

Abstract
Here we describe the synthesis and structure-activity relationship for a class of pyrazoline-containing dihydroquinolone negative allosteric modulators of the NMDA receptor that show strong subunit-selectivity for GluN2C- and GluN2D-containing receptors over GluN2A-and GluN2B-containing receptors. Several members of this class inhibit NMDA receptor responses in the nanomolar range, and are more than 50-fold selective over GluN1/GluN2A and GluN1/GluN2B NMDA receptors, as well as AMPA, kainate, GABA, glycine, nicotinic, serotonin, and purinergic receptors. Analysis of the purified enantiomers of one of the more potent and selective compounds shows that the S-enantiomer is both more potent and more selective than the R-enantiomer. The S-enantiomer had an IC50 value of 0.17–0.22 µM at GluN2D- and GluN2C-containing receptors, respectively, and showed over 70-fold selectivity over other NMDA receptor subunits. The subunit-selectivity of this class of compounds should be useful in defining the role of GluN2C- and GluN2D-containing receptors in specific brain circuits in both physiological and patho-physiological conditions.
Keywords: NMDA receptor, negative allosteric modulator, glutamate, Parkinson’s disease, neuroprotection
Introduction
Glutamatergic neurotransmission through ionotropic-glutamate receptors is the primary means of excitatory synaptic transmission in the mammalian central nervous system (CNS). The receptor family comprises the α-amino-3-hydroxy-5-methyl-4-isoxazolepropionate (AMPA), N-methyl-D-aspartate (NMDA) and kainate receptors.1, 2 NMDA receptors are widely expressed in the CNS and are thought to be involved in a range of important physiological processes including axonal guidance, synaptic plasticity, and memory formation.2–5 NMDA receptors are also thought to play an important role in pathophysiological conditions including Parkinson’s disease, schizophrenia, depression, and ischemia.2, 6–8
NMDA receptors mediate the slow component of excitatory synaptic transmission and require the binding of both glutamate and glycine for channel activation. Glycine binds to the GluN1 subunits, which have eight splice variants encoded by a single gene.9–11 The GluN2 subunits (GluN2A–D) bind glutamate, and are encoded by four distinct genes.12 The GluN2 subunits control many of the functional and pharmacological properties of the receptor, including agonist EC50, single channel open time and open probability, as well as deactivation-time course following removal of glutamate.2, 13–17 NMDA receptor deactivation-time course determines the time course for the slow, Ca2+-permeable component of synaptic transmission.18 Typically, NMDA receptors are blocked by extracellular Mg2+ at resting membrane potentials, and the requirements of the glutamate release and depolarization-induced relief of Mg2+ block have led to the idea that the NMDA receptors act as coincidence detectors in the brain.19, 20 The Mg2+ IC50 and the kinetics of block and unblock also vary according to the GluN2 subunit.21
The GluN1 subunits are expressed throughout the CNS, but GluN2 subunit composition and expression vary both during development and anatomically.8, 22–28 The spatially-restricted expression patterns, together with distinct functional and pharmacological differences imparted by the GluN2 subunits, make NMDA receptor subunit-selective modulators of therapeutic interest for several neurological disorders, including stroke, schizophrenia, treatment-resistant depression and Parkinson’s disease.7, 8, 25, 29 Subunit-selectivity will restrict modulator actions to brain regions that express the subunit of interest, potentially limiting side effects that occur as a result of global NMDA receptor blockade.
We previously have described the discovery, preliminary SAR of 25 compounds and pharmacological mechanism of a representative member of the dihydroquinolone-pyrazoline (DQP) class of GluN2C/D subunit-selective antagonists.30 In our previous study, the most potent analog, DQP-1105, had an IC50 of 2.7 µM at GluN2D-containing NMDA receptors and was 41-fold selective over GluN2B-containing receptors (Figue 1A).30 In this report we provide an extensive exploration of the SAR, confirmation of mechanism of action, off-target analysis, an analysis of the stereoselectivity for a representative member of the class and data regarding metabolic stability and potential for blood-brain barrier (BBB) penetration. These efforts have yielded potent and selective analogues as well as insights into the pharmacophore for these pyrazoline-containing compounds.
Figure 1. Previously reported best in class compound and representative structure for SAR.

A. The structure of the previously reported best in class compound, DQP-110530 B. The structure of a general analogue with numbered substituents is shown.
RESULTS
Chemistry
The structure-activity relationship around the quinolone-pyrazoline core was probed by testing the potency and selectivity of analogues which contained a variety of aromatic substitutions in combination with perturbations of the acyl chain moiety (Figure 1B). A representative synthesis of these analogs is shown in Scheme 1. Briefly, anthranilic acids were reacted with triphosgene under standard conditions to yield the isatoic anhydride derivatives 80. These compounds were then converted, via the Weinreb amide 81 and a subsequent lithium halogen exchange reaction with aryl bromides 82, to the appropriate benzophenones, 83.31 The substituted quinolone core was accessed by condensation of 83 with ethyl acetoacetate using microwave irradiation yielding compounds 84. The resultant methyl ketone underwent base-catalyzed condensation with an appropriate aryl aldehyde, 85, yielding the α,β-unsaturated ketone compounds 86. These intermediates could be treated with hydrazine monohydrate in ethanol, utilizing microwave irradiation, to yield the pyrazoline-containing compounds typified by 87. The pyrazoline amine was then functionalized with succinic anhydride (88), glutaric anhydride (89), or maleic anhydride (90) to yield the fully saturated or cis-double bond acyl chain derivatives (Scheme 1).
Scheme 1. Synthesis of dihydro-quinolone-pyrazoline derivatives.

(a) Anhydrous THF, Triphosgene (warning, triphosgene is toxic, see Methods), reflux. (b) EtOH, Weinreb’s HCl salt, reflux. (c) Anhydrous THF, n-Butyllithium, −78 °C. (d) Ethylacetoacetate, DMF, 4Å molecular sieves, 180 °C, µW. (e) 4:3 EtOH:H2O (0.05 M), 0 °C to r.t. (f) hydrazine monohydrate, EtOH, 110 °C, µW. (g) Anhydrous THF, 4Å molecular sieves, 165 °C, µW.
Standard esterification conditions from compound 4 yielded the saturated mono-methyl ester analog, 56 (Scheme 2). Reduction of the acid in compound 26 with BH3-DMS led to the primary alcohol containing compound, 64; a coupling reaction with compound 26 and NH3 gave the primary amide compound, 65 (Scheme 2). The unsaturated fumaric esters could be accessed under standard amide coupling conditions using (E)-4-methoxy-4-oxobut-2-enoic acid and compound 55f, yielding compound 54g, or with acylation of 22f using (E)-methyl 4-chloro-4-oxobut-2-enoate, yielding compound 60 (Scheme 2). Additionally, the acyl chain was replaced with the alkyl chain by reacting the pyrazoline derivative 26f and methyl 4-oxobutanoate under reductive amination conditions to give compound 66 (Scheme 2). Mono-fluoro butyrate 68c (Scheme S1) was synthesized in three steps and also coupled to compound 26f yielding 68 as an isostere of the hydroxyl-containing compound 64 (Scheme 2).32, 33 The ester containing compounds, 54g, 60 and 66, could be saponified under basic conditions yielding the target scaffolds (54, 58 and 67, Scheme 2). All compounds were assayed for activity using two-electrode voltage clamp recordings from Xenopus laevis oocytes recombinantly expressing the desired NMDA receptor subtypes (see Methods).
Scheme 2. Modifications to the acyl-chain.

(a) HCl, MeOH. (b) BH3-Me2S, Anhydrous THF, 0 °C. (c) EDCI, DMAP, NH3 in dioxane (0.5M), THF. (d) EDCI, DMAP, (E)-4-methoxy-4-oxobut-2-enoic acid. (e) Anhydrous THF, (E)-methyl 4-chloro-4-oxobut-2-enoate, 4Å molecular sieves, 165 °C, µW. (f) methyl 4-oxobutanoate, BH3-Me2S, THF. (g) EDCI, DMAP, 4-fluorobutanoic acid, DCM (h) NaOH, EtOH:H2O.
A-Ring substituent optimization
We first evaluated the effect of substituents on the A-phenyl ring (Figure 1B) by holding the chlorine substitution on the quinolone core constant and evaluating the substitutions shown in Tables 1 and S1. At the A-ring, 4-substituted phenyl derivatives resulted in the best potency and selectivity. This observation led to the identification of 5 (Table 1), with a nitro group at R7, which was the most potent para-substituted analogue at GluN2D-containing receptors compared to the un-substituted A-ring, compound 1 (1.1 µM vs. 88 µM, respectively, Table 1). Realizing the substantial liabilities associated with the nitro group, we explored bio-isosteres of the substitution; replacing this group with a carboxylic acid 6 (Table 1) which showed no activity. By contrast, the methyl ester 7 (Table 1) remained active but had decreased potency at GluN2D-containing receptors (32 µM vs. 1.1 µM). Interestingly, sp3 hybridization is tolerated, but not preferred, as can be seen with the tri-fluoromethyl-containing compound 8 (IC50 4.1 µM, GluN2D, Table 1). Although substitution at any of the three available positions on the A-ring is tolerated, substitution at either the R5 or R6 position showed no improvement in potency or selectivity with any of the analogs tested (12–20, 71; Table S1). Similarly, analogs with the A-ring replaced with furan, thiophene and pyridine substituents were evaluated and were inactive (Table S2).
Table 1.
Evaluation of A-Ring para- and ether substitutions.
 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| DQP- | R6 | R7 |
|
|
GluN2A IC50 (µM) |
GluN2B IC50 (µM) |
GluN2C IC50 (µM) |
GluN2D IC50 (µM) |
||
| 1 | H | H | - | - | NE | NE | 86 | 88 | ||
| 2 | H | F | 9 | 6 | 128 | 87 | 23 | 14 | ||
| 3 | H | Cl | - | - | NE | NE | 5.5 | 4.5 | ||
| 4 | H | Br | - | 7 | NE | 22 | 3.6 | 3.1 | ||
| 5 | H | NO2 | 85 | 42 | 91 | 45 | 0.9 | 1.1 | ||
| 6 | H | COOH | - | - | NE | NE | NE | NE | ||
| 7 | H | COOMe | - | - | NE | NE | 93 | 32 | ||
| 8 | H | CF3 | 20 | 13 | 80 | 54 | 5 | 4.1 | ||
| 9 | H | OMe | 9 | 9 | 197 | 187 | 28 | 21 | ||
| 10 | H | NMe2 | - | - | NE | NE | 39 | 19 | ||
| 11 | -OCH2O- | - | 4 | NE | 90 | 23 | 23 | |||
IC50 values were obtained by fitting the Hill equation to the average composite concentration-effect curves (see Methods). Data are from 7–18 oocytes between 2–4 frogs; NE indicates less than 30% inhibition at 100 µM.
A manual Hansch analysis, similar to that of the Topliss approach, was employed to better understand the physicochemical properties governing potency.34 Analysis of the steric, σ and π substituent effects for the A-ring suggests that only the para-σ contribution is directly associated with the IC50 values (r2=0.75, p < 0.05 Pearson two-tailed correlation analysis) at the GluN2C- and GluN2D-containing NMDA receptors (Compounds 2–5 and 8–10, Table 1 and Figure 2A).34, 35
Figure 2. Evaluation of substituent effects for A- and B-ring modifications.

A. The σ substituent constants of the para-substituted A-ring analogs vs. activity show a correlation for GluN2C- and GluN2D-containing receptors, when the R1 position of the C-ring is substituted with chloro. (GluN2D r2=0.82, p < 0.05 Pearson two-tailed correlation analysis; GluN2C r2=0.84, p < 0.05 Pearson two-tailed correlation analysis; Compounds 2–5 and 8–10). B. The analysis of the para-substituents on the B-ring as a function of activity at GluN2C- and GluN2D-containing receptors appears parabolic with respect to the σ substituent constants, with an optimal value close to that of the chloro- and bromo-substitutions (Compounds 29–33). C. The analysis of the para-substituents at GluN2A-containing receptors shows a similar parabolic relationship as observed at the GluN2C- and GluN2D-containing receptors when the activity is plotted as a function of the σ substituent constants (Compounds 29–33). D. The analysis of the substituent effects appears parabolic with respect to the π substituent constants for B-ring meta-substituted compounds at GluN2A-containing receptors, suggesting an optimal hydrophobicity close to that of the chloro-substitution. Substituent constants were obtained from the same source (Compounds 34–39).35
B-Ring substituent optimization
We next modified the B-ring substituents with the aim of understanding the substituent effects at the meta- and para-positions (Figure 1B).34 The para-bromo substitutions at the A- and B-rings in compound 21 showed enhanced potency at GluN2C- and GluN2D-containing receptors, with IC50 values of 0.71 µM and 0.39 µM respectively (Table 2). Interestingly, this compound also showed less selectivity for GluN2D- over GluN2A-containing receptors (33-fold) as compared to GluN2B-containing receptors (59-fold), suggesting a more favorable interaction with GluN2A-containing receptors had been formed (Table 2).
Table 2.
A- and B-Ring Modifications.
 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| DQP- | R3 | R7 |
|
|
GluN2A IC50 (µM) |
GluN2B IC50 (µM) |
GluN2C IC50 (µM) |
GluN2D IC50 (µM) |
||
| 21 | Br | Br | 33 | 59 | 13 | 23 | 0.71 | 0.39 | ||
| 22 | Br | Cl | 34 | 79 | 10 | 23 | 0.56 | 0.29 | ||
| 23 | Br | F | 12 | 26 | 34 | 75 | 3.8 | 2.9 | ||
| 24 | Br | H | 7 | 12 | 64 | 113 | 10 | 9.1 | ||
| 25 | Cl | Br | 37 | 67 | 19 | 34 | 0.95 | 0.51 | ||
| 26 | Cl | Cl | 48 | 50 | 21 | 22 | 0.77 | 0.44 | ||
| 27 | Cl | F | 14 | 26 | 47 | 90 | 4.1 | 3.4 | ||
| 28 | Cl | H | 4 | 13 | 49 | 143 | 13 | 11 | ||
IC50 values were obtained by fitting the Hill equation to the average composite concentration-effect curves (see Methods). Data are from 8–18 oocytes between 2–3 frogs. Compound 26 is shown in Table 4 for comparison.
Co-varying A-ring para-substituents with the para-bromo B-ring substitution allowed us to determine that the para-chloro A-ring substitution was optimal for potency (22–24, Table 2). A similar trend was observed using the para-chloro-substitution on the B-ring while co-varying the A-ring substituents (25–28, Table 2). Therefore, this para-chloro-substitution on the A-ring was used for further SAR elaboration. We varied substituents at the meta- and para- position of the B-ring which identified numerous analogs that were highly potent and selective (Table 3). Notably, the para-fluoro containing compound 29 and the meta-fluoro containing compound 34 were both potent congeners that showed over 90-fold selectivity for GluN2D- over GluN2B-containing receptors; compound 34 also showed 67-fold selectivity over GluN2A-containing receptors (Table 3).
Table 3.
B-Ring Modifications.
 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| DQP- | R2 | R3 |
|
|
GluN2A IC50 (µM) |
GluN2B IC50 (µM) |
GluN2C IC50 (µM) |
GluN2D IC50 (µM) |
||
| 29 | H | F | 36 | 98 | 21 | 57 | 1.0 | 0.58 | ||
| 26 | H | Cl | 48 | 50 | 21 | 22 | 0.77 | 0.44 | ||
| 30 | H | Me | 24 | 30 | 33 | 42 | 2.5 | 1.4 | ||
| 31 | H | OMe | 27 | 33 | 62 | 75 | 5 | 2.3 | ||
| 32 | H | CN | 22 | - | 156 | NE | 12 | 7 | ||
| 33 | H | CF3 | 10 | 14 | 29 | 39 | 3.6 | 2.8 | ||
| 34 | F | H | 67 | 101 | 46 | 70 | 1.1 | 0.69 | ||
| 35 | Cl | H | 20 | 43 | 20 | 43 | 2.1 | 1.0 | ||
| 36 | Me | H | 13 | 37 | 24 | 71 | 4.0 | 1.9 | ||
| 37 | OMe | H | 24 | 34 | 110 | 152 | 7.8 | 4.5 | ||
| 38 | CN | H | - | - | NE | NE | 19 | 13 | ||
| 39 | CF3 | H | 11 | 18 | 28 | 47 | 3.4 | 2.6 | ||
IC50 values were obtained by fitting the Hill equation to the average composite concentration-effect curves (see Methods). Data are from 6–14 oocytes between 2–3 frogs; NE indicates less than 30% inhibition at 100 µM. Data for 26 is presented in Table 2 and repeated here to facilitate comparisons with data.
The Hansch evaluation of the para-σ substituent effects at the B-ring show a seemingly parabolic relationship when compared to potency for only GluN2A-, GluN2C- and GluN2D-containing receptors (Compounds 26 and 29–33, Table 3, Figure 2B and C), with an optimal σ value corresponding to the bromo- and chloro- substitutions at all three receptors. At the meta-position of the B-ring, the correlation between the potency and the hydrophobic π value for substitutions at GluN2A-containing receptors also appears parabolic (Compounds 34–39, Table 3, Figure 2D). The decrease in potency at GluN2A-containing receptors observed with compound 39, which was meta-substituted with the CF3 group (Table 3 and Figure 2D), could be a result of steric clashes with the receptor, or, as was observed with the para-σ substituents, could suggest that the optimal hydrophobicity at the GluN2A-containing receptors is attained with the meta-chloro substitution.
From this analysis of meta- and para-substitutions, we hypothesized that combining an optimal para-substitution for potency at GluN2D-containing receptors with a meta-substitution that was less active at GluN2A-containing receptors might improve selectivity. Both Cl- and F- substitutions gave optimal potency and selectivity when mono-substituted on the B-ring, leading us to co-vary these groups (Table 4). We synthesized compound 40, which has a meta-fluoro and para-chloro substitution pattern on the B-ring. This compound maintained potency but did not increase selectivity (Table 4). Several other compounds that were di-substituted on the B-ring exhibited submicromolar potency at GluN2D-containing receptors, but all showed modest selectivity over GluN2A-containing receptors (41–45, Table 4).
Table 4.
B-Ring Di-substitution.
 | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| DQP- | R2 | R3 | R4 |
|
|
GluN2A IC50 (µM) |
GluN2B IC50 (µM) |
GluN2C IC50 (µM) |
GluN2D IC50 (µM) |
||
| 40 | F | Cl | H | 23 | 53 | 12 | 28 | 0.91 | 0.53 | ||
| 41 | Cl | F | H | 19 | 34 | 19 | 34 | 1.4 | 1.0 | ||
| 42 | Cl | Cl | H | 8 | 22 | 7.7 | 20 | 0.79 | 0.91 | ||
| 43 | F | F | H | 32 | 108 | 21 | 71 | 0.78 | 0.66 | ||
| 44 | F | H | F | 26 | 123 | 21 | 100 | 1.1 | 0.81 | ||
| 45 | Cl | H | Cl | 8 | 19 | 5.5 | 13 | 0.78 | 0.70 | ||
IC50 values were obtained by fitting the Hill equation to the average composite concentration-effect curves from oocyte recordings (see Methods). Data are from 8–15 oocytes between 2 frogs.
