

Abstract

Bottom-up proteomics database search algorithms used for peptide identification cannot comprehensively identify post-translational modifications (PTMs) in a single-pass because of high false discovery rates (FDRs). A new approach to database searching enables global PTM (G-PTM) identification by exclusively looking for curated PTMs, thereby avoiding the FDR penalty experienced during conventional variable modification searches. We identified over 2200 unique, high-confidence modified peptides comprising 26 different PTM types in a single-pass database search.
Keywords: Morpheus, PTM, post-translational modification, phosphorylation, acetylation, G-PTM, Jurkat, database search, proteomics
Introduction
Protein post-translational modifications (PTMs)1 play essential roles in protein signaling,2 localization,3,4 function,5,6 degradation,7 and other important biological processes. Despite the considerable success of protein identification via liquid chromatography–mass spectrometry (LC–MS), most studies do not provide information regarding PTMs on the identified proteins. This is because identifying even a single type of PTM requires specialized procedures and introduces problems with the increased database size required for the search.8 Extension of such approaches to analysis of multiple PTMs is generally unrealistic, and consequently, there are few examples where multiple types of PTMs are analyzed in a single experiment.1,9 Despite widespread recognition of the importance of PTMs,10 nearly all database search algorithms still rely solely on primary sequence information and ignore all prior knowledge regarding the presence of PTMs. Two notable exceptions are the ProSight software for top-down proteomics, which performs shotgun annotation11 of multiple sources of variation contained within the UniProt repository,12 and X!Tandem, which can make use of annotated PTMs from Swiss-Prot.13 X!Tandem and ProSight use probabilistic algorithms for scoring of correct spectrum matches. Morpheus was designed from the ground up to take advantage of the specificity afforded by currently available high mass accuracy instruments and distinguishes correct and incorrect identifications by counting the number of matching products in conjunction with a target–decoy approach.
The standard strategy for identifying PTMs from peptide MS data is a “variable modification search,” where a particular PTM (e.g., phosphorylation) is allowed to occur on any instance of selected amino acid residues (e.g., serine, threonine, or tyrosine) in all of the theoretical peptides from the entire search database. This process is a useful and effective means to identify unknown modifications in a sample; however, the database search space is expanded enormously even for just a few PTM options to say nothing of including the hundreds of varieties of known PTMs. A two-pass database search strategy14 reduces the search-space expansion but may introduce bias when employed with target–decoy false discovery rate (FDR) calculations.15 All such searches using variable modification lead to longer search times and an increase in the number of false positive identifications, both of which grow with the size of the search space. These issues compromise the utility of the search and cause many researchers to ignore PTMs altogether in their proteomics data. Blind modification searches can reveal unknown modifications but face challenges from increased run time and decreased identification of unmodified peptides, though recent advances in this area are showing promise.16,17 Spectral library searching for identification of post-translationally modified peptides has historically been limited by the availability of tandem mass spectra from modified peptides, especially those outside of the most common PTMs (e.g., phosphorylation). However, new approaches in this area are beginning to address this challenge.18 A further problem with library searching is that library spectra corresponding to all possible modification sites may not be present in the library potentially leading to site misassignment.
Here we report a new strategy for comprehensive identification of protein PTMs in a single-pass database search. This global post-translational modification (G-PTM) search strategy alters the variable modification approach to consider only previously curated PTMs at specific amino acid residue positions. The “variable” aspect of these searches allows for the presence or absence of the PTM at that specific residue. This residue position specificity generates only a modest increase in database search space, unlike variable searches that allow modifications to occur at any selected amino acid residue on all protein sequences in the database.
We have implemented the G-PTM strategy in the open-source search program Morpheus,19 which was specifically designed to accommodate high-resolution mass spectra. Morpheus, much like its predecessors, identifies peptides by comparing tandem mass spectra to theoretical spectra derived in silico from primary sequence data by making use of a simple score similar to the H-score suggested by Savitski et al.20 The advantage conferred by Morpheus through the improvements described herein is the ability to identify a multitude of different PTMs in a single-pass search while maintaining a high level of confidence in the identifications.
We compared three types of searches using the software program Morpheus to characterize the effectiveness of the G-PTM strategy (Figure 1). Two comprehensive sets of proteomics data, one from human Jurkat cell lysate and one from mouse pancreatic islet lysate, were used to demonstrate the approach. The UniProt repository was the source of the protein sequence and PTM data for all database searches. The first two types of searches employed the UniProt FASTA file either without allowing for any PTMs, referred to simply as “FASTA”, or by allowing variable phosphorylation of every serine, threonine, and tyrosine, referred to as “FASTA vP.” The G-PTM search utilized the UniProt XML (extensible markup language) file, which includes the same amino acid sequences as the FASTA file but also contains the curated list of position-specific PTMs for those protein sequences.
Figure 1.
