

Abstract
Objective
The aim of this study was to identify liver proteome changes in a mouse model of severe insulin resistance and markedly decreased leptin levels.
Methods
Two-dimensional differential gel electrophoresis was utilized to identify liver proteome changes in AKT1+/-/AKT2-/- mice. Proteins with altered levels were identified with tandem mass spectrometry. Ingenuity Pathway analysis was performed for the interpretation of the biological significance of the observed proteomic changes.
Results
11 proteins were identified from 2 biological replicates to be differentially expressed by a ratio of at least 1.3 between age-matched insulin resistant (Akt1+/-/Akt2-/-) and wild type mice. Albumin and mitochondrial ornithine aminotransferase were detected from multiple spots, which suggest post-translational modifications. Enzymes of the urea cycle were common members of top regulated pathways.
Conclusion
Our results help to unveil the regulation of the liver proteome underlying altered metabolism in an animal model of severe insulin resistance.
Keywords: mouse, liver, AKT, insulin resistance, proteomics
1. Introduction
Type 2 diabetes is a life-long metabolic disorder characterized by hyperglycemia and insulin resistance. Blood glucose level increases when beta cells are unable to produce sufficient insulin to compensate for insulin resistance in liver, adipose and skeletal muscle.
The mechanisms of insulin resistance in type 2 diabetes remain largely unraveled. Genetic alterations of different components of the IRS-PI3K-Akt pathway help to dissect the complex metabolism of insulin resistance [1-8]. Among the three Akt isoforms, Akt2 is predominantly expressed in insulin-responsive tissues and a missense mutation in Akt2 was reported to result in insulin resistance and diabetes in a Caucasian family [9]. While Akt2 knockout mice display a mild diabetic phenotype with insulin resistance, Akt1 haplodeficiency on an Akt2 null background significantly exacerbates this phenotype and is also characterized by markedly decreased leptin levels [10]. This suggests a compensatory role of Akt1 in maintaining glucose homeostasis in Akt2 knockout mice. Since Akt1-/-/Akt2-/- mice do not survive past the neonatal period, Akt1+/-/Akt2 -/- compound knock-out mice provide a useful model to study the mechanisms and metabolic pathways involved in severe insulin resistance[10, 11].
Hepatic insulin resistance leads to disrupted glucose homeostasis, including decreased glycogen storage and enhanced gluconeogenesis, resulting in increased glucose output. Although multiple studies investigated the essential role of hepatic insulin resistance in the development of type 2 diabetes, the molecular mechanisms underlying the defective insulin signaling remain understudied. Here we investigated the liver proteome profile using two-dimensional differential-gel electrophoresis and tandem-mass-spectrometry in Akt1+/-/Akt2-/- mice as a model of severe insulin resistance. The results of this study may help identify potential biomarkers associated with the development and progression of altered liver metabolism in type 2 diabetes.
2. Materials and Methods
2.1. Mice
All studies were approved by the Institutional Animal Care and Use Committee at The University of California at Irvine and comply with the National Institutes of Health guidelines. The generation of Akt1-/- and Akt2-/- mice has previously been described [5, 6] and were purchased from The Jackson Laboratories (Bar Harbor, ME). Mice have been backcrossed to a C57BL/6 background and were intercrossed to generate the desired genotypes. Briefly, given gender specific decreased fecundity of these strains [12, 13], Akt1-/- females and Akt2-/- males were intercrossed to generate Akt1+/-/Akt2+/-mice. Female AKT1+/-/AKT2+/- mice used for subsequent breeding were generated from this single intercrossing. Akt2-/- males were then crossed with Akt1+/-/Akt2+/- females to generate Akt1+/-/Akt2-/-mice. Mice were housed under a 12:12 hour light-dark cycle. All mice used in experiments were male. 8 mice per group were used for the assessment of body weight, random glucose levels, oral glucose tolerance testing and insulin level determination during the oral glucose tolerance testing.
2.2. Measurement of glucose and insulin levels
Blood was collected by tail vein sampling. Blood glucose levels were determined using an ACCU-CHEK Compact Plus (Roche Diagnostics, Indianapolis, IN) automatic glucometer. Insulin levels of serum samples were determined by using a Mouse Insulin ELISA (ALPCO, Salem, NH) according to the manufacturer's instructions.
2.3. Oral Glucose Tolerance Test
Oral glucose tolerance testing was done after a 6 hour fast and an oral feeding of 2g glucose/kg body weight in 20 week old mice as previously described [14]. Blood was collected from the tail vein. Glucose and insulin levels were determined immediately before the feeding and 15, 30, 60 and 120 minutes after the feeding.
2.4. Liver Histology
Liver samples were fixed in 10% buffered formalin and embedded in paraffin wax. A Leica AutoStainer XL (Leica Biosystems Inc., Buffalo Grove, IL) was used for automated hematoxylin and eosin staining (H&E). The histology of liver tissue (n=3 per group) was examined in 4.5-μm thick H&E stained sections.
2.5. Sample Preparation for 2-D DIGE
All mice were euthanized after a 6 hour fast at the same time of their light-dark cycle (8 hour time point of light during the light-dark cycle). Liver samples were homogenized on ice in DIGE sample labeling buffer (7M urea, 2M thiourea, 30 mM Tris base, 4% CHAPS, pH 9) using a Polytron Homogenizer (Power Gen 125, Thermo Fisher Scientific, Hampton, NH) and incubated at room temperature for 10 min prior to protein quantification by the Bradford assay (Bio-Rad, Hercules, CA). Samples were minimally labeled with CyDye DIGE Fluor dyes Cy2, Cy3 and Cy5 according to the manufacturer's instructions (GE Healthcare, Little Chalfont, United Kingdom) and as per a previously described strategy[15]. Briefly, 50ug of sample from each group (Akt1+/-/Akt2-/- or C57BL/6) was labeled and comprised of an equal amount of protein from each individual animal of the group (4 per group; different mice than from those used for body weight, glucose level and insulin level measurements). The labeling strategy was as follows: Akt1+/-/Akt2-/- and C57BL/6 samples were labeled with Cy3 and Cy5 and for normalization the same amount of protein from a pool of all samples was labeled with Cy2. Differentially labeled samples were mixed prior to rehydration loading. Additionally, a biological replicate of the above was performed and processed through all subsequent steps requisite for protein identification.
