both candidate regulators in vitro for their ability to bind predicted sites, and found that only DVU2956 shows binding (Fig. 4).
Fig. 4.
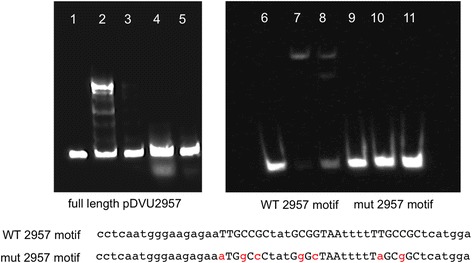
Transcriptional regulator DVU2956, and not DVU2960, binds upstream of target gene DVU2957. Lanes 1–5 show electrophoretic mobility shift assays with full-length (370 bp) upstream region of DVU2957: lane 1 – DNA only; lanes 2 and 3 – with purified DVU2956 protein (260 and 52 pmol, respectively); lanes 4 and 5 – with purified DVU2960 protein (400 and 80 pmol, respectively). Lanes 6–11 show EMSAs with predicted binding site motif upstream of DVU2957 (wild-type motif –lanes 6–8; mutated motif – lanes 9–11). Sequences of the two sites are shown below the gel image; the three conserved half-sites are shown in caps, and substitutions made in the mutated site are shown in red. Lanes 6, 9 – DNA only; lanes 7, 10 – with 390 pmol of DVU2956 protein; lanes 8, 11 – with 260 pmol of DVU2956 protein
All the identified TF binding motifs were compared by TOMTOM [21], since similarity of motifs could suggest a cross-regulation between different EBPs. All-against-all comparison of 33 EBP binding motifs found similarity in only 17 motifs that could be grouped into five based on similarity (Fig. 2). In three of these groups, motifs consist of half sites TGT and ACA separated by 9, 11 or 12 bp. It is interesting that similarity of TF binding motifs do not always reflect similarity of DNA-binding domains, as demonstrated by a comparison of the groups of motifs with a phylogenetic tree based on DNA-binding domains (Fig. 2). Nearly half of the motifs (16 out of 33) did not show similarity to any other motif.
Using the collection of these EBP binding motifs, we reconstructed or confirmed regulons of 34 EBPs by a whole-genome comparative analysis (Additional file 4). For one additional EBP, DVU3220, we had no predicted motif, thus its regulated genes were deduced from experimental results [13]. For the majority of σ54-dependent regulators, the regulons consist of just one to four regulated operons (Fig. 5). Only DVUA0057, DVU1949 and DVU1063 regulators have larger regulons with 6, 9 and 12 operons, respectively.
Fig. 5.
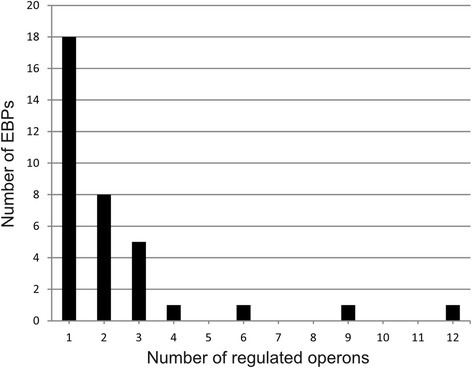
Distribution of sizes of EBP regulons
Functional diversity of σ54-dependent genes in D. vulgaris Hildenborough
We identified the content of σ54 sigmulon and 33 EBP regulons in D. vulgaris Hildenborough by a combination of computational and experimental data analysis (see Methods). We found 87 σ54-dependent promoters upstream of 85 operons that encompass 270 genes from different functional groups (Table 2, Additional files 5 and 6). The largest functional group (37 operons) includes genes involved in flagellar assembly and other functions linked to cell exterior: appendages, cell wall, secreted exopolysaccharides and biofilm components. This is in agreement with results of a recent large-scale analysis that demonstrated a connection of σ54-dependent transcription to the make-up of the bacterial exterior [3].
Table 2.
Summary of functions regulated by σ54-dependent operons. Functional assignment of individual operons shown in Additional file 5
| Category | Function | Operons | Regulators |
|---|---|---|---|
| Motility and cell exterior | Flagella | 19 | 2 |
| Chemotaxis | 2 | ||
| Pili | 3 | 1 | |
| Type III secretion | 4 | 1 | |
| Exopolysaccharide and biofilm synthesis | 7 | 2 | |
| Cell wall | 1 | ||
| Lipid A biosynthesis | 1 | 1 | |
| Metabolism and transport | Nitrogen metabolism and transport | 4 | 3 |
| Amino acid metabolism and transport | 2 | 1 | |
| Pyruvate metabolism and transport | 3 | 2 | |
| Lactate metabolism and transport | 2 | 4 | |
| Amide metabolism | 1 | 1 | |
| Energy metabolism | 2 | 2 | |
| Potassium uptake | 1 | 1 | |
| Other transmembrane transport | 9 | 6 | |
| Regulation | Transcriptional regulation | 4 | 3 |
| Posttranscriptional regulation | 6 | 5 | |
| Stress response | Envelope stress response | 2 | 2 |
| Nitrite stress response | 1 | 1 | |
| Phage shock response | 1 | 1 | |
| Other functions | Cell division | 2 | 1 |
| Function unknown | 8 | 1 |
Another large group of operons from σ54 sigmulon (24 operons) is comprised of metabolic and transport genes. A significant part of this group consists of genes of ammonia assimilation, nitrogen fixation and catabolism of amino acids and amides. Other genes in this group are involved in the metabolism of carboxylic acids, which are important carbon and energy sources for D. vulgaris and related species. σ54-dependent regulation of nitrogen metabolism, as well as the regulation of energy production, has been described in various taxonomic groups of bacteria (reviewed in [2]). However, σ54-dependent transcription of pyruvate and lactate metabolism genes have not been described in any other taxa. Nine operons in this group encode transporters from TSUP (toluene sulfonate uptake permease [22]) or PSE (putative sulfate export [23]) families. There is no data on substrate specificity of these transporting systems, but two of the PSE family transporters have been shown to be co-regulated with lactate metabolism genes [13].
