

Abstract
Background
Bacillus cereus sensu lato comprises eight closely related species including the human pathogens Bacillus anthracis and Bacillus cereus. Within B. cereus sensu lato, chromosomally and plasmid-encoded toxins exist. While plasmid-mediated horizontal gene transfer of the emetic toxin, anthrax and insecticidal toxins is known, evolution of enterotoxin genes within the group has not been studied.
Results
We report draft genome assemblies of 25 strains, a phylogenetic network of 142 strains based on ANI derived from genome sequences and a phylogeny based on whole-genome SNP analysis. The data clearly support subdivision of B. cereus sensu lato into seven phylogenetic groups. While group I, V and VII represent B. pseudomycoides, B. toyonensis and B. cytotoxicus, which are distinguishable at species level (ANI border ≥ 96 %), strains ascribed to the other five species do not match phylogenic groups. The chromosomal enterotoxin operons nheABC and hblCDAB are abundant within B. cereus both isolated from infections and from the environment. While the duplicated hbl variant hbla is present in 22 % of all strains investigated, duplication of nheABC is extremely rare (0.02 %) and appears to be phylogenetically unstable. Distribution of toxin genes was matched to a master tree based on seven concatenated housekeeping genes, which depicts species relationships in B. cereus sensu lato as accurately as whole-genome comparisons. Comparison to the phylogeny of enterotoxin genes uncovered ample evidence for horizontal transfer of hbl, cytK and plcR, as well as frequent deletion of both toxins and duplication of hbl. No evidence for nhe deletion was found and stable horizontal transfer of nhe is rare. Therefore, evolution of B. cereus enterotoxin operons is shaped unexpectedly different for yet unknown reasons.
Conclusions
Frequent exchange of the pathogenicity factors hbl, cytK and plcR in B. cereus sensu lato appears to be an important mechanism of B. cereus virulence evolution, including so-called probiotic or non-pathogenic species, which might have consequences for risk assessment procedures. In contrast, exclusively vertical inheritance of nhe was observed, and since nhe-negative strains appear to be extremely rare, we suggest that fitness loss may be associated with deletion or horizontal transfer of the nhe operon.
Electronic supplementary material
The online version of this article (doi:10.1186/s12862-015-0529-4) contains supplementary material, which is available to authorized users.
Keywords: Bacillus cereus sensu lato, Average nucleotide identity, Enterotoxin, nhe, hbl, cytK, Horizontal gene transfer, gene duplication, Functional constraints of evolution
Background
Bacillus cereus sensu lato comprises eight species of gram-positive, endospore-forming bacteria with greatly varying pathogenic potential. B. cereus sensu stricto, which was discovered in 1887 as a bacterium occurring ubiquitously in nature [1], contains probiotic as well as pathogenic strains. The latter are mostly associated with food-borne illness characterized by diarrhea or vomiting, but occasionally B. cereus is responsible for severe infections, e.g. endophthalmitis or meningitis [2]. The emetic type of B. cereus food poisoning is caused by ingestion of the small, cyclic and heat-stable toxin cereulide [3]. The three most important and well-known enterotoxins are the non-hemolytic enterotoxin (Nhe), hemolysin BL (Hbl) and cytotoxin K (CytK). B. thuringiensis produces insecticidal parasporal protein crystals of Cry (crystal) and/or Cyt (cytolytic) proteins that are mostly encoded on plasmids. Occasionally B. thuringiensis have been found to cause human infections very similar to B. cereus [4, 5]. B. anthracis is the best known human and animal pathogen of the B. cereus group and was demonstrated to be the causative agent of anthrax by Robert Koch in 1876 [6]. The anthrax-associated plasmids pXO1 (encoding anthrax toxin genes pag, lef and cya) and pXO2 (encoding the poly-γ-D-glutamic acid capsule genes cap) have been found in a few B. cereus strains such as B. cereus G9241 and B. cereus biovar anthracis CA [7, 8] with a similar pathogenic potential as B. anthracis. B. weihenstephanensis is psychrotolerant and able to grow below 7 °C [9]. Occasionally, this species houses the emetic toxin cereulide [10, 11]. The psychrotolerant B. mycoides is closely related to the other B. cereus sensu lato species (16S rRNA sequences showed > 99 % identity [12]), but it can easily be distinguished by its rhizoidal colonial growth [13]. To our knowledge, no infections by B. mycoides have been reported, but it carries both nhe and hbl and its cytotoxicity was shown [14]. Within B. mycoides a group of bacteria with a clearly distinguishable fatty acid profile was recognized and described as B. pseudomycoides [15]. In 1998, a highly enterotoxic and rare variant of cytotoxin K, CytK-1, was discovered in B. cereus NVH 391–98, a strain responsible for severe food poisoning. This strain was published in 2013 as the type strain of the new species B. cytotoxicus on the basis of presence of the cytK-1 gene, its thermotolerance (growth at up to 50 °C), a distinctive fatty acid profile, DNA-DNA hybridization and multilocus sequence typing (MLST) [16]. The eighth member of B. cereus sensu lato was isolated in Japan in 1966 but has been described as a separate species B. toyonensis only recently [17]. Interestingly, it is a commercially available probiotic (TOYOCERIN®). B. toyonensis was distinguished from other B. cereus sensu lato type species by pairwise calculations of the average nucleotide identity (ANI).
While the trend in prokaryotic species distinction is moving towards comparison of entire genomes [18, 19], differentiation of the closely related B. cereus species is still based on the presence or absence of phenotypic characters. It has been reported that species affiliation of B. cereus group strains often does not match the phylogenetic relatedness [12, 20, 21]. One reason for such discrepancies may be the exchange of virulence plasmids between species. Plasmids pXO1 and pXO2 encoding the anthrax toxin complex and the poly-γ-D-glutamic acid capsule are found not only in B. anthracis, but also in some B. cereus strains [7, 22, 23]. The B. cereus cereulide synthetase gene cluster is also located on a large pXO1-like plasmid [3] and is not restricted to a single lineage within B. cereus sensu lato [11, 24]. Some of the B. thuringiensis insecticidal cry genes are encoded on plasmids [25] and can spread via horizontal gene transfer (HGT) among B. cereus sensu lato. Therefore, the transfer of a single plasmid from one species to another species may result in a change of species affiliation.
Several studies have addressed the general possibility of HGT among B. cereus sensu lato, obtaining controversial results. B. cereus and B. thuringiensis isolates have been studied by multilocus enzyme electrophoresis (MLEE) resulting in a high variety of closely related electrophoretic types, evidence of extensive recombination between species and a low degree of clonality [26]. MLST analyses revealed that B. anthracis is a homogeneous and clonal cluster within B. cereus sensu lato, while B. cereus and B. thuringiensis are of higher diversity [27]. Similar studies have been conducted with a different set of housekeeping genes for MLST, amplified fragment length polymorphism (AFLP) and genomic comparisons, concluding that HGT involves chromosomal genes and is probably mediated by transposable elements [26, 28, 29]. Other studies conclude from MLST, AFLP, MLEE and genomic data that chromosomal recombination events are generally rare, but appear more often among B. cereus/B. thuringiensis, while only plasmids are transmitted by HGT [30–32]. HGT among distantly related bacteria can be inferred by several approaches [33], but these are difficult to apply when bacteria as closely related as B. cereus sensu lato are investigated [34]. Conflicting results are therefore not surprising.
While horizontal spread of virulence plasmids is not unusual, little is known about the lateral transfer of chromosomal B. cereus virulence factors [27, 29]. Therefore, this work aims to systematically evaluate the prevalence and distribution of enterotoxin genes within B. cereus sensu lato, with a focus on horizontal versus vertical gene transfer as well as toxin gene duplication and loss. As an indispensable prerequisite for this task we inferred the phylogeny of B. cereus sensu lato strains based on 142 genomes (25 of which we sequenced de novo in this work) as well as stable MLSA (multilocus sequence analysis) phylogeny constructed from seven housekeeping genes of the B. cereus core genome.
Results and Discussion
De novo sequencing of 25 B. cereus sensu lato strains
In this study whole genomes of 25 B. cereus sensu lato strains with different enterotoxic potential (for details see [35]) have been sequenced. 21 of them are known members of B. cereus sensu stricto that were either isolated from food or associated with food poisoning cases. B. cereus #17 (#236) has been isolated from mouse gut (T. Clavel, personal communication) and B. cereus IP5832 (#237) is a commercially available probiotic strain (Bactisubtil®) [36]. Additionally, B. mycoides WSBC 10969 (#283), and B. cytotoxicus CVUAS 2833 (#249) [37] were added, because only very few genomes of these species are publicly available. Detailed sequencing and assembly data are given in Additional file 1: Table S1. Assembly sizes of the newly sequenced strains ranged from 4.1 to 6.8 Mbp (Table 1) and were compared to genome sizes of published B. cereus sensu lato strains. It has been discussed controversially whether well-adapted pathogenic bacteria generally contain smaller genomes (due to less variable selection pressure) than environmental isolates [38]. In our study, some B. cereus sensu lato genomes seem to support this hypothesis of a reduced genome size in pathogens, such as B. anthracis (5.0 – 5.5 Mbp), B. cytotoxicus (4.1 Mbp) and some of the enteropathogenic strains we sequenced. These showed smaller genomes than environmental and innocuous strains (B. thuringiensis 5.3 – 6.8 Mbp, B. mycoides 5.6 – 6.1 Mbp, B. pseudomycoides 5.8 Mbp). However, several exceptions like the enterotoxic Nhe reference strain B. cereus NVH 0075–95 (6.1 Mbp) support the notion that genome size does not correlate with pathogenicity.
Table 1.