2.6. IPG-strip Rehydration, IEF and 2-D Gel Electrophoresis
Mixed labeled samples were made up to 425 μl with DeStreak-rehydration solution (GE-Healthcare), including 2% IPG buffer (pH intervals 4-7 or 6-11; GE-Healthcare) and then transferred to a rehydration tray. 24cm Immobiline Dry Strips covering pH intervals 3-7NL and 6-11 (GE Healthcare) were placed into the channel. Strips were then overlayed with mineral oil and rehydrated for 20 hours at room temperature in the dark. The first dimension was performed on an Ettan IPGphor3 (GE Healthcare) and started at 100V and continued until the current was less than 5uA per strip (approximately 12hrs) to desalt the sample and to ensure reaching the highest voltage. Isoelectric focusing the pH 3-7NL dry strips was as follows: 1 hour at 500V, a gradient voltage for 1 hour to 1000V, a gradient voltage for 3 hours to 8000V, 4.5 hours at 8000V, a gradient voltage for 3 hours to 10000V and a final step at 10 000V for 3.25 hours. Isoelectric focusing the pH 6-11 dry strips was as follows: 1 hour at 500V, a gradient voltage for 1 hour to 1000V, a gradient voltage for 3 hours to 8000V, 7 hours at 8000V, a gradient voltage for 3 hours to 10000V and a final step at 10000V for 5.5 hours. Prior to the second dimension, the gel strips were equilibrated and disulfide bonds were reduced and then subsequently alkylated in buffer (6 M urea, 2% SDS, 30% glycerol, and 50 mM Tris–HCl, pH 8.6) containing 1% (w/v) dithiothreitol for the reduction reaction and then 2.5% (w/v) iodoacetamide for the alkylation reactions. Both reactions were performed for 15 minutes at room temperature. Alkylated sample strips were then rinsed with 2× electrophoresis buffer (25 mM Tris, pH 8.3, 192 mM glycine, and 0.2% SDS) and sealed onto 12.5% SDS-PAGE gels with 1% (w/v) agarose solution in 1× SDS-PAGE buffer containing a trace of bromophenol blue (0.002% w/v). Electrophoresis was performed in 1× SDS-PAGE buffer at 20°C with an Ettan Dalt-six system (GE Healthcare) applying 10mA/gel for one hour and then 40mA/gel until the tracking blue dye front reached the bottom of the gel. For preparative gels, the same protocol as above was followed with the exception of 1000ug of unlabeled protein (with an equal portion from all individuals) used for the rehydration process. Preparative gels were stained with Coomassie Brilliant Blue G-250 overnight as per a previously described protocol [16]. Protein spot matching between analytical and preparative gels and spot excisions were performed manually.
2.7. 2-D DIGE Image Analysis
The Cy2, Cy3 and Cy5 labeled images were individually acquired on a Typhoon FLA 9500 (GE Healthcare) at the excitation/emission wavelengths of 473/530 nm, 532/570 nm, 635/665 nm, respectively. Gels were analyzed using DeCyder 2D Differential Analysis Software (version 7.2, GE Healthcare). To optimize comparative gel analysis, spot detection was performed after background subtraction and normalization of a set of images from the same gel. Gels were analyzed in the DIA module with exclusion selection criteria on spot volume (<10,000) and maximum slope (>1.0) then loaded into the BVA module for comparative analysis across multiple gels to identify differential spots between the Akt1+/-/Akt2-/- and C57BL/6 group. Protein spots with abundance changes of ≥+1.3 or ≤-1.3 fold with a p-value ≤ 0.05 by t -test were deemed statistically significant. All spots meeting statistical criteria for a difference in abundance were manually verified for an acceptable and characteristic three dimensional profile.
2.8. In-Gel Digestion and Protein Identification using MALDI TOF MS and MS/MS
Manual in-gel trypsin digestion of spots was performed essentially as previously described [17]. Briefly, excised gel pieces were washed once in 25mM ammonium bicarbonate, followed by two washes in 50%ACN/25mM ammonium bicarbonate and finally dehydrated in 100% ACN. Gel pieces were then dried to completion with a centrifugal evaporator. Gel pieces were incubated with trypsin (Sigma-Aldrich Corporation, St. Louis, MO) at a concentration of 12.5ng/ul overnight at 37°C. Peptides were recovered with three serial extractions with 60% ACN/5% formic acid and resuspended in 0.1% formic acid. All samples were desalted using ZipTipsC18 (EMD Millipore, Billerica, MA) as per the manufacturer's protocol and eluted directly in an α-cyano-4-hydroxycinnamic acid saturated solution (60% ACN/5% formic acid) onto a MALDI target plate with the dried droplet method. Spectra were acquired on an AB SCIEX TOF/TOF 5800 system (Framingham, MA) with spectra from MALDI-TOF acquired in positive ion reflector mode and MS/MS spectra acquired in 1-kV positive mode. A mass accuracy tolerance of 30 ppm for precursors, 0.3 Da for fragments, and one missed trypsin cleavage were permitted for tryptic mass searches of Mus musculus proteins in the UniProt database using the Paragon algorithm (ProteinPilot version 4.0; ABSCIEX). Positive protein identifications were made if more than two peptides with 95% confidence (as per the Paragon algorithm scoring system) mapped to the same protein and the percent sequence coverage was greater than 5%. When possible, spot locations of identified proteins were further validated by comparing the location of our identified spots to those in the SWISS-2D PAGE database [18].
2.9. Cellular Component and Canonical Pathway Analysis
The UniProt Knowledgebase (UniProtKB, http://www.uniprot.org) was used to identify the cellular component localization of the differentially expressed proteins. Ingenuity Pathway Analysis (IPA) software (Ingenuity, QIAGEN Silicon Valley, Redwood City, CA) was used to identify canonical pathways associated with differentially regulated proteins.
2.10. Statistical Analyses
All data presented for body weight, glucose and insulin measurements in the figures were expressed as mean ± SD. Areas under the curve were calculated with the trapezoidal method[19]. Statistical significance between groups for this data was evaluated using Student's t-test. Differences were deemed statistically significant at p<0.05. Statistical analyses for protein spot analysis, peptide/protein identification and pathway analysis were performed using default settings of application specific software (DeCyder, ProteinPilot, and Ingenuity Pathway Analysis, respectively).