Of the remaining 24 operons in σ54 sigmulon, 9 are involved in regulation, 5 in stress response, 2 in cell division and 8 have no assigned function. Regulatory RNAs and coding genes with σ54-dependent promoters are involved in transcriptional and posttranscriptional regulation, thus suggesting an existence of regulatory cascades possibly integrating different extracellular and/or intracellular signals. For some of the functions listed, transcriptional control by σ54 and EBPs has been shown in Deltaproteobacteria and other taxonomic groups (reviewed in [2]). The most studied examples of such functions are flagellar assembly and nitrogen metabolism. Since the homologous EBPs involved in the response to metal stress and phage shock have been documented in very distant bacteria such as E. coli [24], Salmonella enterica [2] and Yersinia enterocolitica [25], the conservation of these functions in σ54–dependent regulome of D. vulgaris Hildenborough emphasizes importance of such systems for bacteria.
In addition to the functions that are commonly regulated by σ54 and EBPs, we observed several EBP regulons that have not been described in any other bacteria. The four most interesting cases of σ54-dependent regulators for type III secretion, pyruvate transport metabolism, electron transfer and alanine dehydrogenase are described in detail below.
Regulator of type III secretion genes (DVUA0100)
The type III secretion system (T3SS) is a large multisubunit complex secreting effector proteins across bacterial membranes [26]. In D. vulgaris Hildenborough, T3SS components are encoded by two divergent operons with σ54-dependent promoters on the native pDV1 plasmid [27], and the gene encoding DVUA0100 EBP that is located in one of these operons. We predicted a putative DVUA0100 binding motif in the common upstream region of these two operons in three Desulfovibrio spp. genomes (Fig. 2). A whole-genome regulon analysis identified additional regulatory sites upstream of DVU4010 gene and its orthologs in two other genomes. These genes have predicted σ54-dependent promoters, and their products are annotated as hypothetical proteins. Using a computational tool for prediction of effector proteins of type III secretion system [28], we demonstrated a high probability for DVU4010 to be a secreted T3SS effector (NaiveBayes Prediction Score = 0.994). Colocalization of DVU4010 gene with DVU2392 gene encoding type III secretion chaperone on the chromosome supports our functional assignment.
Regulators of pyruvate formate-lyases (DVU2275 and DVU2827)
Pyruvate formate-lyase (PFL) catalyzes the reversible nonoxidative dissimilation of pyruvate to acetyl-coenzyme A and formate [29]. D. vulgaris Hildenborough genome contains two genes encoding PFLs, DVU2272 and DVU2824 [30], and both of them are co-transcribed with genes encoding PFL-activating proteins. DVU2824-encoding operon also contains genes for C4-dicarboxylate transporting system, DVU2822 and DVU2823 (Fig. 6). We observed that orthologs of two EBP-encoding genes, DVU2275 and DVU2827, are co-localized with PFL operons in Desulfovibrionales genomes, and these operons had conserved σ54-dependent promoters. We predicted that DVU2275 regulator binds an AT-rich palindromic site upstream of the DVU2272 promoter (Fig. 6). DVU2271 gene demonstrated increased expression during growth on hydrogen and sulfate [31], thus suggesting that the DVU2272-DVU2269 operon may be involved in pyruvate biosynthesis when acetate and CO2 are the only carbon sources available.
Fig. 6.
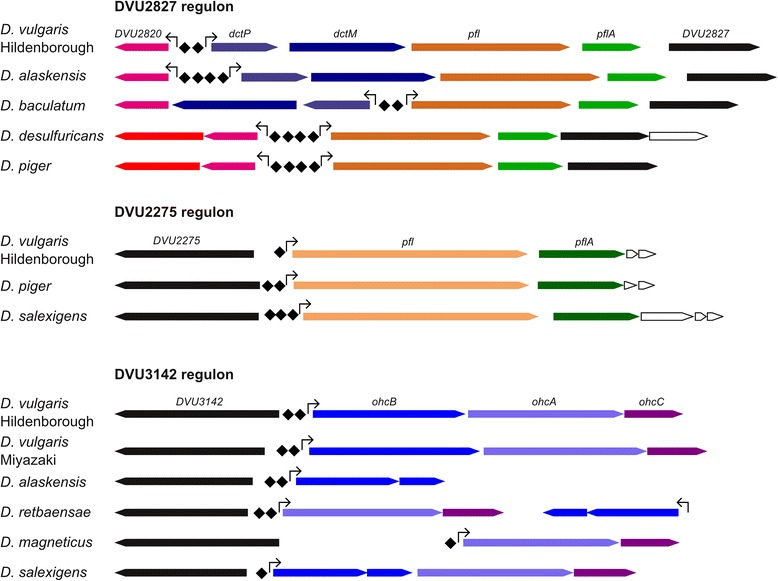
DVU2827, DVU2275, DVU3142 regulons and their conservation in other Desulfovibrionales. EBP-encoding genes are marked black, promoters are shown by arrows, EBP binding sites are shown by diamonds. Genes encoding MFS transporters are marked red, genes encoding amydohydrolase family proteins are marked magenta, genes encoding PFL are marked orange, genes encoding PFL-activating enzymes are marked green
We also predicted DVU2827 binding sites upstream of operons encoding pyruvate-formate lyase DVU2824 and its orthologs in five Desulfovibrionales genomes (Fig. 6). These sites probably activate a neighbor gene DVU2820 encoding amidohydrolase family protein, which also has σ54-dependent promoter. Orthologs of DVU2820 in D. desulfuricans and D. piger (Ddes_11438 and DESPIG_02846) are co-transcribed with genes encoding major facilitator superfamily (MFS) transporters. These two genomes lack C4-dicarboxylate transporters encoded by DVU2822 and DVU2823 orthologs. Search in Transporter Classification Database [PMID: 24225317] identified similar MFS proteins from metabolite:H+ symporter family (2.A.1.6) that can transport citrate, oxoglutarate, dicarboxylates, acetate and shikimate (e-value below e−46). These data suggest that MFS transporters Ddes_1438 and DESPIG_02846 may transport C4-dicarboxylates.