Genome size and pathogenicity of 25 de novo sequenced B. cereus sensu lato strains
| Strain | Assembly size [bp] | Source | Enterotoxicity |
|---|---|---|---|
| B. cytotoxicus CVUAS 2833 | 4127075 | Food poisoning | N.d. |
| B. cereus F4429/71 | 5284967 | Vanilla pudding | High |
| B. cereus HWW 274-2 | 5290159 | Milk powder | Low |
| B. cereus SDA KA 96 | 5335844 | Raw milk | High |
| B. cereus RIVM BC 126 | 5417487 | Human faeces | High |
| B. cereus 7/27/S | 5479572 | Human faeces | High |
| B. cereus 14294–3 (M6) | 5523305 | Ice cream | Middle |
| B. cereus MHI 86 | 5551873 | Infant food | Low |
| B. cereus RIVM BC 90 | 5559670 | Human faeces | Low |
| B. cereus F4430/73 | 5577793 | Pea soup | Middle |
| B. cereus F3162/04 | 5591156 | Human faeces | High |
| B. cereus IP5832 | 5592318 | Probiotic | N.d. |
| B. cereus INRA C3 | 5596453 | Carrot | High |
| B. cereus WSBC 10035 | 5619577 | Milk | High |
| B. weihenstephanensis WSBC 10204 | 5655039 | Milk | N.d. |
| B. cereus F3175/03 | 5733808 | Human faeces | Middle |
| B. cereus RIVM BC 964 | 5815402 | Kebab | High |
| B. cereus #17 | 5852222 | Commensal | N.d. |
| B. cereus F528/94 | 5935300 | Food poisoning | Low |
| B. cereus INRA A3 | 6075647 | Starch | Low |
| B. mycoides WSBC 10969 | 6101972 | Raw milk | N.d. |
| B. cereus NVH 0075-95 | 6112682 | Food poisoning | High |
| B. cereus MHI 226 | 6233017 | Milk | Low |
| B. cereus 6/27/S | 6771128 | Human faeces | Middle |
| B. cereus RIVM BC 934 | 6840916 | Lettuce | Low |
Strains are sorted by assembly size. Classification in high/low enterotoxic strains was determined by using a Vero cell assay [35]. N. d.: not determined
After assembly, the new sequences were screened for the presence and location of virulence determinants and housekeeping genes and used for whole-genome comparison as well as multi-locus sequence analysis.
Whole genome comparison confirms seven phylogenetic groups
For taxonomical purposes, and in order to analyze horizontal gene transfer, the construction of a phylogenetic master tree which depicts the assumed “true” phylogenetic relationships of the organisms studied as correctly as possible is mandatory. Towards this end, we constructed an MLSA tree (Fig. 1) based on concatenated sequences of seven housekeeping genes [39] from the B. cereus sensu lato core genome. The topologies of trees calculated on the basis of individual housekeeping genes are highly similar to this master tree but partially lack resolution due to different levels of conservation (data not shown). The overall MLSA tree topology was confirmed by an analysis of whole-genome pairwise ANI comparison, visualized by a neighbor network (Additional file 2: Figure S1). Pairwise comparison of ANI versus pairwise distances of the seven concatenated housekeeping genes correlated nicely (Fig. 3a). These results were additionally confirmed by whole-genome SNP-based phylogeny (Fig. 2). All three methods showed highly similar tree topologies and confirmed that the MLSA tree should correctly display the strain phylogeny of the 142 B. cereus sensu lato strains included in this study.
Fig. 1.
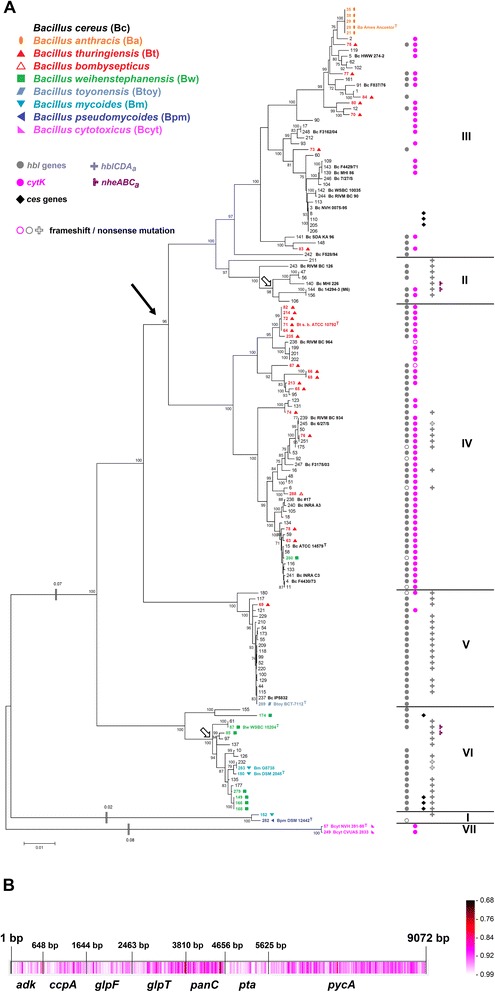
MLSA-based species relationship within B. cereus sensu lato. a: The phylogenetic tree (Maximum Likelihood Method) was calculated using the concatenated sequence of seven housekeeping genes from 142 B. cereus sensu lato strains. Arrow: Suspected first appearance of cytK-2. Empty arrows: Suspected origin of nhe a operons. b: Visualization of the sequence homology derived from a multiple sequence alignment calculated with RDP3. Color ranges (identity score: ≤ 1, 1 = identical in all sequences) from identical (white) to highly dissimilar (black)
Fig. 3.
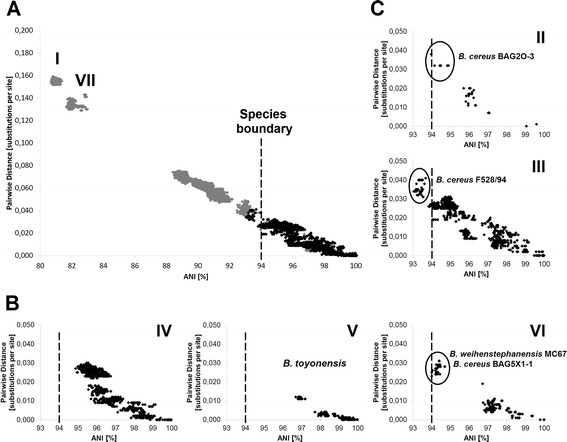
Correlation of pairwise distance of concatenated housekeeping genes with whole genome ANI values. a: Correlation of whole-genome ANI and pairwise distance of seven concatenated housekeeping genes of all 142 B. cereus sensu lato strains. Intra-cluster values (for clusters see Fig. 1) are depicted in black, all inter-cluster values in grey. Pearson’s correlation of all values is r2 = 0.974. b and c: Intra-cluster comparison of whole-genome ANI and pairwise distance of seven concatenated housekeeping. Cluster affiliation (I – VII) according to species tree (Fig. 1). Strains that are difficult to sort into a distinct genomospecies are named and marked by circles. b: Clusters I, IV – VI can be distinguished at an ANI species boundary of ≥ 94 %. Cluster I: ANI 98.3 %, pairwise distance 0.005 substitutions per site. Cluster VII: ANI 99.5 %, pairwise distance 0.001 substitutions per site. c: Clusters II and III cannot be discerned on the basis of the comparisons presented: strain B. cereus F528/94 (#242) is affiliated to Cluster III by MLSA (Fig. 1) and whole-genome SNP analysis (Fig. 2), but to Cluster II by ANI (Additional file 2: Figure S1)
Fig. 2.
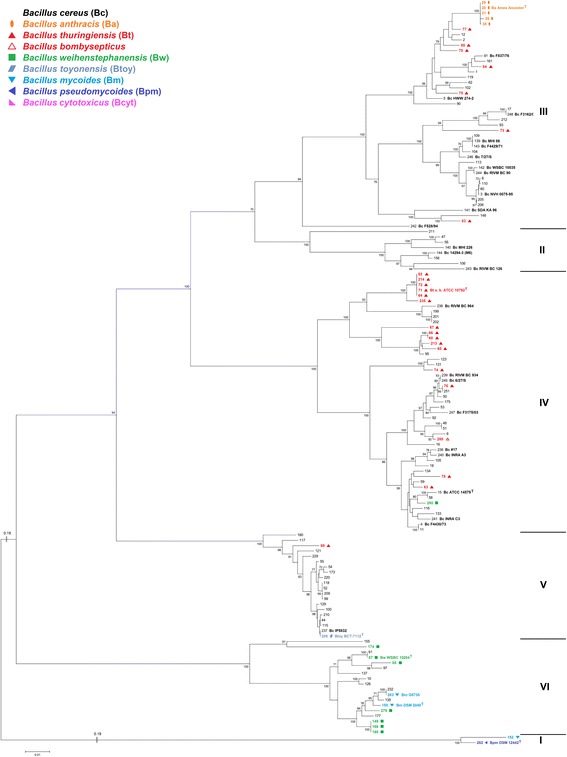
SNP-based species relationship within B. cereus sensu lato. The phylogenetic tree (Maximum Likelihood Method) was calculated using the genome-wide core SNP matrix (SNPs that are present in all of the analyzed genomes) of 140 B. cereus sensu lato genomes. Phylogenetic cluster VII is too divergent for accurate detection of core SNPs using kSNP3 and had to be excluded
Based on a more limited MLST approach, using partial gene sequences only as well as a different set of genes than we applied in our MLSA analysis, Cardazzo et al. [28] concluded that reticulate evolution of housekeeping genes should be an important factor of B. cereus evolution. Based on our whole genome and MLSA analysis we cannot exclude limited reticulate evolution within the seven housekeeping genes used in our study but we suggest that, apparently, this does not mask the strain phylogeny of our B. cereus strain set.
Seven phylogenetic clusters of B. cereus sensu lato have been reported previously [20, 40, 41] and these appear also in our data set. The whole genome analysis reveals that these seven phylogenetic groups are separated by at least 94 % ANI. It has been proposed to use average nucleotide identity between genomes for bacterial species delineation [42]. The current species distinction states that a group of strains belonging to one species must have > 70 % DDH similarity, < 5 °C ΔTm, < 5 % mol G + C difference of total genomic DNA and > 98 % 16S rRNA identity [18]. A boundary of 94–96 % ANI corresponding to ~ 70 % DDH similarity was proposed [42, 43]. Phylogenetic groups within B. cereus sensu lato can be distinguished with a species boundary of 94 % identity (Fig. 3). When comparing strains that do not belong to the same phylogenetic group with each other, ANI values are in the range of 80–94 %. Our data therefore suggest that seven genomospecies exist within B. cereus sensu lato.
B. cereus sensu lato species affiliation reviewed
The distribution of the strains investigated in this study confirms the observation [12, 17, 20, 21] that many existing species affiliations do not match the genomic relationships (Fig. 1). Cluster IV contains mesophilic pathogenic B. cereus and B. thuringiensis and one B. weihenstephanensis strain. The presence of a unique cspA signature is described as being specific for psychrotolerant B. cereus sensu lato (B. mycoides, B. pseudomycoides and B. weihenstephanensis) [20, 44], but the psychrotolerant cspA-signature could not be detected in B. weihenstephanensis FSL R5-860 (#280, IV). Thus, this strain is likely incorrectly classified as B. weihenstephanensis. Cluster III comprises B. anthracis, emetic and non-emetic B. cereus and B. thuringiensis. While it is almost impossible to distinguish between cluster III B. cereus and B. thuringiensis isolates (Fig. 1), B. anthracis is an easily discernable monophyletic group in the species tree. In cluster II potentially pathogenic psychrotolerant strains like B. cereus RIVM BC 126 [20] are found that are closely related to cluster III.