C-Ring substitutions with optimized A- and B-ring substituents
We next made a series of substitutions to the C-ring on the quinolone core (Table S3). Beginning with a methyl group at R1 (Figure 1B) in combination with either the para-chloro- or the meta-fluoro-substitution on the B-ring, we synthesized compounds 46 and 47, which decreased the potency as compared to the more favorable compounds with only B-ring and A-ring substitutions (Table S3). Interestingly, the modifications showed variability with regards to the relative selectivity for GluN2A- over GluN2B-containing receptors, suggesting that there remains room in this portion of the binding pocket for potential optimization of selectivity (Compounds 48–53, Table S3).
Acyl-chain perturbations
We subsequently evaluated a series of perturbations to the acyl chain of the pyrazoline nitrogen (Table 5). Restricting the conformation to a cis-configuration with the maleate derivative maintained similar potency to the parent compound in each instance tested (55 and 59 vs. 4 and 22 respectively, Table 5). The trans-fumaric derivative 58 was the most potent compound identified, but was no more selective over GluN2A- or GluN2B-containing receptors than the saturated derivative 22 (Tables 5 and 2). The succinic ester containing compound, 56, was inactive, as was the fumaric ester, 60 (Table 5). We also evaluated glutaric-containing derivatives such as compound 61 (Table 5), which showed similar potencies to that of the succinic derivative 22 (Tables 5 and 2) at all receptors tested, suggesting that the length of the acyl chain is not crucial for activity.
Table 5.
Acyl chain perturbations.
 | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| DQP- | R1 | R3 | R7 | Acyl Chain |
|
|
GluN2A IC50 (µM) |
GluN2B IC50 (µM) |
GluN2C IC50 (µM) |
GluN2D IC50 (µM) |
||
| 4 | Cl | H | Br | - | 7 | NE | 22 | 3.6 | 3.1 | |||
| 54 | Cl | H | Br | 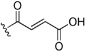 |
- | - | NE | NE | 2.1 | 1.4 | ||
| 55 | Cl | H | Br | 15 | 7 | 74 | 37 | 8.9 | 5 | |||
| 56 | Cl | H | Br | 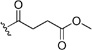 |
- | - | NE | NE | NE | NE | ||
| 57 | Cl | H | Br | 20 | 23 | 78 | 90 | 6.4 | 4.0 | |||
| 22 | H | Br | Cl | 34 | 79 | 10 | 23 | 0.56 | 0.29 | |||
| 58 | H | Br | Cl | 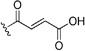 |
23 | 63 | 4.3 | 12 | 0.20 | 0.19 | ||
| 59 | H | Br | Cl | 21 | 30 | 12 | 17 | 1.0 | 0.57 | |||
| 60 | H | Br | Cl | - | - | NE | NE | 59 | 95 | |||
| 61 | H | Br | Cl | 33 | 91 | 10 | 29 | 0.6 | 0.32 | |||
| 5 | Cl | H | NO2 | 85 | 42 | 91 | 45 | 0.9 | 1.1 | |||
| 63 | Cl | H | NO2 | 10 | 8 | 109 | 92 | 10 | 11 | |||
| 26 | H | Cl | Cl | 48 | 50 | 21 | 22 | 0.77 | 0.44 | |||
| 64 | H | Cl | Cl | 90 | 48 | 62 | 33 | 1.7 | 0.69 | |||
| 65 | H | Cl | Cl | 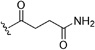 |
31 | 18 | 34 | 20 | 2.0 | 1.1 | ||
| 66 | H | Cl | Cl | - | - | NE | NE | NE | NE | |||
| 67 | H | Cl | Cl | 34 | 31 | 58 | 53 | 3 | 1.7 | |||
| 68 | H | Cl | Cl | - | - | NE | NE | NE | NE | |||
IC50 values were obtained by fitting the Hill equation to the average composite concentration-effect curves (see methods). Data are from 5–24 oocytes between 2–4 frogs; NE indicates less than 30% inhibition at 100 µM. Data for compounds 4, 22, 5 and 26 were presented in preceding tables, and are shown here for comparison.
The primary alcohol containing compound, 64, retained similar activity as that of the parent compound 26 at GluN2C- and GluN2D-containing receptors (IC50 of 1.7 and 0.69 µM, respectively) while improving selectivity over GluN2A-containing receptors to 90-fold (Table 5). The primary amide derivative of the succinate acyl chain in compound 65 retained activity but showed decreased potency and selectivity compared to the alcohol and acid moieties (Table 5). Replacing the amide linkage to the pyrazoline with the alkyl derivative in compound 67 slightly diminished potency at GluN2D-containing receptors over the parent compound 26, but maintained selectivity over the other receptor subtypes (Table 5 and 2). The mono-fluoro isostere of the hydroxyl-group in compound 64 was tested with compound 68 (Table 5). While this compound retained the ability to accept a hydrogen bond, it was inactive.
Stereochemical preference of a representative analog
Finally, we evaluated the selectivity and potency of purified enantiomers for a representative member of this class of compounds. The enantiomers of the racemic final product 26 were separable via reverse phase chiral chromatography using an OD-RH column (Figure 3, see Methods). Absolute stereochemistry of the second peak to elute during the enantiomeric resolution was assigned using X-ray crystallography as the R-enantiomer (70) (Figure 3, see Methods). Evaluation of the purified enantiomers showed that the S-enantiomer, 69, is 11-fold more potent at GluN2D-containing receptors (IC50 0.17 µM) than the R-enantiomer, 70 (IC50 1.9 µM; Figure 4 and Table 6). In addition, 69 shows enhanced selectivity for GluN2C- and GluN2D- over GluN2A- and GluN2B-containing receptors as compared to the racemic 26 and the R-enantiomer, compound 70, due to the enhanced potency at the GluN2D-containing receptors (Figure 4 and Table 6).
Figure 3. Separation of enantiomers.

A. The enantiomers of the final compound, 26, could be separated using reverse phase chiral chromatography (see Methods). B. The crystal structure of the inactive enantiomer, 70 (Table 6) was solved using X-ray diffraction and has the R configuration.
Figure 4. Improvements in selectivity and potency.

A. The potency of the racemic compounds at GluN2D-containing receptors was improved 10-fold over the previous members in the class. B. The potency of the S-enantiomer of compound 26, compound 69, is two-fold more potent than the racemic mixture at GluN2D-containing receptors while the potency at GluN2A- and GluN2B-containing receptors is unaffected, making it more selective for GluN2C- and GluN2D-containing receptors. C. The potency of the R-enantiomer of compound 26, compound 70, at GluN2C- and GluN2D-containing receptors is diminished as compared to the racemate 26, making it less selective over GluN2A- and GluN2B-containing receptors. D. Bar graph showing the fold-selectivity improvements attained through SAR. Data for compounds 997 and DQP-1105 (Panels A and D) are previously published and shown here for comparison.30
Table 6.
Stereo-selectivity for the purified enantiomers of compound 26.
 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| DQP- | R2 | R4 |
|
|
GluN2A IC50 (µM) |
GluN2B IC50 (µM) |
GluN2C IC50 (µM) |
GluN2D IC50 (µM) |
||
| 26 | Cl | Cl | 48 | 50 | 21 | 22 | 0.77 | 0.44 | ||
| 69 (S-26) | Cl | Cl | 78 | 156 | 13 | 26 | 0.22 | 0.17 | ||
| 70 (R-26) | Cl | Cl | 23 | 49 | 45 | 52 | 2.1 | 1.9 | ||
IC50 values were obtained by fitting the Hill equation to the average composite concentration-effect curves (see methods). Data are from 8–17 oocytes between 2–4 frogs; data for compound 26, which was presented in preceding tables, are shown here for comparison.
Mechanism and site of action
A previous study of a representative member suggested that this class of compounds inhibits NMDA receptor function in a voltage-independent and non-competitive manner.30 We confirmed this mechanism for compound 58. Inhibition of GluN1/GluN2D responses by 1 µM 58 was not surmounted by increasing both glutamate and glycine from 30 µM to 3 mM (4.1 ± 0.58% of control in 30 µM, 3.1 ± 0.50% of control in 3 mM; n=6, unpaired t-test), implying that the compound is non-competitive at the glutamate and glycine binding sites. Moreover, inhibition produced by 1 µM 58 was not significantly different at −40 or +30 mV (4.9 + 1.3% and 4.8 + 1.2% of control, respectively, n=8, Student’s t-test, p=0.93), suggesting that the receptor blockade by this more potent compound is also voltage-independent. We also examined whether these more potent analogues interacted with the binding site first identified for both the previously described DQP-1105 (Figure 1A) and the quinazoline-4-one (QNZ), QNZ-46.30, 36 Mutations to the wild type GluN2D receptor (Q801Y, L705F and A752V), previously shown to decrease sensitivity of the GluN2D receptor to blockade by either DQP-1105 and/or QNZ-46 in the membrane proximal region of the bi-lobed ligand binding domain encoded by the S2 region of the polypeptide chain were evaluated.30, 36 A test of the effectiveness of racemic 26 revealed that these mutants each significantly reduced the degree of inhibition, consistent with it acting at a similar site as DQP-1105 and QNZ-46 (Figure S1).
Evaluation of off-target effects
We next evaluated the off-target actions for the racemic compounds 26 and 58 in a series of two-electrode voltage-clamp recordings using recombinant ligand-gated ion channels expressed in Xenopus oocytes (Table 7, see Methods). Compounds 26 and 58 were tested at 3 µM on the AMPA receptors (GluA1–4), kainate receptors (GluK1–2 and GluK2/GluK5), the serotonin receptor (5HT3A), the gamma-amino butyric acid receptors (GABAA and GABAC), the glycine receptor (glycine α1), nicotinic acetylcholine receptors comprised of α1β1δγ, α3β4, α4β2, α7, or α9/α10, and purinergic P2X2 receptors. Of the ion channel classes evaluated, compounds 26 and 58 altered agonist-induced currents by less than 10%, with the exception of the nicotinic acetylcholine receptors, which exhibited 13–28% inhibition (Table 7).
Table 7.
Off-target responses for compound 26 and 58.
| Receptor | Agonist (µM) | (26) ITEST / ICONTROL (mean ± SEM, %) |
N | (58) ITEST / ICONTROL (mean ± SEM, %) |
N |
|---|---|---|---|---|---|
| GluN1/GluN2A | 100 glutamate, 30 glycine | 89± 4.2* | 14 | 89 ± 2.2* | 12 |
| GluN1/GluN2B | 100 glutamate, 30 glycine | 79 ± 2.9* | 12 | 98 ± 1.9 | 12 |
| GluN1/GluN2C | 100 glutamate, 30 glycine | 21 ± 1.2* | 14 | 26 ± 1.5* | 11 |
| GluN1/GluN2D | 100 glutamate, 30 glycine | 14 ± 1.6* | 13 | 14 ± 1.6* | 13 |
| GluA1 | 100 glutamate | 97 ± 2.2 | 6 | 99 ± 1.1 | 6 |
| GluA2 | 100 glutamate | 98 ± 0.8 | 3 | 96 ± 0.9* | 4 |
| GluA3 | 100 glutamate | 99 ± 0.3 | 4 | 100 ± 1.1 | 4 |
| GluA4 | 100 glutamate | 96 ± 1.3 | 3 | 97 ± 1.5 | 4 |
| GluK1 | 100 glutamate | 97 ± 1.0 | 3 | 100 ± 4.4 | 3 |
| GluK2 | 100 glutamate | 97 ± 1.1 | 4 | 97 ± 0.6* | 4 |
| GluK2/GluK5 | 100 glutamate | 97 ± 1.3 | 3 | 95 ± 1.8 | 3 |
| Serotonin 5-HT3A | 3 serotonin | 95 ± 1.5* | 4 | 95 ± 1.2* | 4 |
| GABAA αβ1β2γ2s | 20 GABA | 97 ± 2.7 | 4 | 95 ± 3.1 | 4 |
| GABAc(ρ1) (human) | 2 GABA | 99 + 2.1 | 4 | 97 + 0.6* | 4 |
| Glycine α1 | 50 glycine | 101 ± 1.6 | 4 | 99 ± 1.0 | 4 |
| Nicotinic α1β1γδ(mouse) | 1 acetylcholine | 94 ± 1.2* | 6 | 98 ± 0.7 | 7 |
| Nicotinic α4β2(humn) | 10 acetylcholine | 79 ± 4.2* | 6 | 82 ± 1.7* | 5 |
| Nicotinic α3β4(humn) | 10 acetylcholine | 77 ± 2.7* | 7 | 87 ± 2.2* | 5 |
| Nicotinic α7(humn) | 300 acetylcholine | 82 ± 9.4 | 3 | 64 ± 7.3* | 3 |
| Nicotinic α9α10 | 100 acetylcholine | 67 ± 4.6* | 3 | 72 ± 4.7* | 4 |
| Purinergic P2x2(human) | 9 ATP | 113 ± 1.8* | 5 | 100 ± 0.9 | 4 |
| Purinergic P2x2(rat) | 9 ATP | 97 + 1.2* | 5 | 96 + 1.0* | 4 |
Agonist-evoked currents were recorded from the receptors listed using the Xenopus laevis oocyte expression system under two-electrode voltage clamp (VHOLD=−30 to −60 mV) in the absence and presence of 3 µM 26 or 3 µM 58. The cDNA origin used was rat unless otherwise indicated (paired t-test),
p<0.05.
We also tested the actions of racemic compounds 26 and 58 at 5 µM on 42 different ion channels, G-protein coupled receptors, and transporters via the National Institutes of Mental Health (NIMH) psychoactive drug screening program (PDSP; Supplemental Table S4). The primary binding assay demonstrated that compounds 26 and 58 had a minimal effect on the receptors and transporters, with initial screens showing inhibition of three receptors by 26 (5HT6, H2, kappa-opioid) and four receptors by 58 (5-HT1E, 5-HT6, kappa-opioid, mu-opioid). For both compounds, the Ki values at these receptors were greater than 10 µM on all receptors. The data collected from both the two-electrode voltage-clamp experiments and the PDSP demonstrate the utility of this class of compounds as selective inhibitors of the GluN2C- and GluN2D-containing NMDA receptors.
Plasma stability, aqueous solubility, BBB penetration and human liver microsomal stability
Three of the more potent analogs, 58, 26 and 64 were evaluated for plasma stability. The compounds showed minimal degradation in human, rat or mouse plasma over a two-hour time-course (Figure S2). The aqueous solubility of compound 26 was evaluated in oocyte recording buffer using nephelometry and assessed to be soluble at > 80 µM (see Methods and Supplemental pg. S8).
The topological polar surface area (TPSA) of the carboxylic acid compounds was calculated to be outside the optimal range (< 90 Å2) for blood-brain barrier (BBB) penetration.37 However, reduction of the acid to the alcohol moves the properties of this class closer to a typical range for CNS penetration ((64); 102.0 Å2, QikProp).37 In order to assess the potential for BBB penetration, compounds 26 and 64 were selected for evaluation in the MDR1-MDCK permeability assay which has been demonstrated to accurately predict BBB penetration due to the overexpression of p-glycoprotein (P-gp) and high transepithelial electrical resistance of the cell line (Table 8).38 As was anticipated with the carboxylic acid containing 26, the potential for BBB penetration was low (Table 8). The results for the hydroxyl-containing compound 64 also suggested low BBB potential; however, the permeability coefficient (Papp (A–B)) was much closer to the recommended 3.0 × 10−6 cm/s ((64), (Papp A–B; 2.46 × 10−6 cm/s)) than that of the carboxylic acid-containing compound ((26), (Papp A–B; 0.47 × 10−6 cm/s)), suggesting that efflux may be problematic with this congener.38 In order to evaluate an analog with lower TPSA, the mono-fluoro-containing compound, 68, (Table 5, TPSA 79.08 Å2; QikProp) was assessed in the MDR1-MDCK assay and was classified as being highly brain penetrable (Papp A–B; 3.88 × 10−6 cm/s; Papp B-A; 9.52 × 10−6 cm/s; Table 8).38
Table 8.
MDR1-MDCK permeability.
| Test Compound | Direction | Recovery (%) |
Papp(10−6cm/s) | Efflux Ratio | ||
|---|---|---|---|---|---|---|
| 1 | 2 | Avg | ||||
| 64 | A-to-B | 43 | 2.45 | 2.48 | 2.46 | 26 |
| B-to-A | 73 | 66.2 | 62.7 | 64.5 | ||
| 26 | A-to-B | 73 | 0.47 | 0.47 | 0.47 | 55 |
| B-to-A | 76 | 33.4 | 17.9 | 25.6 | ||
| 68 | A-to-B | 43 | 4.51 | 3.24 | 3.88 | 2.5 |
| B-to-A | 67 | 9.32 | 9.72 | 9.52 | ||
The Papp and efflux ratio were calculated as described in the Methods. Compounds displaying a Papp < 3.0 X 10−6 cm/s and an efflux ratio > 10 are interpreted to have a low potential for crossing the BBB. Compounds with Papp > 3.0 × 10−6 cm/s and an efflux ratio < 10 are expected to have high brain penetration.38
The same compounds were also evaluated for metabolic stability using human liver microsomes. While the carboxylic acid containing 26 showed minimal degradation over the 60 minute assay, the hydroxyl-containing derivative 64 had a half-life of 13 minutes (Table 9). The half-life of the mono-fluoro compound, 68, was determined to be 35 minutes in the human liver microsomal assay (Table 9). These data suggest that the acyl-chain is a candidate for further optimization of desirable pharmacokinetic properties.
Table 9.
Human microsomal stability.
| Test Compound | % Remaining of initial | Half-life | CLintb (ml/min/mg protein) |
||||
|---|---|---|---|---|---|---|---|
| 0 min |
10 min |
20 min |
30 min |
60 min |
|||
| 64 | 100 | 52 | 31 | 24 | 6.5 | 13 | 0.110 |
| 26 | 100 | 101 | 100 | 115 | 86 | >60 | <0.02 |
| 68 | 100 | 67 | 49 | 49 | 39 | 35 | 0.040 |
Half-life was calculated based on t1/2 = 0.693/k, where k is the elimination rate constant based on the slope of the natural logarithm percent remaining versus incubation time.
Intrinsic clearance (CLint) was calculated on CLint = k/P, where k is the elimination rate constant and P is the protein concentration in the incubation.