Regulator of electron transport system (DVU3142)
It was previously demonstrated that octaheme cytochrome complex (Ohc) transports electrons from a periplasmic octahemic c-type cytochrome OhcA to an iron-sulfur membrane protein OhcB and then to a haem b-containing membrane protein OhcC [32]. In D. vulgaris Hildenborough Ohc complex proteins are encoded by ohcBAC operon, and one or several ohc genes are present in other Desulfovibrio spp. genomes as well [33]. We observed that a regulatory gene DVU3142 and its orthologs are divergently transcribed from the ohc genes in six Desulfovibrionales genomes (Fig. 6). A comparative analysis identified σ54-dependent promoters upstream of ohc genes, thus we predicted that DVU3142 EBP is a transcriptional regulator of Ohc system. We detected DVU3142 binding sites upstream of all ohc operons. Remarkably, D. alaskensis G20 lacks ohcA and ohcC genes but the last remaining gene of the Ohc system, ohcB, is still regulated by a DVU3142 ortholog since we found σ54-dependent promoter and DVU3142 binding site upstream of the ohcB gene (Fig. 6).
Regulator of alanine dehydrogenase (DVU0569)
L-Alanine dehydrogenase (Ald) catalyzes the oxidative deamination of L-alanine and the reductive amination of pyruvate. Various regulators of alanine dehydrogenase have been characterized in Bacillus subtilis (PucR family TF [34]), Amycolatopsis mediterranei (GlnR family TF [35]), Rhizobium leguminozarum and Mycobacterium smegmatis (AsnC family TFs [36, 37]), but no such regulators have been studied in Deltaproteobacteria. A gene encoding Ald enzyme in D. vulgaris (DVU0571) is located downstream from the regulatory gene DVU0569 as well as in the six other Desulfovibrionales genomes. A conserved regulatory motif (Fig. 2) and σ54-dependent promoters were found upstream of ald genes.
Evolutionary conservation of σ54-dependent regulome in Desulfovibrionales species
To study phylogenetic distribution of the reconstructed σ54-dependent regulons, we performed two-dimensional hierarchical clustering based on the presence/absence of reconstructed regulons among Desulfovibrionales genomes (Fig. 7). The largest group (20 D. vulgaris Hildenborough regulons) is almost exclusively conserved in free-living (i. e. inhabiting such environments as soils, hotsprings, sediments and lakes) bacteria. Many regulons controlling metabolism, extracellular structures, non-coding RNAs and stress response belong to this group. A group of four D. vulgaris Hildenborough regulons is conserved in most species, regardless of their lifestyle. This group includes regulons controlling flagellar genes, stress response and nitrogen metabolism. The third group consists of 13 low-conserved regulons that sporadically occur in different genomes.
Fig. 7.
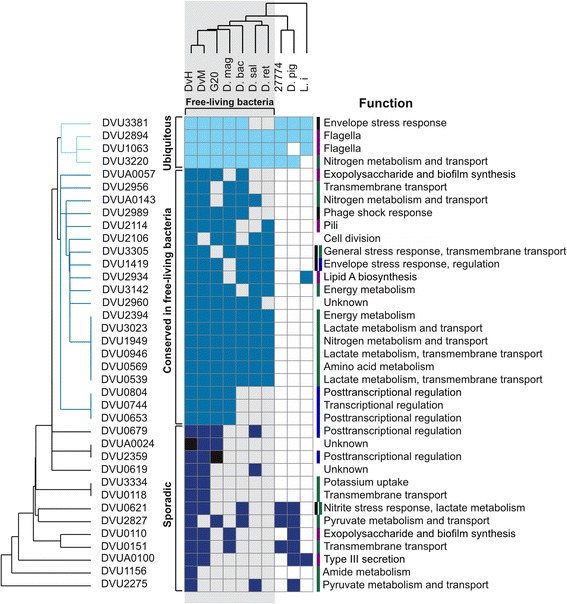
Two-dimensional hierarchical clustering based on conservation of reconstructed σ54-dependent regulons across Desulfovibrionales genomes. Free-living bacteria are marked with grey background. Pseudogenes of EBPs are marked black. Species abbreviations used: DvH, Desulfovibrio vulgaris Hildenborough; DvM: Desulfovibrio vulgaris Miyazaki; G20: Desulfovibrio alaskensis G20; D. mag: Desulfovibrio magneticus; D. bac: Desulfomicrobium baculatum; D.sal: Desulfovibrio salexigens; D. ret: Desulfohalobium retbaensae; 27774: D. desulfuricans 27774; D. pig: Desulfovibrio piger; L. i: Lawsonia intracellularis
Clustering results demonstrate that bacterial species are grouped by their lifestyle rather than by their phylogenetic relatedness (see Fig. 3 for comparison). Free-living species are clustered separately from animal host-associated species. The variation in the numbers of σ54-dependent regulons among Desulfovibrionales species suggest that σ54-dependent regulation may play an important role in the adaptation of free-living species to constantly changing environmental conditions. Based on this analysis we speculate that the relative higher abundance of EBPs and associated σ54-dependent regulons in free-living Deltaproteobacteria is a consequence of their life style. However, neither functional category is strictly limited to free-living bacteria: EBPs regulating metabolism and transport and EBPs regulating extracellular functions may be ubiquitous or be restricted to free-living bacteria. Only regulators of non-coding RNAs were found to have a propensity toward free-living species.
Co-occurrence of EBP binding sites and σ54-dependent promoters in D. vulgaris Hildenborough
Our reconstruction of σ54-dependent regulome includes UASs upstream of 64 operons with σ54-dependent promoters (Fig. 8a). In addition to these operons, EBP binding sites were found upstream of nine operons that have no σ54-dependent promoters. The biggest fraction of σ54-dependent promoters were found within 100 nucleotides from the closest EBP binding site (Fig. 8b), while in four cases, the promoter was found at more than 300 bp downstream from the cognate UAS. Further, while EBP binding sites are typically located upstream of the σ54-dependent promoter, a small number of UASs were found at a distance of 40–70 bp downstream from the promoters, and most of these were binding sites for the flagellar regulator DVU1063.
Fig. 8.