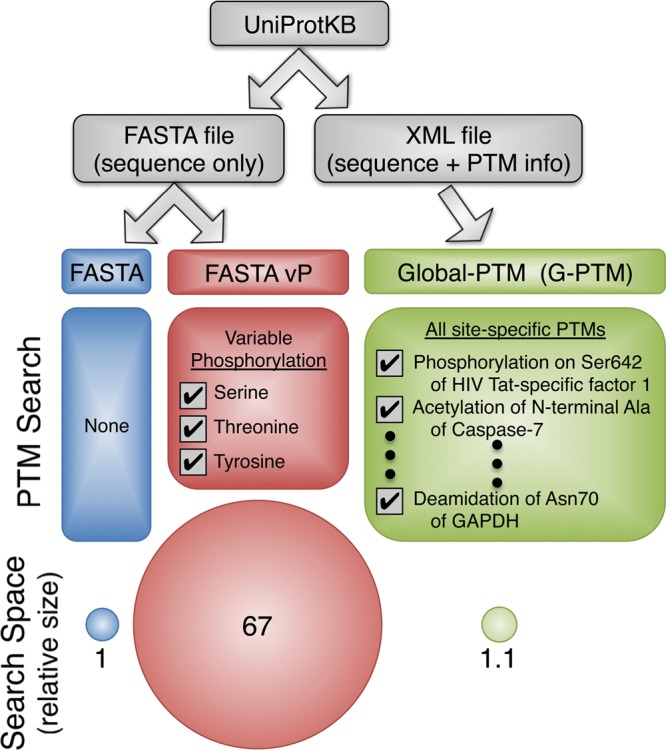
Comparison of three proteomics database search strategies: no PTMs considered (FASTA), conventional variable modification for phosphorylation (FASTA vP), and consideration of a long list of residue- and protein-specific PTMs of numerous different types (G-PTM). Search space size increases by 67-fold for FASTA vP, but only by 10% for G-PTM.
Experimental Procedures
Human Data
Sample preparation and MS analysis of the Jurkat cells were previously reported21 and similar to that described further for mouse. The MS raw files for the Jurkat cell lysate samples are available via FTP from the PeptideAtlas data repository22 by accessing the following link: http://www.peptideatlas.org/PASS/PASS00215.
Mouse Sample Preparation and MS Analysis
Male C57BL/6J (B6) and CAST/EiJ (CAST) mice were purchased from The Jackson Laboratories (Bar Harbor, Maine) and housed in an environmentally controlled vivarium at the University of Wisconsin Biochemistry Department. The mice were provided standard rodent chow (Purina no. 5008) and water ad libitum and were maintained on a 12-h light/dark cycle (6 AM–6 PM). At 10 weeks of age, the mice were sacrificed by CO2 asphyxiation. All animal procedures were preapproved by the University of Wisconsin Animal Care and Use Committee.
Intact pancreatic islets were isolated using a collagenase digestion procedure as previously described.23 A 5 mL volume of collagenase type XI (Sigma), dissolved in Hanks’ balanced salt solution (Gibco), was injected into the pancreas via the common bile duct (0.45 mg/mL with 0.02% BSA). The pancreas was removed and incubated at 37 °C for 16 min with intermittent agitation. A Ficoll gradient was used to partially purify islets from the digested pancreas, followed by further purification by manual picking under a stereomicroscope. During the purification procedure, islets were maintained in Krebs-Ringer bicarbonate buffer containing (mM): 118.41 NaCl, 4.69 KCl, 2.52 CaCl2, 1.18 MgSO4, 1.18 KH2PO4, 25 NaHCO3, and 5 HEPES supplemented with 0.2% BSA and 16.7 mM glucose. Islets from two B6 mice (and separately two CAST mice) were pooled and then apportioned for protein analyses (400 B6 islets, 470 CAST islets). Islets were washed three times in ice-cold PBS, gently pelleted, and snap frozen in liquid nitrogen.
Protein was extracted and then digested into peptides using an adaptation of the filter aided sample preparation (FASP) procedure.21,24 Islets were thawed on ice, and the protein extracted with 90 μL of SDT lysis buffer, consisting of 4% sodium dodecyl sulfate (SDS, Bio-Rad), 0.1 M Tris-HCl (pH 7.6, Teknova), and 0.1 M dithiothreitol (DTT, Sigma). The mixtures were incubated at 95 °C for 6 min with intermittent vortexing. The resulting lysates were cooled in an ice bath, followed by bath sonication (Fisher Scientific FS20) for three cycles of 20 s with ice bath cooling during 20 s rest periods. Debris was pelleted by centrifugation at 16 000g for 5 min, and 85 μL of supernatant was collected.
Removal of detergents and salts was accomplished by FASP using multiple washes in a 30K MWCO filter (Vivacon 500 from Sartorius). The 85 μL protein extract was diluted with 570 μL of 8 M urea (Sigma), 0.1 M Tris-HCl pH 8.0. Half of this solution was centrifuged at 14 000g through the filter (25 min), followed by the other half (25 min), and then 0.2 mL of urea/Tris wash (20 min). The proteins were alkylated in the filter with 0.1 mL of 0.05 M iodoacetamide (Sigma) in urea/Tris for 20 min at room temperature in the dark, followed by centrifugation (15 min). The proteins were washed three times with 0.1 mL of urea/Tris and three times with 0.1 mL of 0.05 M ammonium bicarbonate (ABC, Fluka) with 15 min centrifugations for each wash. Tryptic digestion was performed at 37 °C overnight on the proteins in the filter by addition of 2 μg of trypsin (Promega) in 75 μL of 0.05 M ABC. Peptides were collected by centrifugation (10 min) into new collection tubes, followed by washing the remaining peptides out of the filter with 40 μL of ABC, 10 min centrifugation, 50 μL of 0.5 M NaCl, and a final 10 min centrifugation.