However, three phylogenetic groups can be matched easily to three species. Cluster V is clearly separated from all other phylogenetic groups and includes the type strain of the recently described species B. toyonensis BCT-7112 which differs from other B. cereus sensu lato type strains (ANI < 92 % and a distinct peptidoglycan diamino acid pattern [17]). All isolates belonging to the phylogenetic group V should be renamed as members of B. toyonensis [17]. Both B. toyonensis BCT-7112 and the newly sequenced B. cereus IP5832 are commercially available probiotics, Toyocerin® and Bactisubtil® [36], respectively. Thus, the almost clonally related Bacillus strains of cluster V might also be feasible as probiotics for animal feeding but the distribution of enterotoxin genes (see below) probably renders this species also a human pathogen. Cluster I is constituted by B. pseudomycoides [20]. Phylogenetic analysis of housekeeping genes clustered B. mycoides Rock3-17 (#152, I) together with B. pseudomycoides DSM 12442 (type strain, #282, I) in cluster I. Based on these data we suggest that this strain should be renamed as B. pseudomycoides after confirmation by fatty acid profiling. Strains of clusters I and VII (B. cytotoxicus) show the greatest distance to all other members of B. cereus sensu lato.
Occurrence of virulence genes
To gain an overview over the distribution of virulence genes, 218 B. cereus sensu lato strains, including our de novo sequenced strains, were analyzed (Additional file 1: Table S2). All B. cereus strains possess the nhe genes. We analyzed the presence of the toxin genes hbl, cytK and ces and found that 64 % of the 218 strains contained hbl and 34 % of these possess a second hbl operon. CytK-2 appears in 40 %, cytK-1 in 1 % and the emetic gene cluster ces in 5 % of all strains. CytK-2 is far more frequent in B. thuringiensis strains (75 %) than in the rest of B. cereus sensu lato (35 %). These results match a study from 2006 that investigated 74 uncharacterized B. thuringiensis strains. All of them harbored the nhe genes, 74 % hbl and 73 % cytK-2, displaying about the same potential to cause diarrhea as B. cereus [45]. The combined presence of nhe, hbl and cytK occurs more often among diarrheal (63 %) than among food-borne B. cereus sensu stricto strains (33 %) [46]. We find that 30 % of the 218 B. cereus sensu lato strains contain all three enterotoxins.
Evolution of hbl
Massive horizontal transfer of hbl
Hbl is not an essential operon, since only 64 % of 218 strains contain hbl, which appear rarely in cluster III and are absent from B. cytotoxicus (cluster VII) (Fig. 1). A comparison of the concatenated hblCDAB gene tree with a species tree consisting of all 101 hbl-containing strains shows vastly different topologies (Additional file 2: Figure S2). All phylogenetic clusters except cluster V are mixed which is evidence for massive horizontal transfer of the enterotoxin operon hblCDAB, both between and within phylogenetic groups.
It has been speculated that hblCDAB is part of a large 18 kb transposon [29, 47, 48]. A detailed comparison of putative transposon regions including hbl or hbla of the 16 hbl-containing strains sequenced de novo in this work is shown in Additional file 2: Figure S3. However, while half of the hbl operons are inserted within the uvrC gene as described earlier [29], in the other strains neither insertion sites nor length of the inserted region or adjacent genes are conserved.
Ancient origin of hblCDA
Phylogenetic trees represent the species evolution only if orthologous genes are compared which have not been transferred laterally. In contrast, paralogs result from gene duplications [49]. After several generations it becomes increasingly difficult to recognize paralogs, especially when gene loss is involved. It has been known that two distinct homologs of hbl exist in B. cereus [50]. The more abundant version of hblCDAB occurs in 64 % while the second, truncated hbla operon (hblCDA) is present in 22 % of the investigated strains. Six of the de novo sequenced and assembled strains contain two versions of hbl and/or nhe. We confirmed the existence of duplicated enterotoxin operons by comparison of coverage depths over operons and their respective background contigs as described in (Additional file 1: Tables S5 and S6). The duplication of hblCDAB as well as the subsequent loss of hblB must be an ancient and unique event which occurred early in the evolution of B. cereus sensu lato since all hblCDAa genes cluster together and are clearly separated from hblCDA (Fig. 4b) despite the fact that they are scattered over five phylogenetic clusters. Subsequently, hblCDAa appears to have been deleted in various lines of the species group (Fig. 1 and Additional file 2: Figure S2). While hblCDAB was subject to extensive horizontal gene transfer (see Fig. 1 and Additional file 2: Figure S2), hblCDAa seems to have been transferred vertically due to a much more conserved tree topology (Fig. 4a and Additional file 2: Figure S2). It has been shown that hblB is not essential for enterotoxic activity, because hblCDAB mRNA synthesis appears to terminate within the hblB gene [51], which might be a pseudogene [52]. HblCDAa is located on plasmid in the four strains B. thuringiensis serovar kurstaki YBT-1520 (pBMB293), B. thuringiensis serovar kurstaki HD-1 (pBMB299), B. thuringiensis serovar chinensis CT-43 (pCT281) and B. thuringiensis serovar thuringiensis IS5056 (pIS56-285), but the detection of plasmid-location may increase with an ongoing completion of genomes.
Fig. 4.
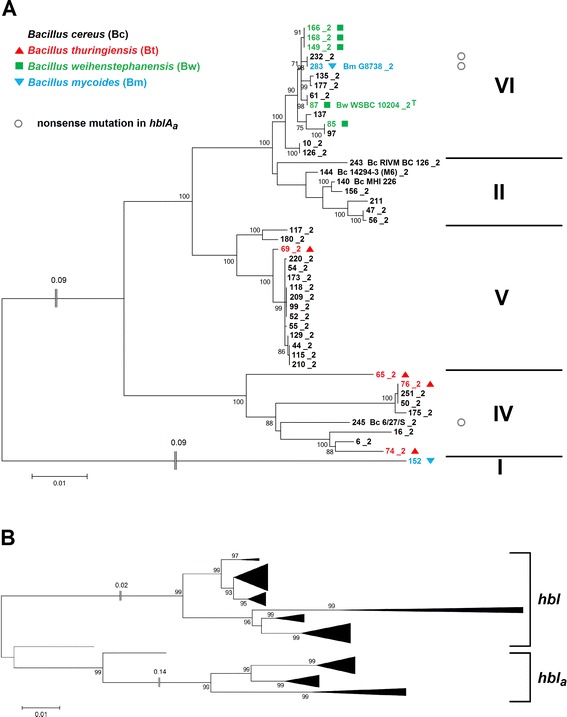
HblCDA a in B. cereus sensu lato. a: Phylogenetic hblCDA a tree (Maximum Likelihood Method) based on the concatenated sequence from 46 B. cereus sensu lato strains. b: Evidence for an ancient origin of hbl a based on the concatenated sequence of hblCDA and hblCDA a from 101 B. cereus sensu lato strains
Functional constraints in the evolution of hbl
While horizontal transfer of the hblCDAB operon is frequent, intra-operon recombinations are extremely rare. Only five statistically significant recombination events within the set of 142 strains could be detected (Additional file 1: Table S4) and all of these occurred exclusively within their respective phylogenetic group. Intra-operon recombinations between different phylogenetic groups have probably been removed by negative selection. DNA sequence identities of hbl between B. cereus sensu lato strains are quite high (93–100 %). Thus, hbl is more conserved than nhe or even housekeeping genes in B. cereus sensu lato (Figs. 1, 6 and Additional file 2: Figure S2). Both, high sequence conservation and rare intra-operon recombination, suggest that the interaction between Hbl components might be quite specific, thus constraining sequence variation. This assumption is supported by experimental studies of the interaction of Hbl and Hbla proteins. Both operons encode a functional toxin. However, despite their high similarity, not all Hbl/Hbla components are exchangeable to form functional toxins [50].
Fig. 6.
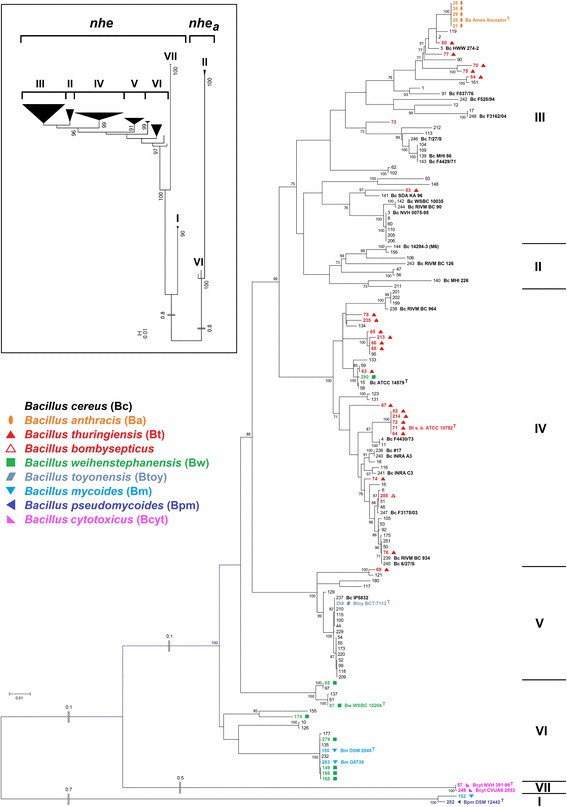
Nhe in B. cereus sensu lato. Phylogenetic nheABC tree (Maximum Likelihood Method) based on the concatenated sequences from 142 B. cereus sensu lato strains. Inset: Phylogenetic tree based on the concatenated sequences of nheABC and nheABC a from 142 B. cereus sensu lato strains, indicating an ancient origin of nheABC a
Horizontal transfer of cytK
The third diarrhea causing agent is the single-component chromosomally encoded toxin CytK, which is a hemolytic, dermonecrotic and β-barrel pore-forming enterotoxin [53]. Two variants of CytK are known. 89 % of the amino acid sequence of CytK-2 is identical to CytK-1 and CytK-2 is also able to form pores in planar lipid bilayers, but it shows only 20 % of CytK-1 toxicity [54]. The gene cytK-2 occurs in 47 % of the strainset and only the two B. cytotoxicus strains (#57, #249, VII) possess cytK-1. We detected cytK-2 in strains of clusters II – V, which might have acquired this toxin gene via lateral transfer prior to splitting into clusters II, III and IV (Fig. 1, marked by an arrow) from the B. cytotoxicus phylogenetic line where the CytK ancestor may have originated. It may have been lost subsequently in some strains of clusters II and III. In contrast, a recent horizontal transfer of cytK-2 to a few cluster V strains seems to have occurred (Fig. 1). Lateral transfer of cytK can also be inferred from a direct comparison of the cytK tree with the species tree of all 68 cytK-containing strains (Additional file 2: Figure S4).