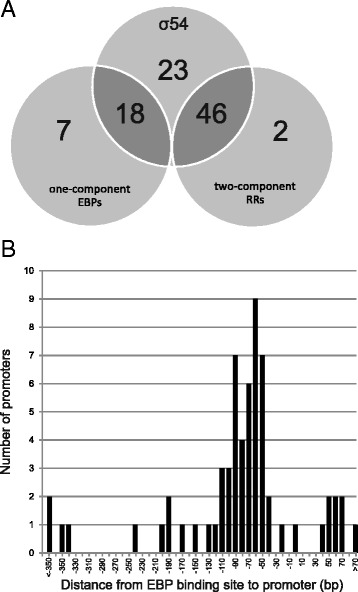
a Number of operons in σ54-dependent sigmulon, one-component EBP regulome and two-component EBP regulome. Dark grey areas correspond to operons co-regulated by both EBP and σ54 protein. Light grey areas correspond to either operons with EBP binding sites or σ54-dependent promoters only. b Distribution of distances between σ54-dependent promoter and closest EBP binding site
For 23 operons with σ54-dependent promoters, we could not find predicted UASs (Fig. 8b). These promoters belong to operons from different functional groups, but a majority of them are involved in the composition of the bacterial exterior. In particular, almost half of these operons are predicted to be involved in flagellar biogenesis and motility: five operons encode flagellar components and six operons are co-expressed with flagellar genes (data not shown). The remaining seven promoters without predicted UASs are linked with chemotaxis, exopolysaccharide biosynthesis, biofilm formation, cell wall metabolism, pili and type III secretion. We cannot exclude the possibility that some of these operons may belong to the unidentified DVUA0024 and DVU2960 regulons. However, they also may be regulated by EBP binding sites located far upstream or downstream from the promoter, beyond the limits of our search or within coding regions. For example, an NtrC-dependent enhancer retains its activity even being positioned up to 15 kb away from σ54-dependent glnAp2 promoter of E. coli [38]. A high conservation of coding regions precludes us from prediction of such enhancers by computational methods, thus further experimental studies of such cases are needed.
Conclusions
In this study we comprehensively characterized the genome-wide σ54-dependent regulome of D. vulgaris Hildenborough. The results include: the σ54-dependent EBPs (Table 1), the EBP binding sites (Additional file 4), the σ54-dependent promoters (Additional file 6) and the σ54-dependent genes and operons (Additional file 5).
Our results demonstrate that the σ54-dependent regulome controls a wide variety of cellular functions in D. vulgaris Hildenborough. We identified regulatory elements that activate σ54-dependent transcription of several dozens of operons. In particular, we inferred several σ54-dependent regulons not known before (including those associated with type III secretion, electron transport, pyruvate and formate metabolism). Many of these elements are conserved between D. vulgaris Hildenborough and related sulfate-reducing Deltaproteobacteria that makes possible the semi-automated inference of σ54-dependent regulome in newly sequenced Deltaproteobacteria by projection of the collection of σ54-dependent regulons reconstructed in this study.
Methods
Data sources
In this work we used genome and protein sequences from NCBI GenBank [39] database and MicrobesOnline [10], and the protein domain annotations from MicrobesOnline.
Identification of enhancer binding proteins (EBPs)
EBPs were identified as proteins with PF00158 domain, AAA+ domain involved in ATP-dependent interaction with σ54 protein.
To check for EBP regulatory function, we verified the presence of the peptide ‘GAFTGA’ motif required for the EBP interaction with σ54. We used alignments from MicrobesOnline database [10] for comparison with the PF00158 model from PFAM [40].
We identified a set of orthologs of D.vulgaris Hildenborough EBPs in other Desulfovibrionales genomes using tree-orthologs computed by MicrobesOnline by examining the pre-computed domain-based gene trees [10]. Cluster 3.0 was used for hierarchical two-dimensional clustering [41].
Identification of σ54-dependent promoters
To identify σ54-dependent transcripts in D. vulgaris Hildenborough genome, we used both the results of experimental analysis and comparative genomics reconstruction. The presence of σ54-dependent promoters in the D. vulgaris Hildenborough genome have been shown in several studies [15, 42], but the results of computational and experimental analyses were substantially different. A computational analysis [42] identified 70 candidate σ54-dependent promoters. A genome-wide experimental analysis of transcription by 5′-RNA-seq [15] identified another set of 70 putative σ54-dependent promoters. However, only 19 promoters are present in both lists.
For comparative analysis we constructed a new, taxon-specific motif for recognition of σ54-dependent promoters in Desulfovibrio spp. The initial motif was deduced from the upstream regions of DVU0316, DVU0318, DVU1032 and DVU1441 genes and their orthologs from five Desulfovibrio genomes. These four genes were selected because they are regulated by σ54-dependent DVU1063 response regulator [13], and their predicted σ54-dependent promoters perfectly fit 5′-RNA-seq data [15]. We used this motif for initial search of σ54-dependent promoters in the ten Desulfovibrionales genomes using RegPredict [43]. From this search, to refine the σ54-dependent promoter motif, we selected those D. vulgaris Hildenborough promoters that are located close to the mapped RNA 5′-ends [15] and orthologous promoters from the other nine species. The final motif, based on comparison of the ten genomes, is highly similar to the σ54-dependent promoter motif identified by 5′-RNA-seq analysis in D. vulgaris Hildenborough [15]. It has two promoter elements similar to −24 and −12 elements from other taxonomic groups [44], with consensus sequences tGGcacg and tTGCt, respectively.
A comparative analysis with the refined σ54-dependent promoter motif identified 84 conserved promoters in the D. vulgaris Hildenborough genome. Additional three high-score non-conserved σ54-dependent promoters were added to our dataset from the 5′-RNA-seq results [15]. Thus, our reconstruction of σ54-dependent sigmulon of D. vulgaris Hildenborough produced 87 promoters (Additional file 6).
Reconstruction of regulons for one-component EBPs
To identify binding sites for the one-component σ54-dependent regulatory proteins, we applied comparative genomics approach. It assumes that TF binding sites upstream of regulated orthologous genes are conserved in different species [45]. An important requirement is the presence of orthologous TFs in these species. We considered σ54-dependent operons co-localized with EBP-encoding genes the most probable targets of these EBPs since bacterial regulatory genes are often co-localized with their regulated genes [46].