3. Results
3.1. Growth and glucose homeostasis in AKT1+/-/AKT2-/- mice
AKT1+/-/AKT2-/- mice on a mixed genetic background have previously been shown to have a modestly reduced birth weight (8%) in comparison to wild type littermates [11]. To determine the subsequent effect of this genotype on body weight on a C57BL/6 background, age-matched wild type and AKT1+/-/AKT2-/- mice were weighed serially. The relative average body weight of AKT1+/-/AKT2-/- mice in comparison to wild type mice was 74-81% percent from 8 to 20 weeks of age (Figure 1A). To assess the diabetic phenotype in AKT1+/-/AKT2-/- mice, blood glucose levels were measured in both the fed and fasted state (Figures 1B and 1C). These mice demonstrated overt hyperglycemia regardless of their nutritional status. Additionally, the response to oral glucose tolerance testing (OGTT) was impaired in these mice and consistent with a diabetic phenotype (Figure 1C). During OGTT, AKT1+/-/AKT2-/- mice blood glucose levels were significantly elevated at all measured time points in comparison to wild type mice. As expected, the incremental area under the curve for glucose (mmol/L in 120 minutes) during OGTT was also increased in AKT1+/-/AKT2-/- mice in comparison to wild type mice (mean ± SD. wild type 432.9 ± 60.6 vs. AKT1+/-/AKT2-/- 809.7 ± 237.0; p<0.001). Interestingly, the insulin level during the fasted state is increased in AKT1+/-/AKT2-/- mice in comparison to wild type mice but the insulin level in response to oral feeding in AKT1+/-/AKT2-/- mice was not significantly increased (p-value=0.46 for 0 minutes vs. 15 minutes in AKT1+/-/AKT2-/- mice) (Figure 1D), similar to the inadequate insulin response seen in type 2 diabetes. To assess for hepatic insulin resistance as a contributor to the impaired glucose tolerance testing, we calculated the product of the area under the curve for glucose and insulin during the first 30 minutes of the OGTT as a previous study demonstrated a positive Pearson's correlation coefficient for this product and hepatic insulin resistance[20]. This value was 6.3-fold greater in the AKT1+/-/AKT2-/- mice in comparison to wild type mice and implies hepatic insulin resistance in AKT1+/-/AKT2-/- mice (mean ± SEM. wild type 13,787 ± 1,148 vs. AKT1+/-/AKT2-/- 87,250 ± 15,159; p<0.001). At 5-months of age, the liver parenchyma from AKT1+/-/AKT2-/- mice was normal in comparison to age matched wild type mice and did not demonstrate evidence of lipid accumulation (Figure 1E). This is consistent with other studies which have demonstrated a pivotal role of Akt2 in fatty liver disease[21, 22]. These results recapitulate the insulin resistant phenotype previously observed on a mixed genetic background and allowed us to pursue proteomic profiling that could otherwise be confounded by a mixed genetic background [10, 23].
Figure 1. Analyses of body weights, random serum glucose levels, OGTT and liver histology.
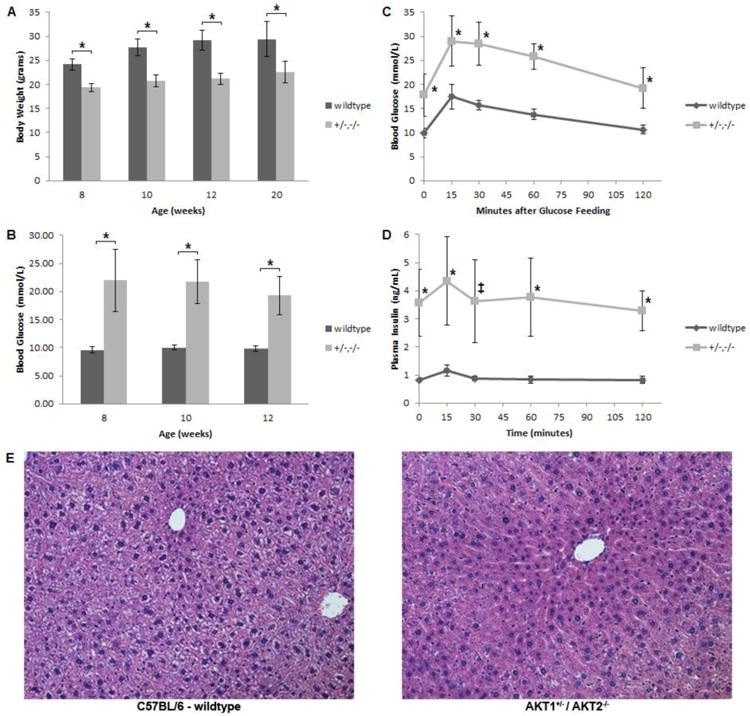
(A) Adult body weights of wild type and Akt1+/-/Akt2-/- mutant mice. (B) Random blood glucose levels of wild type and Akt1+/-/Akt2-/- mutant mice. (C) Blood glucose levels during OGTT of 20-week- old of wild type and Akt1+/-/Akt2-/- mutant mice. (D) Plasma insulin levels during OGTT of 20-week- old of wild type and Akt1+/-/Akt2-/- mutant mice. (E) Representative photomicrographs (×200) of liver sections stained with H&E from 20-week-old wild type and Akt1+/-/Akt2-/- mutant mice. All results are mean +/- SD. The markings indicate statistical significance in comparison to wild type mice at the same time point (*p<0.001, ‡p<0.005).
3.2. Identification of differentially expressed liver proteins of AKT1+/-/AKT2 -/- mice using 2D-DIGE
To determine the effects of insulin resistance on protein expression in AKT1+/-/AKT2-/- mice, we performed a proteomic analysis using 2D-DIGE on liver samples from 20 week old mice. To increase overall protein spot resolution and proteome coverage, labelled protein extracts were analyzed on two separate 2D-DIGE gels with overlapping isoelectric points (pI 3-7 and 6-11). Each group (AKT1+/-/AKT2-/-or wild type mice) was comprised of an equal protein pool from 4 individual mice and a biological replicate was performed. Only matched spots demonstrating a statistical cut off point with a p-value ≤0.05 and an intensity fold change ≤-1.3 or ≥+1.3 on both replicates were deemed to be significantly changed. An overlay of channel images with differentially expressed spots from the 2D-DIGE for proteins with a pI 3-7 is shown in Figure 2. As per DeCyder software (version 7.2), approximately 2100 and 500 spots were identified on the gels with the pI ranges from 3-7 and 6-11, respectively. The 2D-DIGE analysis revealed 17 differentially expressed spots that met our statistical criteria across both replicates. These spots were excised from a Coomassie Brilliant Blue G-250 stained preparative gel and identified through the use of MALDI-TOF/TOF and database interrogation. All 17 of these spots were identified and resulted in 11 unique and differentially expressed proteins (Table 1). Peptide information used for identification is provided in Supplemental Table 1. 2 of these 11 proteins (albumin and ornithine aminotransferase, mitochondrial) were identified in multiple spots in the same molecular weight range and thus likely represent post-translational modifications or unique isoforms. Of note, 4 proteins demonstrated an increased abundance and 7 demonstrated a decreased abundance in the AKT1+/-/AKT2-/- liver samples in comparison to wild type liver samples.