Discussion and Conclusion
This study describes the development of potent, selective, and soluble negative allosteric modulators for GluN2C- and GluN2D-containing NMDA receptors that act on the membrane proximal lobe of the GluN2 glutamate binding domain. We describe here several compounds with IC50 values in the 100–500 nanomolar range that show 50–200 fold selectivity over GluN2A- and GluN2B-containing receptors. We have taken a classical approach to the SAR, allowing proposal of features for a hypothetical pharmacophore. The A-ring substituents that were explored directly correlate potency with the para-σ substituent coefficients at only GluN2C- and GluN2D-containing receptors, when R1 was substituted with chloro (Figure 2A and 5A). While we maintained selectivity with many of the analogs, our results suggest there is a conserved portion of the binding pocket among GluN2A-, GluN2C- and GluN2D-containing receptors with respect to the para-position of the B-ring; the electronic effects relative to the σ substituent coefficient show similar trends when plotted as a function of potency (Figure 2B, C and 5A). We visualized the electron density of these analogs by carrying out Hartree-Fock calculations using the 6-31G* basis set. While several of the analogs substituted at the para-position of the B-ring could theoretically accept a hydrogen bond, only the bromo- and chloro-containing compounds (26 and 21) exhibit an accessible deficiency of electron density at the terminal position of the substitutions. We hypothesize that the apparent sigma hole found at the bromo and chloro atoms could enhance potency by forming a halogen bond with an oxygen electron donor at the receptors (Figure 5B).39, 40 Furthermore, the SAR has revealed that rigidifying the acyl-chain in the trans-conformation can enhance potency at the GluN2D-containing receptors, while the length of the chain was found not to be crucial for activity (Figure 5A). The finding that the hydroxyl-containing compound 64 (Table 5) retains both potency and selectivity suggests that the charge on the carboxylic acid is not crucial for either property. While it would be ideal to interpret these data in the context of the receptor, no high quality crystallographic data exists for the region of the LBD where these compounds are thought to act, making docking studies challenging.
Figure 5. Pharmacophore model and electrostatic potential maps of para-B-ring modifications.

A. The para-substitution of the A-ring shows correlation between the σ substituent constants and activity at GluN2C/D-containing receptors, when R1 is a chlorine. The length and configuration of the acyl-chain is flexible, with the trans-configuration improving potency; B-ring modification shows an optimal para-sigma coefficient close to that of chloro- and bromo-substitutions for GluN2A-, GluN2C- and GluN2D-containing receptors, suggesting a conserved nature of the binding interaction at each of the three receptors. The C-ring substitutions explored are consistent with this portion of the molecule interacting with a hydrophobic pocket and could allow for improvements is selectivity. B. The electrostatic potential maps of the para-B-ring modifications evaluated are shown. Only the Cl-and Br-substituents show significant electron deficiency at the termini of the substituents, suggesting a potential halogen bond could be responsible for the improved potency of these compounds.
We expect a reduction in molecular weight in conjunction with further optimization of the topological polar surface area will be required to obtain optimal BBB penetration and pharmacokinetic properties. However, the improvements in potency and selectivity suggest this class of compounds should be useful as pharmacologic probes to evaluate the contributions of the GluN2C- and GluN2D-containing NMDA receptors in normal and patho-physiologic processes in isolated systems.
The GluN2C- and GluN2D-NMDA receptor subunits remain understudied, largely because of a lack of potent and selective pharmacological tools. However, these NMDA receptor subunits reside in a number of brain regions that are highly relevant for neurological disease. For example, expression of functional GluN2D in the subthalamic nuclei raises the possibility that GluN2D-selective inhibitors could attenuate neuronal firing rate and alter firing patterns in subthalamic neurons, which could be of utility in Parkinson’s disease.22, 24, 26, 28, 41–44 In addition, expression of GluN2D-containing receptors in substantia nigra pars compacta neurons raises the possibility that GluN2D-selective antagonists might possess neuro-protective properties in Parkinson’s disease by diminishing Ca2+ influx into the dopaminergic substantia nigra pars comapcta neurons, which may lead to neuronal death.8, 45 GluN2C is expressed widely in the cerebellum, and has also been suggested to have a role in both emotional learning and schizophrenia.46–48 The compounds described here could therefore be tools with which to evaluate GluN2C- and GluN2D-containing receptor function in specific circuits implicated in these conditions.
BIOLOGY EXPERIMENTAL
Two-electrode voltage-clamp electrophysiology
The Emory University Institutional Animal Care and Use Committee approved all protocols involving the use of animals. Xenopus laevis oocytes were isolated and maintained as previously described.36 The cDNAs for the desired NMDA receptor subunits (GenBank accession numbers U11418 and U08261; GluN1, D13211; GluN2A, U11419; GluN2B, M91563; GluN2C, L31611; GluN2D) were obtained from Drs. S. Heinemann (Salk Institute), S. Nakanishi (Kyoto University), and P. Seeburg (University of Heidelberg). Preparation of cRNA, injection of oocytes with RNA, and two-electrode voltage clamp recordings have been described elsewhere.30, 36, 49 Briefly, oocytes were placed in a perfusion chamber with recording solution comprised of (in mM) 90 NaCl, 1 KCl, 0.5 BaCl2, 0.005 EDTA and 10 HEPES, pH adjusted to 7.4 with NaOH at 23 °C. The glass electrodes used had tip resistances of 0.5–2.0MΩ and were filled with 0.3–3.0 M KCl. Compounds were made as 20 mM stock solutions in DMSO and diluted to final concentrations in recording solution (final DMSO was 0.1–0.05% vol/vol). The current recordings were performed using a Warner OC-725B or C amplifier at a holding potential of −40 mV.
Subunit selectivity was determined by recording from various ligand-gated ion channels expressed in Xenopus laevis oocytes as previously described.50 The cDNA encoding GABAA, GABAC and glycine receptor cDNAs were provided by Dr. Weiss (University of Texas Health Science Center San Antonio), nicotinic acetylcholine and 5-HT3A serotonin receptor cDNAs were provided by Drs. Papke and Heinemann (University of Florida and Salk Institute), and purinergic receptor cDNA were provided by Dr. Hume (University of Michigan). The glutamate receptors GluA1–4, GluK1, and GluK2 were activated by 100 µM glutamate. GluK1 and GluK2 expressing oocytes were incubated for 5 minutes in 1 mg/ml of concanavalin A prior to recording. The GluK2/5 receptor was activated with 100 µM AMPA (2-amino-3-(3-hydroxy-5-methyl-isoxazol-4-yl)propanoic acid). The GABAA and GABAC receptors were activated by 20 µM and 2 µM gamma-aminobutyric acid, respectively. Acetylcholine was used activate the nicotinic acetylcholine α1β1δγ (1 µM), α3β4 (10 µM), α2β4 (10 µM), α7 (300 µM), α9α10 (10 µM) receptors. The glycine α1 and 5-HT3A currents were evoked by 50 µM glycine receptor and 3 µM serotonin, respectively. The human and rat P2X2 purinergic receptors were activated with 9 µM adenosine tri-phosphate.
The receptor binding profiles (Supplemental Table S2) and Ki determinations for compounds 26 and 58 was generously provided by the National Institute of Mental Health Psychoactive Drug Screening Program, Contract # HHSN-271-2008-025C (NIMH PDSP). The NIMH PDSP is directed by Bryan L. Roth MD, Ph.D. at the University of North Carolina at Chapel Hill and Project Officer Jamie Driscoll at NIMH, Bethesda MD, USA. Data was collected using 5 µM of compounds 26 and 58.
MDR1-MDCK Permeability
Cell monolayers were grown to confluence on collagen-coated, microporous, polycarbonate membranes in 12-well Costar Transwell® plates. The permeability assay buffer was Hanks Balanced Salt Solution containing 10 mM HEPES and 15 mM glucose at pH 7.4. The buffer in the receiver chamber also contained 1% bovine serum albumin. The dosing solution concentration in the assay buffer was 5 µM for each compound tested. The cell monolayers were dosed on the apical side (A-to-B) or basolateral side (B-to-A) and incubated at 37 °C with 5% CO2 in a humidified incubator. Samples were taken from the donor and receiver chambers at 120 minutes. Each determination was performed in duplicate. All samples were assayed by LC-MS/MS using eletrospray ionization. The apparent permeability (Papp) and percent recovery were calculated as follows.
| (1) |
| (2) |
where dCr/dt is the slope of the cumulative concentration in the receiver compartment versus time in µM s−1; Vr is the volume in the receiver compartment in cm3; Vd is the volume in the donor compartment in cm3; A is the area of the insert (1.13 cm2 for12-well Transwell®); CA is the average of the nominal dosing concentration and the measured 120 minute donor concentration in µM; CN is the nominal concentration of the dosing solution in µM; Crfinal is the cumulative receiver concentration in µM at the end of the incubation period; Cdfinal is the concentration of the donor in µM at the end of the incubation period. The MDR1-MDCK permeability assays were performed by Absorption Systems.
Human liver microsomal stability
Human liver microsomes were obtained from XenoTech. The reaction mixture was prepared with 0.5 mg/mL human liver microsomes, 100 mM potassium phosphate (pH 7.4), 5 mM magnesium chloride, 1 µM test compound. The reaction mixture was incubated in a shaking water bath at 37 °C for 3 minutes prior to the addition of NADPH (1 mM). Testosterone was run simultaneously in a separate vessel as a control. 100 µl aliquots were taken at 0, 10, 20, 30 and 60 minutes for both test compound and testosterone. The aliquots were combined immediately with 400 µl of ice cold 50/50 acetonitrile/deionizd H2O containing 0.1% formic acid and internal standard to terminate the reaction. The samples were then mixed and centrifuged to precipitate microsomal proteins. All samples were assayed by LC-MS/MS using electrospray ionization and multiple reaction monitoring and the peak area responses to internal standard of the compounds at each time point was compared to the peak area response at time 0 to determine the percent compound remaining. The human liver microsomal stability assays were performed by Absorption Systems.
Data analysis
Potency of compounds was assessed by fitting the composite concentration-response curve obtained from the average of multiple recordings with the equation
| (3) |
where IC50 is the concentration of compound that is required to inhibit the response half-maximally and N is the hill slope. Saturating conditions were assumed to give complete inhibition. Data for compounds that did not inhibit the steady-state current by at least 30% were not fit by the above equation and are designated as NE in the data tables.
CHEMISTRY EXPERIMENTAL
Commercial vendors provided compounds 4, 5, 9–11, 14, 16, 17, 20, 57 and 62, that are not described below, which were ≥ 90% purity, as provided by the vendor, or determined independently as below. All reagents were obtained from commercial suppliers and used without further purification unless otherwise noted. Reaction progress was monitored by thin layer chromatography (TLC) on pre-coated glass plates (silica gel 60 F254, 0.25 mm). Proton, carbon and fluorine NMR spectra were recorded on INOVA-400 (400 MHz), VNMRS 400 (400 MHz), UNITY-600 (600 MHz), or INOVA-600 (600 MHz) instruments. Proton and carbon spectra were referenced to the residual solvent peak while fluorine spectra were referenced to trifluoroacetic acid residual peak. The Emory University Mass Spectrometry Center collected mass spectral data on either a VG 70-S Nier Johnson or JEOL instrument. Compound purity was assessed by reverse phase liquid chromatography using an Agilent Zorbax, 3.5 µm, XD B-C18 column, 4.6 × 50 mm (254 nm), or by elemental analyses, performed by Atlantic Microlab Inc. Purity for all compounds synthesized and tested was at or above 95% unless otherwise noted. Flash chromatography was performed on a Teledyne ISCO Combiflash Companion with pre-packaged disposable normal phase silica columns.
Computational Analysis
Energy minimized conformations of the compounds being analyzed were generated using the OPLS_2005 force field in MacroModel (MacroModel, version 9.9, Schrödinger, LLC, New York, NY, 2011). The TPSA approximations obtained from QikProp (QikProp, version 3.4, Schrödinger, LLC, New York, NY, 2011) using energy minimized conformations, as above. The Hartree-Fock calculations were carried out using the neutral compounds and the 6-31G* basis set in Spartan ’10 (Spartan ’10, Wavefunction, Inc., Irvine, CA). The equilibrium geometry at the ground state in vacuum was calculated. For visualization purposes, the energy range in the electrostatic potential maps was limited from −100.00 to 280.00 kJ/mol.
Separation and X-Ray Crystallography of Enantiomers
Separation of the final compounds used for biological testing from the racemic 26 was obtained using a ChiralPak OD-RH 30 mm X 250 mm, 5µm column using the following conditions: Flow rate 10 ml/min, injection volume 4–6 ml (2 mg/ml), 60% acetonitrile (0.1% Formic acid) : 40% H2O (0.1% Formic acid); 69 Rt, 21.8; 70 Rt, 25.1 minutes. The enantiomeric excess (e.e.) of the enantiomers, 69 and 70, was determined using an Agilent 1200 HPLC pump on a ChiralPak OD-RH column (4.6 mm × 150 mm, 5µm) using the following conditions: flow rate 0.5 ml/min, injection volume 10 µl, 60% acetonitrile (0.1% Formic acid) : 40% H2O (0.1% Formic acid); 69 (S-26, [α]D20 −34.0 (c = 0.32 mg/ml, chloroform), Rt, 7.47 min, 100% e.e. 70 (R-26) [α]D20 + 36.0 (c = 0.245 mg/ml, MeOH), Rt, 8.79 min, 98% e.e. Optical rotation data were collected using a Perkin-Elmer 314 instrument. The proton NMR spectrum was identical to that of racemic 26 for each enantiomer.
Single crystals of the second peak to elute from the separation of racemic 26 (70; retention time: 25.1 min.) were grown by slow evaporation of a solution of the compound in a mixture of methanol and water. Crystal data: C28H23Cl2N3O5, (M =552.39): 1.124 × 0.087 × 0.056, orthorhombic, space group P 212121 (no.19), a = 8.0529(5) Å, b = 10.2097(5) Å, c = 31.2978(13) Å, V = 2573.2(2) Å3, Z = 4, µ(MoKα) = 0.315 mm−1 , Dcalc=1.490 g/mm ; temperature 173 K. Intensity data were collected on a Bruker APEX II CCD diffractometer with monochromated MoKα radiation (λ = 0.71073 Å) at 173 K, in the 2θ range 2.6–53.4°. The user interface Olex2 was used for the crystallographic calculations and crystal structure visualization.51 The structure was solved with Superflip by charge flipping and refined by least squares minimization using Shelxl.52,53 A total of 15745 reflections were measured (2.602 ≤ 2θ ≤ 53.41) while 5408 unique data (Rint=0.124) were used in the refinements. The final R1 was 0.0590 (I > 2σ(I)) and the weighted R value wR2 was 0.0874 (all data).
General procedure for the synthesis of acylated quinolone-pyrazoline products (Procedure G)
In an appropriately sized microwaveable vessel, the pyrazol-3-yl-quinolin-2(1H)-one intermediate (1.00 equiv) was dissolved in anhydrous tetrahydrofuran (THF) (0.15 M) with 4 Å molecular sieves present. The appropriate anhydride (1.00 equiv) was added. The solution was microwaved (Biotage Initiator) with stirring for 20 minutes at 165 °C. The THF was removed under vacuum, and the organics were dissolved in dichloromethane (DCM), washed three times with acidified (pH 2, HCl) brine, dried over magnesium sulfate, filtered, concentrated under reduced pressure and subjected to flash column chromatography using a 0–10% MeOH:DCM gradient unless otherwise noted.
4-[3-(6-Chloro-2-oxo-4-phenyl-1,2-dihydroquinolin-3-yl)-5-(4-fluorophenyl)-4,5-dihydro-1H-pyrazol-1-yl]-4-oxobutanoic acid (2)
Compound 2 was prepared according to general procedure G using succinic anhydride (88) (0.062 g, 0.622 mmol) and 2f (0.260 g, 0.622 mmol). After removal of the THF, the residue was dissolved in hot EtOAc and small portions of hexanes were added until a solid began to form. The solid was filtered and column chromatographed using 0–10% MeOH:DCM and the title compound was obtained as a yellow solid. Yield 0.236 g, 73.2%. 1H NMR (400 MHz, DMSO-d 6) δ 12.42 (s, 1H), 12.15 (s, 1H), 7.68 – 7.38 (m, 6H), 7.27 (d, J = 7.4 Hz, 1H), 7.04 (td, J = 8.8, 2.4 Hz, 2H), 6.93 (d, J = 2.5 Hz, 1H), 6.85 – 6.76 (m, 2H), 5.35 - 5.30 (m, 1H), 3.80 – 3.67 (m, 1H), 2.83 – 2.74 (m, 1H), 2.48 – 2.39 (m, 2H), 2.34 – 2.25 (m, 2H). 13C NMR (100 MHz, DMSO-d6) δ 173.59, 168.66, 160.13, 152.45, 149.96, 138.39, 137.34, 134.56, 131.25, 129.47, 128.51, 127.55, 126.09, 124.64, 120.68, 117.65, 115.26, 115.05, 58.24, 45.23, 28.92, 28.59, 28.22. 19F NMR (376 MHz, DMSO-d6) δ−116.13 – −116.20 (m). HRMS (m/z): [M-H]− calcd for C28H20ClN3O4F, 516.11319; found, 516.11246. Anal. Calcd for C28H21ClN3O4F: C, 64.93; H, 4.09; N, 8.11. Found C, 61.01; H, 4.22; N, 7.15. HPLC 85% MeOH:H2O (0.1% Formic Acid) Rt 0.95 min; > 95% purity; 75% ACN:H2O (0.1% Formic Acid) Rt 0.72 min; > 95% purity.
4-[3-(6-Chloro-2-oxo-4-phenyl-1,2-dihydroquinolin-3-yl)-5-(4-chlorophenyl)-4,5-dihydro-1H-pyrazol-1-yl]-4-oxobutanoic acid (3)
Compound 3 was prepared according to general procedure G using 87 (0.046 g, 0.460 mmol) and 3f (0.200 g, 0.460 mmol). The title compound was obtained by removing the THF, dissolving the crude mixture into hot EtOAc and adding small portions of hot hexanes until a yellow precipitate formed. The mixture was allowed to cool and filtered to give the title compound as a yellow solid. Yield 0.187 g, 76.0 %. 1H NMR (400 MHz DMSO-d6) δ 12.42 (s, 1H), 12.15 (s, 1H), 7.64 (dd, J = 8.7, 2.5 Hz, 1H), 7.61 – 7.38 (m, 5H), 7.27 (d, J = 7.9 Hz, 3H), 6.94 (d, J = 2.5 Hz, 1H), 6.79 (d, J = 8.2 Hz, 2H), 5.33 (dd, J = 12.1, 4.7 Hz, 1H), 3.75 (dd, J = 18.5, 12.0 Hz, 1H), 2.78 (dd, J = 18.4, 4.7 Hz, 1H), 2.49 – 2.23 (m, 4H). 13C NMR (100 MHz, DMSO-d6) δ 173.55, 168.67, 160.09, 152.42, 149.96, 141.13, 137.32, 134.54, 131.59, 131.24, 129.47, 128.57, 128.46, 128.37, 127.38, 126.13, 126.06, 124.56, 120.66, 117.64, 58.29, 45.12, 28.57, 28.23. HRMS (m/z): [M+H]+ calcd for C28H22Cl2N3O4, 534.09819, found; 534.09774. Anal. Calcd for C28H21Cl2N3O4·0.40H2O: C, 62.09; H, 4.06; N: 7.76. Found C, 61.90; H, 4.13; N: 7.64.