Massive horizontal transfer and sequence variation of the virulence regulator plcR
The expression of many virulence factors in B. cereus is regulated by the pleiotropic transcriptional activator PlcR (Phospholipase C Regulator) [52]. The promoters of genes regulated by PlcR show a highly conserved palindromic recognition site (TATGNAN4TNCATA), which occurs in the promoter regions of nhe, hbl [52] and cytK [53]. Several other proteins were discovered that are under control of PlcR, such as two-component sensors, chemotaxis proteins, transporters, cytoplasmic regulators and cell wall biogenesis proteins. This indicates that PlcR is a global regulator and a key component in adaptation to (host) environment [55]. The gene phylogeny of plcR hints to lateral transfer within 52 B. cereus, B. anthracis and B. thuringiensis strains [27]. Our comparison of the plcR gene phylogeny of 142 strains of all species of the B. cereus group (Fig. 5) with the B. cereus sensu lato species tree (Fig. 1) confirms a low degree of conservation of plcR (DNA sequence identities range from 70 % up to 100 %) and a surprisingly low similarity of both trees. These grossly conflicting topologies may be a result of both frequent horizontal transfer and rapid divergent evolution of plcR driven by a variety of environmental selection pressures. The papR gene is encoded less than 100 bp downstream of plcR and shows a phylogeny highly similar to plcR (data not shown). PapR is the quorum sensing peptide necessary for activation PlcR and part of the PlcR regulon [56]. The genomes of B. pseudomycoides DSM 12442 (#282, I), B. mycoides Rock3-17 (#152, I) and B. mycoides Rock1-4 (#151) do not contain papR, which might cause reduced pathogenicity of Cluster I strains.
Fig. 5.
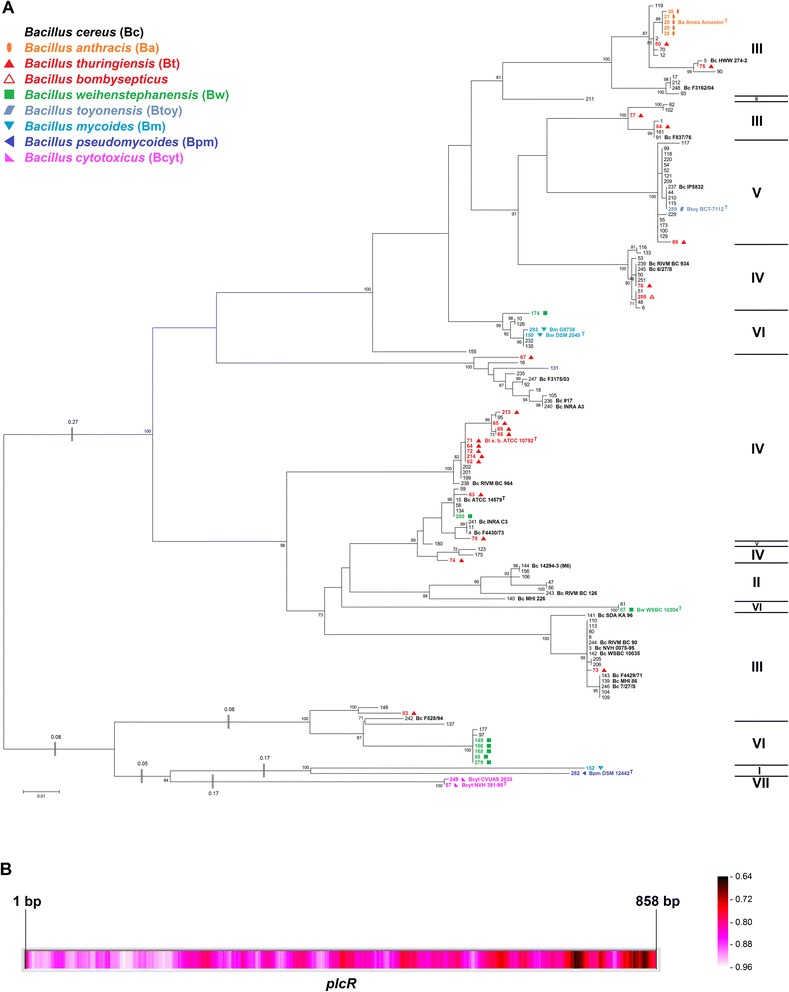
PlcR in B. cereus sensu lato. a: Phylogenetic plcR tree (Maximum Likelihood Method) based on the sequence from 142 B. cereus sensu lato strains. b: Visualization of the sequence homology derived from a multiple plcR sequence alignment calculated with RDP3. Color ranges (identity score: ≤ 1, 1 = identical in all sequences) from identical (white) to highly dissimilar (black)
PlcR was experimentally inactivated in B. cereus and B. thuringiensis by a number of mutations [57] including deletions, additions, nonsense mutations, high diversity, and mutations in papR or opp, genes necessary for PlcR activation. Inactivation of the PlcR regulon and a subsequent non-hemolytic phenotype also occur naturally. In B. anthracis a nonsense mutation after 642 bp leads to a truncated protein, while in B. cereus biovar anthracis a C-terminal frameshift results in a four amino acids longer protein, both completely inactivate the plcR regulon [52, 58]. The B. cereus biovar anthracis plcR mutation also occurs in B. cereus ISP3191 (#102, III). C-terminal plcR gene extensions appear in B. cytotoxicus (+9 bp), B. pseudomycoides (+33 bp) and B. weihenstephanensis (+6 bp), but their function is yet unknown. The DNA binding domain helix-turn-helix motif is located in the N-terminal part of the active protein, the regulatory domain in the C-terminal region [59]. The greatest sequence variation is found in the 3′ end of plcR (Fig. 5b). While binding to the recognition site requires a highly conserved protein structure, the regulatory function obviously tolerates a higher sequence variation. Hence, this might be a way of allowing fast adaptation to changing environmental or host conditions by modulating transcriptional activation of specific PlcR-controlled genes without unnecessary inactivation of the entire regulon.
Evolution of nheABC enterotoxin genes
Ancient origin of nhe and recent horizontal gene transfer of the nhe variant nhea
While no duplications of cytK or plcR could be found, a very rare second nheABC operon was noticed in four of the 142 B. cereus sensu lato strains investigated (Fig. 6). Two B. cereus strains (#140, #144, II) and two B. weihenstephanensis strains (#85, #87, VI) possess a second nheABC variant which we term nhea. In B. weihenstephanensis KBAB4 (#85, VI) the nhea operon is part of the 417 kb plasmid pBWB401. The other three nhea copies could not be located on a similar plasmid, but were shown to be genuine duplications (Additional file 1: Table S5). The nhea operon contains all three nhe genes, is actively transcribed, albeit not in all strains (Fig. 7), and we found a putative PlcR recognition site upstream of nheAa (data not shown). Nhea operons differ greatly from all known nhe (76–88 % sequence identity), including B. cytotoxicus’ nheABC that was until now considered the only major and most distantly related variant. Nhea might have resulted from two relatively recent, but separate HGT events into two strains of clusters II and VI, since cluster II and cluster VI nhea are two clearly distinct variants. The donor strains harboring the two nhea versions have not yet been identified, but sequence comparison shows that their nhea must have separated very early in the evolution of the B. cereus group (Fig. 6, inset). Apparently, the nhea operon is not stably integrated in the genome since several strains in both phylogenetic groups II and VI seem to have lost it shortly after acquisition (Fig. 1). Nhea and hbla might be starting points for the evolution of new pore-forming enterotoxins in B. cereus sensu lato, analogous to the suspected development of hbl and nhe themselves from an ancient ancestor by gene duplication [48].
Fig. 7.
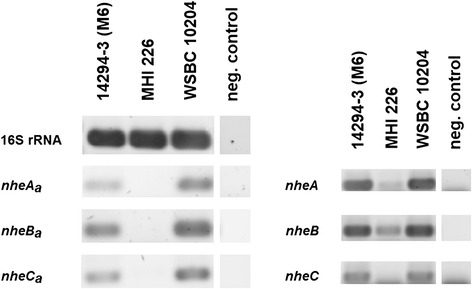
Transcriptional analysis of nhe and nhe a operons. Transcription of nhe and nhe a in B. cereus 14294–3 (M6), MHI 226 and B. weihenstephanensis WSBC 10204. Nhe a is transcribed in B. cereus 14294–3 (M6) and B. weihenstephanensis WSBC 10204, but not in B. cereus MHI 226. The latter showed also a weak transcription of nhe
Strictly vertical transmission of nheABC
A phylogenetic tree of the concatenated nheABC genes was calculated (Fig. 6) and compared with the MLSA species tree (Fig. 1). Surprisingly, their topology is almost identical. Some strains contain more distantly related nhe sequences, such as B. mycoides and B. pseudomycoides (74–77 % sequence identity) or B. cytotoxicus NVH 391–98 with only 72–88 % DNA sequence identity to other nhe operons [60] (Fig. 6). We suggest that no significant recombination of distantly related nhe operons has occurred. After discovering the strictly vertical transmission pattern of nheABC, recombination within the nhe operon was investigated in more detail. In the set of 142 concatenated nheABC genes 21 statistically proven intra-operon recombination events could be detected, but these occurred only between closely related nhe regions, which keeps recombination derived variation small (Additional file 1: Table S4). Furthermore, and most significantly, nhe occurs in all known B. cereus strains without exception, which may be evidence for an important function of the operon. It is noticable that nheA is the most highly conserved gene of the nhe operon (data not shown).
While Nhe components and their interactions are currently analyzed extensively [61–64] all studies have been focused on the pore-forming activity. However, in NheA an enlarged β-tongue structure was found [64] which, despite its similarity to ClyA and HblB, might be involved in a protein-protein interaction associated with an unknown function. If an unknown function exists, balancing selection may have resulted in a co-evolution of nhe with a (yet unknown) interacting factor. Such an interaction would impose certain constraints on the NheA structure, allowing recombination between closely related nhe copies but leading to the observed absence of horizontal gene transfer between distantly related strains in order to prevent a fitness loss. We speculate that the strictly vertical evolution, the rare and unstable occurrence of nhe copies and high sequence conservation may be evidence for an additional function of Nhe. While the existence of such a hypothetical function can be predicted based on our phylogenetic analysis, further experimental investigation certainly is necessary.