The tryptic peptide digests were fractionated using high-pH reverse-phase chromatography on a Shimadzu HPLC system (LC-10AD, SCL-10A VP, SPD-10A VP, Shimadzu, Columbia, MD) and a Phenomenex C18 Gemini 3 μ, 110 Å, 3.0 × 150 mm2 column (Phenomenex, Torrance, CA). The high-pH method was adapted from Gilar et al.25 Mobile phase A (MPA) was 20 mM ammonium formate, pH 10, and B (MPB) was 20 mM ammonium formate, pH 10, in 70% acetonitrile. The HPLC flow was 1.0 mL/min, and the gradient was 0% MPB isocratic for 10 min (trapping step), linear ramp to 100% MPB over 20 min, hold at 100% MPB for 5 min, to 0% MPB over 1 min, and equilibration at 0% MPB for 10 min. Fractions were collected every minute using a Gilson 203 fraction collector (Gilson, Middleton, WI) for a total of 15 fractions collected during the range of peptide elution as discernible from the UV–vis trace. Because of the relatively low amounts near the beginning and the end of the gradient, the first two fractions were combined, as were the last six fractions, leading to a total of nine fractions. By comparison of the UV–vis trace to that of a standard, it was estimated that the B6 and CAST samples contained 34 and 33 μg of peptides, respectively. Fractions were dried down using vacuum centrifugal concentration (Savant Speed- Vac, Thermo, Pittsburgh, PA) and stored at −80 °C.
Each fraction was reconstituted in 10 μL of 5% acetonitrile and 1% formic acid in water, and then between 4 and 9.5 μL of each fraction was analyzed by LC–MS (using the earlier UV–vis trace to estimate peptide content of each fraction and avoid injecting more than 2.5 μg of peptides per run). The HPLC–ESI–MS/MS system consisted of a Waters nanoAcquity HPLC (Milford, MA) connected to an electrospray ionization (ESI) ion-trap/orbitrap mass spectrometer (LTQ Orbitrap Velos, Thermo Scientific, San Jose, CA). The LC column was prepared by packing 20 cm of 3 μm MAGIC aqC18 beads (Bruker-Michrom, Auburn, CA) into a 100 μm i.d. capillary whose tip was pulled to ∼1 μm with a P-2000 laser puller (Sutter Instruments, Novato, CA). The full HPLC method was 195 min long at a flow rate of 0.3 μL/min, and it included a 124 min gradient from 2% to 30%, with a brief ramp to 70%, MPB (0.1% formic acid in acetonitrile) with the remainder being MPA (0.1% formic acid in water). A full-mass scan (300–1500 m/z) was performed in the orbitrap at a resolution of 60 000. The ten most intense peaks with z > 1 from the full scan were selected for fragmentation by higher-energy collisional dissociation (HCD, collision energy = 42). The isolation width for the precursor ions was 3.0 m/z, and the product ions from fragmentation were analyzed in the orbitrap detector at a resolution of 7500. Dynamic exclusion was enabled with a repeat count of two over 30 s and an exclusion duration of 120 s. Xcaliber software version 2.1.0 was used for data collection. MS raw files for the mouse samples are available via FTP from the PeptideAtlas data repository22 by accessing the following link: http://www.peptideatlas.org/PASS/PASS00470.
Protein Search Databases
Protein FASTA and XML files were obtained from the UniProt repository.26 We chose to use a subset of the available human protein sequences for the Jurkat sample analyses, including only those with a matching mRNA transcript above 0.1 transcripts per million (TPM).21 We used the complete set of available mouse protein sequences for the B6 and CAST islet proteomics. Both sets of protein accession numbers (14 138 entries for Jurkat and 43 238 entries for mouse) included in the databases are supplied in Supplemental Table S1, and the summarized list of target PTM types from the XML file is in Supplemental Table S2. The database used for the Jurkat samples was the Homo sapiens (Human) reference proteome from UniProt release 2013_12 (downloaded February 25, 2015), limited to those proteins with mRNA transcript abundances exceeding 0.1 TPM.21 The database used for the mouse samples was the Mus musculus (Mouse) reference proteome from UniProt release 2015_02 (downloaded February 25, 2015).
Database Searching
The software program Morpheus (revision 142) was used for all database searching. It can be obtained at http://morpheus-ms.sourceforge.net/. For this work, it was modified to accept UniProt XML in addition to FASTA protein databases. When a UniProt XML database is specified, all curated modifications are extracted. Details of each modification (name, mass shift, target) are read from a local copy of http://www.uniprot.org/docs/ptmlist. All valid modifications are added to the variable modifications box in the Morpheus graphical user interface with the prefix “UniProt” and selected by default. During each search, all protein sequences are read along with the locations of selected UniProt variable modifications. The order of the unmodified and modified amino acid residues is reversed during on-the-fly generation of decoy protein sequences. PTMs move with their companion amino acid. For example, the phosphorylated tetrapeptide, TES(UniProt: Phosphoserine)Q, becomes QS(UniProt: Phosphoserine)ET.
A critical routine in the code takes a base peptide sequence and generates all of the isoform combinations possible given the variable modifications selected, up to a user-defined limit (1024 by default). This code was modified slightly to consider UniProt modifications only at their location as reported in the database. Otherwise, the logic for combinatorially generating all possible peptide isoforms is identical.