4-[1-(3-Carboxypropanoyl)-3-(6-chloro-2-oxo-4-phenyl-1,2-dihydroquinolin-3-yl)-4,5-dihydro-1H-pyrazol-5-yl]benzoic acid (6)
Compound 6 was prepared according to general procedure G using 87 (0.045 g, 0.451 mmol) and 6f (0.200 g, 0.451 mmol). The solvent was removed and the product was obtained by dissolving in hot EtOAc and adding hexanes until a solid began to precipitate. The solution was cooled and the product filtered to yield a white solid. Yield 0.040 g, 16.3 %. 1H NMR (400 MHz, DMSO-d6) δ 12.42 (bs, 3H), 7.79 (d, J = 8.0 Hz, 2H), 7.64 (dd, J = 8.7, 2.4 Hz, 1H), 7.60 - 7.41 (m, 5H), 7.27 (d, J = 7.4 Hz, 1H), 6.96 - 6.82 (m, 3H), 5.39 (dd, J = 12.0, 4.8 Hz, 1H), 3.79 (dd, J = 18.4, 12.2 Hz, 1H), 2.79 (dd, J = 18.4, 4.7 Hz, 1H), 2.61 - 2.37 (m, 2H), 2.30 (t, J = 6.9 Hz, 2H). 13C NMR (150 MHz, DMSO-d6) δ 173.51, 172.19, 168.72, 167.04, 160.09, 152.41, 149.99, 146.92, 137.33, 134.56, 131.25, 129.68, 129.55, 129.51, 128.52, 128.48, 128.38, 126.12, 126.07, 125.54, 124.53, 120.68, 117.65, 58.71, 45.12, 28.53, 28.22. HRMS (m/z): [M+Na]+ calcd for C29H22ClN3O6Na, 566.10893; found, 566.10923. HPLC 85% MeOH:H2O (0.1% Formic Acid) Rt 0.71 min; > 95% purity; 75% ACN:H2O (0.1% Formic Acid) Rt 0.53 min; > 95% purity.
4-{3-(6-Chloro-2-oxo-4-phenyl-1,2-dihydroquinolin-3-yl)-5-[4-(methoxycarbonyl)phenyl]-4,5-dihydro-1H-pyrazol-1-yl}-4-oxobutanoic acid (7)
Compound 7 was prepared according to general procedure G using 87 (0.055 g, 0.546 mmol) and 7f (0.250 g, 0.546 mmol). The title compound was purified using flash chromatography (2–10% MeOH:DCM), followed by precipitation from hot EtOAc using hot hexanes. Yield 0.040 g, 13.1%. 1H NMR (400 MHz, DMSO-d6) δ 12.38 (s, 1H), 12.06 (s, 1H), 7.84 – 7.72 (m, 2H), 7.64 – 7.35 (m, 6H), 7.22 (d, J = 7.4 Hz, 1H), 6.87 (d, J = 8.4 Hz, 3H), 5.36 (dd, J = 12.3, 4.6 Hz, 1H), 3.90 – 3.69 (m, 4H), 2.74 (dd, J = 18.5, 4.6 Hz, 1H), 2.51 – 2.33 (m, 2H), 2.30 – 2.21 (m, 2H). 13C NMR (100 MHz, DMSO-d6) δ 173.53, 168.77, 165.96, 160.10, 152.43, 150.02, 147.43, 137.34, 134.59, 131.29, 129.44, 128.41, 126.14, 125.76, 124.50, 120.67, 117.67, 58.68, 52.16, 45.09, 28.52, 28.22. HRMS (m/z): [M+Na]+ calcd for C30H24ClN3O6Na, 580.12458; found; 580.12484. Anal. Calcd for C30H24ClN3O6·0.80H2O: C, 62.95; H, 4.51; N, 7.34. Found C, 63.04; H, 4.55; N, 7.36.
4-{3-(6-Chloro-2-oxo-4-phenyl-1,2-dihydroquinolin-3-yl)-5-[4-(trifluoromethyl)phenyl]-4,5-dihydro-1H-pyrazol-1-yl}-4-oxobutanoic acid (8)
Compound 8 was prepared according to general procedure G using 87 (0.043 g, 0.427 mmol) and 8f (0.200 g, 0.427 mmol). The title compound was obtained after flash chromatography (2–10% MeOH:DCM) followed by trituration from EtOAc as a yellow solid. Yield 0.030 g, 12.4 %. 1H NMR (400 MHz, DMSO-d6) δ 12.42 (s, 1H), 12.04 (s, 1H), 7.65 (dd, J = 8.7, 2.4 Hz, 1H), 7.61 – 7.40 (m, 7H), 7.26 (d, J = 7.4 Hz, 1H), 6.99 (d, J = 8.0 Hz, 2H), 6.93 (d, J = 2.3 Hz, 1H), 5.43 (dd, J = 12.2, 4.7 Hz, 1H), 3.79 (dd, J = 18.5, 12.2 Hz, 1H), 2.80 (dd, J = 18.6, 4.7 Hz, 1H), 2.60 – 2.40 (m, 2H), 2.29 (t, J = 6.8 Hz, 2H). 13C NMR (150 MHz, DMSO-d6) δ 174.14, 169.46, 160.72, 153.09, 150.68, 147.32, 138.01, 135.23, 131.92, 130.09, 129.22, 129.12, 129.05, 126.88, 126.79, 126.72, 126.05, 125.13, 121.31, 118.31, 59.19, 45.74, 29.19, 28.89. HRMS (m/z): [M+H]+ calcd for C29H22ClN3O4F3, 568.12455; found, 568.12554. C29H21ClN3O4F3·0.07EtOAc; HPLC 85% MeOH:H2O (0.1% Formic Acid) Rt 1.1 min; > 95% purity; 75% ACN: H2O (0.1% Formic Acid) Rt 0.86 min; > 95% purity.
4-[3-(6-Chloro-2-oxo-4-phenyl-1,2-dihydroquinolin-3-yl)-5-(4-methoxyphenyl)-4,5-dihydro-1H-pyrazol-1-yl]-4-oxobutanoic acid (9)
Compound 9 was prepared according to general procedure G using 87 (0.058 g, 0.582 mmol) and 9f (0.250 g, 0.582 mmol). After removal of the THF, the title compound was obtained by dissolving the crude mixture into hot EtOAc and adding small portions of hot hexanes until a yellow precipitate formed, the mixture was allowed to cool and filtered to give the title compound as a yellow solid. Yield 0.132 g, 42.8%. 1H NMR (400 MHz, DMSO-d6) δ 12.40 (s, 1H), 12.16 (s, 1H), 7.64 (dd, J = 8.9, 2.4 Hz, 1H), 7.60 - 7.49 (m, 3H), 7.45 (d, J = 8.7 Hz, 1H), 7.43 −7.38 (m, 1H), 7.29 (d, J = 7.0 Hz, 1H), 6.93 (d, J = 2.5 Hz, 1H), 6.82-6.66 (m, 4H), 5.24 (dd, J = 12.1, 4.6 Hz, 1H), 3.89 - 3.62 (m, 4H), 2.78 (dd, J = 18.3, 4.6 Hz, 1H), 2.45 (t, J = 7.0 Hz, 2H), 2.27 (t, J = 6.9 Hz, 2H). 13C NMR (150 MHz, DMSO-d6) δ 173.58, 168.49, 160.14, 158.22, 152.39, 149.86, 137.30, 134.58, 134.31, 131.22, 129.42, 128.57, 128.43, 128.35, 126.77, 126.09, 126.02, 124.74, 120.71, 117.63, 113.71, 58.47, 55.10, 45.27, 28.63, 28.24. HRMS (m/z): [M+Na]+ calcd for C29H24ClN3O5Na, 552.12967; found, 552.13018. Anal. Calcd for C29H24ClN3O5·1.00H2O: C, 63.56; H, 4.78; N, 7.67. Found C, 63.68; H, 4.57; N, 7.59.
4-[3-(6-Chloro-2-oxo-4-phenyl-1,2-dihydroquinolin-3-yl)-5-(3-fluorophenyl)-4,5-dihydro-1H-pyrazol-1-yl]-4-oxobutanoic acid (12)
Compound 12 was prepared according to general procedure G using 87 (0.120 g, 1.20 mmol) and 12f (0.500 g, 1.20 mmol). After removal of the THF, the title compound was obtained by precipitation from EtOAc using hexanes, as a yellow solid. Yield 0.380 g, 62%. 1H NMR (400 MHz, DMSO-d6) δ 12.42 (s, 1H), 12.15 (s, 1H), 7.65 (dd, J = 8.7, 2.6 Hz, 1H), 7.53 (m, 2H), 7.44 (m, 3H), 7.26 (d, J = 7.0 Hz, 2H), 7.03 (t, J = 8.7 Hz, 1H), 6.93 (d, J = 2.3 Hz, 1H), 6.64 (dd, J = 12.3, 8.8 Hz, 2H), 5.35 (dd, J = 12.3, 4.4 Hz, 1H), 3.75 (dd, J = 18.6, 12.1 Hz, 1H), 2.81 (dd, J = 17.7, 4.7 Hz, 1H), 2.60 – 2.23 (m, 4H). 13C NMR (100 MHz, DMSO-d6) δ 173.57, 168.75, 163.36, 160.94, 160.12, 152.47, 149.98, 144.95, 144.88, 137.33, 134.56, 131.25, 130.52, 130.45, 129.40, 128.56, 128.42, 128.38, 128.29, 126.14, 126.07, 124.53, 121.50, 120.65, 117.65, 114.00, 113.79, 112.34, 112.13, 58.41, 45.14, 28.53, 28.20. HRMS (m/z): [M-H]− calcd for C28H20ClN3O4F; 516.11319; found, 516.11239. Anal. Calcd for C28H21ClN3O4F·0.40H2O: C, 64.93; H, 4.09; N, 8.11. Found C, 64.11; H, 4.03; N, 7.94. HPLC 85% MeOH:H2O (0.1% Formic Acid) Rt 0.97 min; > 95% purity; 75% ACN:H2O (0.1% Formic Acid) Rt 0.73 min; > 95% purity.
4-[3-(6-Chloro-2-oxo-4-phenyl-1,2-dihydroquinolin-3-yl)-5-(3-chlorophenyl)-4,5-dihydro-1H-pyrazol-1-yl]-4-oxobutanoic acid (13)
Compound 13 was prepared according to general procedure G using 87 (0.046 g, 0.460 mmol) and 13f (0.200 g, 0.460 mmol). After removal of the THF, the title compound was obtained by precipitating from EtOAc using hexanes, as a yellow solid. Yield 0.156 g, 63.4%. 1H NMR (400 MHz, DMSO-d6) δ 12.39 (s, 1H), 12.04 (s, 1H), 7.64 – 7.57 (m, 1H), 7.56 – 7.34 (m, 5H), 7.27 – 7.17 (m, 3H), 6.97 – 6.87 (m, 2H), 6.68 (d, J = 6.8 Hz, 1H), 5.29 (dd, J = 12.2, 4.8 Hz, 1H), 3.71 (dd, J = 18.5, 12.0 Hz, 1H), 2.84 – 2.73 (m, 1H), 2.50 – 2.32 (m, 2H), 2.25 (t, J = 6.7 Hz, 2H). 13C NMR (100 MHz, DMSO-d6) δ 173.55, 168.78, 152.53, 150.00, 144.54, 137.35, 134.62, 133.05, 130.47, 129.37, 128.60, 128.45, 127.14, 126.15, 125.51, 124.51, 124.11, 120.64, 58.40, 45.10, 28.53, 28.18. HRMS (m/z): [M+Na]+ calcd for C28H21Cl2N3O4Na; 556.08013; found, 556.07988. Anal. Calcd for C28H21Cl2N3O4·0.20H2O: C, 61.68; H, 4.10; N, 7.71. Found C, 61.74; H, 4.27; N, 7.32.
4-[3-(6-Chloro-2-oxo-4-phenyl-1,2-dihydroquinolin-3-yl)-5-(3-methoxyphenyl)-4,5-dihydro-1H-pyrazol-1-yl]-4-oxobutanoic acid (15)
Compound 15 was prepared according to general procedure G using 87 (0.029 g, 0.29 mmol) and 15f (0.13 g, 0.29 mmol). The title compound was purified using flash chromatography (2–10% MeOH:DCM) and isolated as a yellow solid. Yield 0.090 g, 58.7%. 1H NMR (400 MHz, DMSO-d6) δ 12.41 (s, 1H), 12.08 (s, 1H), 7.64 (dt, J = 8.8, 2.1 Hz, 1H), 7.58 – 7.42 (m, 4H), 7.39 (d, J = 7.3 Hz, 1H), 7.27 (d, J = 7.3 Hz, 1H), 7.13 (t, J = 7.9 Hz, 1H), 6.93 (d, J = 2.2 Hz, 1H), 6.77 (dd, J = 8.2, 2.4 Hz, 1H), 6.59 (s, 1H), 6.32 (d, J = 7.7 Hz, 1H), 5.27 (dd, J = 12.0, 4.7 Hz, 1H), 3.77 – 3.63 (m, 4H), 2.85 (dd, J = 18.4, 4.7 Hz, 1H), 2.48 – 2.36 (m, 2H), 2.28 (t, J = 6.8 Hz, 2H). 13C NMR (150 MHz, DMSO-d6) δ 173.54, 168.64, 160.16, 159.25, 152.47, 149.91, 143.75, 137.32, 134.64, 131.21, 129.59, 129.15, 128.59, 128.48, 128.37, 128.30, 126.09, 124.56, 120.68, 117.63, 117.39, 112.37, 111.35, 58.88, 54.98, 45.23, 28.56, 28.22. HRMS (m/z): [M-H]− calcd for C29H23ClN3O5, 528.13317; found, 528.13351. Anal. Calcd for C29H23Cl1N3O5·0.70H2O: C, 64.32; H, 4.54; N, 7.76. Found C, 64.38; H, 4.67; N: 7.67.
4-[3-(6-Chloro-2-oxo-4-phenyl-1,2-dihydroquinolin-3-yl)-5-(2-chlorophenyl)-4,5-dihydro-1H-pyrazol-1-yl]-4-oxobutanoic acid (18)
Compound 18 was prepared according to general procedure G using 87 (0.046 g, 0.460 mmol) and 18f (0.200 g, 0.460 mmol). After removal of the THF, the title compound was obtained by precipitation from EtOAc using hexanes, as a yellow solid. Yield 0.136 g, 55.3%. 1H NMR (400 MHz, DMSO-d6) δ 12.40 (s, 1H), 12.13 (s, 1H), 7.63 (dd, J= 8.6, 1.6 Hz, 1H), 7.56-7.49 (m, 2H), 7.45-7.41 (m, 4H), 7.26 (t, J = 7.6 Hz, 1H), 7.19-7.15 (m, 2H), 6.93 (s, 1H), 6.41 (d, J = 7.4 Hz, 1H), 3.87 (dd, J=18.4, 12.4 Hz, 1H), 2.75 (dd, J = 18.2, 4.7 Hz, 1H), 2.60-2.54 (m, 1H), 2.46-2.32 (m, 2H), 2.39-2.30 (m, 2H). 13C NMR (100 MHz, DMSO-d6) δ 173.55, 168.78, 160.02, 152.56, 150.10, 138.66, 134.68, 131.27, 130.52, 129.36, 128.78, 128.37, 128.58, 126.13, 125.90, 124.33, 120.66, 117.64, 56.40, 44.20, 28.43, 28.22. HRMS (m/z): [M+Na]+ calcd for C28H21Cl2N3O4Na, 556.08013; found 556.08038. Anal. Calcd for C28H21Cl2N3O4·0.30H2O: C, 62.30; H, 4.03; N, 7.78. Found C, 62.27; H, 4.32; N, 7.49.
4-[5-(2-Bromophenyl)-3-(6-chloro-2-oxo-4-phenyl-1,2-dihydroquinolin-3-yl)-4,5-dihydro-1H-pyrazol-1-yl]-4-oxobutanoic acid (19)
Compound 19 was prepared according to general procedure G using 87 (0.063 g, 0.627 mmol) and 19f (0.300 g, 0.627 mmol). After removal of the THF, the title compound was obtained by precipitation from EtOAc and hexanes, as a yellow solid. Yield 0.140 g, 38.6 %. 1H NMR (400 MHz, DMSO-d6) δ 12.41 (s, 1H), 12.05 (s, 1H), 7.64 (dd, J = 8.9, 2.5 Hz, 1H), 7.59 (d, J = 7.5 Hz, 1H), 7.53 (p, J = 7.6 Hz, 2H), 7.42 (dd, J = 11.1, 7.9 Hz, 3H), 7.26 – 7.15 (m, 3H), 6.93 (d, J = 2.3 Hz, 1H), 6.39 – 6.32 (m, 1H), 5.48 (dd, J = 12.1, 4.6 Hz, 1H), 3.87 (dd, J = 18.3, 12.0 Hz, 1H), 2.73 (dd, J = 18.4, 4.6 Hz, 1H), 2.62 – 2.51 (m, 1H), 2.49 – 2.28 (m, 3H). 13C NMR (100 MHz, DMSO-d6) δ 173.53, 168.76, 160.06, 152.42, 150.11, 140.19, 137.33, 132.58, 131.25, 129.08, 128.41, 126.12, 124.30, 120.64, 117.63, 58.69, 44.33, 28.41, 28.21. HRMS (m/z): [M+Na]+ calcd for C28H21ClBrN3O4Na; 600.02962, found 600.02945. Anal. Calcd for C28H21ClBrN3O4: C, 58.10; H, 3.66; N, 7.26. Found C, 54.48; H, 3.54; N, 6.55. HPLC 85% MeOH:H2O (0.1% Formic Acid) Rt 1.17 min; > 95% purity; 75% ACN:H2O (0.1% Formic Acid) Rt 1.17 min; > 95% purity.