Figure 2. Representative 2D-DIGE gel of liver proteins.

The gel depicts proteins with an pI 3-7 further separated by molecular weight in a 12.5% polyacrylamide gel. Statistically significant, differentially expressed proteins from both replicates are circled on the above gel. Red circles indicate increased and blue circles indicate decreased normalized protein levels in AKT1+/-/AKT2-/- mice in comparison to wild type mice. Information on their identities and differential expression is in Table 1.
Table 1. 2D-DIGE Analysis.
List of proteins identified in spots when comparing liver samples from wild type and AKT1+/-/AKT2-/- mice. All variant protein spots have a fold cutoff change of ≤ or ≥ 1.3 and a p-value <0.05 in both replicates. Mean fold change is for AKT1+/-/AKT2-/- mice in comparison to wild type mice. The spot numbers refer to spot number in Figure 2. Carbonic anhydrase 3 was identified on the gel for the basic range (data not shown). Additional information can be found in Supplemental Table 1.
| Spot # | Protein Name | UniProt ID | Mean Fold Change |
|---|---|---|---|
| 1 | Annexin A6 | ANXA6_MOUSE | -1.37 |
| 2,3,4,5,6 | Serum albumin | ALBU_MOUSE | -2.04, -2.28, -2.36, -2.24, -2.05 |
| 7 | Carboxylesterase 3A | EST3A_MOUSE | -1.5 |
| 8 | Aldehyde dehydrogenase, mitochondrial | ALDH2_MOUSE | 1.36 |
| 9,10,11 | Ornithine aminotransferase, mitochondrial | OAT_MOUSE | +1.71,+2.21, +2.20 |
| 12 | S-adenosylrnethionine synthase isoform type-1 | METK1_MOUSE | +1.48 |
| 13 | Annexin A5 | ANXA5_MOUSE | +1.95 |
| 14 | Arginase-1 | ARGI1_MOUSE | +1.71 |
| 15 | Regucalcin | RGN_MOUSE | -1.69 |
| 16 | Phenazine biosynthesis-like domain-containing protein 2 | PBLD2_MOUSE | -1.38 |
| - | Carbonic anhydrase 3 | CAH3_MOUSE | -1.98 |
3.3. Subcellular localizations and functional networks of differentially regulated proteins
Significantly altered proteins were classified based on their cellular component localization according to UniProt database searches (Figure 3A.) The differentially expressed proteins localized to the cytoplasm, mitochondrial matrix, endoplasmic reticulum, plasma membrane and secreted fraction with the majority located in the cytoplasm (6/11). For the interpretation of the biological significance of the proteins from Table 1, they were analyzed using IPA software. Canonical pathways of proteins that were up- or down regulated are shown in Figure 3B. The top regulated pathways involve the metabolism of intermediates of the urea cycle and underscore the significant up-regulation of the urea cycle in the liver of AKT1+/-/AKT2-/- mice [24]. The S-adenosyl-L-methionine (SAM) biosynthesis pathway was also up-regulated and may suggest the dependency in an insulin resistant state to generate a methyl group donor (SAM) for a multitude of reactions needed to maintain homeostasis.
Figure 3. Classification of identified proteins and biological pathway analysis.
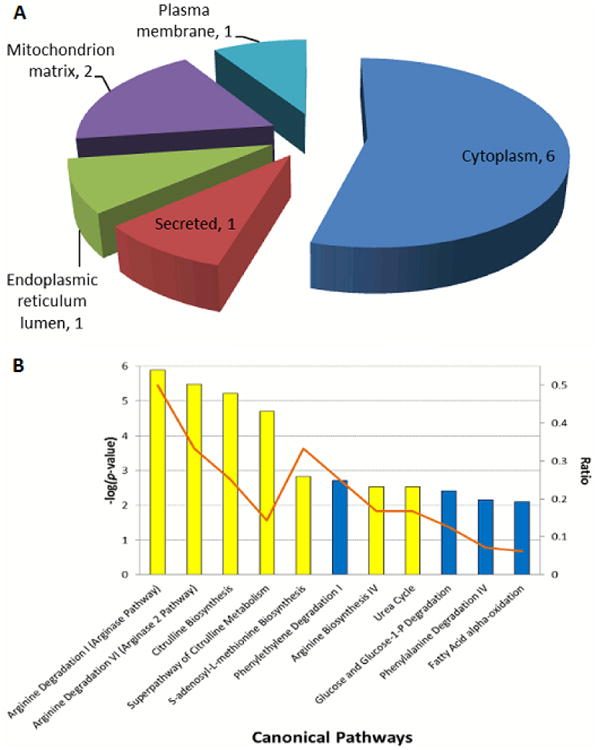
(A) Cellular component localization of the 11 differentially expressed proteins identified as per UniProt. For each cellular component, the number of proteins in each class is given. (B) Enriched biological process networks by IPA software for the 11 differentially expressed proteins. The –log(p-value) for the enriched networks is as is per the height of the bars along the y-axis on the left. Upregulated pathways are colored yellow and downregulated pathways are colored blue. The ratio of the number of enriched proteins from our dataset in comparison to the total number of proteins within a pathway is indicated by the orange line and as per the y-axis on the right.
4. Discussion
Hereditary forms of lipodystrophy are often associated with insulin resistance, diabetes and hepatic steatosis. AKT1+/-/AKT2-/- mice are similar to other murine models of lipodystrophy in that they demonstrate an overt diabetic phenotype that is largely secondary to a leptin deficiency and resulting insulin resistance[25, 26]. However, unlike these other models, AKT1+/-/AKT2-/- mice do not demonstrate accumulation of fat in the liver. This is most likely secondary to Akt2 being requisite for hepatic lipogenesis and the development of fatty liver disease[21, 22]. Mice which are deficient for the insulin receptor substrates (IRS) IRS-1 and IRS-3 also demonstrate this unique phenotype of leptin deficient insulin resistance without hepatic steatosis[27]. AKT1+/-/AKT2-/- mice highlight the nonredundant and complementary roles of signaling molecules downstream of the insulin receptor and this study, to our knowledge, is the first to investigate proteomic changes in such a model.