Searches were performed on a Dell Precision T7610 workstation with a Xeon 2.70 GHz processor and 32.0 GB of RAM using 12 cores. The following settings were used in all searches: Protease = trypsin (no proline rule); Maximum Missed Cleavages = 2; Initiator Methionine Behavior = variable; Fixed Modifications = carbamidomethylation of C; Variable Modifications = oxidation of M; Maximum Variable Modification Isoforms Per Peptide = 1024; Precursor Mass Tolerance = ±10.0 ppm (monoisotopic); Precursor Monoisotopic Peak Correction = disabled; Product Mass Tolerance = ±0.01 Da (monoisotopic);27 Maximum False Discovery Rate = 1%. FASTA vP searches used additionally variable phosphorylation of S, T, and Y. G-PTM searches used XML database files rather than FASTA files. Counts of post-translationally modified peptides do not include the oxidation of methionine or the carbamidomethylation of cysteine as these occur during sample handling and therefore are somewhat uninteresting. Search results are available via FTP using the aforementioned PeptideAtlas data repository hyperlinks. Summary lists of identified proteins including numbers of PSMs (total and modified) are provided in Supplemental Table S3 for both Jurkat and mouse, where B6 and CAST mouse data are segregated to allow for their comparison. Total search times are as follows: Jurkat FASTA, 46 min.; Jurkat G-PTM, 30 min.; Jurkat variable phosphorylation, 367 min.; B6 and CAST mouse FASTA, 63 min.; B6 and CAST mouse G-PTM, 62 min.; B6 and CAST mouse variable phosphorylation, 471 min.
Results and Discussion
The G-PTM search strategy limits the expansion of the target and decoy databases, as shown by the sizes of the circles in Figure 1 and by the data in Supplemental Table S4. Despite including 22 540 site-specific human PTMs from 104 different PTM types, the search space for G-PTM increased by only 10% compared to the FASTA search, which did not consider any PTMs. In contrast, the FASTA vP search, with only three variable modifications (phosphorylation of serine, threonine, and tyrosine) increased the search space by 67-fold (see Figure 1). The number of variable modifications per peptide isoforms was limited to 1024 for this comparison, which is the value often allowed in database searches of this type. This massive, 67-fold expansion of the database for variable phosphorylation substantially increases both the search time (6- to 10-fold) and the error rate for phosphopeptide identification.
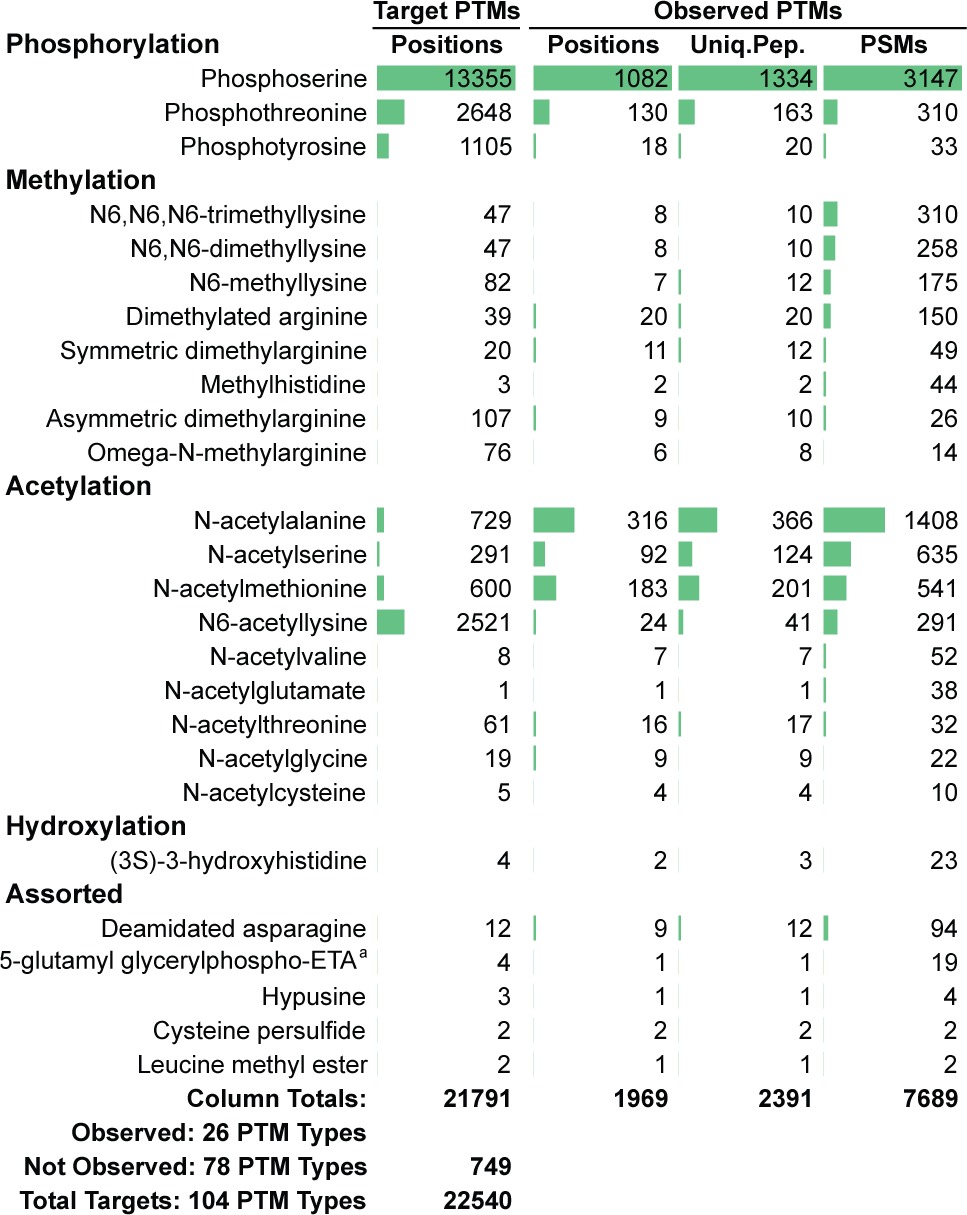
We applied the G-PTM search strategy to a large proteomic data set obtained from human Jurkat cells. About 490 000 tandem mass spectra were obtained from a highly fractionated sample of Jurkat cell lysate, and counts of the modified peptides resulting from the G-PTM search are listed in Table 1. Within this single-pass database search, over 2200 unique post-translationally modified peptides were identified, encompassing 26 different types of PTMs from five categories (phosphorylation, methylation, acetylation, hydroxylation, and assorted). These modified peptides would have gone undetected using a typical proteomics database search, but the G-PTM search readily revealed this rich array of PTMs present in the Jurkat cells.