4-{5-(4-Bromophenyl)-3-[4-(4-bromophenyl)-2-oxo-1,2-dihydroquinolin-3-yl]-4,5-dihydro-1H-pyrazol-1-yl}-4-oxobutanoic acid (21)
Compound 21 was prepared according to general procedure G using 87 (0.057 g, 0.573 mmol) and 21f (0.300 g, 0.573 mmol). There was a yellow solid present in the reaction vessel which was filtered, dried and determined to be the title compound. Yield 0.320 g, 90%. 1H NMR (400 MHz, DMSO-d6) δ 12.30 (s, 1H), 12.18 (s, 1H), 7.75 (d, J = 8.2 Hz, 1H), 7.66 (d, J = 8.2 Hz, 1H), 7.58 (t, J = 7.8 Hz, 1H), 7.48 – 7.39 (m, 3H), 7.36 (d, J = 8.1 Hz, 1H), 7.22 (d, J = 8.1 Hz, 1H), 7.15 (t, J = 7.7 Hz, 1H), 7.03 (d, J = 8.2 Hz, 1H), 6.74 (d, J = 7.9 Hz, 2H), 5.33 (dd, J = 11.8, 4.4 Hz, 1H), 3.74 (dd, J = 18.7, 12.1 Hz, 1H), 2.76 (dd, J = 18.5, 4.4 Hz, 1H), 2.65 – 2.39 (m, 2H), 2.39 – 2.30 (m, 2H). 13C NMR (100 MHz, DMSO-d6) δ 173.52, 168.65, 160.14, 152.69, 149.89, 141.52, 138.55, 134.40, 131.73, 131.45, 131.24, 130.82, 127.63, 127.28, 123.41, 122.41, 121.82, 120.09, 119.07, 115.59, 58.28, 45.14, 28.49, 28.23. HRMS (m/z): [M-H]− calcd for C28H20Br2N3O4, 619.98260; found, 619.98231. Anal. Calcd for C28H21Br2N3O4: C, 53.96; H, 3.40; N, 6.74. Found C, 52.23; H, 3.67; N, 5.55. HPLC 85% MeOH:H2O 0.1% Formic Acid) Rt 1.2 min; > 95% purity; 75% ACN:H2O (0.1% Formic Acid) Rt 0.84 min; > 94% purity.
4-{3-[4-(4-Bromophenyl)-2-oxo-1,2-dihydroquinolin-3-yl]-5-(4-chlorophenyl)-4,5-dihydro-1H-pyrazol-1-yl}-4-oxobutanoic acid (22)
Compound 22 was prepared according to general procedure G using 87 (0.105 g, 1.04 mmol) and 22f (0.500 g, 1.044 mmol). After removal of the THF, the residue was partitioned between EtOAc and acidified brine. The organics were washed three times, dried over magnesium sulfate and concentrated under vacuum. The title compound was obtained after column chromatography using 10% MeOH:DCM. Yield 0.240 g, 39.7 %. 1H NMR (400 MHz, DMSO-d6) δ 12.29 (s, 1H), 12.05 (s, 1H), 7.74 (dd, J = 8.1, 2.2 Hz, 1H), 7.65 (dd, J = 8.1, 2.2 Hz, 1H), 7.58 (t, J = 7.6 Hz, 1H), 7.42 (d, J = 8.3 Hz, 1H), 7.36 (dd, J = 8.1, 2.3 Hz, 1H), 7.34 - 7.25 (m, 2H), 7.22 (dd, J = 8.2, 2.3 Hz, 1H), 7.15 (t, J = 7.6 Hz, 1H), 7.03 (d, J = 8.1 Hz, 1H), 6.85 - 6.75 (m, 2H), 5.35 (dd, J = 11.9, 4.4 Hz, 1H), 3.73 (dd, J =18.4, 12.1 Hz, 1H), 2.76 (dd, J = 18.6, 4.4 Hz, 1H), 2.65 - 2.42 (m, 2H), 2.34 (t, J = 6.7 Hz, 2H). 13C NMR (100 MHz, DMSO-d6) δ 173.54, 168.69, 160.17, 152.70, 149.90, 141.10, 138.58, 134.42, 131.61, 131.45, 131.27, 130.84, 128.34, 127.30, 123.44, 122.42, 119.08, 115.60, 58.23, 45.16, 28.48, 28.23. HRMS (m/z): [M-H]− calcd for C28H20BrClN3O4, 576.03312; found, 576.03267. Anal. Calcd for C28H21BrClN3O4·0.50H2O: C, 57.21; H, 3.77; N, 7.15. Found C, 57.02; H, 3.72; N, 7.05.
4-{3-[4-(4-Bromophenyl)-2-oxo-1,2-dihydroquinolin-3-yl]-5-(4-fluorophenyl)-4,5-dihydro-1H-pyrazol-1-yl}-4-oxobutanoic acid (23)
Compound 23 was prepared according to general procedure G using 87 (0.054 g, 0.541 mmol) and 23f (0.250 g, 0.541 mmol). The title compound, a yellow solid, was obtained after purifying using flash chromatography (0–10% MeOH:DCM). Yield 0.100 g, 32.9%. 1H NMR (400 MHz, DMSO-d6) δ 12.29 (s, 1H), 12.11 (s, 1H), 7.74 (d, J = 8.0 Hz, 1H), 7.66 (d, J = 8.2 Hz, 1H), 7.58 (t, J = 7.7 Hz, 1H), 7.43 (d, J = 8.3 Hz, 1H), 7.36 (d, J = 8.0 Hz, 1H), 7.23 (d, J = 8.2 Hz, 1H), 7.15 (t, J = 7.6 Hz, 1H), 7.05 (t, J = 8.9 Hz, 3H), 6.83 (dd, J = 8.4, 5.2 Hz, 2H), 5.35 (dd, J = 12.0, 4.4 Hz, 1H), 3.72 (dd, J = 18.5, 12.0 Hz, 1H), 2.78 (dd, J = 18.4, 4.4 Hz, 1H), 2.65 - 2.43 (m, 2H), 2.34 (t, J = 6.7 Hz, 2H). 13C NMR (100 MHz, DMSO-d6) δ 173.56, 168.64, 160.16, 152.71, 149.87, 138.56, 138.36, 134.42, 131.69, 131.46, 131.24, 130.86, 127.46, 127.38, 127.29, 123.47, 122.43, 121.80, 119.08, 115.59, 115.18, 114.96, 58.15, 45.26, 28.50, 28.23. 19F NMR (376 MHz, DMSO-d6) δ −116.06 – −116.19 (m). HRMS (m/z): [M+H]+ calcd for C28H22BrFN3O4, 562.07722, found; 562.07669. Anal. Calcd for C28H21BrFN3O4: C, 59.80; H, 3.76; N, 7.47. Found C, 50.97; H, 3.55; N, 6.00. HPLC 75–95% MeOH:H2O (0.1% Formic Acid) Rt 0.89 min; > 95% purity; 75% ACN: H2O (0.1% Formic Acid) Rt 0.67 min; > 95% purity.
4-{3-[4-(4-Bromophenyl)-2-oxo-1,2-dihydroquinolin-3-yl]-5-phenyl-4,5-dihydro-1H-pyrazol-1-yl}-4-oxobutanoic acid (24)
Compound 24 was prepared according to general procedure G using 87 (0.023 g, 0.23 mmol) and 24f (0.100 g, 0.23 mmol). The title compound was obtained after purifying using flash chromatography (0–10% MeOH:DCM) as an off-white solid. Yield 0.110 g, 86%. 1H NMR (400 MHz, DMSO-d6) δ 12.29 (s, 1H), 12.20 (s, 1H), 7.76 (d, J = 8.0 Hz, 1H), 7.67 – 7.54 (m, 2H), 7.47 – 7.34 (m, 2H), 7.23 (d, J = 5.4 Hz, 4H), 7.15 (t, J = 7.6 Hz, 1H), 7.03 (d, J = 8.3 Hz, 1H), 6.77 (d, J = 6.7 Hz, 2H), 5.32 (dd, J = 11.8, 4.7 Hz, 1H), 3.75 (dd, J = 18.2, 11.9 Hz, 1H), 2.77 (dd, J = 18.3, 4.5 Hz, 1H), 2.62 – 2.41 (m, 2H), 2.33 (t, J = 6.7 Hz, 2H). 13C NMR (150 MHz, DMSO-d6) δ 173.54, 168.57, 160.17, 152.68, 149.84, 142.25, 138.54, 134.42, 131.74, 131.40, 131.25, 131.16, 130.84, 128.36, 127.28, 127.03, 125.33, 123.55, 122.39, 121.77, 119.11, 115.57, 58.87, 45.39, 28.54, 28.24. HRMS (m/z): [M-H]− calcd for C28H21BrN3O4, 542.07209; found, 542.07235. Anal. Calcd for C28H22BrN3O4: C, 61.89; H, 3.89; N, 7.73. Found C, 56.02; H, 4.02; N, 6.78. HPLC 95% MeOH:H2O (0.1% Formic Acid) Rt 0.91 min; > 95% purity; 65% ACN: H2O (0.1% Formic Acid) Rt 0.70 min; > 95% purity.
4-{5-(4-Bromophenyl)-3-[4-(4-chlorophenyl)-2-oxo-1,2-dihydroquinolin-3-yl]-4,5-dihydro-1H-pyrazol-1-yl}-4-oxobutanoic acid (25)
Compound 25 was prepared according to general procedure G using 87 (0.031 g, 0.31 mmol) and 25f (0.150 g, 0.31 mmol). The title compound was obtained after purifying using flash chromatography (0–10% MeOH:DCM) as an off-white solid. Yield 0.048 g, 27%. 1H NMR (400 MHz, DMSO-d6) δ 12.30 (s, 1H), 7.64 – 7.48 (m, 2H), 7.42 (t, J = 7.9 Hz, 5H), 7.28 (d, J = 8.2 Hz, 1H), 7.15 (t, J = 7.6 Hz, 1H), 7.02 (d, J = 8.1 Hz, 1H), 6.74 (d, J = 8.0 Hz, 2H), 5.33 (dd, J = 11.9, 4.4 Hz, 1H), 3.72 (dd, J = 18.5, 12.0 Hz, 1H), 2.79 – 2.69 (m, 1H), 2.53 – 2.36 (m, 2H), 2.32 (t, J = 6.9 Hz, 2H). 13C NMR (150 MHz, DMSO-d6) δ 174.14, 169.65, 160.19, 152.36, 149.87, 141.71, 138.60, 133.96, 133.25, 131.59, 131.44, 131.23, 130.56, 128.37, 127.64, 127.26, 123.65, 122.43, 120.06, 119.18, 115.66, 58.23, 45.12, 30.27, 29.69. HRMS (m/z): [M+H]+ calcd for C28H22BrClN3O4, 578.04767; found, 578.04719. Anal. Calcd for C28H21BrClN3O4·1.20H2O: C, 56.01; H, 3.93; N, 7.00. Found C, 56.02; H, 4.02; N, 6.78.
4-{5-(4-Chlorophenyl)-3-[4-(4-chlorophenyl)-2-oxo-1,2-dihydroquinolin-3-yl]-4,5-dihydro-1H-pyrazol-1-yl}-4-oxobutanoic acid (26)
Compound 26 was prepared according to general procedure G using 87 (0.046 g, 0.460 mmol) and 26f (0.200 g, 0.460 mmol). The title compound was obtained after removing the THF under vacuum, precipitating from EtOAc with hexanes and further purification using flash chromatography (2–10% MeOH:DCM) as a yellow solid. Some compound was lost due to spillage. Yield 0.054 g, 22%. 1H NMR (400 MHz, DMSO-d6) δ 12.29 (s, 1H), 12.06 (s, 1H), 7.65 – 7.55 (m, 2H), 7.52 (dd, J = 8.2, 2.3 Hz, 1H), 7.48 – 7.38 (m, 2H), 7.34 – 7.24 (m, 3H), 7.15 (t, J = 7.6 Hz, 1H), 7.03 (d, J = 8.1 Hz, 1H), 6.82 (d, J = 8.5 Hz, 2H), 5.35 (dd, J = 11.9, 4.5 Hz, 1H), 3.73 (dd, J = 18.5, 12.0 Hz, 1H), 2.77 (dd, J = 18.4, 4.4 Hz, 1H), 2.65 – 2.43 (m, 2H), 2.34 (t, J = 6.7 Hz, 2H). 13C NMR (100 MHz, DMSO-d6) δ 173.54, 168.67, 160.17, 152.72, 149.89, 141.09, 138.57, 134.04, 133.22, 131.59, 131.46, 130.55, 128.32, 127.31, 122.42, 119.15, 115.59, 58.21, 45.16, 28.47, 28.20. HRMS (m/z): [M+H]+ calcd for C28H22Cl2N3O4, 534.09819; found, 534.09787. Anal. Calcd for C28H21Cl2N3O4: C, 62.93; H; 3.96; N, 7.86. Found C, 62.38; H, 4.03; N, 7.73. HPLC 85% MeOH:H2O (0.1% Formic Acid) Rt 0.95 min; > 95% purity; 75% ACN: H2O (0.1% Formic Acid) Rt 0.61 min; > 95% purity.
4-{3-[4-(4-Chlorophenyl)-2-oxo-1,2-dihydroquinolin-3-yl]-5-(4-fluorophenyl)-4,5-dihydro-1H-pyrazol-1-yl}-4-oxobutanoic acid (27)
Compound 27 was prepared according to general procedure G using 87 (0.029 g, 0.287 mmol) and 27f (0.120 g, 0.287 mmol). The title compound was obtained after removal of the residual solvent, dissolving the crude material in DCM and washing 3X with acidified brine. The organics were collected, dried over magnesium sulfate and concentrated to yield the title compound as a brown solid. Yield 0.085 g, 57%. 1H NMR (400 MHz, DMSO-d6) δ 12.30 (s, 1H), 12.08 (s, 1H), 7.65 – 7.49 (m, 3H), 7.47 – 7.34 (m, 2H), 7.34 – 7.26 (m, 1H), 7.15 (t, J = 7.7 Hz, 1H), 7.10 – 7.00 (m, 3H), 6.88 – 6.80 (m, 2H), 5.35 (dd, J = 11.7, 4.3 Hz, 1H), 3.72 (dd, J = 18.1, 12.0 Hz, 1H), 2.78 (dd, J = 18.5, 4.3 Hz, 1H), 2.64 – 2.42 (m, 2H), 2.33 (t, J = 6.6 Hz, 2H). 13C NMR (150 MHz, DMSO-d6) δ 174.19, 169.29, 160.82, 153.38, 150.51, 139.22, 138.99, 134.70, 133.85, 132.08, 131.24, 128.97, 128.11, 128.05, 127.94, 124.20, 123.06, 119.81, 116.25, 115.76, 115.62, 58.80, 45.91, 29.16, 28.87. HRMS (m/z): [M-H]− calcd for C28H20ClFN3O4, 516.11319; found, 516.11362. Anal. Cacld for C28H21ClFN3O4·0.70DCM: C, 59.70; H, 3.91; N, 7.28. Found C, 59.54; H, 4.15; N, 7.20.
4-{3-[4-(4-Chlorophenyl)-2-oxo-1,2-dihydroquinolin-3-yl]-5-phenyl-4,5-dihydro-1H-pyrazol-1-yl}-4-oxobutanoic acid (28)
Compound 28 was prepared according to general procedure G using 87 (0.055 g, 0.550 mmol) and 28f (0.220 g, 0.550 mmol). The title compound was obtained by filtering from DCM after removal of the THF in vacuo. Yield 0.204 g, 74%. 1H NMR (400 MHz, DMSO-d6) δ 12.29 (s, 1H), 12.13 (s, 1H), 7.66 – 7.54 (m, 2H), 7.54 – 7.39 (m, 3H), 7.32 – 7.11 (m, 5H), 7.03 (d, J = 8.1 Hz, 1H), 6.77 (dd, J = 6.5, 3.0 Hz, 2H), 5.32 (dd, J = 11.9, 4.3 Hz, 1H), 3.75 (dd, J = 18.4, 12.1 Hz, 1H), 2.77 (dd, J = 18.3, 4.3 Hz, 1H), 2.64 – 2.39 (m, 2H), 2.33 (t, J = 6.6 Hz, 2H). 13C NMR (150 MHz, DMSO-d6) δ 173.55, 168.57, 160.17, 152.70, 149.84, 142.23, 138.54, 134.03, 133.17, 131.48, 131.40, 130.57, 128.33, 128.25, 127.28, 127.03, 125.33, 123.62, 122.39, 119.18, 115.57, 58.86, 45.39, 28.53, 28.23. HRMS (m/z): [M-H]− calcd for C28H21ClN3O4, 498.12261; found, 498.12276. HPLC 85% MeOH:H2O (0.1% Formic Acid) Rt 0.87 min; > 95% purity; 75% ACN: H2O (0.1% Formic Acid) Rt 0.69 min; > 95% purity.
4-{5-(4-Chlorophenyl)-3-[4-(4-fluorophenyl)-2-oxo-1,2-dihydroquinolin-3-yl]-4,5-dihydro-1H-pyrazol-1-yl}-4-oxobutanoic acid (29)
Compound 29 was prepared according to general procedure G using 87 (0.072 g, 0.718 mmol) and 29f (0.300 g, 0.718 mmol). The title compound was obtained by filtering from DCM after removal of the THF in vacuo. Yield 0.210 g, 57%. 1H NMR (400 MHz, DMSO-d6) δ 12.27 (s, 1H), 12.09 (s, 1H), 7.58 (t, J = 7.7 Hz, 1H), 7.43 (d, J = 7.6 Hz, 2H), 7.40 - 7.25 (m, 5H), 7.15 (t, J = 7.7 Hz, 1H), 7.04 (d, J = 8.2 Hz, 1H), 6.86 (d, J = 8.2 Hz, 2H), 5.34 (dd, J = 11.9, 4.6 Hz, 1H), 3.74 (dd, J = 18.5, 11.9 Hz, 1H), 2.79 (dd, J = 18.4, 4.6 Hz, 1H), 2.64 - 2.42 (m, 2H), 2.33 (t, J = 6.8 Hz, 2H). 13C NMR (100 MHz, DMSO-d6) δ 173.54, 168.67, 160.21, 152.82, 150.17, 141.11, 138.53, 131.57, 131.44, 130.80, 128.30, 127.36, 123.62, 122.38, 119.40, 115.57, 115.31, 115.10, 58.21, 45.16, 28.50, 28.20. HRMS (m/z): [M+Na]+ calcd for C28H21ClFN3O4Na, 540.10968; found, 540.10938. Anal. Calcd for C28H21ClFN3O4·0.90H2O: C, 62.96; H, 4.30; N, 7.86. Found C, 62.80; H, 4.06; N: 7.80.