Tissue based proteomic studies have highlighted tissue specific changes, the influence of the model utilized and technical aspects of such work. Meta-analyses of these studies have also demonstrated a surprisingly low concordance of such datasets [28, 29]. To this end, we utilized a genetic model of insulin resistance and an experimental approach designed to detect reproducible changes. As such, we used inbred mice of the same gender and age and of a similar nutritional status at the time of sacrifice. Additionally, mice were euthanized at the same time of their entrained light-dark cycle to control for potential diurnal variation of protein levels. In this study, only male mice were used as female AKT1+/-/AKT2-/- mice do not demonstrate an overt diabetic phenotype and random blood glucose levels are not significantly different from wild type mice. Sexual dimorphism has been observed in other mouse models of diabetes and insulin resistance and is likely in part due to gender differences in basal and fed leptin levels in this mouse strain[2, 4, 30-32]. Liver samples from insulin resistant and wild type animals were pooled and a biological replicate of the 2D-DIGE and downstream spot analysis was performed. Only protein spots determined to be in matching locations with statistically significant changes in abundance were considered to be differentially regulated in this model. This stringent approach yielded 11 proteins. The majority of the differentially regulated proteins (54%) are typically localized to the cytoplasm. While our study replicates demonstrated variability, subsequent pathway analysis of altered proteins revealed highly similar affected pathways between the replicates when analyzed individually, as well as in comparison to the proteins identified on both replicates (data not shown).
It is important to put the identified proteins in the context of the pathophysiology of insulin resistance and previous studies of insulin resistance and diabetes.
In multiple studies and ours, hepatic regucalcin levels have been shown to be decreased in the setting of diabetes [33-35]. Importantly, previous work has also shown that a subcutaneous injection of insulin increases regucalcin mRNA levels and that insulin stimulation of a human hepatoma cell line increases regucalcin protein abundance [36, 37]. This implies that regucalcin gene expression and protein levels are directly regulated by insulin signaling. The usefulness of individual serum regucalcin levels as a surrogate for insulin resistance remains to be elucidated.
The physiological role of carbonic anhydrase 3 (CAH3) remains unclear and it has been proposed to have functions other than of a hydratase and esterase given its minimal activity levels in comparison to other isoforms [38]. To this point, the abundance and activity of hepatic CAH3 have previously been shown to be decreased in the streptozotocin induced diabetes model in rats [39, 40]. This is of interest in the context of that downregulation of CAH3 leads to significantly increased PPARγ2 gene expression in vitro [41]. In a separate study, insulin receptor substrate-1 (IRS-1) levels were decreased in PPARγ2 knockout mice and these animals demonstrated impaired insulin sensitivity [42]. Therefore, the down regulation of CAH3 levels in our dataset is perhaps part of a compensatory mechanism to increase insulin sensitivity in the overt model of insulin resistance that we utilized.
Annexins are a broadly expressed, evolutionarily conserved group of proteins that bind phospholipids in a calcium dependent manner. The function of these proteins in the liver remains unknown, but they are typically implicated to play roles in development, proliferation, apoptosis and inflammation[43]. Annexin A5 levels have been positively correlated with many different tumor types, including hepatocellular cancer[44]. In our study, annexin A5 was shown to be upregulated and annexin A6 was shown to be downregulated. Other proteomic studies in models of insulin resistance and liver injury have demonstrated similar changes with regards to abundance and direction of change of these annexins [45-48]. Phenazine biosynthesis-like domain-containing protein 2, found to be of decreased abundance in the AKT1+/-/AKT2-/- - mice in our dataset, has previously been detected at reduced levels in hepatocellular tumors [49]. This pattern of changes of annexin A5, annexin A6 and phenazine biosynthesis-like domain-containing protein 2 implies a pro-inflammatory state and liver dysfunction in this model of insulin resistance.
S-adenosylmethionine synthase isoform type-1 is the rate limiting step of the methionine cycle and catalyzes the formation of S-adenosylmethionine (SAM) from methionine and ATP. SAM donates its methyl group in transmethylation reactions to secondary metabolites, lipids, proteins and DNA. In the context of metabolism, SAM has been implicated in the development of obesity and diabetes and is the methyl donor in the synthesis of carnitine which is required to shuttle long chain acyl groups into the mitochondria for beta-oxidation [50]. In insulin resistant AKT1+/-/AKT2-/- mice, one would hypothesize that fatty acid oxidation would be the primary source of energy for the liver and thus sufficient levels of carnitine would be needed to meet the metabolic demands of the liver. Consistent with this, a previous study has shown decreased fatty acid accumulation in the livers of diabetic mice upon carnitine supplementation [51]. This proposed mechanism of carnitine facilitated fatty acid oxidation has led to multiple clinical trials with mixed results on metabolic parameters including blood glucose and lipid levels [52-54].
Liver carboxylesterases are major hydrolases that play an important role in drug and lipid metabolism [55]. It was reported recently that the expression of carboxylesterase 1d (Ces1d) and carboxylesterase 1e (Ces1e) decreased in type 2 diabetic mice [56]. Another study showed that carboxylesterase expression and activity were inhibited by lipopolysaccharide or interleukin-6 under hyperglycemic conditions in primary mouse hepatocytes [57]. Our results show that mouse liver carboxylesterases 3A is downregulated in AKT1+/-/AKT2-/- mice, which implicates possible drug and lipid metabolism disorder in this model of insulin-resistance.
Aldehyde dehydrogenase catalyzes the oxidation of aldehyde during alcohol metabolism. Aldehyde dehydrogenase protein abundance was decreased by a high fat diet in mice [58]. It is one of the most common proteins to display a significant quantitative change in liver mitochondria proteomes in different pathological states, implying it can be a biomarker for mitochondrial dysfunction[29]. The decrease of aldehyde dehydrogenase protein in our dataset might be an indicator of liver mitochondrial dysfunction in response to severe insulin resistance.
Ornithine aminotransferase is a mitochondrial enzyme of the urea cycle that catalyzes the formation of glutamate from ornithine. It is reported to be a potential target for treating hyperammonia [59]. Consistent with our findings, a previous study showed that ornithine aminotransferase was upregulated by a high-fat diet and served as a biomarker for liver injury and increased gluconeogenesis via amino acid catabolism [60]. Glucagon levels are abnormally elevated in type 2 diabetes and dramatically increase the synthesis rate of ornithine aminotransferase [61, 62]. Interestingly, our two-dimensional gel electrophoresis detected this enzyme in 3 different spots with different isoelectric points and the same molecular weight, consistent with post-translational modifications such as acetylation. It is noteworthy that high fat feeding was reported to induce mitochondrial protein hyperacetlyation due to downregulation of NAD-dependent deacetylase sirtuin-3 (SIRT3) [63, 64].