Table 1. Numbers of Target and Identified PTMs Using the G-PTM Search Strategy. These Data Are for Human Jurkat Cells; The Analogous Data for Mouse Are in Supplemental Table S5. The “Database PTM Positions” Column Shows That a Total of 22 540 Residue Positions from 104 PTM Types Were Included in the Human UniProt XML File. The G-PTM Search Identified Peptides with Modifications at 1969 of Those Positions, across the 26 Observed PTM Types Listed Here (See Supplementary Table S2 for Entire List). aETA Is an Abbreviation for “Ethanolamine”.
We also applied the G-PTM search strategy to proteomics data sets from mouse pancreatic islets. About 430 000 tandem mass spectra were obtained from a highly fractionated samples of CAST and B6 mouse islet lysate, and the counts of modified peptides resulting from the G-PTM search are listed in Supplemental Table S5. Within this single-pass database search, ∼1100 unique post-translationally modified peptides were identified, encompassing 32 different types of PTMs from five categories (phosphorylation, methylation, acetylation, hydroxylation, and assorted). These modified peptides would also have gone undetected using a typical proteomics database search, but the G-PTM search readily revealed this rich array of PTMs present in the mouse islets.
These same tandem mass spectra were searched using Morpheus with the two other search strategies, FASTA and FASTA vP. The results for the three types of searches, for both human and mouse data, are shown in Figure 2. FDR is calculated with the target–decoy approach. The tabular data within the figure lists the total number of identified proteins, unique peptides, and peptide spectral matches (PSMs). The “All” columns (orange) include both unmodified and post-translationally modified peptides. These total identifications show similar results across the three search strategies, although the G-PTM search consistently produced more identifications than either the FASTA or FASTA vP searches. As expected for samples such as these without a PTM enrichment step, the majority of identified peptides are unmodified (100%, 97%, and 97% for the FASTA, FASTA vP, and G-PTM search results, respectively; see Supplemental Table S6, which categorizes the identifications by the number of PTMs per peptide). Nonetheless, there are hundreds of modified peptides identified in these samples; see Figure 2 (purple data columns) and Supplemental Tables S7 and S8. The G-PTM search produces substantially more modified peptide spectral matches than the FASTA vP search, and furthermore, these PTM identifications are of much higher confidence (vide infra).
Figure 2.

Database search results and measures of their confidences for PTM peptide assignments. Results are shown for the three search types for both human and mouse proteomics data sets. The tabulated results are given for “All” (unmodified and modified peptides) and for “Modified” only peptides. The FDR for the “Modified” peptides was calculated from the numbers of decoy and target identifications meeting the global 1% FDR cutoff (i.e., the FDR for “All” peptide identifications). The FDR values for FASTA vP are >1%, indicating substantially poorer confidence, whereas the FDRs are <1% for the G-PTM searches, indicating high confidence in these PTM peptide assignments. The PEP plots corroborate this result by showing higher error probabilities for phosphorylated peptides in the FASTA vP search but lower error probabilities for modified peptides identified with G-PTM.
The results for all peptide identifications were uploaded to Protein Prospector’s MS-Viewer tool (http://prospector2.ucsf.edu/prospector/cgi-bin/msform.cgi?form=msviewer), and Supplemental Table S9 provides hyperlinks to the MS-Viewer report for each sample. For ease of examining the PTM identifications, hyperlinks to the MS-Viewer report containing each modified peptide are provided in Supplemental Tables S7 and S8 along with detailed instructions on how to access a specific spectrum or to filter the data to look at all peptides containing a particular PTM. Note that Morpheus, in its current implementation, does not export peak lists, but this capability may be added in future versions.