4-[5-(4-Chlorophenyl)-3-(2-oxo-4-p-tolyl-1,2-dihydroquinolin-3-yl)-4,5-dihydro-1H-pyrazol-1-yl]-4-oxobutanoic acid (30)
Compound 30 was prepared according to general procedure G using 87 (0.060 g, 0.604 mmol) and 30f (0.250 g, 0.604 mmol). The title compound was obtained as a yellow solid after purifying using flash chromatography (2–10% MeOH:DCM) followed by precipitation from EtOAc using hexanes. Yield 0.260 g, 84%. 1H NMR (400 MHz, DMSO-d6) δ 12.24 (s, 1H), 12.08 (s, 1H), 7.57 (t, J = 7.5 Hz, 1H), 7.42 (d, J = 8.2 Hz, 1H), 7.36 – 7.21 (m, 5H), 7.18 – 7.04 (m, 3H), 6.84 (dd, J = 8.5, 2.3 Hz, 2H), 5.32 (dd, J = 11.9, 4.4 Hz, 1H), 3.68 (dd, J = 18.4, 12.0 Hz, 1H), 2.74 (dd, J = 18.4, 4.5 Hz, 1H), 2.63 – 2.39 (m, 5H), 2.31 (t, J = 6.7 Hz, 2H). 13C NMR (150 MHz, DMSO-d6) δ 173.55, 168.65, 160.25, 152.86, 151.26, 141.13, 138.55, 137.66, 132.18, 131.51, 131.28, 129.24, 128.79, 128.69, 128.54, 128.26, 127.45, 123.25, 122.23, 119.43, 115.52, 58.17, 45.20, 28.79, 28.60, 28.24, 20.90. HRMS (m/z): [M+H]+ calcd for C29H25ClN3O4, 514.15281; found, 514.15260. Anal. Cacld for C29H24ClN3O4·0.80H2O: C, 65.92; H, 4.88; N, 7.95. Found C, 65.86; H, 4.84; N, 7.75.
4-{5-(4-Chlorophenyl)-3-[4-(4-methoxyphenyl)-2-oxo-1,2-dihydroquinolin-3-yl]-4,5-dihydro-1H-pyrazol-1-yl}-4-oxobutanoic acid (31)
Compound 31 was prepared according to general procedure G using 87 (0.047 g, 0.465 mmol) and 31f (0.200 g, 0.465 mmol). The title compound was obtained as a white solid by filtering from DCM after removal of the THF in vacuo. Yield 0.165 g, 67%. 1H NMR (400 MHz, DMSO-d6) δ 14.05 (s, 1H), 12.24 (s, 1H), 7.57 (t, J = 7.7 Hz, 1H), 7.42 (d, J = 8.3 Hz, 2H), 7.38 – 7.06 (m, 6H), 7.01 (d, J = 8.5 Hz, 1H), 6.83 (d, J = 7.8 Hz, 3H), 5.33 (dd, J = 11.8, 4.4 Hz, 1H), 3.85 (s, 3H), 3.67 (dd, J = 18.4, 12.1 Hz, 1H), 2.78-2.53 (m, 2H), 2.41 – 2.22 (m, 2H). 13C NMR (100 MHz, DMSO-d6) δ 175.22, 173.68, 168.75, 160.42, 160.28, 159.20, 152.91, 151.06, 141.19, 138.54, 131.50, 131.26, 131.01, 130.92, 130.00, 128.39, 128.24, 127.68, 127.44, 127.00, 123.52, 122.23, 119.66, 115.54, 113.68, 113.56, 58.16, 55.17, 45.24, 32.60, 28.76, 28.44. HRMS (m/z): [M+K]+ calcd for C29H24ClN3O5, 568.10361; found, 568.10341. Anal. Calcd for C29H24ClN3O5: C, 65.72; H, 4.56; N, 7.92. Found C, 57.27; H, 4.20; N, 6.50. HPLC 85% MeOH:H2O (0.1% Formic Acid) Rt 0.89 min; > 95% purity; 75% ACN: H2O (0.1% Formic Acid) Rt 0.67 min; > 95% purity.
4-{5-(4-Chlorophenyl)-3-[4-(4-cyanophenyl)-2-oxo-1,2-dihydroquinolin-3-yl]-4,5-dihydro-1H-pyrazol-1-yl}-4-oxobutanoic acid (32)
Compound 32 was prepared according to general procedure G using 87 (0.059 g, 0.588 mmol) and 32f (0.150 g, 0.588 mmol). The title compound was obtained as a yellow solid after precipitation from EtOAc using hexanes followed by purification using flash chromatography with 10% MeOH:DCM as a yellow solid. Yield 0.150 g, 48.6%. 1H NMR (400 MHz, DMSO-d6) δ 12.33 (s, 1H), 12.09 (s, 1H), 7.97 (ddd, J = 11.8, 7.7, 1.7 Hz, 2H), 7.64 – 7.56 (m, 2H), 7.50 (dd, J = 7.8, 1.7 Hz, 1H), 7.43 (d, J = 8.1 Hz, 1H), 7.35 – 7.27 (m, 2H), 7.14 (t, J = 7.7 Hz, 1H), 6.95 (dd, J = 8.2, 1.2 Hz, 1H), 6.89 – 6.84 (m, 2H), 5.35 (dd, J = 12.0, 4.4 Hz, 1H), 3.79 (dd, J = 18.5, 12.0 Hz, 1H), 2.89 (dd, J = 18.5, 4.5 Hz, 1H), 2.57 – 2.23 (m, 4H). 13C NMR (150 MHz, DMSO-d6) δ 173.45, 168.70, 160.12, 152.50, 149.37, 140.99, 140.55, 138.61, 132.20, 131.62, 130.59, 130.50, 129.79, 128.37, 128.25, 127.32, 127.23, 123.18, 122.52, 118.75, 118.63, 115.65, 111.18, 58.30, 45.00, 28.77, 28.22. HRMS (m/z): [M-H]− calcd for C29H20ClN4O4, 523.11786; found, 523.11828. HPLC 85% MeOH:H2O (0.1% Formic Acid) Rt 0.67 min; 87% purity; 75% ACN: H2O (0.1% Formic Acid) Rt 0.89 min; 85% purity.
4-[5-(4-Chlorophenyl)-3-{2-oxo-4-(4-(trifluoromethyl)phenyl)-1,2-dihydroquinolin-3-yl}-4,5-dihydro-1H-pyrazol-1-yl]-4-oxobutanoic acid (33)
Compound 33 was prepared according to general procedure G using 87 (0.039 g, 0.385mmol) and 33f (0.180 g, 0.220 mmol). The title compound was obtained after flash column chromatography using 0–10% MeOH:DCM and precipitation from EtOAc using hexanes as a yellow solid. Yield 0.125 g, 57.2%. 1H NMR (400 MHz, DMSO-d6) δ 12.34 (s, 1H), 12.07 (s, 1H), 7.90 (d, J = 8.1 Hz, 1H), 7.85 (d, J = 8.1 Hz, 1H), 7.65 (d, J = 8.1 Hz, 1H), 7.65 (d, J = 8.1 Hz, 1H), 7.59 (t, J = 7.6 Hz, 1H), 7.53 (d, J = 8.1 Hz, 1H), 7.45 (d, J = 8.2 Hz, 1H), 7.26 (d, J = 8.2 Hz, 2H), 7.15 (t, J = 7.7 Hz, 1H), 6.98 (d, J = 8.1 Hz, 1H), 6.86 (d, J = 8.2 Hz, 2H), 5.36 (dd, J = 12.0, 4.6 Hz, 1H), 3.80 (dd, J = 18.4, 12.0 Hz, 1H), 2.91 (dd, J = 18.5, 4.8 Hz, 1H), 2.52 - 2.21 (m, 4H). 13C NMR (100 MHz, DMSO-d6) δ 173.42, 168.68, 160.19, 152.58, 149.61, 141.06, 139.85, 138.62, 131.62, 130.35, 129.69, 128.31, 127.27, 125.20, 123.28, 122.54, 118.95, 115.63, 58.23, 45.04, 28.32, 28.05. 19F NMR (376 MHz, DMSO-d6) δ −61.595 (s). HRMS (m/z): [M-H]− calcd for C29H20ClF3N3O4, 566.10999; found, 566.11036. Anal. Clacd for C29H21ClF3N3O4·0.40H2O: C, 60.56; H, 3.82; N, 7.31. Found C, 60.57; H, 4.00; N: 7.23.
4-{5-(4-Chlorophenyl)-3-[4-(3-fluorophenyl)-2-oxo-1,2-dihydroquinolin-3-yl]-4,5-dihydro-1H-pyrazol-1-yl}-4-oxobutanoic acid (34)
Compound 34 was prepared according to general procedure G using 87 (0.060 g, 0.598 mmol) and 34f (0.250 g, 0.367 mmol). The title compound was obtained after precipitating from EtOAc using hexanes followed by flash column chromatography using 10% MeOH:DCM, as a yellow solid. Yield 0.190 g, 61.3%. 1H NMR (400 MHz, DMSO-d6) δ 12.24 (s, 1H), 12.08 (s, 1H), 7.57 (t, J = 7.5 Hz, 1H), 7.42 (d, J = 8.2 Hz, 1H), 7.36 - 7.21 (m, 5H), 7.18 - 7.04 (m, 3H), 6.84 (d, J = 8.2 Hz, 2H), 5.32 (dd, J = 12.0, 4.5 Hz, 1H), 3.68 (dd, J = 18.5, 12.0 Hz, 1H), 2.74 (dd, J = 18.5, 4.5 Hz, 1H), 2.52 - 2.39 (m, 4H). 13C NMR (100 MHz DMSO-d6) δ 173.48, 168.60, 160.19, 149.62, 141.15, 138.53, 131.53, 131.45, 130.35, 128.37, 127.33, 127.16, 122.44, 119.07, 115.57, 58.27, 45.11, 28.79, 28.47, 28.18. 19F NMR (376 MHz, DMSO-d6) δ −113.29 – −113.63 (m). HRMS (m/z): [M+Na]+ calcd for C28H21ClFN3O4Na, 540.10968; found, 540.11014. Anal. Calcd for C28H21ClFN3O4·0.90H2O: C, 62.96; H, 4.30; N, 7.87. Found C, 63.04; H, 4.38; N, 7.55.
4-{5-(4-Chlorophenyl)-3-[4-(3-chlorophenyl)-2-oxo-1,2-dihydroquinolin-3-yl]-4,5-dihydro-1H-pyrazol-1-yl}-4-oxobutanoic acid (35)
Compound 35 was prepared according to general procedure G using 87 (0.069 g, 0.691 mmol) and 35f (0.300 g, 0.691 mmol). The title compound was obtained as a yellow solid after flash column chromatography using 2–10% MeOH:DCM. Yield 0.170 g, 46.1%. 1H NMR (600 MHz, DMSO-d6) δ 12.25 (s, 1H), 12.04 (s, 1H), 7.58 – 7.51 (m, 2H), 7.51 – 7.46 (m, 1H), 7.39 (d, J = 8.2 Hz, 1H), 7.36 – 7.32 (m, 1H), 7.25 (ddt, J = 7.1, 4.9, 2.5 Hz, 2H), 7.19 (d, J = 7.5 Hz, 1H), 7.12 (t, J = 7.7 Hz, 1H), 7.01 – 6.94 (m, 1H), 6.81 – 6.73 (m, 2H), 5.34 – 5.28 (m, 1H), 3.83 – 3.74 (m, 1H), 2.84 – 2.70 (m, 1H), 2.52 – 2.34 (m, 2H), 2.31 – 2.24 (m, 2H). 13C NMR (100 MHz, DMSO-d6) δ 173.50, 168.64, 160.20, 152.66, 149.46, 141.16, 138.53, 137.44, 137.28, 133.18, 131.56, 131.47, 130.15, 130.00, 129.59, 128.49, 128.40, 127.29, 127.16, 123.43, 122.48, 119.17, 115.58, 58.30, 45.10, 28.78, 28.49, 28.19. HRMS (m/z): [M+Na]+ calcd for C28H21Cl2N3O4Na, 556.08013; found, 556.07990. Anal. Calcd for C28H21Cl2N3O4·0.70H2O: C, 61.48; H, 4.13; N, 7.68. Found C, 61.51; H, 4.11; N: 7.48.
4-{5-(4-Chlorophenyl)-3-[2-oxo-4-(m-tolyl)-1,2-dihydroquinolin-3-yl]-4,5-dihydro-1H-pyrazol-1-yl}-4-oxobutanoic acid (36)
Compound 36 was prepared according to general procedure G using 87 (0.060 g, 0.604 mmol) and 36f (0.250 g, 0.604 mmol). The title compound was obtained as a yellow solid after flash column chromatography using 0–10% MeOH:DCM followed by precipitation from hot EtOAc and hexanes. Yield 0.240 g, 77.0%.1H NMR (400 MHz, DMSO-d6) δ 12.21 (s, 1H), 12.05 (s, 1H), 7.53 (t, J = 7.6 Hz, 1H), 7.42 – 7.27 (m, 3H), 7.27 – 7.17 (m, 2H), 7.16 – 7.06 (m, 2H), 7.00 (q, J = 10.2, 8.9 Hz, 2H), 6.80 – 6.67 (m, 2H), 5.33 – 5.23 (m, 1H), 3.80 – 3.62 (m, 1H), 2.76 (dd, J = 18.4, 4.6 Hz, 0.5H), 2.64 (dd, J = 18.5, 4.6 Hz, 0.5H), 2.57 – 2.34 (m, 5H), 2.31 – 2.18 (m, 2H). 13C NMR (150 MHz, DMSO-d6) δ 173.61, 173.54, 168.63, 168.57, 160.29, 152.88, 152.70, 151.33, 151.24, 141.25, 141.20, 138.52, 137.49, 137.43, 135.17, 135.03, 131.53, 131.30, 130.25, 128.84, 128.36, 128.32, 128.13, 127.50, 127.41, 127.34, 127.26, 126.39, 125.46, 123.16, 122.28, 119.42, 115.55, 115.51, 58.28, 58.23, 45.25, 28.90, 28.79, 28.67, 28.57, 28.35, 28.24, 28.14, 21.09, 21.02, 20.97, 20.91. HRMS (m/z): [M+H]+ calcd for C29H25ClN3O4, 514.15281; found, 514.15263. Anal. Calcd for C29H24ClN3O4·1.50H2O·0.06EtOAc: C, 64.29; H, 5.07; N, 7.69. Found C, 64.65; H, 4.90; N, 7.29.
4-{5-(4-Chlorophenyl)-3-[4-(3-methoxyphenyl)-2-oxo-1,2-dihydroquinolin-3-yl]-4,5-dihydro-1H-pyrazol-1-yl}-4-oxobutanoic acid (37)
Compound 37 was prepared according to general procedure G using 87 (0.070 g, 0.698 mmol) and 37f (0.300g, 0.698 mmol). The title compound was obtained as a yellow solid after flash column chromatography using 0–10% MeOH:DCM. Yield 0.090 g, 24.3%. 1H NMR (600 MHz, DMSO-d6) δ 12.23 (s, 1H), 12.03 (s, 1H), 7.57 (t, J = 7.8 Hz, 1H), 7.48 – 7.35 (m, 2H), 7.32 – 7.21 (m, 2H), 7.18 – 7.12 (m, 1H), 7.09 (t, J = 8.3 Hz, 2H), 6.95 (d, J = 9.9 Hz, 1H), 6.85 – 6.72 (m, 3H), 5.37 – 5.30 (m, 1H), 3.84 – 3.64 (m, 4H), 2.83 – 2.72 (m, 1H), 2.60 – 2.51 (m, 2H), 2.31 (t, J = 7.1 Hz, 2H). 13C NMR (150 MHz, DMSO-d6) δ 173.52, 168.60, 160.26, 159.03, 152.90, 150.95, 141.24, 141.19, 138.52, 138.50, 136.50, 136.46, 131.56, 131.48, 131.33, 131.31, 129.43, 129.38, 128.35, 128.21, 128.14, 127.46, 127.43, 127.19, 123.24, 123.14, 122.32, 121.65, 120.70, 119.31, 119.27, 115.51, 115.13, 114.02, 113.88, 113.58, 58.25, 55.26, 54.99, 45.30, 45.19, 28.59, 28.22. HRMS (m/z): [M+H]+ calcd for C29H25ClN3O5, 530.14773; found, 530.14715. Anal. Calcd for C29H24ClN3O5: C, 65.72; H, 4.56; N, 7.93. Found C, 65.52; H, 4.72; N, 7.97.
4-{5-(4-Chlorophenyl)-3-[4-(3-cyanophenyl)-2-oxo-1,2-dihydroquinolin-3-yl]-4,5-dihydro-1H-pyrazol-1-yl}-4-oxobutanoic acid (38)
Compound 38 was prepared according to general procedure G using 87 (0.021g, 0.212mmol) and 38f (0.090 g, 0.212 mmol). The title compound was obtained as a yellow solid after flash column chromatography using 0–10% MeOH:DCM. Yield 0.034 g, 31.0%. 1H NMR (400 MHz, DMSO-d6) δ 12.35 (s, 1H), 12.10 (s, 1H), 8.04 – 7.91 (m, 1H), 7.84 – 7.67 (m, 2H), 7.65 – 7.55 (m, 2H), 7.43 (d, J = 8.2 Hz, 1H), 7.31 (d, J = 8.3 Hz, 2H), 7.16 (t, J = 7.7 Hz, 1H), 6.97 (d, J = 8.2 Hz, 1H), 6.82 (m, 2H), 5.40 – 5.32 (m, 1H), 3.96 – 3.81 (m, 1H), 2.87 (dt, J = 18.3, 4.1 Hz, 1H), 2.42 (d, J = 7.6 Hz, 2H), 2.35 – 2.26 (m, 2H). 13C NMR (150 MHz, DMSO-d6) δ 173.58, 173.42, 168.61, 160.13, 152.58, 148.90, 141.02, 138.54, 136.80, 136.70, 134.66, 133.48, 133.05, 132.18, 132.09, 131.73, 131.57, 129.51, 129.32, 128.47, 128.41, 127.34, 127.25, 127.04, 123.64, 123.53, 122.54, 119.03, 118.52, 115.59, 111.60, 111.40, 58.27, 45.03, 28.75, 28.32, 28.10. HRMS (m/z): [M-H]− calcd for C29H20ClN4O4, 523.11786; found, 523.11778. Anal. Calcd for C29H21ClN4O4: C, 66.35; H, 4.03; N, 10.67. Found C, 63.97; H, 4.59; N, 9.39. HPLC 85% MeOH:H2O (0.1% Formic Acid) Rt 0.68 min; > 95% purity; 75% ACN: H2O (0.1% Formic Acid) Rt 0.59 min; > 95% purity.