Evolution of enterotoxin genes and risk assessment
While B. anthracis is considered non-enteropathogenic due to the deactivated PlcR-regulon [57], B. cereus and B. thuringiensis are known to be enterotoxic [48, 65]. B. weihenstephanensis strains also carry enterotoxin genes. While this psychrotolerant species is not known to be involved in outbreaks – probably due to reduced toxicity at elevated temperatures [66–68] – it is listed as a risk level 2 organism in Germany. The enterotoxicity of B. mycoides and B. pseudomycoides is unknown, but our results and other studies confirm that nhe, hbl and cytK can be found in strains of both species [39, 69, 70]. B. toyonensis and other ‘probiotic’ B. cereus strains have been used as feed additive. However, their toxic potential is controversially discussed and some are not licensed in the EU [71–73]. Our results confirm the presence of nhe and hbl in probiotic strains B. toyonensis BCT-7112 (#289, V) and B. cereus IP5832 (#237, V), as well as cytK in two other strains of the species. The pathogenicity of a B. cereus sensu lato strain is, most probably, influenced by additional virulence factors such as sphingomyelinase, hemolysin II and metalloproteases [35, 74–76]. Some B. toyonensis strains contain nprA (neutral protease A), sph (sphingomyelinase) and inhA (immune inhibitor A) virulence genes. Additional virulence factors can also be found in B. mycoides and B. pseudomycoides strains. Since HGT is wide-spread among B. cereus sensu lato, we consider it difficult to categorize a species of this group as non-pathogenic per se. The evolutionary analysis of pathogenicity factors may give reason to initiate a reconsideration of the current enteropathogenicity risk assessment strategies of the whole B. cereus group.
Conclusions
Our results show that seven concatenated housekeeping gene sequences depict species relationships in B. cereus sensu lato as accurately as whole-genome comparisons. Many potentially probiotic B. cereus strains form a clearly distinguishable phylogenetic line within B. cereus sensu lato (Cluster V, ANI boundary > 96 %), which we propose to designate as B. toyonensis named according to the type strain. However, species affiliation of strains is contradictory in many cases. The phylogenetic analysis of this study, therefore, calls for a reassessment of this group’s taxonomy. Lateral transfer of virulence genes hbl, cytK, nhe and plcR within B. cereus sensu lato appears to be constrained only by preservation of gene function, which leads us to hypothesize that the strictly vertical transmission of nhe operons is caused by a second, unknown but fitness relevant function of nhe. We note that evolution of the three B. cereus enterotoxin operons is shaped unexpectedly different. Furthermore, ancient diversification of nhe and hbl operons and propagation of hbla suggest a potential to develop new enterotoxin variants. The distribution of pathogenicity factors and frequent recombination among B. cereus sensu lato phylogenetic groups should be taken into account during risk assessment of the currently valid species of this group, especially concerning probiotic B. toyonensis and B. mycoides strains.
Methods
B. cereus strains and growth conditions
The origin of Bacillus cereus sensu lato strains is given in the Additional file 1: Table S2. B. cereus strains were grown in either plate count (PC) liquid medium (2.5 g/l yeast extract, 5 g/l peptone from casein and 1 g/l glucose) at 150 rpm or on PC plates at 30 °C. Overnight cultures were inoculated from agar plates or angular agar and grown under shaking (150 rpm) in 3 ml PC medium for 15 h.
Isolation of genomic DNA
Genomic DNA from B. cereus was isolated using a modified CTAB (cetyl trimethylammonium bromide) DNA preparation method [77]. B. cereus overnight cultures were pelleted and solved in 567 μl TE buffer (pH 8.0, sterile filtrated). Cells were then lysed using a Fastprep 24 instrument (M. P. Biomedicals, 0.1 mm zirconia beads). Proteins in the supernatant were denatured by adding 30 μl 10 % (w/v) SDS and 3 μl proteinase K (20 mg/ml) and incubated at 37 °C for 3 h. Then, 100 μl 5 M NaCl were added and carefully mixed. 80 μl CTAB/NaCl (10 % CTAB in 0.7 M NaCl) were added, mixed carefully and incubated at 65 °C for 30 min to complex nucleic acids. To separate proteins from nucleic acids phenol chloroform extraction was performed. Nucleic acids were precipitated by adding 0.6 volumes of ice-cold isopropyl alcohol at 4 °C for at least 15 min. Samples were washed twice in ice-cold 70 % (v/v) ethanol, air-dried and dissolved in sterile ddH2O at 4 °C overnight. RNA digestion with 20 μg RNAse for 30 min at 37 °C followed by a second phenol chloroform extraction ensured that only genomic DNA remained. Samples were washed twice in ice-cold 70 % (v/v) ethanol, air-dried and dissolved in sterile ddH2O at 4 °C overnight. Quality of DNA isolation was tested by agarose gel electrophoresis and spectroscopy (Nanodrop photometer). DNA concentration was determined using the Qubit dsDNA High Sensitivity Assay (Qubit® 2.0 fluorometer).
NGS sample preparation and sequencing
DNA libraries were prepared using the TruSeq® DNA PCR-Free Sample Preparation Kit. The TruSeq® protocol was optimized with respect to DNA shearing and fragment size selection to improve assembly quality (Huptas et al., in preparation). DNA libraries were sequenced on the Illumina MiSeq® system according to manufacturer’s instructions. The reagent kits used for sequencing of respective strains are indicated in Additional file 1: Table S1.
Quality control of read data and assembly
Quality of raw sequencing data was assessed by FastQC (www.bioinformatics.bbsrc.ac.uk/projects/fastqc/), followed by quality filtering and trimming of reads with the NGS QC Toolkit v.2.3.2 [78]. Individual settings for each sequenced genome are summarized in Additional file 1: Table S1. The program KmerGenie v.1.5924 [79] was used to calculate the best k-mer value for each assembly from k = 23 to the maximal possible k = 223 in the default two passes. Contigs produced by the short read sequence assembler ABySS v.1.3.7 [80] (minimum scaffold and contig length 500 bp) were further quality controlled by QUAST v.2.2 [81] (using only contigs > 500 bp). Results of data processing, coverage, k-mer values and contig assembly are shown in Additional file 1: Table S1.
Identification of virulence genes in de novo sequenced genomes
The newly generated contigs were aligned against reference genomes (B. cereus ATCC 14579, B. cereus F837/76 and B. cytotoxicus NVH 391/98) with progressiveMauve v.2.3.1 [82] and nheABC, hblCDAB, cytK, plcR, adk, ccpA, glpF, glpT, panC, pta and pycA were identified according to already annotated features. A second comparison of single contigs with CloneManager Suite 7 (Sci-Ed Software) was used to confirm gene locations and to control start, end and length of genes of interest.
Collection of data from databases
Further gene sequences and genomes were downloaded from NCBI and Patric databases All available B. cereus sensu lato genomic sequences were scanned for the nhe operon. Since B. anthracis is known to be highly clonal we decided to include only five representative B. anthracis strains. A total of 218 strains containing nheABC were found and further analyzed for the presence of seven housekeeping genes. The genes adk (adenylate kinase), ccpA (catabolite control protein A), glpF (glycerol uptake facilitator), glpT (glycerol-3-phosphate transporter), panC (pantoate-β-alanine ligase), pta (phosphate acetyltransferase) and pycA (pyruvate carboxylase) were chosen to calculate the species tree. These housekeeping genes have already been selected for MLST [39], because they are scattered over the entire chromosome (http://mlstoslo.uio.no/cgi-bin/mlstdb/mlstdbnet4.pl?dbase=optimized&page=scheme-optimized&file=bcereusgrp_isolates.xml). Due to draft status and partially insufficient sequence quality of genome sequences from the databases, selected housekeeping gene sequences could not be identified for all 218 B. cereus sensu lato strains. Hence, the final set of strains was reduced to 142 B. cereus sensu lato strains (Additional file 1: Table S2). Sequences of the final set are available in the NCBI database either completed or as draft genome. Accession numbers for the 25 genomes reported in this work are given in Additional file 1: Table S1.
Phylogenetic relationships based on single gene and whole genome sequences
Pairwise average nucleotide identity (ANIb) of 142 B. cereus sensu lato genomes was calculated for all possible distinct pairs according to the algorithm described before [43]. For calculation the script ANI.pl (by Jiapeng Chen) available at https://github.com/chjp/ANI was used taking one strain of a pair as query and the other one as reference and vice versa. The resulting two ANIb values for each pair were averaged, which yielded 10,011 similarity values. Similarities were converted into distances by applying the formula 1 – (pairwise avg. ANIb/100). The resulting distance matrix in nexus file format served as input for the neighbor-network computed by SplitsTree4 (version 4.13.1) [83]. Single nucleotide polymorphisms (SNPs) were detected in the entire genomes of the B. cereus sensu lato strain set using the program kSNP3 v.3.0 [84] at k-mer size 21 (determined by Kchooser, implemented in kSNP3). The most distant cluster, the two B. cytotoxicus strains (phylogenetic cluster VII) had to be excluded to obtain an FCK (fraction of k-mers present in all genomes) value > 0.1, which is necessary for adequate SNP detection efficiency. The resulting core SNP matrix was basis for the calculation of a phylogenetic tree using MEGA6 [85] (Maximum likelihood method, Tamura-Nei model [86], uniform rates, using all sites, bootstrap 1000).
Multiple DNA sequence alignments were generated online with ClustalΩ [87] and used to compare genes. Alignments of (concatenated) genes served as input for MEGA6. The species tree of B. cereus sensu lato strains was built from concatenated DNA sequences of seven housekeeping genes. The genes adk, ccpA, glpF, glpT, panC, pta, and pycA were taken from the optimized MLST scheme developed by Tourasse et al. [39], because they are evenly distributed over the entire B. cereus chromosome. In contrast to the MLST-approach, we decided to use the entire genes in our comparison to display relationships as accurately as possible. To compare the operons nheABC and hblCDAB concatenated sequences without intergenic regions were used. The order of the genes within the operons was conserved in all strains, thus a rearrangement of genes was not necessary. Phylogenetic trees of genes or concatenated genes were calculated in MEGA6 using the maximum likelihood method based on the Tamura-Nei model with a discrete Gamma distribution, permitting some invariant sites (TN93 + G + I). This was the ideal substitution model determined by the ‘Find Best DNA/Protein Models (ML)’ function of MEGA6 [85] for the species tree dataset containing seven concatenated housekeeping genes of 142 strains and was applied for the calculation of all phylogenetic trees. Sequences were first aligned and then shortened to the ‘lowest common denominator’ to ensure that all sequences of an alignment start and end together, because protruding ends within an alignment are automatically cut during calculation of the phylogenetic tree (gaps within sequences remained intact). While the housekeeping genes were of the same length in all strains, the toxin genes did not show uniform lengths in alignments and required cutting of sequence ends. All sites (including gaps) were used to calculate the phylogenetic trees and reliability was tested by 1000 bootstrap repeats. Branching points with bootstrap values ≥ 70 are considered reliable. The phylogenetic trees are not rooted to a certain outgroup to allow comparison of subsets and toxin genes that only occur in some strains. To ensure comparability, we used the same settings for the calculation of all phylogenetic trees. To further control the reliability of the maximum likelihood approach, the phylogenetic tree calculations were repeated using the neighbor joining and minimum evolution methods implemented in MEGA6. Results were almost identical albeit with lower resolutions (data not shown).