FDR is a common measure of the confidence of peptide identifications. For any given score cutoff, the FDR is the ratio of decoy peptide assignments to target peptide assignments. It is common to set the score threshold at a 1% FDR level where 1% of the assignments are decoys. As an example, the G-PTM search of the human data (upper right panel of Figure 2) shows 2104 decoy and 210 307 target PSMs resulting in an FDR of 1%. This is considered a “global” FDR because it encompasses all PSMs from that database search. One can take a subset of the PSMs meeting the 1% global FDR cutoff and calculate an FDR for just this subset of PSMs. Since the PTM-containing peptides are of particular interest, we used the 17 decoy and 7278 target PSMs assigned to modified peptides to calculate an FDR of 0.23% for modified peptides. Thus, the G-PTM search actually identified peptides with PTMs more confidently than peptides without PTMs, as indicated by 0.23% being less than the 1% global FDR. In contrast, the FASTA vP search yielded much lower confidence for modified peptides (11.0% FDR) than for all peptides (1% global FDR). It is likely that the FDR of 0.23% for modified peptides underestimates the actual error rate. Certain labile PTMs such as phosphorylation or GlcNAcylation present the possibility that the localization of PTMs on high scoring peptides may be incorrect. We performed a second search of the Jurkat data that allowed variable phosphorylation on all S, T, and Y amino acid residues of phosphopeptides identified in the original G-PTM search. We found 127 instances (out of 3425 possible) on 88 different sequences where the Morpheus score improved with the phosphorylation at an alternative position (see Supplemental Table S10). The FDR for the positioning of the PTM on phosphopeptides considered in this manner is thus 127/3425 = 3.7%. This source of error is reduced as soon as these positions of phosphorylation get added to the search database.
The posterior error probability (PEP) is another measure of peptide identification confidence.28 PEP represents the probability that an individual peptide identification is false. PEP is often referred to as a local FDR because it is calculated from spectral matches to target and decoy peptides having the same or nearly the same identification score. Plots of PEP values as a function of score are shown in Figure 2 for the list of peptides meeting the 1% global FDR cutoff. High-scoring peptides have PEP values of zero because there were no decoy peptides with these high scores. Low-scoring peptides have PEP values that rise above zero. For example, unmodified peptides in the G-PTM search of human data (orange dots in upper right panel of Figure 2) have a PEP value of 0.04 for the relatively low score of 9, indicating that any given PSM with a score of 9 has a 4% chance of being incorrect. Modified peptides (purple dots) from that same G-PTM search were identified with the same or lower PEP scores (2% chance of being incorrect at a score of 9), meaning their confidence is at least as high as for the unmodified peptides. In contrast, modified peptides from the variable phosphorylation search (middle panels in Figure 2) have dramatically higher PEP scores (60% chance of being incorrect at a score of 9) than their unmodified counterparts, indicating lower confidence. The observation that PEP values for modified peptides were consistently lower at all scores for both mouse and human samples in the G-PTM approach (right panels in Figure 2) suggests that correct identifications of modified peptides are statistically more likely than for unmodified peptides. This phenomenon was previously described for phosphopeptides by Marx et al.29
Both FDR and PEP calculations reveal that the G-PTM search strategy identifies modified peptides with high confidence, whereas the variable modification approach identifies modified peptides with much lower confidence. This is due to the dramatic differences in search space. The 67-fold increase in search space for variable phosphorylation (Supplemental Table S4) means that 98.5% of peptide targets in the database are modified, which lies in stark contrast to the actual low level of phosphorylation in typical unenriched mammalian samples. Consequently, the set of target peptides in the database used for spectral matching is a poor reflection of reality, and the probability of producing false spectral identifications of phosphorylated peptides is quite high. The G-PTM strategy, however, employs site-specific addition of PTMs to the peptide target database, thereby avoiding a large increase in search space and resulting in a similar or even lower level of false identifications for modified as for unmodified peptides.
G-PTM searches deliver confident PTM identifications from proteomics data, but they currently have a few limitations. The identified PTM location within a peptide is limited to the one(s) assigned in the XML file. Thus, it is possible to get a relatively confident assignment of a modified peptide where the residue position of the PTM is erroneously assigned because its true position was not a considered option. It is also possible that a mass shift assigned to a rare PTM in the XML file also corresponds to a more common PTM not listed within the file. For example, citrulline is an uncommon PTM that has the same mass shift as asparagine deamidation, which commonly occurs during sample handling and thus is not usually listed in the XML database.
We believe that there is substantial value in the ability to identify a multitude of PTMs in a single-pass search, despite the possibility that a few PTM locations or identities will be incorrectly assigned within the correct peptide sequences. Prior to assigning biological importance to a particular post-translationally modified peptide, researchers should evaluate whether alternative possibilities are more likely. Of course, the best solution to this issue is a more complete and accurate compilation of PTM sites. Several PTM repositories exist, but the current incarnation of G-PTM within Morpheus requires an XML version of the FASTA database, formatted according to UniProt guidelines. In an excellent recent review of large-scale analysis of PTMs, Olsen and Mann described the current status of the major PTM repositories and called upon the community “to establish more refined ways of integrating and presenting the PTM data that it generates”.10 We believe G-PTM searching is a significant step in this direction.
Since the curated list of PTMs used in G-PTM searches remains incomplete at present, there is still a need for PTM discovery experiments employing sample enrichment or variable modification searches. Over time, the curated list of PTMs will continue to expand and improve, especially with the advent of tools such as this G-PTM search that enable easy and effective use of that data. Researchers are encouraged to provide data supporting identification of novel PTMs to UniProt (http://www.uniprot.org/update) to the benefit of the entire proteomics community.