4-[5-(4-Chlorophenyl)-3-{2-oxo-4-[3-(trifluoromethyl)phenyl]-1,2-dihydroquinolin-3-yl}-4,5-dihydro-1H-pyrazol-1-yl]-4-oxobutanoic acid (39)
Compound 39 was prepared according to general procedure G using 87 (0.054 g, 0.53mmol) and 39f (0.250 g, 0.53 mmol). The title compound was obtained as a yellow solid using a 0–8% MeOH:DCM. Yield 0.073 g, 24%. 1H NMR (400 MHz, DMSO-d6) δ 12.34 (s, 1H), 12.06 (s, 1H), 7.96-7.84 (m, 1H), 7.82 – 7.66 (m, 2H), 7.59 (t, J = 8.4 Hz, 2H), 7.44 (d, J = 8.3 Hz, 1H), 7.28 (d, J = 8.2 Hz, 1H), 7.23 (d, J = 8.1 Hz, 1H), 7.20 – 7.13 (m, 1H), 6.98 (d, J = 8.2 Hz, 0.5H), 6.92 (d, J = 8.2 Hz, 0.5H), 6.84 (d, J = 8.1 Hz, 1H), 6.76 (d, J = 8.0 Hz, 1H), 5.34 (dt, J = 12.2, 4.3 Hz, 1H), 3.95 – 3.70 (m, 1H), 3.00 – 2.74 (m, 1H), 2.54 – 2.23 (m, 4H). 13C NMR (150 MHz, DMSO-d6) δ 173.40, 173.34, 168.62, 168.52, 160.20, 160.17, 152.67, 152.61, 149.38, 141.10, 141.03, 138.57, 136.57, 136.47, 133.59, 132.57, 131.60, 131.51, 129.50, 129.33, 129.15, 128.92, 128.49, 128.38, 128.30, 127.67, 127.25, 127.17, 127.13, 126.47, 125.07, 123.62, 123.45, 122.54, 122.36, 119.26, 119.11, 115.61, 58.26, 58.21, 45.12, 45.01, 28.37, 28.29, 28.19, 28.08. HRMS (m/z): [M+H]+ calcd for C29H22ClF3N3O4, 568.12455; found, 568.12417. Anal. Calcd for C29H21ClF3N3O4·0.20H2O; C, 60.94; H, 3.77; N, 7.35. Found C, 60.79; H, 3.96; N, 7.33.
4-{3-[4-(4-Chloro-3-fluorophenyl)-2-oxo-1,2-dihydroquinolin-3-yl]-5-(4-chlorophenyl)-4,5-dihydro-1H-pyrazol-1-yl}-4-oxobutanoic acid (40)
Compound 40 was prepared according to general procedure G using 87 (0.040 g, 0.398 mmol) and 40f (0.180 g, 0.398 mmol). The title compound was obtained after flash column chromatography using 0–10%MeOH:DCM as a yellow solid followed by precipitation from EtOAc and hexanes. Yield 0.096 g, 43.7%. 1H NMR (400 MHz, DMSO-d6) δ 12.33 (s, 1H), 12.12 (s, 1H), 7.79 – 7.64 (m, 1H), 7.62 – 7.51 (m, 2H), 7.46 – 7.38 (m, 2H), 7.33 – 7.23 (m, 2H), 7.21 – 7.00 (m, 2H), 6.93 – 6.78 (m, 2H), 5.39 (dt, J = 12.1, 4.4 Hz, 1H), 3.85 – 3.72 (m, 1H), 2.91 – 2.79 (m, 1H), 2.66 – 2.39 (m, 2H), 2.34 (td, J = 7.0, 2.2 Hz, 2H). 13C NMR (150 MHz, DMSO-d6) δ 173.61, 173.47, 173.44, 168.68, 168.66, 168.09, 168.05, 160.11, 157.84, 156.19, 152.77, 152.70, 152.63, 148.67, 141.03, 140.99, 138.55, 136.45, 136.36, 136.30, 131.63, 131.57, 130.63, 130.53, 128.34, 128.28, 127.32, 127.21, 127.12, 127.00, 126.21, 123.57, 123.45, 122.54, 119.69, 119.58, 118.92, 117.28, 115.58, 58.28, 58.17, 45.15, 28.77, 28.39, 28.17. 19F NMR (376 MHz, DMSO-d6) δ −116.32 – −116.51 (m). HRMS (m/z): [M+K]+ calcd for C28H20Cl2FN3O4K, 590.04465; found, 590.04631. Anal. Calcd for C28H20Cl2FN3O4 ·0.80H2O·0.10EtOAc: C, 59.26; H, 3.92; N, 7.30. Found C, 59.16; H, 4.21; N, 7.14.
4-{3-[4-(3-Chloro-4-fluorophenyl)-2-oxo-1,2-dihydroquinolin-3-yl]-5-(4-chlorophenyl)-4,5-dihydro-1H-pyrazol-1-yl}-4-oxobutanoic acid (41)
Compound 41 was prepared according to general procedure G using 87 (0.040 g, 0.398 mmol) and 41f (0.180 g, 0.398 mmol). The title compound was obtained after purifying using flash column chromatography using 0–10%MeOH:DCM as a yellow solid. Yield 0.120 g, 54.6%. 1H NMR (400 MHz, DMSO-d6) δ 12.32 (s, 1H), 12.12 (s, 1H), 7.73 – 7.50 (m, 3H), 7.44 (dd, J = 8.2, 1.7 Hz, 2H), 7.29 (td, J = 8.8, 2.0 Hz, 2H), 7.17 (t, J = 7.6 Hz, 1H), 7.09 – 7.01 (m, 1H), 6.88 (dt, J = 8.6, 2.4 Hz, 2H), 5.38 (dd, J = 10.4, 4.6 Hz, 1H), 3.90 – 3.76 (m, 1H), 2.93 – 2.77 (m, 1H), 2.66 – 2.40 (m, 2H), 2.40 – 2.28 (m, 2H). 13C NMR (100 MHz, DMSO-d6) δ 173.63, 173.48, 173.44, 168.68, 160.15, 155.93, 152.68, 152.60, 148.76, 148.72, 141.01, 138.50, 133.00, 132.90, 131.93, 131.62, 131.57, 131.50, 130.63, 130.51, 129.53, 128.39, 128.34, 127.38, 127.23, 127.11, 123.74, 123.66, 122.50, 119.77, 119.46, 119.24, 116.87, 116.66, 115.56, 58.25, 45.10, 28.78, 28.43, 28.38, 28.15. HRMS (m/z): [M+H]+ calcd for C28H21Cl2FN3O4, 552.08877; found, 552.09030. Anal. Calcd for C28H20Cl2FN3O4: C, 60.88; H, 3.64; N, 7.61. Found C, 58.29; H, 4.04; N, 6.66. HPLC 85% MeOH:H2O (0.1% Formic Acid) Rt 0.93 min; > 95% purity; 75% ACN: H2O (0.1% Formic Acid) Rt 0.55 min; > 95% purity.
4-{5-(4-Chlorophenyl)-3-[4-(3,4-dichlorophenyl)-2-oxo-1,2-dihydroquinolin-3-yl]-4,5-dihydro-1H-pyrazol-1-yl}-4-oxobutanoic acid (42)
Compound 42 was prepared according to general procedure G using 87 (0.043 g, 0.427 mmol) and 42f (0.200 g, 0.427 mmol). The title compound was obtained after purifying flash column chromatography using 0–10% MeOH:DCM as a yellow solid. Yield 0.102 g, 42.0%. 1H NMR (400 MHz, DMSO-d6) δ 12.31 (s, 1H), 12.10 (s, 1H), 7.83 – 7.70 (m, 1H), 7.63 – 7.54 (m, 2H), 7.46 – 7.37 (m, 2H), 7.32 – 7.22 (m, 2H), 7.16 (t, J = 7.7 Hz, 1H), 7.04 (t, J = 7.0 Hz, 1H), 6.87 – 6.80 (m, 2H), 5.37 (dd, J = 12.0, 4.3 Hz, 1H), 3.88 – 3.74 (m, 1H), 2.90 – 2.74 (m, 1H), 2.53 – 2.39 (m, 2H), 2.38 – 2.29 (m, 2H). 13C NMR (150 MHz, DMSO-d6) δ 174.04, 169.33, 160.76, 153.24, 149.13, 141.66, 139.20, 136.68, 132.40, 132.27, 132.19, 131.96, 131.08, 130.76, 129.73, 129.05, 128.98, 127.99, 127.83, 127.74, 123.18, 119.69, 116.23, 58.93, 45.75, 29.62, 29.11, 29.07, 28.86. HRMS (m/z): [M+H]+ calcd for C28H21Cl3N3O4, 568.05922; found, 568.06168. Anal. Calcd for C28H20Cl3N3O4 ·0.80H2O: C, 57.66; H, 3.73; N, 7.20. Found C, 57.70; H, 3.65; N: 6.94.
4-{5-(4-Chlorophenyl)-3-[4-(3,4-difluorophenyl)-2-oxo-1,2-dihydroquinolin-3-yl]-4,5-dihydro-1H-pyrazol-1-yl}-4-oxobutanoic acid (43)
Compound 43 was prepared according to general procedure G using 87 (0.0427 g, 0.427 mmol) and 43f (0.200 g, 0.427 mmol). The title compound was obtained after flash column chromatography using 0–10% MeOH:DCM as a yellow solid. Yield 0.110 g, 68.8%. 1H NMR (400 MHz, DMSO-d6) δ 12.31 (s, 1H), 12.13 (s, 1H), 7.63 – 7.52 (m, 3H), 7.46 – 7.39 (m, 1H), 7.35 – 7.28 (m, 1H), 7.29 – 7.23 (m, 2H), 7.20 – 7.10 (m, 1H), 7.06 (d, J = 8.2 Hz, 1H), 6.95 – 6.84 (m, 2H), 5.43 – 5.33 (m, 1H), 3.88 – 3.73 (m, 1H), 2.86 (dd, J = 18.5, 4.4 Hz, 1H), 2.66 – 2.29 (m, 4H). 13C NMR (150 MHz, DMSO-d6) δ 174.08, 169.36, 160.79, 153.45, 149.53, 141.69, 139.20, 139.20, 132.28, 132.14, 128.99, 128.91, 127.98, 127.80, 124.26, 123.12, 119.83, 116.22, 58.92, 45.79, 29.07, 28.84. 19F NMR (376 MHz, DMSO-d6) δ −5.88 – −6.23 (m), −6.44 – −6.75 (m). HRMS (m/z): [M-H]− calcd for C28H19ClF2N3O4, 534.10376; found, 534.10358. Anal. Calcd for C28H20ClF2N3O4 ·0.40H2O: C, 61.92; H, 3.86; N, 7.74. found C, 61.76; H, 4.15; N, 7.52.
4-{5-(4-Chlorophenyl)-3-[4-(3,5-difluorophenyl)-2-oxo-1,2-dihydroquinolin-3-yl]-4,5-dihydro-1H-pyrazol-1-yl}-4-oxobutanoic acid (44)
Compound 44 was prepared according to general procedure G using 87 (0.041 g, 0.413 mmol) and 44f (0.180 g, 0.413 mmol). The title compound as a yellow solid was obtained after purifying using flash column chromatography with 0–10% MeOH:DCM. Yield 0.133 g, 56.5%. 1H NMR (400 MHz, DMSO-d6) δ 12.33 (s, 1H), 12.07 (s, 1H), 7.59 (t, J = 7.7 Hz, 1H), 7.47 - 7.33 (m, 2H), 7.29 (d, J = 8.2 Hz, 2H), 7.23 (d, J = 8.5 Hz, 1H), 7.17 (t, J = 7.6 Hz, 1H), 7.08 (d, J = 8.0 Hz, 2H), 6.89 (d, J = 8.1 Hz, 2H), 5.40 (dd, J = 12.0, 4.6 Hz, 1H), 3.86 (dd, J = 18.4, 12.0 Hz, 1H), 2.90 (dd, J = 18.5, 4.6 Hz, 1H), 2.59 - 2.41 (m, 2H), 2.33 (t, J = 6.8 Hz, 2H). 13C NMR (100 MHz, DMSO-d6) δ 173.65, 173.46, 168.64, 163.55, 163.43, 161.11, 160.95, 160.11, 152.68, 148.60, 141.09, 139.05, 138.95, 138.52, 131.58, 128.55, 128.40, 127.26, 127.13, 123.41, 122.57, 118.79, 115.58, 113.31, 113.12, 112.32, 112.12, 103.85, 58.30, 45.02, 28.80, 28.37, 28.15. 19F (376 MHz, DMSO-d6) δ −109.98 - −110.05 (m). HRMS (m/z): [M-H]− calcd for C28H19ClF2N3O4, 534.10376; found, 534.10402. Anal. Calcd for C28H20ClF2N3O4·0.50H2O: C, 61.71, H: 3.88, N: 7.71; C: 61.52, H: 4.05, N: 7.42.
4-{5-(4-Chlorophenyl)-3-[4-(3,5-dichloro(phenyl)-2-oxo-1,2-dihydroquinolin-3-yl]-4,5-dihydro-1H-pyrazol-1-yl}-4-oxobutanoic acid (45)
Compound 45 was prepared according to general procedure G using 87 (0.038 g, 0.384 mmol) and 45f (0.180 g, 0.384 mmol). The title compound was obtained as a yellow solid after purifying using flash column chromatography with 0–10% MeOH:DCM. Yield 0.080 g, 36.6%. 1H NMR (400 MHz, DMSO-d6) δ 12.33 (s, 1H), 12.10 (s, 1H), 7.84 - 7.71 (m, 1H), 7.64 −7.56 (m, 2H), 7.51 - 7.39 (m, 2H), 7.29 (t, J = 7.3 Hz, 2H), 7.17 (t, J = 7.6 Hz, 1H), 7.06 (t, J = 6.9 Hz, 1H), 6.84 (d, J = 8.1 Hz, 2H), 5.39 (dd, J =12.0, 4.3 Hz, 1H), 3.89 - 3.75 (m, 1H), 2.85 – 2.67 (m, 1H), 2.64 - 2.30 (m, 4H). 13C NMR (100 MHz, DMSO-d6) δ 173.46, 168.66, 160.11, 152.60, 152.53, 148.48, 141.01, 138.52, 136.01, 131.77, 131.59, 131.31, 131.11, 130.39, 130.13, 129.07, 128.40, 128.33, 127.35, 127.17, 127.06, 122.55, 119.02, 115.57, 58.24, 45.09, 28.43, 28.38, 28.17. HRMS calcd for C28H19Cl3N3O4 [M-H]−; 566.04466, found; 566.04483. Anal. C28H20Cl3N3O4·0.30H2O; C, 58.57; H, 3.62; N, 7.32. Found C, 58.58; H, 3.75; N, 7.23.
4-{5-(4-Chlorophenyl)-3-[4-(4-chlorophenyl)-6-methyl-2-oxo-1,2-dihydroquinolin-3-yl]-4,5-dihydro-1H-pyrazol-1-yl}-4-oxobutanoic acid (46)
Compound 46 was prepared according to general procedure G using 87 (0.039 g, 0.39 mmol) and 46f (0.175 g, 0.39 mmol). The title compound was obtained after flash column chromatography using 0–8% MeOH:DCM as a yellow solid. Yield 0.114 g, 53.3%. 1H NMR (400 MHz, DMSO-d6) δ 12.18 (s, 1H), 11.79 (s, 1H), 7.56 (dt, J = 8.2, 2.3 Hz, 1H), 7.48 (dt, J = 8.2, 2.3 Hz, 1H), 7.41 – 7.34 (m, 2H), 7.30 (dd, J = 8.3, 2.2 Hz, 1H), 7.27 – 7.20 (m, 3H), 6.80 – 6.72 (m, 3H), 5.35 – 5.25 (m, 1H), 3.75 – 3.59 (m, 1H), 2.76 – 2.65 (m, 1H), 2.61 – 2.33 (m, 2H), 2.26 – 2.32 (m, 2H), 2.19 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δ 173.54, 168.64, 160.00, 152.85, 149.66, 141.10, 136.63, 134.07, 133.16, 132.79, 131.56, 131.48, 131.40, 130.53, 128.37, 128.30, 127.30, 126.53, 123.47, 119.05, 115.59, 58.18, 45.18, 28.47, 28.19, 20.58. HRMS (m/z): [M-H]− calcd for C29H22Cl2N3O4, 546.09929; found, 546.09872. Anal. Calcd for C29H23Cl2N3O4·0.50H2O: C, 62.49; H, 4.34; N, 7.54. Found C, 62.44; H, 4.55; N 7.39.
4-{5-(4-Chlorophenyl)-3-[4-(3-fluorophenyl)-6-methyl-2-oxo-1,2-dihydroquinolin-3-yl]-4,5-dihydro-1H-pyrazol-1-yl}-4-oxobutanoic acid (47)
Compound 47 was prepared according to general procedure G using 87 (0.037 g, 0.37 mmol) and 47f (0.160 g, 0.37 mmol). The title compound was obtained after flash column chromatography using 0–10% MeOH:DCM as a brown solid. Yield 0.094 g, 47.7%. 1H NMR (400 MHz, DMSO-d6) δ 12.24 (s, 1H), 12.12 (s, 1H), 7.64 – 7.47 (m, 1H), 7.42 (d, J = 8.5 Hz, 1H), 7.40 – 7.32 (m, 2H), 7.32 – 7.22 (m, 3H), 7.11 (dd, J = 23.8, 8.5 Hz, 1H), 6.86 – 6.77 (m, 3H), 5.35 (dt, J = 12.5, 3.4 Hz, 1H), 3.80 (dd, J = 18.6, 12.1 Hz, 1H), 2.81 (dt, J = 18.5, 5.2 Hz, 1H), 2.53 – 2.39 (m, 2H), 2.31 (t, J = 6.8 Hz, 2H), 2.23 (s, 3H). 13C NMR (150 MHz, DMSO-d6) δ 173.46, 168.57, 162.67, 161.11, 160.03, 152.88, 152.81, 149.39, 141.15, 137.59, 136.61, 132.77, 131.58, 131.52, 131.38, 130.25, 128.36, 128.20, 127.33, 127.15, 126.57, 126.52, 125.83, 124.79, 123.38, 123.30, 118.98, 116.66, 116.52, 115.57, 115.19, 115.05, 58.25, 45.16, 28.98, 28.47, 28.19, 20.61. 19F NMR (376 MHz, DMSO-d6) δ −113.28 – −113.50 (m). HRMS (m/z): [M-H]− calcd for C29H22ClFN3O4, 530.12884; found, 530.12883. Anal. Calcd for C29H23ClFN3O4·1.20H2O: C, 62.92; H, 4.62; N, 7.59. Found C, 62.82; H, 4.37; N, 7.34.