We found that ten out of thirteen genes encoding one-component EBPs are co-localized with σ54-dependent promoters. Three of these genes, DVU2827, DVU2960 and DVUA0100, are located in σ54-dependent operons. Six other genes, DVU0151, DVU2275, DVU2359, DVU2956, DVU2989, DVU3142, are divergently transcribed from σ54-dependent promoters. And for one gene encoding EBP, DVU0569, σ54-dependent promoter was observed downstream of the gene. In other Desulfovibrionales genomes, orthologs of these EBP-encoding genes are co-localized with σ54-dependent promoters as well. We used the “Discover profile” tool of the RegPredict web-server [43] for reconstruction of EBP binding motifs. Sets of upstream sequences of orthologous genes (from −650 to +50 with respect to the translation start) were selected and a common motif with highest information content was identified in each set. We identified candidate binding motifs for all ten EBPs upstream of the respective orthologous σ54-dependent promoters.
However, for three regulatory genes, DVU1949, DVU2894 and DVUA0143, we could not find conserved σ54-dependent promoters nearby and thus used other evidences such as co-expression and genome context analysis as described below.
For DVU2894 gene we found evidence of its co-expression with the genes encoding flagella assembly proteins by analyzing transcriptomics data from MicrobesOnline. In addition, this protein is similar to the flagellar regulators FlrA and FleQ based upon phylogenetic analysis with AAA+ domain-based phylogenetic tree. We proposed that DVU2894 gene is an auto-regulated member of the flagellar regulatory cascade, since it is not regulated by the flagellar regulator DVU1063 [13]. We also found a conserved inverted repeat with consensus yGTCA-N6-TGACr upstream of all DVU2894 orthologs. Since a similar inverted repeat GTCA-N6-TGGC has been observed in the FlrA binding region upstream of flrBC operon promoter in V. cholerae [47], we predict that this repeat constitutes DVU2894 binding motif.
For the DVUA0143 regulatory gene, genome context analysis did not provide a clue for possible targets, but the following three evidences suggest that DVUA0143 may be a regulator of nitrogenase operon DVUA0015-DVUA0007. First, phylogenetic analysis indicates similarity of its N-terminal domain to AnfA and VnfA, regulators of nitrogenases from Azotobacter vinelandii [48]. Second, phylogenetic distribution of the DVUA0143 orthologs in Desulfovibrionales corresponds to the distribution of nitrogenase operons: all five of the ten genomes that have nifHDK genes also possess DVUA0143 homologs. And, finally, DVUA0143 ortholog is co-localized with nitrogenase operon in Syntrophobacter fumaroxidans. Following these conclusions, we found a candidate DVUA00143 binding motif in upstream regions of nitrogenase operons from five Desulfovibrionales genomes.
For the gene encoding DVU1949 regulator, genome context analysis indicated a co-transcription with orthologs of DVU1952 gene encoding a hypothetical protein in seven Desulfovibrionales genomes. In four genomes, these genes are also co-transcribed with iorA and iorB genes encoding alpha and beta subunits of indolepyruvate ferredoxin oxidoredctase (an enzyme involved in the metabolism of aromatic amino acids [49]). We proposed that DVU1949 gene is autoregulated and identified a conserved sequence motif upstream of this operon.
The next steps of regulon reconstruction were done with RegPredict, a web-tool for comparative genomic analysis (21). Briefly, EBP binding motifs identified in this work were used for a whole-genome search in upstream regions of coding genes (from −650 to +50 with respect to the translation start) with a threshold equal to a minimal score among all sites reported by the “Discover profile” tool for the motif.
Refinement of regulon data for two-component σ54-dependent regulatory systems
In our previous study [13], we identified target genes of two-component response regulators in D. vulgaris Hildenborough by a combination of experimental and computational approaches. In that study we found target genes for 18 of 22 σ54-dependent response regulators and assigned TF binding motifs to 13 of them. We could not find TF binding motifs for the rest of TFs since those proteins are conserved in just four or less Desulfovibrio genomes available at that time [10]. As more genome sequences of Desulfovibrionales spp., both complete and incomplete, became available in GenBank [39], we added them to characterize the TF binding motifs. For the incomplete genomes that have orthologs of TF and target genes previously found by DAP-chip, we extracted upstream regions of the respective orthologs for search of conserved sequence motifs. It allowed us to identify TF binding motifs for DVU0110, DVU0621, DVU0653, DVU0744, DVU0804 and DVUA0057 regulatory proteins. For DVU1156 regulator that has no orthologs in Desulfovibrio spp., we applied a specific approach for reconstruction of singleton regulons described earlier [50].
Our whole-genome regulon reconstruction with RegPredict demonstrated that TF binding motif predictions fit our existing DAP-chip data [13] and correlate with the predictions of σ54-dependent promoters (Additional file 6). However, we could not find TF binding sites upstream of several genes that lack σ54-dependent promoters, despite the fact that the binding was demonstrated by DAP-chip (for example, DVU3131 for DVU0804 protein, DVU0736 for DVU0744 protein and DVU0328, DVU0330, DVU1639, DVU2842, DVU1017 for DVUA0057 protein).
For DVU0110 and DVUA0057 EBPs, we predict a common binding motif. Previously, we demonstrated that these two EBPs bind upstream of DVU0132-DVU0133 operon encoding TSUP family permease [13]. Here, we found a conserved motif upstream of this operon in six Desulfovibrionales genomes. Two of these genomes have only DVU0110 orthologs (D. magneticus and D. piger), two genomes have only DVUA0057 orthologs (D. alaskensis and D. baculatum) and two genomes have both EBPs (D. vulgaris strains Hildenborough and Miyazaki). Thus, the found motif cannot be attributed to either of these regulators but only to both of them. Similar binding sites were found upstream of other known DVUA0057 target genes [13] with σ54-dependent promoters (DVUA0030, DVUA0032, DVUA0036, DVUA0089), thus suggesting that DVUA0057 binds them.
We also assigned binding sites for DVU0619, a response regulator not studied by DAP-chip in our previous work. Comparative analysis demonstrated that the DVU0617-DVU0616 operon and its homologs in several genomes are co-localized with DVU0619 orthologs, and σ54-dependent promoters are present upstream of the operons. A putative DVU0619 binding motif was identified upstream of all these operons.