The G-PTM search strategy enables researchers to confidently identify numerous types of PTMs in bottom-up proteomics data sets. We have made G-PTM available in the open-source Morpheus software; in future work, we hope to see the strategy implemented in other search packages as well to increase accessibility and ease-of-use to the broader research community. The G-PTM approach helps to reveal the myriad ways by which PTMs define and control biological systems. Implementing this approach in standard proteomics search algorithms will greatly improve comprehensive identification of peptide PTMs.
Acknowledgments
We thank Donnie Stapleton for supplying the mouse islet samples, Bosco Ho for providing the software Peptagram for spectrum annotation, and Peter Baker for uploading the data to MS-Viewer. This work was supported by National Institutes of Health (NIH) Grant Nos. P50HG004952, U54DK093467, P01GM081629, and R01GM103315. G.M.S. was supported by the NIH Genomic Sciences Training Program T32HG002760. M.P.K. and A.D.A. were supported by National Institute of Diabetes and Digestive Kidney Diseases Grant Nos. R01DK058037, R24DK091207, and R01DK066369.
Glossary
Abbreviations
- PTM
post-translational modification
- G-PTM
global post-translational modification
- FDR
false discovery rate
- LC–MS
liquid chromatography–mass spectrometry
- XML
extensible markup language
- PSM
peptide spectral match
- PEP
posterior error probability
- RNA-seq
RNA-sequencing
- FASP
filter aided sample preparation
- DTT
dithiothreitol
- SDS
sodium dodecyl sulfate
- HCD
higher-energy collisional dissociation
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jproteome.5b00599.
Human and mouse accession numbers (XLSX)
Target PTMs included in the searches (XLSX)
Lists of identified proteins and numbers of total and modified PSMs for human and mouse (XLSX)
Search space sizes (XLSX)
Numbers of target and identified PTMs using the G-PTM search strategy (XLSX)
Number of PTMs per peptide (XLSX)
List of modified peptides identified in Morpheus G-PTM searches of Jurkat data (XLSX)
List of modified peptides identified in Morpheus G-PTM searches of mouse data (XLSX)
MS-viewer links for each fraction (XLSX)
Phosphoptides with increased score at alternative phosphosites (XLSX)
Author Contributions
M.R.S. and C.D.W. contributed equally to this work. M.R.S. conceived of the concept of using curated PTMs in database search, performed all database search analyses, and processed the results for publication. C.D.W. implemented the concept in the Morpheus search software. M.P.K. and A.D.A. helped in the design of the mouse studies and designed protocols for islet isolation and lysate preparation. B.L.F. performed experimental sample preparation and MS analyses of the mouse samples. G.M.S. performed experimental sample preparation, RNA-seq, and MS analyses of the human Jurkat cell samples. L.M.S. directed the entire project. M.R.S., C.D.W., B.L.F., and M.S. wrote the manuscript with support from all authors.
The authors declare no competing financial interest.
Author Status
§ No affiliation.
Supplementary Material
References
- Doerr A. Making PTMs a priority. Nat. Methods 2012, 9, 862–3 10.1038/nmeth.2153. [DOI] [PubMed] [Google Scholar]
- Deribe Y. L.; Pawson T.; Dikic I. Post-translational modifications in signal integration. Nat. Struct. Mol. Biol. 2010, 17, 666–72 10.1038/nsmb.1842. [DOI] [PubMed] [Google Scholar]
- Sirover M. A. Subcellular dynamics of multifunctional protein regulation: mechanisms of GAPDH intracellular translocation. J. Cell. Biochem. 2012, 113, 2193–200 10.1002/jcb.24113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Steen T.; Tindall D. J.; Huang H. Posttranslational modification of the androgen receptor in prostate cancer. Int. J. Mol. Sci. 2013, 14, 14833–59 10.3390/ijms140714833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gould N.; Doulias P. T.; Tenopoulou M.; Raju K.; Ischiropoulos H. Regulation of protein function and signaling by reversible cysteine S-nitrosylation. J. Biol. Chem. 2013, 288, 26473–9 10.1074/jbc.R113.460261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cousin C.; Derouiche A.; Shi L.; Pagot Y.; Poncet S.; Mijakovic I. Protein-serine/threonine/tyrosine kinases in bacterial signaling and regulation. FEMS Microbiol. Lett. 2013, 346, 11–9 10.1111/1574-6968.12189. [DOI] [PubMed] [Google Scholar]
- Ahner A.; Gong X.; Frizzell R. A. Cystic fibrosis transmembrane conductance regulator degradation: cross-talk between the ubiquitylation and SUMOylation pathways. FEBS J. 2013, 280, 4430–8 10.1111/febs.12415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y.; Jensen O. N. Modification-specific proteomics: strategies for characterization of post-translational modifications using enrichment techniques. Proteomics 2009, 9, 4632–41 10.1002/pmic.200900398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo J.; Jeong J.; Kim Y. M.; Hwang N.; Paek E.; Lee K. J. Strategy for comprehensive identification of post-translational modifications in cellular proteins, including low abundant modifications: application to glyceraldehyde-3-phosphate dehydrogenase. J. Proteome Res. 2008, 7, 587–602 10.1021/pr700657y. [DOI] [PubMed] [Google Scholar]