Disease-based proteomic studies have inherent strengths and weaknesses that are largely determined by the pathology of the disease and the approach taken to investigate the proteome. We feel that relative strengths of this study were our attempt to control for potential confounders (genetic background, gender, age, nutritional status, diurnal fluctuations and statistical power) and the use of 2D-DIGE. While the use of gel-based proteomics comes with limitations, in particular decreased sensitivity for low abundance and membrane proteins, it does allow for the simultaneous detection of proteins with an absolute change in abundance and/or those that have undergone post-translational modifications. This is of clinical relevance as particular post-translational modifications may play a role in the pathogenesis of heterogeneous diseases such as diabetes. While the animal model of insulin resistance utilized in this study is not widely studied, our approach did identify proteomic changes previously identified in other models of insulin resistance, diabetes and hepatocellular injury. In fact, several of the proteins we identified have previously been studied in humans as biomarkers of disease activity[65-67]. Importantly, it is becoming evident that biomarker panels comprised of several proteins can result in improved clinical management as they can have greater sensitivity, specificity and predictive value than a single biomarker[68, 69]. Further work is needed to determine if such a panel would be clinical useful in the management of diabetes and its complications.
In summary, our proteomic analysis in a murine model of severe insulin resistance detected 11 liver proteins with significant changes. These changes can be explained in the context of previous studies utilizing models of a high fat diet, insulin resistance and overt diabetes. The stringent approach of using two biological replicates of pooled samples facilitates the identification of potential biomarkers for liver dysfunction in patients suffering from insulin resistance.
Supplementary Material
Acknowledgments
Sources of Support: This work is supported in part by National Heart, Lung, and Blood Institute Grant R01-HL-096987 (to PW); the Paul and Marybelle Musco Fellowship; and the UCI Diabetes Center.
Abbreviations
- IRS
insulin receptor substrate
- PI3K
phosphoinositide 3-kinase
- 2-D
DIGE-two-dimensional differential gel electrophoresis
- pI
isoelectric point
- MALDI
matrix-assisted laser desorption/ionization
- TOF
time of flight
Footnotes
Authors' Contributions: BP, WW and PW planned the study design; BP, WW and JT prepared, drafted, and revised the manuscript; BP, WW, JT, OK and YC contributed to data collection; BP, WW, JT and PY performed and revised statistical analyses; RE examined the histology; PY and PW provided critical revision of the manuscript.
Conflict of Interest: The authors have no relevant conflicts of interest to disclose.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Araki E, et al. Alternative pathway of insulin signalling in mice with targeted disruption of the IRS-1 gene. Nature. 1994;372(6502):186–90. doi: 10.1038/372186a0. [DOI] [PubMed] [Google Scholar]
- 2.Withers DJ, et al. Disruption of IRS-2 causes type 2 diabetes in mice. Nature. 1998;391(6670):900–4. doi: 10.1038/36116. [DOI] [PubMed] [Google Scholar]
- 3.Liu SC, et al. Insulin receptor substrate 3 is not essential for growth or glucose homeostasis. J Biol Chem. 1999;274(25):18093–9. doi: 10.1074/jbc.274.25.18093. [DOI] [PubMed] [Google Scholar]
- 4.Fantin VR, et al. Mice lacking insulin receptor substrate 4 exhibit mild defects in growth, reproduction, and glucose homeostasis. Am J Physiol Endocrinol Metab. 2000;278(1):E127–33. doi: 10.1152/ajpendo.2000.278.1.E127. [DOI] [PubMed] [Google Scholar]
- 5.Cho H, et al. Insulin resistance and a diabetes mellitus-like syndrome in mice lacking the protein kinase Akt2 (PKB beta) Science. 2001;292(5522):1728–31. doi: 10.1126/science.292.5522.1728. [DOI] [PubMed] [Google Scholar]
- 6.Cho H, et al. Akt1/PKBalpha is required for normal growth but dispensable for maintenance of glucose homeostasis in mice. J Biol Chem. 2001;276(42):38349–52. doi: 10.1074/jbc.C100462200. [DOI] [PubMed] [Google Scholar]
- 7.Foukas LC, et al. Critical role for the p110alpha phosphoinositide-3-OH kinase in growth and metabolic regulation. Nature. 2006;441(7091):366–70. doi: 10.1038/nature04694. [DOI] [PubMed] [Google Scholar]
- 8.Jia S, et al. Essential roles of PI(3)K-p110beta in cell growth, metabolism and tumorigenesis. Nature. 2008;454(7205):776–9. doi: 10.1038/nature07091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.George S, et al. A family with severe insulin resistance and diabetes due to a mutation in AKT2. Science. 2004;304(5675):1325–8. doi: 10.1126/science.1096706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen WS, et al. Leptin deficiency and beta-cell dysfunction underlie type 2 diabetes in compound Akt knockout mice. Mol Cell Biol. 2009;29(11):3151–62. doi: 10.1128/MCB.01792-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Peng XD, et al. Dwarfism, impaired skin development, skeletal muscle atrophy, delayed bone development, and impeded adipogenesis in mice lacking Akt1 and Akt2. Genes Dev. 2003;17(11):1352–65. doi: 10.1101/gad.1089403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim ST, Omurtag K, Moley KH. Decreased spermatogenesis, fertility, and altered Slc2A expression in Akt1-/- and Akt2-/- testes and sperm. Reprod Sci. 2012;19(1):31–42. doi: 10.1177/1933719111424449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Restuccia DF, Hynx D, Hemmings BA. Loss of PKBbeta/Akt2 predisposes mice to ovarian cyst formation and increases the severity of polycystic ovary formation in vivo. Dis Model Mech. 2012;5(3):403–11. doi: 10.1242/dmm.008136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Andrikopoulos S, et al. Evaluating the glucose tolerance test in mice. Am J Physiol Endocrinol Metab. 2008;295(6):E1323–32. doi: 10.1152/ajpendo.90617.2008. [DOI] [PubMed] [Google Scholar]
- 15.Unlu M, Morgan ME, Minden JS. Difference gel electrophoresis: a single gel method for detecting changes in protein extracts. Electrophoresis. 1997;18(11):2071–7. doi: 10.1002/elps.1150181133. [DOI] [PubMed] [Google Scholar]