Electrophoretic mobility shift assay
For the putative regulatory motif upstream of DVU2957 promoter, we had two neighbor candidate regulatory proteins, DVU2956 and DVU2960. DVU2956 gene is divergently transcribed from the DVU2957-DVU2967 operon, while DVU2960 gene is a part of the operon. The final assignment of a regulatory protein for this motif was done by EMSA. DVU2956 and DVU2960 genes were amplified from D. vulgaris Hildenborough genomic DNA and each cloned into vector pSKB3 with a N-terminal His-tag. The resulting constructs were transformed into E. coli BL21 (DE3), and His-tagged proteins were expressed and purified as described previously [13]. Full-length EMSA substrate was prepared by amplifying a 370 bp region upstream of DVU2957 with a biotin-labeled reverse primer and an unlabeled forward primer, followed by gel purification. Predicted motif substrates for EMSA were prepared by annealing biotinylated oligonucleotides containing the binding site as described previously [13]. EMSA reactions (20 μl) were set up at room temperature for 20 min with 100 fmol of DNA and purified His-tagged protein (amounts shown in Fig. 4) in 10 mM Tris HCl pH 7.5, 1 mM DTT, 50 mM KCl, 5 mM MgCl2 and 1 μg/ml poly dI.dC. Electrophoresis, blotting, and chemiluminescent detection were performed as described previously [13]. Final imaging of the blot was done using the Fluor Chem Q system (Protein Simple, Santa Clara, CA).
Availability of supporting data
The data sets supporting the results of this article are included within the article and its additional files. The collection of EBP regulons reconstructed with the RegPredict web-server [43] is available in the RegPrecise database, http://regprecise.lbl.gov [51].
Acknowledgements
The authors are grateful to Dmitry Rodionov for useful discussions. This material by ENIGMA- Ecosystems and Networks Integrated with Genes and Molecular Assemblies (http://enigma.lbl.gov), a Scientific Focus Area Program at Lawrence Berkeley National Laboratory is based upon work supported by the U.S. Department of Energy, Office of Science, Office of Biological & Environmental Research under contract number DE-AC02-05CH11231.
Abbreviations
- EBP
Enhancer binding protein
- MFS
Major facilitator superfamily
- OCS
One-component system
- PFL
Pyruvate formate-lyase
- TF
Transcription factor
- TCS
Two-component system
- T3SS
Type III secretion system
- UAS
Upstream activating sequence
Additional files
EBP taxonomic distribution. (XLSX 78 kb)
EBP DNA-binding domains. (XLSX 9 kb)
{kind=link}
Multiple sequence alignment of homologous regions corresponding to the SSF46785 domain in DVU0151 and its orthologs. (PNG 22 kb)
EBP binding sites. (XLSX 41 kb)
σ 54 -dependent genes and operons. (XLSX 18 kb)
σ 54 -dependent promoters. (XLSX 19 kb)
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
AEK performed the bioinformatical analysis and drafted the manuscript. LR, EGL and AC carried out the cloning, protein purification and mobility shift assays. ID discussed the results and helped to draft the manuscript. AM participated in the design and coordination of the study. PSN conceived of the study, directed the whole research and drafted the manuscript. All authors read and approved the final manuscript.
Contributor Information
Alexey E. Kazakov, Email: aekazakov@lbl.gov
Lara Rajeev, Email: lrajeev@lbl.gov.
Amy Chen, Email: amychenn@gmail.com.
Eric G. Luning, Email: eluning4@gmail.com
Inna Dubchak, Email: ildubchak@lbl.gov.
Aindrila Mukhopadhyay, Email: amukhopadhyay@lbl.gov.
Pavel S. Novichkov, Email: psnovichkov@lbl.gov
References
- 1.Buck M, Gallegos MT, Studholme DJ, Guo Y, Gralla JD. The bacterial enhancer-dependent sigma(54) (sigma(N)) transcription factor. J Bacteriol. 2000;182:4129–36. doi: 10.1128/JB.182.15.4129-4136.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bush M, Dixon R. The role of bacterial enhancer binding proteins as specialized activators of σ54-dependent transcription. Microbiol Mol Biol Rev. 2012;76:497–529. doi: 10.1128/MMBR.00006-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Francke C, Groot Kormelink T, Hagemeijer Y, Overmars L, Sluijter V, Moezelaar R, et al. Comparative analyses imply that the enigmatic Sigma factor 54 is a central controller of the bacterial exterior. BMC Genomics. 2011;12:385. doi: 10.1186/1471-2164-12-385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Keseler IM, Kaiser D. Sigma54, a vital protein for Myxococcus xanthus. Proc Natl Acad Sci U S A. 1997;94:1979–84. doi: 10.1073/pnas.94.5.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Leang C, Krushkal J, Ueki T, Puljic M, Sun J, Juárez K, et al. Genome-wide analysis of the RpoN regulon in Geobacter sulfurreducens. BMC Genomics. 2009;10:331. doi: 10.1186/1471-2164-10-331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Reitzer L, Schneider BL. Metabolic context and possible physiological themes of sigma(54)-dependent genes in Escherichia coli. Microbiol Mol Biol Rev. 2001;65:422–44. doi: 10.1128/MMBR.65.3.422-444.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Salazar E, Díaz-Mejía JJ, Moreno-Hagelsieb G, Martínez-Batallar G, Mora Y, Mora J, et al. Characterization of the NifA-RpoN regulon in Rhizobium etli in free life and in symbiosis with Phaseolus vulgaris. Appl Environ Microbiol. 2010;76:4510–20. doi: 10.1128/AEM.02007-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cases I, Ussery DW, de Lorenzo V. The sigma54 regulon (sigmulon) of Pseudomonas putida. Environ Microbiol. 2003;5:1281–93. doi: 10.1111/j.1462-2920.2003.00528.x. [DOI] [PubMed] [Google Scholar]