- Olsen J. V.; Mann M. Status of large-scale analysis of post-translational modifications by mass spectrometry. Mol. Cell. Proteomics 2013, 12, 3444–52 10.1074/mcp.O113.034181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pesavento J. J.; Kim Y. B.; Taylor G. K.; Kelleher N. L. Shotgun annotation of histone modifications: a new approach for streamlined characterization of proteins by top down mass spectrometry. J. Am. Chem. Soc. 2004, 126, 3386–7 10.1021/ja039748i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth M. J.; Forbes A. J.; Boyne M. T. 2nd; Kim Y. B.; Robinson D. E.; Kelleher N. L. Precise and parallel characterization of coding polymorphisms, alternative splicing, and modifications in human proteins by mass spectrometry. Mol. Cell. Proteomics 2005, 4, 1002–8 10.1074/mcp.M500064-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenyo D.; Beavis R. C. A method for assessing the statistical significance of mass spectrometry-based protein identifications using general scoring schemes. Anal. Chem. 2003, 75, 768–774 10.1021/ac0258709. [DOI] [PubMed] [Google Scholar]
- Craig R.; Beavis R. C. A method for reducing the time required to match protein sequences with tandem mass spectra. Rapid Commun. Mass Spectrom. 2003, 17, 2310–6 10.1002/rcm.1198. [DOI] [PubMed] [Google Scholar]
- Gupta N.; Bandeira N.; Keich U.; Pevzner P. A. Target-decoy approach and false discovery rate: when things may go wrong. J. Am. Soc. Mass Spectrom. 2011, 22, 1111–20 10.1007/s13361-011-0139-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chick J. M.; Kolippakkam D.; Nusinow D. P.; Zhai B.; Rad R.; Huttlin E. L.; Gygi S. P. A mass-tolerant database search identifies a large proportion of unassigned spectra in shotgun proteomics as modified peptides. Nat. Biotechnol. 2015, 33, 743–9 10.1038/nbt.3267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Na S.; Bandeira N.; Paek E. Fast Multi-blind Modification Search through Tandem Mass Spectrometry. Mol. Cell. Proteomics 2012, 11, M111.010199. 10.1074/mcp.M111.010199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma C. W. M.; Lam H. Hunting for unexpected post-translational modifications by spectral library searching with tier-wise scoring. J. Proteome Res. 2014, 13, 2262–2271 10.1021/pr401006g. [DOI] [PubMed] [Google Scholar]
- Wenger C. D.; Coon J. J. A proteomics search algorithm specifically designed for high-resolution tandem mass spectra. J. Proteome Res. 2013, 12, 1377–86 10.1021/pr301024c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savitski M. M.; Mathieson T.; Becher I.; Bantscheff M. H-score, a mass accuracy driven rescoring approach for improved peptide identification in modification rich samples. J. Proteome Res. 2010, 9, 5511–6 10.1021/pr1006813. [DOI] [PubMed] [Google Scholar]
- Sheynkman G. M.; Shortreed M. R.; Frey B. L.; Smith L. M. Discovery and mass spectrometric analysis of novel splice-junction peptides using RNA-Seq. Mol. Cell. Proteomics 2013, 12, 2341–53 10.1074/mcp.O113.028142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desiere F.; Deutsch E. W.; King N. L.; Nesvizhskii A. I.; Mallick P.; Eng J.; Chen S.; Eddes J.; Loevenich S. N.; Aebersold R. The PeptideAtlas project. Nucleic Acids Res. 2006, 34, D655–8 10.1093/nar/gkj040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabaglia M. E.; Gray-Keller M. P.; Frey B. L.; Shortreed M. R.; Smith L. M.; Attie A. D. Alpha-Ketoisocaproate-induced hypersecretion of insulin by islets from diabetes-susceptible mice. Am. J. Physiol. Endocrinol. Metab. 2005, 289, E218–24 10.1152/ajpendo.00573.2004. [DOI] [PubMed] [Google Scholar]
- Wisniewski J. R.; Zougman A.; Nagaraj N.; Mann M. Universal sample preparation method for proteome analysis. Nat. Methods 2009, 6, 359–62 10.1038/nmeth.1322. [DOI] [PubMed] [Google Scholar]
- Gilar M.; Olivova P.; Daly A. E.; Gebler J. C. Orthogonality of separation in two-dimensional liquid chromatography. Anal. Chem. 2005, 77, 6426–34 10.1021/ac050923i. [DOI] [PubMed] [Google Scholar]
- Consortium U. Activities at the Universal Protein Resource (UniProt). Nucleic Acids Res. 2014, 42, D191–8 10.1093/nar/gkt1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zubarev R.; Mann M. On the proper use of mass accuracy in proteomics. Mol. Cell. Proteomics 2006, 6, 377–381 10.1074/mcp.M600380-MCP200. [DOI] [PubMed] [Google Scholar]
- Kall L.; Storey J. D.; MacCoss M. J.; Noble W. S. Posterior error probabilities and false discovery rates: two sides of the same coin. J. Proteome Res. 2008, 7, 40–4 10.1021/pr700739d. [DOI] [PubMed] [Google Scholar]
- Marx H.; Lemeer S.; Schliep J. E.; Matheron L.; Mohammed S.; Cox J.; Mann M.; Heck A. J.; Kuster B. A large synthetic peptide and phosphopeptide reference library for mass spectrometry-based proteomics. Nat. Biotechnol. 2013, 31, 557–64 10.1038/nbt.2585. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.