Detection of potential recombination events
Putative recombination events within enterotoxin operons and a graphical representation of differences within multiple alignments were calculated by RDP3 [88]. In a very conservative approach only statistically proven recombination events according to the following criteria were considered: a recombination event i) was detected by at least three of the programs implemented in RDP3, ii) showed a maximum average p-value of < 0.05 and iii) both parental sequences are part of the investigated strainset.
Analysis of nhe and nhea transcription
Bacteria were cultured in CGY medium [89] supplemented with 1 % (w/v) glucose as described previously [35] to an OD600 of 4. Six ml of the culture were harvested, the cell pellets were snap-frosted in liquid nitrogen and stored at −80 °C. Total RNA was extracted by resuspending the pellet in 1 ml TRIreagent and cells were disrupted as described above. DNase I digestion and RNA isolation were performed as previously described [90]. First strand synthesis was performed using the qScript™ cDNA Supermix (Quanta Biosciences). Subsequent PCR (annealing temperature 56 °C) contained 50 pmol of each primer (Additional file 1: Table S3), 5 μl green GoTaq® G2 reaction buffer, 0.05 mM of each dNTP, 62.5 mM MgCl2 and 0.5 U GoTaq® G2 DNA Polymerase (Promega) in a volume of 25 μl. Amplification of the 16S rRNA gene rrn transcript served as a positive control, nuclease-free H2O as a negative control. RT-PCR results were visualized on 2 % (w/v) agarose gels.
Availability of supporting data
Draft genome assemblies are deposited in NCBI’s GenBank, accession numbers are listed in Additional file 1: Table S1. All data supporting the results of this article are included within the article and its Additional files 1 and 2.
Acknowledgements
This research project was supported by the German Ministry of Economics and Energy (via AiF) and the FEI (Forschungskreis der Ernährungsindustrie e.V., Bonn), Project AiF 17506 N.
We thank Dr. Mareike Wenning and Dr. Thomas Clavel for providing the strains B. mycoides WSBC 10969 and B. cereus #17, respectively. We would also like to thank Johanna Reiter and Scharifa Bornschier for their excellent technical assistance in compiling the nucleotide sequences.
Abbreviations
- Adk
Gene encoding adenylate kinase
- ANI
Average nucleotide identity
- ANIb
Calculation of ANI based on the BLAST algorithm
- AFLP
Amplified fragment length polymorphism
- Cap
Operon encoding B. anthracis poly-γ-D-glutamic acid capsule
- CcpA
Gene encoding catabolite control protein A
- Ces
Seven gene operon encoding among others both cereulide synthetase subunits A and B
- ClyA
Gene encoding the pore-forming hemolysin cytolysin A
- Cry
Genes encoding B. thuringiensis insecticidal crystal endotoxins
- CspA
Gene encoding cold shock protein A
- Cya
Gene encoding B. anthracis edema factor
- Cyt
Genes encoding B. thuringiensis cytolysins
- CytK
Gene encoding cytotoxin K, exists in the variants CytK-1 and CytK-2
- DDH
DNA-DNA Hybridization
- FCK
Fraction of core k-mers
- GlpF
Gene encoding glycerol uptake facilitator protein
- GlpT
Gene encoding glycerol-3-phosphate transporter
- Hbl
Operon encoding the tripartite enterotoxin hemolysin BL, exists in the variants Hbl and Hbla
- HGT
Horizontal gene transfer
- Lef
Gene encoding B. anthracis lethal factor
- ML
Maximum likelihood
- MLEE
Multi locus enzyme electrophoresis
- MLSA
Multi locus sequence analysis
- MLST
Multi locus sequence typing
- Nhe
Operon encoding the tripartite non-hemolytic enterotoxin, exists in the variants Nhe and Nhea
- PagA
Gene encoding B. anthracis protective antigen A
- PanC
Gene encoding pantoate-β-alanine ligase
- PapR
Gene encoding the quorum sensing peptide PapR necessary for activation of PlcR
- PlcR
Gene encoding the Phospholipase C virulence regulator
- Pta
Gene encoding phosphotransacetylase
- pXO1
B. anthracis virulence mega-plasmid containing the pathogenicity island encoding among others PagA, Lef and Cya
- pXO2
B. anthracis virulence mega-plasmid containing among others the cap operon
- PycA
Gene encoding pyruvate carboxylase subunit A
- SNP
Single nucleotide polymorphism
- Tm
Melting temperature
Additional files
Contains Tables S1-S6. (PDF 561 kb)
Contains Figures S1-S4. (PDF 1084 kb)
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
M-E. B., S. S. and V. M. K. designed the study. M-E. B. carried out the phylogenetic and transcriptional analysis. C. H. performed calculations of pairwise average ANI values, kSNP3 calculations and FASTQ read remapping. M-E. B. and S. S. wrote the manuscript. All authors read and approved the final manuscript.
Contributor Information
Maria-Elisabeth Böhm, Email: marlies.boehm@wzw.tum.de.
Christopher Huptas, Email: christopher.huptas@wzw.tum.de.
Viktoria Magdalena Krey, Email: vk@borreliosecentrum.de.
Siegfried Scherer, Email: siegfried.scherer@wzw.tum.de.
References
- 1.Frankland GC, Frankland PF. Studies on some new micro-organisms obtained from air. Philos Trans R Soc B: Biol Sci. 1887;178:257–87. doi: 10.1098/rstb.1887.0011. [DOI] [Google Scholar]
- 2.Drobniewski FA. Bacillus cereus and related species. Clin Microbiol Rev. 1993;6(4):324–38. doi: 10.1128/cmr.6.4.324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ehling-Schulz M, Fricker M, Grallert H, Rieck P, Wagner M, Scherer S. Cereulide synthetase gene cluster from emetic Bacillus cereus: structure and location on a mega virulence plasmid related to Bacillus anthracis toxin plasmid pXO1. BMC Microbiol. 2006;6:20. doi: 10.1186/1471-2180-6-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Damgaard PH, Granum PE, Bresciani J, Torregrossa MV, Eilenberg J, Valentino L. Characterization of Bacillus thuringiensis isolated from infections in burn wounds. FEMS Immunol Med Microbiol. 1997;18(1):47–53. doi: 10.1111/j.1574-695X.1997.tb01026.x. [DOI] [PubMed] [Google Scholar]
- 5.Kuroki R, Kawakami K, Qin L, Kaji C, Watanabe K, Kimura Y, et al. Nosocomial bacteremia caused by biofilm-forming Bacillus cereus and Bacillus thuringiensis. Intern Med. 2009;48(10):791–6. doi: 10.2169/internalmedicine.48.1885. [DOI] [PubMed] [Google Scholar]
- 6.Koch R. Die Ätiologie der Milzbrand-Krankheit, begründet auf die Entwicklungsgeschichte des Bacillus Anthracis. 1876. [Google Scholar]
- 7.Hoffmaster AR, Ravel J, Rasko DA, Chapman GD, Chute MD, Marston CK, et al. Identification of anthrax toxin genes in a Bacillus cereus associated with an illness resembling inhalation anthrax. Proc Natl Acad Sci U S A. 2004;101(22):8449–54. doi: 10.1073/pnas.0402414101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brezillon C, Haustant M, Dupke S, Corre JP, Lander A, Franz T, et al. Capsules, toxins and AtxA as virulence factors of emerging Bacillus cereus Biovar anthracis. PLoS Negl Trop Dis. 2015;9(4) doi: 10.1371/journal.pntd.0003455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lechner S, Mayr R, Francis KP, Pruss BM, Kaplan T, Wiessner-Gunkel E, et al. Bacillus weihenstephanensis sp. nov. is a new psychrotolerant species of the Bacillus cereus group. Int J Syst Bacteriol. 1998;48 Pt 4:1373–82. doi: 10.1099/00207713-48-4-1373. [DOI] [PubMed] [Google Scholar]
- 10.Thorsen L, Hansen BM, Nielsen KF, Hendriksen NB, Phipps RK, Budde BB. Characterization of emetic Bacillus weihenstephanensis, a new cereulide-producing bacterium. Appl Environ Microbiol. 2006;72(7):5118–21. doi: 10.1128/AEM.00170-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hoton FM, Fornelos N, N’Guessan E, Hu X, Swiecicka I, Dierick K, et al. Family portrait of Bacillus cereus and Bacillus weihenstephanensis cereulide-producing strains. Environ Microbiol Rep. 2009;1(3):177–83. doi: 10.1111/j.1758-2229.2009.00028.x. [DOI] [PubMed] [Google Scholar]
- 12.Ash C, Farrow JA, Dorsch M, Stackebrandt E, Collins MD. Comparative analysis of Bacillus anthracis, Bacillus cereus, and related species on the basis of reverse transcriptase sequencing of 16S rRNA. Int J Syst Bacteriol. 1991;41(3):343–6. doi: 10.1099/00207713-41-3-343. [DOI] [PubMed] [Google Scholar]
- 13.Nakamura LK, Jackson MA. Clarification of the taxonomy of Bacillus mycoides. Int J Syst Bacteriol. 1995;45(1):4. [Google Scholar]
- 14.Beattie SH, Williams AG. Detection of toxigenic strains of Bacillus cereus and other Bacillus spp. with an improved cytotoxicity assay. Lett Appl Microbiol. 1999;28(3):221–5. doi: 10.1046/j.1365-2672.1999.00498.x. [DOI] [PubMed] [Google Scholar]
- 15.Nakamura LK. Bacillus pseudomycoides sp. nov. Int J Syst Bacteriol. 1998;48 Pt 3:1031–5. doi: 10.1099/00207713-48-3-1031. [DOI] [PubMed] [Google Scholar]
- 16.Guinebretiere MH, Auger S, Galleron N, Contzen M, De Sarrau B, De Buyser ML, et al. Bacillus cytotoxicus sp. nov. is a novel thermotolerant species of the Bacillus cereus Group occasionally associated with food poisoning. Int J Syst Evol Microbiol. 2013;63(Pt 1):31–40. doi: 10.1099/ijs.0.030627-0. [DOI] [PubMed] [Google Scholar]
- 17.Jimenez G, Urdiain M, Cifuentes A, Lopez-Lopez A, Blanch AR, Tamames J, et al. Description of Bacillus toyonensis sp. nov., a novel species of the Bacillus cereus group, and pairwise genome comparisons of the species of the group by means of ANI calculations. Syst Appl Microbiol. 2013;36(6):383–91. doi: 10.1016/j.syapm.2013.04.008. [DOI] [PubMed] [Google Scholar]