- 9.Stevens MJA, Molenaar D, de Jong A, De Vos WM, Kleerebezem M. Sigma54-Mediated control of the mannose phosphotransferase sytem in Lactobacillus plantarum impacts on carbohydrate metabolism. Microbiology. 2010;156(Pt 3):695–707. doi: 10.1099/mic.0.034165-0. [DOI] [PubMed] [Google Scholar]
- 10.Dehal PS, Joachimiak MP, Price MN, Bates JT, Baumohl JK, Chivian D, et al. MicrobesOnline: an integrated portal for comparative and functional genomics. Nucleic Acids Res. 2010;38(Database issue):D396–400. doi: 10.1093/nar/gkp919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jakobsen JS, Jelsbak L, Jelsbak L, Welch RD, Cummings C, Goldman B, et al. Sigma54 enhancer binding proteins and Myxococcus xanthus fruiting body development. J Bacteriol. 2004;186:4361–8. doi: 10.1128/JB.186.13.4361-4368.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Heidelberg JF, Seshadri R, Haveman SA, Hemme CL, Paulsen IT, Kolonay JF, et al. The genome sequence of the anaerobic, sulfate-reducing bacterium Desulfovibrio vulgaris Hildenborough. Nat Biotechnol. 2004;22:554–9. doi: 10.1038/nbt959. [DOI] [PubMed] [Google Scholar]
- 13.Rajeev L, Luning EG, Dehal PS, Price MN, Arkin AP, Mukhopadhyay A. Systematic mapping of two component response regulators to gene targets in a model sulfate reducing bacterium. Genome Biol. 2011;12:R99. doi: 10.1186/gb-2011-12-10-r99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fiévet A, My L, Cascales E, Ansaldi M, Pauleta SR, Moura I, et al. The anaerobe-specific orange protein complex of Desulfovibrio vulgaris hildenborough is encoded by two divergent operons coregulated by σ54 and a cognate transcriptional regulator. J Bacteriol. 2011;193:3207–19. doi: 10.1128/JB.00044-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Price MN, Deutschbauer AM, Kuehl JV, Liu H, Witkowska HE, Arkin AP. Evidence-based annotation of transcripts and proteins in the sulfate-reducing bacterium Desulfovibrio vulgaris Hildenborough. J Bacteriol. 2011;193:5716–27. doi: 10.1128/JB.05563-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Aravind L, Ponting CP. The GAF domain: an evolutionary link between diverse phototransducing proteins. Trends Biochem Sci. 1997;22:458–9. doi: 10.1016/S0968-0004(97)01148-1. [DOI] [PubMed] [Google Scholar]
- 17.Ponting CP, Aravind L. PAS: a multifunctional domain family comes to light. Curr Biol. 1997;7:R674–7. doi: 10.1016/S0960-9822(06)00352-6. [DOI] [PubMed] [Google Scholar]
- 18.Elderkin S, Jones S, Schumacher J, Studholme D, Buck M. Mechanism of action of the Escherichia coli phage shock protein PspA in repression of the AAA family transcription factor PspF. J Mol Biol. 2002;320:23–37. doi: 10.1016/S0022-2836(02)00404-7. [DOI] [PubMed] [Google Scholar]
- 19.Vidangos N, Maris AE, Young A, Hong E, Pelton JG, Batchelor JD, et al. Structure, function, and tethering of DNA-binding domains in σ(54) transcriptional activators. Biopolymers. 2013;99:1082–96. doi: 10.1002/bip.22333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wilson D, Pethica R, Zhou Y, Talbot C, Vogel C, Madera M, et al. SUPERFAMILY--sophisticated comparative genomics, data mining, visualization and phylogeny. Nucleic Acids Res. 2009;37(Database issue):D380–6. doi: 10.1093/nar/gkn762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gupta S, Stamatoyannopoulos JA, Bailey TL, Noble WS. Quantifying similarity between motifs. Genome Biol. 2007;8:R24. doi: 10.1186/gb-2007-8-2-r24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shlykov MA, Zheng WH, Chen JS, Saier MH., Jr Bioinformatic characterization of the 4-Toluene Sulfonate Uptake Permease (TSUP) family of transmembrane proteins. Biochim Biophys Acta. 1818;2012:703–17. doi: 10.1016/j.bbamem.2011.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rein U, Gueta R, Denger K, Ruff J, Hollemeyer K, Cook AM. Dissimilation of cysteate via 3-sulfolactate sulfo-lyase and a sulfate exporter in Paracoccus pantotrophus NKNCYSA. Microbiology. 2005;151(Pt 3):737–47. doi: 10.1099/mic.0.27548-0. [DOI] [PubMed] [Google Scholar]
- 24.Jovanovic G, Weiner L, Model P. Identification, nucleotide sequence, and characterization of PspF, the transcriptional activator of the Escherichia coli stress-induced psp operon. J Bacteriol. 1996;178:1936–45. doi: 10.1128/jb.178.7.1936-1945.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Green RC, Darwin AJ. PspG, a new member of the Yersinia enterocolitica phage shock protein regulon. J Bacteriol. 2004;186:4910–20. doi: 10.1128/JB.186.15.4910-4920.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hueck CJ. Type III protein secretion systems in bacterial pathogens of animals and plants. Microbiol Mol Biol Rev. 1998;62:379–433. doi: 10.1128/mmbr.62.2.379-433.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.T3DB, a database for bacterial type III secretion system: Desulfovibrio. http://biocomputer.bio.cuhk.edu.hk/T3DB/Desulfovibrio.php. Accessed May 18, 2015.