- 16.Kang DH, et al. Highly sensitive and fast protein detection with coomassie brilliant blue in sodium dodecyl sulfate-polyacrylamide gel electrophoresis. Bulletin of the Korean Chemical Society. 2002;23(11):1511–1512. [Google Scholar]
- 17.Shevchenko A, et al. In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat Protoc. 2006;1(6):2856–60. doi: 10.1038/nprot.2006.468. [DOI] [PubMed] [Google Scholar]
- 18.Sanchez JC, et al. The mouse SWISS-2D PAGE database: a tool for proteomics study of diabetes and obesity. Proteomics. 2001;1(1):136–63. doi: 10.1002/1615-9861(200101)1:1<136::AID-PROT136>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 19.Purves RD. Optimum numerical integration methods for estimation of area-under-the-curve (AUC) and area-under-the-moment-curve (AUMC) J Pharmacokinet Biopharm. 1992;20(3):211–26. doi: 10.1007/BF01062525. [DOI] [PubMed] [Google Scholar]
- 20.Abdul-Ghani MA, et al. Muscle and liver insulin resistance indexes derived from the oral glucose tolerance test. Diabetes Care. 2007;30(1):89–94. doi: 10.2337/dc06-1519. [DOI] [PubMed] [Google Scholar]
- 21.Leavens KF, et al. Akt2 is required for hepatic lipid accumulation in models of insulin resistance. Cell Metab. 2009;10(5):405–18. doi: 10.1016/j.cmet.2009.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.He L, et al. The critical role of AKT2 in hepatic steatosis induced by PTEN loss. Am J Pathol. 2010;176(5):2302–8. doi: 10.2353/ajpath.2010.090931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sabido E, et al. Targeted proteomics reveals strain-specific changes in the mouse insulin and central metabolic pathways after a sustained high-fat diet. Mol Syst Biol. 2013;9:681. doi: 10.1038/msb.2013.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kramer A, et al. Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics. 2014;30(4):523–30. doi: 10.1093/bioinformatics/btt703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Klebanov S, et al. Adipose tissue transplantation protects ob/ob mice from obesity, normalizes insulin sensitivity and restores fertility. J Endocrinol. 2005;186(1):203–11. doi: 10.1677/joe.1.06150. [DOI] [PubMed] [Google Scholar]
- 26.Shimomura I, et al. Leptin reverses insulin resistance and diabetes mellitus in mice with congenital lipodystrophy. Nature. 1999;401(6748):73–6. doi: 10.1038/43448. [DOI] [PubMed] [Google Scholar]
- 27.Laustsen PG, et al. Lipoatrophic diabetes in Irs1(-/-)/Irs3(-/-) double knockout mice. Genes Dev. 2002;16(24):3213–22. doi: 10.1101/gad.1034802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Petrak J, et al. Deja vu in proteomics. A hit parade of repeatedly identified differentially expressed proteins. Proteomics. 2008;8(9):1744–9. doi: 10.1002/pmic.200700919. [DOI] [PubMed] [Google Scholar]
- 29.Peinado JR, et al. Mitochondria in metabolic disease: getting clues from proteomic studies. Proteomics. 2014;14(4-5):452–66. doi: 10.1002/pmic.201300376. [DOI] [PubMed] [Google Scholar]
- 30.Goren HJ, Kulkarni RN, Kahn CR. Glucose homeostasis and tissue transcript content of insulin signaling intermediates in four inbred strains of mice: C57BL/6, C57BLKS/6, DBA/2, and 129×1. Endocrinology. 2004;145(7):3307–23. doi: 10.1210/en.2003-1400. [DOI] [PubMed] [Google Scholar]
- 31.Casteels KM, et al. Sex difference in resistance to dexamethasone-induced apoptosis in NOD mice: treatment with 1,25(OH)2D3 restores defect. Diabetes. 1998;47(7):1033–7. doi: 10.2337/diabetes.47.7.1033. [DOI] [PubMed] [Google Scholar]
- 32.Dummler B, et al. Life with a single isoform of Akt: mice lacking Akt2 and Akt3 are viable but display impaired glucose homeostasis and growth deficiencies. Mol Cell Biol. 2006;26(21):8042–51. doi: 10.1128/MCB.00722-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Johnson DT, et al. Proteomic changes associated with diabetes in the BB-DP rat. Am J Physiol Endocrinol Metab. 2009;296(3):E422–32. doi: 10.1152/ajpendo.90352.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Isogai M, Kurota H, Yamaguchi M. Hepatic calcium-binding protein regucalcin concentration is decreased by streptozotocin-diabetic state and ethanol ingestion in rats. Mol Cell Biochem. 1997;168(1-2):67–72. doi: 10.1023/a:1006822606198. [DOI] [PubMed] [Google Scholar]
- 35.Edvardsson U, et al. Hepatic protein expression of lean mice and obese diabetic mice treated with peroxisome proliferator-activated receptor activators. Proteomics. 2003;3(4):468–78. doi: 10.1002/pmic.200390061. [DOI] [PubMed] [Google Scholar]
- 36.Murata T, Shinya N, Yamaguchi M. Expression of calcium-binding protein regucalcin mRNA in the cloned human hepatoma cells (HepG2): stimulation by insulin. Mol Cell Biochem. 1997;175(1-2):163–8. doi: 10.1023/a:1006844815743. [DOI] [PubMed] [Google Scholar]
- 37.Yamaguchi M, Oishi K, Isogai M. Expression of hepatic calcium-binding protein regucalcin mRNA is elevated by refeeding of fasted rats: involvement of glucose, insulin and calcium as stimulating factors. Mol Cell Biochem. 1995;142(1):35–41. doi: 10.1007/BF00928911. [DOI] [PubMed] [Google Scholar]
- 38.Sanyal G, Pessah NI, Maren TH. Kinetics and inhibition of membrane-bound carbonic anhydrase from canine renal cortex. Biochim Biophys Acta. 1981;657(1):128–37. doi: 10.1016/0005-2744(81)90136-4. [DOI] [PubMed] [Google Scholar]
- 39.Dodgson SJ, Watford M. Differential regulation of hepatic carbonic anhydrase isozymes in the streptozotocin-diabetic rat. Arch Biochem Biophys. 1990;277(2):410–4. doi: 10.1016/0003-9861(90)90597-r. [DOI] [PubMed] [Google Scholar]
- 40.Nishita T, Igarashi S, Asari M. Determination of carbonic anhydrase-III by enzyme-immunoassay in liver, muscle and serum of male rats with streptozotocin-induced diabetes mellitus. Int J Biochem Cell Biol. 1995;27(4):359–64. doi: 10.1016/1357-2725(94)00090-x. [DOI] [PubMed] [Google Scholar]