- 18.Thompson CC, Chimetto L, Edwards RA, Swings J, Stackebrandt E, Thompson FL. Microbial genomic taxonomy. BMC Genomics. 2013;14:913. doi: 10.1186/1471-2164-14-913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kim M, Oh HS, Park SC, Chun J. Towards a taxonomic coherence between average nucleotide identity and 16S rRNA gene sequence similarity for species demarcation of prokaryotes. Int J Syst Evol Microbiol. 2014;64(Pt 2):346–51. doi: 10.1099/ijs.0.059774-0. [DOI] [PubMed] [Google Scholar]
- 20.Guinebretiere MH, Thompson FL, Sorokin A, Normand P, Dawyndt P, Ehling-Schulz M, et al. Ecological diversification in the Bacillus cereus Group. Environ Microbiol. 2008;10(4):851–65. doi: 10.1111/j.1462-2920.2007.01495.x. [DOI] [PubMed] [Google Scholar]
- 21.Helgason E, Okstad OA, Caugant DA, Johansen HA, Fouet A, Mock M, et al. Bacillus anthracis, Bacillus cereus, and Bacillus thuringiensis - one species on the basis of genetic evidence. Appl Environ Microbiol. 2000;66(6):2627–30. doi: 10.1128/AEM.66.6.2627-2630.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hu X, Swiecicka I, Timmery S, Mahillon J. Sympatric soil communities of Bacillus cereus sensu lato: population structure and potential plasmid dynamics of pXO1- and pXO2-like elements. FEMS Microbiol Ecol. 2009;70(3):344–55. doi: 10.1111/j.1574-6941.2009.00771.x. [DOI] [PubMed] [Google Scholar]
- 23.Hu X, Van der Auwera G, Timmery S, Zhu L, Mahillon J. Distribution, diversity, and potential mobility of extrachromosomal elements related to the Bacillus anthracis pXO1 and pXO2 virulence plasmids. Appl Environ Microbiol. 2009;75(10):3016–28. doi: 10.1128/AEM.02709-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Thorsen L, Budde BB, Henrichsen L, Martinussen T, Jakobsen M. Cereulide formation by Bacillus weihenstephanensis and mesophilic emetic Bacillus cereus at temperature abuse depends on pre-incubation conditions. Int J Food Microbiol. 2009;134(1–2):133–9. doi: 10.1016/j.ijfoodmicro.2009.03.023. [DOI] [PubMed] [Google Scholar]
- 25.Gonzalez JM, Jr, Brown BJ, Carlton BC. Transfer of Bacillus thuringiensis plasmids coding for delta-endotoxin among strains of B. thuringiensis and B. cereus. Proc Natl Acad Sci U S A. 1982;79(22):6951–5. doi: 10.1073/pnas.79.22.6951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Helgason E, Caugant DA, Lecadet MM, Chen Y, Mahillon J, Lovgren A, et al. Genetic diversity of Bacillus cereus/B. thuringiensis isolates from natural sources. Curr Microbiol. 1998;37(2):80–7. doi: 10.1007/s002849900343. [DOI] [PubMed] [Google Scholar]
- 27.Ko KS, Kim JW, Kim JM, Kim W, Chung SI, Kim IJ, et al. Population structure of the Bacillus cereus group as determined by sequence analysis of six housekeeping genes and the plcR Gene. Infect Immun. 2004;72(9):5253–61. doi: 10.1128/IAI.72.9.5253-5261.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cardazzo B, Negrisolo E, Carraro L, Alberghini L, Patarnello T, Giaccone V. Multiple-locus sequence typing and analysis of toxin genes in Bacillus cereus food-borne isolates. Appl Environ Microbiol. 2008;74(3):850–60. doi: 10.1128/AEM.01495-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Han CS, Xie G, Challacombe JF, Altherr MR, Bhotika SS, Brown N, et al. Pathogenomic sequence analysis of Bacillus cereus and Bacillus thuringiensis isolates closely related to Bacillus anthracis. J Bacteriol. 2006;188(9):3382–90. doi: 10.1128/JB.188.9.3382-3390.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ticknor LO, Kolsto AB, Hill KK, Keim P, Laker MT, Tonks M, et al. Fluorescent amplified fragment length polymorphism analysis of Norwegian Bacillus cereus and Bacillus thuringiensis soil isolates. Appl Environ Microbiol. 2001;67(10):4863–73. doi: 10.1128/AEM.67.10.4863-4873.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Helgason E, Tourasse NJ, Meisal R, Caugant DA, Kolsto AB. Multilocus sequence typing scheme for bacteria of the Bacillus cereus group. Appl Environ Microbiol. 2004;70(1):191–201. doi: 10.1128/AEM.70.1.191-201.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sorokin A, Candelon B, Guilloux K, Galleron N, Wackerow-Kouzova N, Ehrlich SD, et al. Multiple-locus sequence typing analysis of Bacillus cereus and Bacillus thuringiensis reveals separate clustering and a distinct population structure of psychrotrophic strains. Appl Environ Microbiol. 2006;72(2):1569–78. doi: 10.1128/AEM.72.2.1569-1578.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ragan MA. Detection of lateral gene transfer among microbial genomes. Curr Opin Genet Dev. 2001;11(6):620–6. doi: 10.1016/S0959-437X(00)00244-6. [DOI] [PubMed] [Google Scholar]
- 34.Lawrence JG, Ochman H. Reconciling the many faces of lateral gene transfer. Trends Microbiol. 2002;10(1):1–4. doi: 10.1016/S0966-842X(01)02282-X. [DOI] [PubMed] [Google Scholar]
- 35.Jeßberger N, Krey VM, Rademacher C, Böhm M-E, Mohr A-K, Ehling Schulz M, et al. From genome to toxicity: a combinatory approach highlights the complexity of enterotoxin production in Bacillus cereus. Front Microbiol. 2015;6. [DOI] [PMC free article] [PubMed]
- 36.Hoa NT, Baccigalupi L, Huxham A, Smertenko A, Van PH, Ammendola S, et al. Characterization of Bacillus species used for oral bacteriotherapy and bacterioprophylaxis of gastrointestinal disorders. Appl Environ Microbiol. 2000;66(12):5241–7. doi: 10.1128/AEM.66.12.5241-5247.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Böhm M-E, Huptas C, Krey VM, Scherer S. Draft genome sequence of Bacillus cytotoxicus CVUAS 2833, a very close relative to type strain NVH 391–98 isolated from a different location. Genome Announc 2015, 3(4). pii: e00901-15. doi:10.1128/genomeA.00901-15 [DOI] [PMC free article] [PubMed]
- 38.Novichkov PS, Wolf YI, Dubchak I, Koonin EV. Trends in prokaryotic evolution revealed by comparison of closely related bacterial and archaeal genomes. J Bacteriol. 2009;191(1):65–73. doi: 10.1128/JB.01237-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tourasse NJ, Helgason E, Okstad OA, Hegna IK, Kolsto AB. The Bacillus cereus group: novel aspects of population structure and genome dynamics. J Appl Microbiol. 2006;101(3):579–93. doi: 10.1111/j.1365-2672.2006.03087.x. [DOI] [PubMed] [Google Scholar]
- 40.Bavykin SG, Lysov YP, Zakhariev V, Kelly JJ, Jackman J, Stahl DA, et al. Use of 16S rRNA, 23S rRNA, and gyrB gene sequence analysis to determine phylogenetic relationships of Bacillus cereus group microorganisms. J Clin Microbiol. 2004;42(8):3711–30. doi: 10.1128/JCM.42.8.3711-3730.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tourasse NJ, Okstad OA, Kolsto AB. HyperCAT: an extension of the SuperCAT database for global multi-scheme and multi-datatype phylogenetic analysis of the Bacillus cereus group population. Database. 2010;2010:baq017. doi: 10.1093/database/baq017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Richter M, Rossello-Mora R. Shifting the genomic gold standard for the prokaryotic species definition. Proc Natl Acad Sci U S A. 2009;106(45):19126–31. doi: 10.1073/pnas.0906412106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Goris J, Konstantinidis KT, Klappenbach JA, Coenye T, Vandamme P, Tiedje JM. DNA-DNA hybridization values and their relationship to whole-genome sequence similarities. Int J Syst Evol Microbiol. 2007;57(Pt 1):81–91. doi: 10.1099/ijs.0.64483-0. [DOI] [PubMed] [Google Scholar]
- 44.Francis KP, Mayr R, von Stetten F, Stewart GS, Scherer S. Discrimination of psychrotrophic and mesophilic strains of the Bacillus cereus group by PCR targeting of major cold shock protein genes. Appl Environ Microbiol. 1998;64(9):3525–9. doi: 10.1128/aem.64.9.3525-3529.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Swiecicka I, Van der Auwera GA, Mahillon J. Hemolytic and nonhemolytic enterotoxin genes are broadly distributed among Bacillus thuringiensis isolated from wild mammals. Microb Ecol. 2006;52(3):544–51. doi: 10.1007/s00248-006-9122-0. [DOI] [PubMed] [Google Scholar]
- 46.Guinebretiere MH, Broussolle V, Nguyen-The C. Enterotoxigenic profiles of food-poisoning and food-borne Bacillus cereus strains. J Clin Microbiol. 2002;40(8):3053–6. doi: 10.1128/JCM.40.8.3053-3056.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Okstad OA, Gominet M, Purnelle B, Rose M, Lereclus D, Kolsto AB. Sequence analysis of three Bacillus cereus loci carrying PIcR-regulated genes encoding degradative enzymes and enterotoxin. Microbiology. 1999;145(Pt 11):3129–38. doi: 10.1099/00221287-145-11-3129. [DOI] [PubMed] [Google Scholar]
- 48.Stenfors Arnesen LP, Fagerlund A, Granum PE. From soil to gut: Bacillus cereus and its food poisoning toxins. FEMS Microbiol Rev. 2008;32(4):579–606. doi: 10.1111/j.1574-6976.2008.00112.x. [DOI] [PubMed] [Google Scholar]
- 49.Koonin EV. Orthologs, paralogs, and evolutionary genomics. Annu Rev Genet. 2005;39:309–38. doi: 10.1146/annurev.genet.39.073003.114725. [DOI] [PubMed] [Google Scholar]
- 50.Beecher DJ, Wong AC. Tripartite haemolysin BL: isolation and characterization of two distinct homologous sets of components from a single Bacillus cereus isolate. Microbiology. 2000;146(Pt 6):1371–80. doi: 10.1099/00221287-146-6-1371. [DOI] [PubMed] [Google Scholar]
- 51.Lindback T, Okstad OA, Rishovd AL, Kolsto AB. Insertional inactivation of hblC encoding the L2 component of Bacillus cereus ATCC 14579 haemolysin BL strongly reduces enterotoxigenic activity, but not the haemolytic activity against human erythrocytes. Microbiology. 1999;145(Pt 11):3139–46. doi: 10.1099/00221287-145-11-3139. [DOI] [PubMed] [Google Scholar]
- 52.Agaisse H, Gominet M, Okstad OA, Kolsto AB, Lereclus D. PlcR is a pleiotropic regulator of extracellular virulence factor gene expression in Bacillus thuringiensis. Mol Microbiol. 1999;32(5):1043–53. doi: 10.1046/j.1365-2958.1999.01419.x. [DOI] [PubMed] [Google Scholar]