- 28.Tay DMM, Govindarajan KR, Khan AM, Ong TYR, Samad HM, Soh WW, et al. T3SEdb: data warehousing of virulence effectors secreted by the bacterial Type III Secretion System. BMC Bioinformatics. 2010;11(Suppl 7):S4. doi: 10.1186/1471-2105-11-S7-S4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Knappe J, Sawers G. A radical-chemical route to acetyl-CoA: the anaerobically induced pyruvate formate-lyase system of Escherichia coli. FEMS Microbiol Rev. 1990;6:383–98. doi: 10.1111/j.1574-6968.1990.tb04108.x. [DOI] [PubMed] [Google Scholar]
- 30.Tang Y, Pingitore F, Mukhopadhyay A, Phan R, Hazen TC, Keasling JD. Pathway confirmation and flux analysis of central metabolic pathways in Desulfovibrio vulgaris hildenborough using gas chromatography–mass spectrometry and Fourier transform-ion cyclotron resonance mass spectrometry. J Bacteriol. 2007;189:940–9. doi: 10.1128/JB.00948-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pereira PM, He Q, Valente FMA, Xavier AV, Zhou J, Pereira IAC, et al. Energy metabolism in Desulfovibrio vulgaris Hildenborough: insights from transcriptome analysis. Antonie Van Leeuwenhoek. 2008;93:347–62. doi: 10.1007/s10482-007-9212-0. [DOI] [PubMed] [Google Scholar]
- 32.Pereira IAC, Haveman SA, Voordouw G. Biochemical, genetic and genomic characterization of anaerobic electron transport pathways in sulphate-reducing Delta proteobacteria. In: Barton LL, Hamilton WA, editors. Sulphate-Reducing Bacteria. Cambridge: Cambridge University Press; 2007. pp. 215–40. [Google Scholar]
- 33.Pereira IAC, Ramos AR, Grein F, Marques MC, da Silva SM, Venceslau SS. A comparative genomic analysis of energy metabolism in sulfate reducing bacteria and archaea. Front Microbiol. 2011;2:69. doi: 10.3389/fmicb.2011.00069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lin T-H, Wei G-T, Su C-C, Shaw G-C. AdeR, a PucR-type transcription factor, activates expression of L-alanine dehydrogenase and is required for sporulation of Bacillus subtilis. J Bacteriol. 2012;194:4995–5001. doi: 10.1128/JB.00778-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang Y, Li C, Duan N, Li B, Ding X-M, Yao Y-F, et al. GlnR negatively regulates the transcription of the alanine dehydrogenase encoding gene ald in Amycolatopsis mediterranei U32 under nitrogen limited conditions via specific binding to its major transcription initiation site. PLoS One. 2014;9:e104811. doi: 10.1371/journal.pone.0104811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lodwig E, Kumar S, Allaway D, Bourdes A, Prell J, Priefer U, et al. Regulation of L-alanine dehydrogenase in Rhizobium leguminosarum bv. viciae and its role in pea nodules. J Bacteriol. 2004;186:842–9. doi: 10.1128/JB.186.3.842-849.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jeong J-A, Baek E-Y, Kim SW, Choi J-S, Oh J-I. Regulation of the ald gene encoding alanine dehydrogenase by AldR in Mycobacterium smegmatis. J Bacteriol. 2013;195:3610–20. doi: 10.1128/JB.00482-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Reitzer LJ, Magasanik B. Transcription of glnA in E. coli is stimulated by activator bound to sites far from the promoter. Cell. 1986;45:785–92. doi: 10.1016/0092-8674(86)90553-2. [DOI] [PubMed] [Google Scholar]
- 39.Benson DA, Clark K, Karsch-Mizrachi I, Lipman DJ, Ostell J, Sayers EW. GenBank. Nucleic Acids Res. 2014;42(Database issue):D32–7. doi: 10.1093/nar/gkt1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Finn RD, Bateman A, Clements J, Coggill P, Eberhardt RY, Eddy SR, et al. Pfam: the protein families database. Nucleic Acids Res. 2014;42(Database issue):D222–30. doi: 10.1093/nar/gkt1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.De Hoon MJL, Imoto S, Nolan J, Miyano S. Open source clustering software. Bioinformatics. 2004;20:1453–4. doi: 10.1093/bioinformatics/bth078. [DOI] [PubMed] [Google Scholar]
- 42.Chhabra SR, He Q, Huang KH, Gaucher SP, Alm EJ, He Z, et al. Global analysis of heat shock response in Desulfovibrio vulgaris Hildenborough. J Bacteriol. 2006;188:1817–28. doi: 10.1128/JB.188.5.1817-1828.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Novichkov PS, Rodionov DA, Stavrovskaya ED, Novichkova ES, Kazakov AE, Gelfand MS, et al. RegPredict: an integrated system for regulon inference in prokaryotes by comparative genomics approach. Nucleic Acids Res. 2010;38(Web Server issue):W299–307. doi: 10.1093/nar/gkq531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Barrios H, Valderrama B, Morett E. Compilation and analysis of sigma(54)-dependent promoter sequences. Nucleic Acids Res. 1999;27:4305–13. doi: 10.1093/nar/27.22.4305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sahota G, Stormo GD. Novel sequence-based method for identifying transcription factor binding sites in prokaryotic genomes. Bioinformatics. 2010;26:2672–7. doi: 10.1093/bioinformatics/btq501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hershberg R, Yeger-Lotem E, Margalit H. Chromosomal organization is shaped by the transcription regulatory network. Trends Genet. 2005;21:138–42. doi: 10.1016/j.tig.2005.01.003. [DOI] [PubMed] [Google Scholar]
- 47.Srivastava D, Hsieh M-L, Khataokar A, Neiditch MB, Waters CM. Cyclic di-GMP inhibits Vibrio cholerae motility by repressing induction of transcription and inducing extracellular polysaccharide production. Mol Microbiol. 2013;90:1262–76. doi: 10.1111/mmi.12432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Walmsley J, Toukdarian A, Kennedy C. The role of regulatory genes nifA, vnfA, anfA, nfrX, ntrC, and rpoN in expression of genes encoding the three nitrogenases of Azotobacter vinelandii. Arch Microbiol. 1994;162:422–9. doi: 10.1007/BF00282107. [DOI] [PubMed] [Google Scholar]
- 49.Mai X, Adams MW. Indolepyruvate ferredoxin oxidoreductase from the hyperthermophilic archaeon Pyrococcus furiosus. A new enzyme involved in peptide fermentation. J Biol Chem. 1994;269:16726–32. [PubMed] [Google Scholar]
- 50.Kazakov AE, Rodionov DA, Price MN, Arkin AP, Dubchak I, Novichkov PS. Transcription factor family-based reconstruction of singleton regulons and study of the Crp/Fnr, ArsR, and GntR families in Desulfovibrionales genomes. J Bacteriol. 2013;195:29–38. doi: 10.1128/JB.01977-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Novichkov PS, Kazakov AE, Ravcheev DA, Leyn SA, Kovaleva GY, Sutormin RA, et al. RegPrecise 3.0--a resource for genome-scale exploration of transcriptional regulation in bacteria. BMC Genomics. 2013;14:745. doi: 10.1186/1471-2164-14-745. [DOI] [PMC free article] [PubMed] [Google Scholar]