- 41.Mitterberger MC, et al. Carbonic anhydrase III regulates peroxisome proliferator-activated receptor-gamma2. Exp Cell Res. 2012;318(8):877–86. doi: 10.1016/j.yexcr.2012.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang J, et al. Selective disruption of PPARgamma 2 impairs the development of adipose tissue and insulin sensitivity. Proc Natl Acad Sci U S A. 2004;101(29):10703–8. doi: 10.1073/pnas.0403652101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gerke V, Moss SE. Annexins: from structure to function. Physiol Rev. 2002;82(2):331–71. doi: 10.1152/physrev.00030.2001. [DOI] [PubMed] [Google Scholar]
- 44.Peng B, et al. Annexin A5 as a potential marker in tumors. Clin Chim Acta. 2014;427:42–8. doi: 10.1016/j.cca.2013.09.048. [DOI] [PubMed] [Google Scholar]
- 45.Jia X, et al. A dynamic plasma membrane proteome analysis of alcohol-induced liver cirrhosis. Proteome Sci. 2012;10(1):39. doi: 10.1186/1477-5956-10-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fella K, et al. Use of two-dimensional gel electrophoresis in predictive toxicology: identification of potential early protein biomarkers in chemically induced hepatocarcinogenesis. Proteomics. 2005;5(7):1914–27. doi: 10.1002/pmic.200401067. [DOI] [PubMed] [Google Scholar]
- 47.Liao CC, Lin YL, Kuo CF. Effect of high-fat diet on hepatic proteomics of hamsters. J Agric Food Chem. 2015;63(6):1869–81. doi: 10.1021/jf506118j. [DOI] [PubMed] [Google Scholar]
- 48.Kim GH, et al. Proteomic and bioinformatic analysis of membrane proteome in type 2 diabetic mouse liver. Proteomics. 2013;13(7):1164–79. doi: 10.1002/pmic.201200210. [DOI] [PubMed] [Google Scholar]
- 49.Long J, et al. Glutamine synthetase as an early marker for hepatocellular carcinoma based on proteomic analysis of resected small hepatocellular carcinomas. Hepatobiliary Pancreat Dis Int. 2010;9(3):296–305. [PubMed] [Google Scholar]
- 50.Kraus D, et al. Nicotinamide N-methyltransferase knockdown protects against diet-induced obesity. Nature. 2014;508(7495):258–62. doi: 10.1038/nature13198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Xia Y, et al. L-carnitine ameliorated fatty liver in high-calorie diet/STZ-induced type 2 diabetic mice by improving mitochondrial function. Diabetol Metab Syndr. 2011;3:31. doi: 10.1186/1758-5996-3-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Liang Y, et al. The effects of oral L-carnitine treatment on blood lipid metabolism and the body fat content in the diabetic patient. Asia Pac J Clin Nutr. 1998;7(2):192–5. [PubMed] [Google Scholar]
- 53.Rahbar AR, et al. Effect of L-carnitine on plasma glycemic and lipidemic profile in patients with type II diabetes mellitus. Eur J Clin Nutr. 2005;59(4):592–6. doi: 10.1038/sj.ejcn.1602109. [DOI] [PubMed] [Google Scholar]
- 54.Zhang JJ, et al. L-carnitine ameliorated fasting-induced fatigue, hunger, and metabolic abnormalities in patients with metabolic syndrome: a randomized controlled study. Nutr J. 2014;13:110. doi: 10.1186/1475-2891-13-110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ross MK, Streit TM, Herring KL. Carboxylesterases: Dual roles in lipid and pesticide metabolism. J Pestic Sci. 2010;35(3):257–264. doi: 10.1584/jpestics.R10-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chen R, et al. Decreased carboxylesterases expression and hydrolytic activity in type 2 diabetic mice through Akt/mTOR/HIF-1alpha/Stra13 pathway. Xenobiotica. 2015:1–12. doi: 10.3109/00498254.2015.1020353. [DOI] [PubMed] [Google Scholar]
- 57.Xiong J, et al. Glucose dominates the regulation of carboxylesterases induced by lipopolysaccharide or interleukin-6 in primary mouse hepatocytes. Life Sci. 2014;112(1-2):41–8. doi: 10.1016/j.lfs.2014.07.019. [DOI] [PubMed] [Google Scholar]
- 58.Eccleston HB, et al. Chronic exposure to a high-fat diet induces hepatic steatosis, impairs nitric oxide bioavailability, and modifies the mitochondrial proteome in mice. Antioxid Redox Signal. 2011;15(2):447–59. doi: 10.1089/ars.2010.3395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Seiler N. Ornithine aminotransferase, a potential target for the treatment of hyperammonemias. Curr Drug Targets. 2000;1(2):119–53. doi: 10.2174/1389450003349254. [DOI] [PubMed] [Google Scholar]
- 60.Luo M, et al. High Fat Diet-Induced Changes in Hepatic Protein Abundance in Mice. Journal of Proteomics & Bioinformatics. 2012;05(03) doi: 10.4172/jpb.1000214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mueckler MM, Merrill MJ, Pitot HC. Translational and pretranslational control of ornithine aminotransferase synthesis in rat liver. J Biol Chem. 1983;258(10):6109–14. [PubMed] [Google Scholar]
- 62.D'Alessio D. The role of dysregulated glucagon secretion in type 2 diabetes. Diabetes Obes Metab. 2011;13 Suppl 1:126–32. doi: 10.1111/j.1463-1326.2011.01449.x. [DOI] [PubMed] [Google Scholar]
- 63.Hirschey MD, et al. SIRT3 deficiency and mitochondrial protein hyperacetylation accelerate the development of the metabolic syndrome. Mol Cell. 2011;44(2):177–90. doi: 10.1016/j.molcel.2011.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Green MF, Hirschey MD. SIRT3 weighs heavily in the metabolic balance: a new role for SIRT3 in metabolic syndrome. J Gerontol A Biol Sci Med Sci. 2013;68(2):105–7. doi: 10.1093/gerona/gls132. [DOI] [PubMed] [Google Scholar]
- 65.Wang S, et al. Assessment of serum arginase I as a type 2 diabetes mellitus diagnosis biomarker in patients. Exp Ther Med. 2014;8(2):585–590. doi: 10.3892/etm.2014.1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Schurgers LJ, et al. Circulating annexin A5 predicts mortality in patients with heart failure. J Intern Med. 2015 doi: 10.1111/joim.12396. [DOI] [PubMed] [Google Scholar]
- 67.Yamaguchi M, Isogai M, Shimada N. Potential sensitivity of hepatic specific protein regucalcin as a marker of chronic liver injury. Mol Cell Biochem. 1997;167(1-2):187–90. doi: 10.1023/a:1006859121897. [DOI] [PubMed] [Google Scholar]
- 68.Curtis JR, et al. Validation of a novel multibiomarker test to assess rheumatoid arthritis disease activity. Arthritis Care Res (Hoboken) 2012;64(12):1794–803. doi: 10.1002/acr.21767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ware Miller R, et al. Performance of the American College of Obstetricians and Gynecologists' ovarian tumor referral guidelines with a multivariate index assay. Obstet Gynecol. 2011;117(6):1298–306. doi: 10.1097/AOG.0b013e31821b1d80. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.