- 53.Lund T, De Buyser ML, Granum PE. A new cytotoxin from Bacillus cereus that may cause necrotic enteritis. Mol Microbiol. 2000;38(2):254–61. doi: 10.1046/j.1365-2958.2000.02147.x. [DOI] [PubMed] [Google Scholar]
- 54.Fagerlund A, Ween O, Lund T, Hardy SP, Granum PE. Genetic and functional analysis of the cytK family of genes in Bacillus cereus. Microbiology. 2004;150(Pt 8):2689–97. doi: 10.1099/mic.0.26975-0. [DOI] [PubMed] [Google Scholar]
- 55.Gohar M, Faegri K, Perchat S, Ravnum S, Okstad OA, Gominet M, et al. The PlcR virulence regulon of Bacillus cereus. PLoS One. 2008;3(7) doi: 10.1371/journal.pone.0002793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Slamti L, Lereclus D. A cell-cell signaling peptide activates the PlcR virulence regulon in bacteria of the Bacillus cereus group. The EMBO journal. 2002;21(17):4550–9. doi: 10.1093/emboj/cdf450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Slamti L, Perchat S, Gominet M, Vilas-Boas G, Fouet A, Mock M, et al. Distinct mutations in PlcR explain why some strains of the Bacillus cereus group are nonhemolytic. J Bacteriol. 2004;186(11):3531–8. doi: 10.1128/JB.186.11.3531-3538.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Klee SR, Brzuszkiewicz EB, Nattermann H, Bruggemann H, Dupke S, Wollherr A, et al. The genome of a Bacillus isolate causing anthrax in chimpanzees combines chromosomal properties of B. cereus with B. anthracis virulence plasmids. PLoS One. 2010;5(7):e10986. doi: 10.1371/journal.pone.0010986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Declerck N, Bouillaut L, Chaix D, Rugani N, Slamti L, Hoh F, et al. Structure of PlcR: insights into virulence regulation and evolution of quorum sensing in gram-positive bacteria. Proc Natl Acad Sci U S A. 2007;104(47):18490–5. doi: 10.1073/pnas.0704501104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Fagerlund A, Brillard J, Furst R, Guinebretiere MH, Granum PE. Toxin production in a rare and genetically remote cluster of strains of the Bacillus cereus group. BMC Microbiol. 2007;7:43. doi: 10.1186/1471-2180-7-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lindback T, Hardy SP, Dietrich R, Sodring M, Didier A, Moravek M, et al. Cytotoxicity of the Bacillus cereus Nhe enterotoxin requires specific binding order of its three exoprotein components. Infect Immun. 2010;78(9):3813–21. doi: 10.1128/IAI.00247-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Phung D, Granum PE, Dietrich R, Martlbauer E, Hardy SP. Inhibition of cytotoxicity by the Nhe cytotoxin of Bacillus cereus through the interaction of dodecyl maltoside with the NheB component. FEMS Microbiol Lett. 2012;330(2):98–104. doi: 10.1111/j.1574-6968.2012.02538.x. [DOI] [PubMed] [Google Scholar]
- 63.Heilkenbrinker U, Dietrich R, Didier A, Zhu K, Lindback T, Granum PE, et al. Complex formation between NheB and NheC is necessary to induce cytotoxic activity by the three-component Bacillus cereus Nhe enterotoxin. PLoS One. 2013;8(4) doi: 10.1371/journal.pone.0063104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ganash M, Phung D, Sedelnikova SE, Lindback T, Granum PE, Artymiuk PJ. Structure of the NheA component of the Nhe toxin from Bacillus cereus: implications for function. PLoS One. 2013;8(9) doi: 10.1371/journal.pone.0074748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gaviria Rivera AM, Granum PE, Priest FG. Common occurrence of enterotoxin genes and enterotoxicity in Bacillus thuringiensis. FEMS Microbiol Lett. 2000;190(1):151–5. doi: 10.1111/j.1574-6968.2000.tb09278.x. [DOI] [PubMed] [Google Scholar]
- 66.Stenfors LP, Mayr R, Scherer S, Granum PE. Pathogenic potential of fifty Bacillus weihenstephanensis strains. FEMS Microbiol Lett. 2002;215(1):47–51. doi: 10.1111/j.1574-6968.2002.tb11368.x. [DOI] [PubMed] [Google Scholar]
- 67.Rejasse A, Gilois N, Barbosa I, Huillet E, Bevilacqua C, Tran S, et al. Temperature-dependent production of various PlcR-controlled virulence factors in Bacillus weihenstephanensis strain KBAB4. Appl Environ Microbiol. 2012;78(8):2553–61. doi: 10.1128/AEM.07446-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Stenfors Arnesen LP, O’Sullivan K, Granum PE. Food poisoning potential of Bacillus cereus strains from Norwegian dairies. Int J Food Microbiol. 2007;116(2):292–6. doi: 10.1016/j.ijfoodmicro.2006.12.021. [DOI] [PubMed] [Google Scholar]
- 69.Oltuszak-Walczak E, Walczak P. PCR detection of cytK gene in Bacillus cereus group strains isolated from food samples. J Microbiol Methods. 2013;95(2):295–301. doi: 10.1016/j.mimet.2013.09.012. [DOI] [PubMed] [Google Scholar]
- 70.Swiecicka I, Mahillon J. Diversity of commensal Bacillus cereus sensu lato isolated from the common sow bug (Porcellio scaber, Isopoda) FEMS Microbiol Ecol. 2006;56(1):132–40. doi: 10.1111/j.1574-6941.2006.00063.x. [DOI] [PubMed] [Google Scholar]
- 71.Kantas D, Papatsiros VG, Tassis PD, Giavasis I, Bouki P, Tzika ED. A feed additive containing Bacillus toyonensis (Toyocerin((R))) protects against enteric pathogens in postweaning piglets. J Appl Microbiol. 2015;118(3):727–38. doi: 10.1111/jam.12729. [DOI] [PubMed] [Google Scholar]
- 72.Hong HA, le Duc H, Cutting SM. The use of bacterial spore formers as probiotics. FEMS Microbiol Rev. 2005;29(4):813–35. doi: 10.1016/j.femsre.2004.12.001. [DOI] [PubMed] [Google Scholar]
- 73.Duc LH, Hong HA, Barbosa TM, Henriques AO, Cutting SM. Characterization of bacillus probiotics available for human use. Appl Environ Microbiol. 2004;70(4):2161–71. doi: 10.1128/AEM.70.4.2161-2171.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Doll VM, Ehling-Schulz M, Vogelmann R. Concerted action of sphingomyelinase and non-hemolytic enterotoxin in pathogenic Bacillus cereus. PLoS One. 2013;8(4) doi: 10.1371/journal.pone.0061404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Guillemet E, Cadot C, Tran SL, Guinebretiere MH, Lereclus D, Ramarao N. The InhA metalloproteases of Bacillus cereus contribute concomitantly to virulence. J Bacteriol. 2010;192(1):286–94. doi: 10.1128/JB.00264-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Cadot C, Tran SL, Vignaud ML, De Buyser ML, Kolsto AB, Brisabois A, et al. InhA1, NprA, and HlyII as candidates for markers to differentiate pathogenic from nonpathogenic Bacillus cereus strains. J Clin Microbiol. 2010;48(4):1358–65. doi: 10.1128/JCM.02123-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Murray MG, Thompson WF. Rapid isolation of high molecular weight plant DNA. Nucleic Acids Res. 1980;8(19):4321–5. doi: 10.1093/nar/8.19.4321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Patel RK, Jain M. NGS QC toolkit: a toolkit for quality control of next generation sequencing data. PLoS One. 2012;7(2) doi: 10.1371/journal.pone.0030619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Chikhi R, Medvedev P. Informed and automated k-mer size selection for genome assembly. Bioinformatics. 2014;30(1):31–7. doi: 10.1093/bioinformatics/btt310. [DOI] [PubMed] [Google Scholar]
- 80.Simpson JT, Wong K, Jackman SD, Schein JE, Jones SJ, Birol I. ABySS: a parallel assembler for short read sequence data. Genome Res. 2009;19(6):1117–23. doi: 10.1101/gr.089532.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Gurevich A, Saveliev V, Vyahhi N, Tesler G. QUAST: quality assessment tool for genome assemblies. Bioinformatics. 2013;29(8):1072–5. doi: 10.1093/bioinformatics/btt086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Darling AE, Mau B, Perna NT. progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS One. 2010;5(6) doi: 10.1371/journal.pone.0011147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Huson DH, Bryant D. Application of phylogenetic networks in evolutionary studies. Mol Biol Evol. 2006;23(2):254–67. doi: 10.1093/molbev/msj030. [DOI] [PubMed] [Google Scholar]
- 84.Gardner SN, Hall BG. When whole-genome alignments just won’t work: kSNP v2 software for alignment-free SNP discovery and phylogenetics of hundreds of microbial genomes. PLoS One. 2013;8(12) doi: 10.1371/journal.pone.0081760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol. 2013;30(12):2725–9. doi: 10.1093/molbev/mst197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Tamura K, Nei M. Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol Biol Evol. 1993;10(3):512–26. doi: 10.1093/oxfordjournals.molbev.a040023. [DOI] [PubMed] [Google Scholar]
- 87.Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol. 2011;7:539. doi: 10.1038/msb.2011.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Martin DP, Lemey P, Lott M, Moulton V, Posada D, Lefeuvre P. RDP3: a flexible and fast computer program for analyzing recombination. Bioinformatics. 2010;26(19):2462–3. doi: 10.1093/bioinformatics/btq467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Beecher DJ, Wong AC. Improved purification and characterization of hemolysin BL, a hemolytic dermonecrotic vascular permeability factor from Bacillus cereus. Infect Immun. 1994;62(3):980–6. doi: 10.1128/iai.62.3.980-986.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Dommel MK, Frenzel E, Strasser B, Blochinger C, Scherer S, Ehling-Schulz M. Identification of the main promoter directing cereulide biosynthesis in emetic Bacillus cereus and its application for real-time monitoring of ces gene expression in foods. Appl Environ Microbiol. 2010;76(4):1232–40. doi: 10.1128/AEM.02317-09. [DOI] [PMC free article] [PubMed] [Google Scholar]