4-{3-[6-Chloro-4-(4-chlorophenyl)-2-oxo-1,2-dihydroquinolin-3-yl]-5-(4-chlorophenyl)-4,5-dihydro-1H-pyrazol-1-yl}-4-oxobutanoic acid (48)
Compound 48 was prepared according to general procedure G using 87 (0.038 g, 0.38 mmol) and 48f (0.180 g, 0.38 mmol). The title compound was obtained as a yellow solid after flash column chromatography using 0–8% MeOH:DCM. Yield 0.033 g, 15.1%. 1H NMR (400 MHz, DMSO-d6) δ 12.44 (s, 1H), 12.08 (s, 1H), 7.69 – 7.58 (m, 2H), 7.54 (dd, J = 8.2, 2.3 Hz, 1H), 7.44 (dd, J = 8.5, 2.7 Hz, 2H), 7.35 – 7.23 (m, 3H), 6.93 (d, J = 2.3 Hz, 1H), 6.85 – 6.76 (m, 2H), 5.35 (dd, J = 12.0, 4.4 Hz, 1H), 3.72 (dd, J = 18.5, 12.0 Hz, 1H), 2.76 (dd, J = 18.5, 4.4 Hz, 1H), 2.64 – 2.38 (m, 2H), 2.33 (t, J = 6.7 Hz, 2H). 13C NMR (150 MHz, DMSO-d6) δ 173.50, 168.71, 159.96, 152.33, 148.76, 141.00, 137.33, 133.52, 133.36, 131.61, 131.48, 131.38, 130.52, 128.52, 128.32, 127.27, 126.25, 125.91, 124.80, 120.43, 117.67, 58.25, 45.08, 28.47, 28.19. HRMS (m/z): [M+H]+ calcd for C28H21Cl3N3O4, 568.05922; found, 568.05881. Anal. Calcd for C28H20Cl3N3O4·0.40H2O: C, 58.38; H, 3.64; N, 7.29. Found C, 58.29; H, 3.73; N, 7.24.
4-{3-[6-Chloro-4-(3-fluorophenyl)-2-oxo-1,2-dihydroquinolin-3-yl]-5-(4-chlorophenyl)-4,5-dihydro-1H-pyrazol-1-yl}-4-oxobutanoic acid (49)
Compound 49 was prepared according to general procedure G using 87 (0.035 g, 0.35 mmol) and 49f (0.160 g, 0.35 mmol). The title compound was obtained as a yellow solid after flash column chromatography using 0–8% MeOH:DCM. Yield 0.039 g, 20.0%. 1H NMR (600 MHz, DMSO-d6) δ 12.45 (s, 1H), 12.05 (s, 1H), 7.65 (dd, J = 8.8, 2.5 Hz, 1H), 7.63 – 7.53 (m, 1H), 7.45 (d, J = 8.8 Hz, 1H), 7.42 – 7.32 (m, 2H), 7.30 (d, J = 8.2 Hz, 1H), 7.29 – 7.25 (m, 1H), 7.19 (dd, J = 9.2, 2.1 Hz, 1H), 7.12 (d, J = 7.5 Hz, 1H), 6.94 (d, J = 2.4 Hz, 1H), 6.83 (dd, J = 11.9, 8.3 Hz, 2H), 5.36 (dt, J = 12.2, 4.6 Hz, 1H), 2.88 – 2.80 (m, 1H), 2.51 – 2.44 (m, 2H), 2.31 (t, J = 6.9 Hz, 2H). 13C NMR (150 MHz, DMSO-d6) δ 174.13, 169.33, 163.45, 161.82, 160.66, 153.02, 152.95, 149.14, 141.73, 137.95, 137.52, 132.03, 131.21, 129.05, 127.96, 127.78, 126.91, 126.57, 125.39, 125.30, 121.01, 118.31, 116.19, 58.96, 45.71, 29.12, 28.85. HRMS (m/z): [M-H]− calcd for C28H19Cl2FN3O4, 550.07421; found, 550.07485. Anal. Calcd for C28H20Cl2FN3O4·1.00H2O: C, 58.96; H, 3.89; N, 7.37. Found C, 58.98; H, 3.78; N, 7.06.
4-{5-(4-Chlorophenyl)-3-[4-(4-chlorophenyl)-6-fluoro-2-oxo-1,2-dihydroquinolin-3-yl]-4,5-dihydro-1H-pyrazol-1-yl}-4-oxobutanoic acid (50)
Compound 50 was prepared according to general procedure G using 87 (0.035 g, 0.35 mmol) and 50f (0.19 g, 0.42 mmol). The title compound was obtained after flash column chromatography using 0–10% MeOH:DCM as a yellow solid. Yield 0.151 g, 65.1%. 1H NMR (600 MHz, DMSO-d6) δ 12.37 (s, 1H), 12.07 (s, 1H), 7.61 (dd, J = 8.2, 2.4 Hz, 1H), 7.56 – 7.41 (m, 4H), 7.34 – 7.25 (m, 3H), 6.85 – 6.78 (m, 2H), 6.71 (dd, J = 9.7, 2.9 Hz, 1H), 5.36 (dd, J = 12.0, 4.5 Hz, 1H), 3.73 (dd, J = 18.5, 12.1 Hz, 1H), 2.78 (dd, J = 18.5, 4.4 Hz, 1H), 2.62 – 2.54 (m, 1H), 2.53 – 2.40 (m, 1H), 2.33 (t, J = 6.7 Hz, 2H). 13C NMR (150 MHz, DMSO-d6) δ 173.48, 168.69, 159.89, 157.84, 156.25, 152.46, 149.03, 141.01, 135.30, 133.52, 133.45, 131.59, 131.40, 130.47, 128.48, 128.30, 127.28, 124.74, 119.94, 119.78, 119.61, 117.67, 117.62, 111.93, 111.77, 58.24, 45.07, 28.46, 28.18. 19F NMR (376 MHz, DMSO-d6) δ −120.13 – −120.29 (m). HRMS (m/z): [M-H]−calcd for C28H19Cl2FN3O4, 550.07421; found, 550.07419. Anal. Calcd for C28H20Cl2FN3O4: C, 60.88; H, 3.65; N, 7.61. Found C, 60.16; H, 3.98; N, 7.28. HPLC 85% MeOH:H2O (0.1% Formic Acid) Rt 1.10 min; > 95% purity; 75% ACN: H2O (0.1% Formic Acid) Rt 0.81 min; > 95% purity.
4-{5-(4-Chlorophenyl)-3-[6-fluoro-4-(3-fluorophenyl)-2-oxo-1,2-dihydroquinolin-3-yl]-4,5-dihydro-1H-pyrazol-1-yl}-4-oxobutanoic acid (51)
Compound 51 was prepared according to general procedure G using 87 (0.041 g, 0.41 mmol) and 51f (0.180 g, 0.41 mmol). The title compound was obtained after flash column chromatography using 0–10% MeOH:DCM as a yellow solid. Yield 0.076 g, 34.3%. 1H NMR (600 MHz, DMSO-d6) δ 12.34 (s, 1H), 12.01 (s, 1H), 7.59 – 7.39 (m, 3H), 7.37 – 7.19 (m, 3H), 7.14 (t, J = 7.8 Hz, 1H), 7.07 (t, J = 7.2 Hz, 1H), 6.83 – 6.75 (m, 1H), 6.74 – 6.63 (m, 1H), 5.40-5.33 (m, 1H), 3.81 (dd, J = 18.5, 12.1 Hz, 2H), 2.85 – 2.75 (m, 1H), 2.46 – 2.36 (m, 2H), 2.27 (t, J = 7.0 Hz, 2H). 13C NMR (150 MHz, DMSO-d6) δ 173.47, 168.66, 162.80, 161.17, 161.11, 159.93, 157.85, 156.26, 152.52, 152.44, 148.77, 141.08, 137.08, 137.02, 136.97, 135.29, 131.62, 131.56, 130.55, 128.39, 127.39, 127.25, 127.21, 127.08, 125.75, 124.69, 124.59, 119.90, 119.83, 117.66, 116.65, 115.50, 115.36, 111.87, 58.36, 58.24, 45.07, 28.46, 28.18. 19F NMR (376 MHz, DMSO-d6) δ −112.89 – −113.28 (m), −120.04 – −120.29 (m). HRMS (m/z): [M-H]− calcd for C28H19ClF2N3O4, 534.10376; found, 534.10345. Anal. Calcd for C28H20ClF2N3O4: C, 62.75; H, 3.76; N, 7.84. Found C, 61.88; H, 3.98; N: 7.67.
4-{5-(4-Chlorophenyl)-3-[4-(4-chlorophenyl)-6-methoxy-2-oxo-1,2-dihydroquinolin-3-yl]-4,5-dihydro-1H-pyrazol-1-yl}-4-oxobutanoic acid (52)
Compound 52 was prepared according to general procedure G using 87 (0.039 g, 0.39 mmol) and 52f (0.180 g, 0.39 mmol). The title compound was obtained as a yellow solid after flash column chromatography using 0–10% MeOH:DCM. Yield 0.095 g, 43.4%. 1H NMR (600 MHz, DMSO-d6) δ 12.19 (s, 1H), 12.09 (s, 1H), 7.61 (dd, J = 8.2, 2.4 Hz, 1H), 7.52 (dd, J = 8.2, 2.4 Hz, 1H), 7.42 (dd, J = 8.2, 2.3 Hz, 1H), 7.39 (d, J = 8.8 Hz, 1H), 7.32 – 7.25 (m, 4H), 6.82 (d, J = 8.5 Hz, 2H), 6.42 (d, J = 2.8 Hz, 1H), 5.34 (dd, J = 11.9, 4.5 Hz, 1H), 3.73 (dd, J = 18.4, 12.0 Hz, 1H), 3.60 (s, 3H), 2.77 (dd, J = 18.4, 4.4 Hz, 1H), 2.63 – 2.44 (m, 2H), 2.33 (t, J = 6.6 Hz, 2H). 13C NMR (150 MHz, DMSO-d6) δ 173.52, 168.66, 159.69, 154.24, 152.88, 149.28, 141.09, 134.02, 133.23, 133.15, 131.57, 131.42, 130.51, 128.39, 128.29, 128.12, 127.31, 123.95, 120.17, 119.68, 117.01, 109.05, 58.20, 55.32, 45.15, 28.48, 28.21. HRMS (m/z): [M-H]− calcd for C29H22Cl2N3O5, 562.09420; found, 562.09430. Anal. Calcd for C29H23Cl2N3O5: C, 61.71; H, 4.11; N, 7.44. Found C, 61.47; H, 4.06; N, 7.36.
4-{5-(4-Chlorophenyl)-3-[4-(3-fluorophenyl)-6-methoxy-2-oxo-1,2-dihydroquinolin-3-yl]-4,5-dihydro-1H-pyrazol-1-yl}-4-oxobutanoic acid (53)
Compound 53 was prepared according to general procedure G using 87 (0.027 g, 0.27 mmol) and 53f (0.120 g, 0.27 mmol). The title compound was obtained as a yellow solid after flash column chromatography using 0–10% MeOH:DCM. Yield 0.040 g, 27.7%. 1H NMR (400 MHz, DMSO-d6) δ 12.21 (s, 1H), 7.64 – 7.48 (m, 1H), 7.43 – 7.07 (m, 6H), 6.87 – 6.77 (m, 2H), 6.45 – 6.40 (m, 1H), 5.40 – 5.30 (m, 1H) 3.87 – 3.74 (m, 2H), 3.60 (s, 3H), 2.83 (dt, J = 18.4, 5.2 Hz, 1H), 2.54 – 2.39 (m, 2H), 2.31 (t, J = 6.9 Hz, 2H). 13C NMR (150 MHz, DMSO-d6) δ 173.55, 168.62, 162.77, 162.71, 161.15, 161.08, 159.73, 154.23, 152.93, 152.85, 149.03, 141.16, 137.56, 137.51, 133.12, 131.59, 131.52, 130.38, 128.41, 128.32, 127.44, 127.26, 127.07, 125.78, 124.73, 123.87, 123.78, 120.06, 119.63, 116.98, 115.17, 109.18, 58.35, 58.17, 55.38, 55.20, 45.10, 28.49, 28.23. 19F NMR (376 MHz, DMSO-d6) δ −113.15 – −113.37 (m). HRMS (m/z): [M-H]− calcd for C29H22ClFN3O5, 546.12375; found, 546.12384. Anal. Calcd for C29H23ClFN3O5: C, 63.56; H, 4.23; N, 7.67. Found C, 52.15; H, 3.91; N, 5.95. HPLC 85% MeOH:H2O (0.1% Formic Acid) Rt 0.809 min; > 95% purity; 75% ACN: H2O (0.1% Formic Acid) Rt 0.625 min; > 95% purity.
(E)-4-[5-(4-Bromophenyl)-3-(6-chloro-2-oxo-4-phenyl-1,2-dihydroquinolin-3-yl)-4,5-dihydro-1H-pyrazol-1-yl]-4-oxobut-2-enoic acid (54)
Compound 54 was prepared from 54g (0.190 g, 0.322 mmol) and 1M NaOH (1.22 mL, 1.22 mmol), which were stirred to give a yellow solution. After four hours, 1M HCl (1.22 mL) was added and a yellow solid precipitated. The solid was filtered and rinsed with water to give the title compound as a yellow solid. Yield 0.170 g, 92.0%. 1H NMR (400 MHz, DMSO-d6) δ 13.03 (s, 1H), 12.46 (s, 1H), 7.66 (dd, J = 8.8, 2.4 Hz, 1H), 7.61 – 7.38 (m, 7H), 7.31 (dt, J = 5.6, 2.5 Hz, 1H), 7.26 (d, J = 15.7 Hz, 1H), 6.94 (d, J = 2.4 Hz, 1H), 6.78 (d, J = 8.3 Hz, 2H), 6.45 (d, J = 15.7 Hz, 1H), 5.45 (dd, J = 11.8, 4.5 Hz, 1H), 3.80 (dd, J = 18.7, 11.9 Hz, 1H), 2.88 (dd, J = 18.7, 4.5 Hz, 1H). 13C NMR (150 MHz, DMSO-d6) δ 167.81, 166.11, 160.30, 159.96, 155.82, 154.41, 150.24, 140.70, 137.39, 134.48, 132.67, 131.83, 131.42, 129.19, 128.63, 128.51, 128.36, 127.93, 127.83, 126.17, 126.05, 124.09, 120.60, 120.44, 117.66, 58.69, 58.63, 45.11. HRMS (m/z): [M-H]−calcd for C28H18ClBrN3O4, 574.01747; found, 574.01750. Anal. Calcd for C28H19ClBrN3O4·1.00H2O: C, 56.54; H, 3.56; N, 7.06. Found C, 56.66; H, 3.76; N: 6.93.
(Z)-4-[5-(4-Bromophenyl)-3-(6-chloro-2-oxo-4-phenyl-1,2-dihydroquinolin-3-yl)-4,5-dihydro-1H-pyrazol-1-yl]-4-oxobut-2-enoic acid (55)
Compound 55 was prepared according to general procedure G using furan-2,5-dione (90) (0.061 g, 0.63 mmol) and 55f (0.300 g, 0.63 mmol). The title compound was obtained after filtration from the cooled reaction medium and rinsed with THF. Yield 0.200 g, 55.3%. 1H NMR (400 MHz, DMSO-d6) δ 12.69 (s, 1H), 12.41 (s, 1H), 7.64 (d, J = 8.7 Hz, 1H), 7.60– 7.36 (m, 7H), 7.25 (d, J = 7.2 Hz, 1H), 6.92 (s, 1H), 6.78 (d, J = 8.1 Hz, 2H), 6.40 (d, J = 12.1 Hz, 1H), 6.16 (d, J = 12.1 Hz, 1H), 5.37 (dd, J = 12.0, 4.9 Hz, 1H), 3.78 (dd, J = 18.5, 12.0 Hz, 1H), 2.79 (dd, J = 18.7, 4.8 Hz, 1H). 13C NMR (100 MHz, DMSO-d6) δ 166.77, 161.77, 160.01, 153.58, 150.11, 140.93, 137.36, 134.39, 131.31, 129.57, 129.46, 128.64, 128.43, 127.96, 126.16, 126.08, 124.26, 120.62, 120.29, 117.67, 58.43, 45.17. HRMS (m/z): [M-H]− calcd for C28H18ClBrN3O4, 574.01747; found, 574.01654. Anal. Calcd for C28H19BrClN3O4: C, 58.30; H, 3.32; N, 7.28. Found C, 43.78; H, 3.00; N: 5.57. HPLC 85% MeOH:H2O (0.1% Formic Acid) Rt 1.18 min; > 95% purity; 75% ACN: H2O (0.1% Formic Acid) Rt 1.06 min; > 95% purity.
Methyl 4-[5-(4-bromophenyl)-3-(6-chloro-2-oxo-4-phenyl-1,2-dihydroquinolin-3-yl)-4,5-dihydro-1H-pyrazol-1-yl]-4-oxobutanoate (56)
Compound 56 was prepared from 56g (0.400 g, 0.69 mmol) in the following manner. 56g (0.400 g, 0.69 mmol) was dissolved in 6.9 mL THF and freshly prepared HCl (Acetyl chloride added to methanol) in MeOH was added dropwise to the reaction vessel with stirring until TLC indicated completion. Upon completion, the THF was removed under vacuum, the residue dissolved in DCM, washed 3X with acidified brine and column chromatographed using a 0–10% MeOH gradient in DCM to give the title compound as a yellow solid. Yield 0.100 g, 24.4%. 1H NMR (400 MHz, DMSO-d6) δ 12.42 (s, 1H), 7.64 (dd, J = 8.7, 2.4 Hz, 1H), 7.54 – 7.49 (m, 3H), 7.48 – 7.38 (m, 4H), 7.31 – 7.24 (m, 1H), 6.93 (d, J = 2.3 Hz, 1H), 6.77 – 6.70 (m, 2H), 5.31 (dd, J = 12.0, 4.6 Hz, 1H), 3.76 (dd, J = 18.5, 12.2 Hz, 1H), 3.59 (s, 1H), 3.54 (s, 2H), 2.79 (dd, J = 18.5, 4.6 Hz, 1H), 2.61 – 2.42 (m, 2H), 2.37 (t, J = 6.7 Hz, 2H). 13C NMR (150 MHz, DMSO-d6) δ 172.44, 168.37, 168.08, 160.09, 152.81, 152.61, 149.98, 141.49, 137.32, 134.58, 131.30, 129.45, 128.48, 128.36, 127.78, 127.67, 126.13, 126.01, 124.50, 120.65, 120.14, 117.65, 58.43, 58.30, 51.35, 51.24, 45.23, 45.08, 28.46, 27.96. HRMS (m/z): [M-H]− calcd for C29H22ClBrN3O4, 590.04877; found, 590.04851. Anal. Calcd for C29H23ClBrN3O4: C, 58.75; H, 3.91; N, 7.09. Found C, 58.35; H, 4.08; N, 6.66.
(E)-4-{3-[4-(4-Bromophenyl)-2-oxo-1,2-dihydroquinolin-3-yl]-5-(4-chlorophenyl)-4,5-dihydro-1H-pyrazol-1-yl}-4-oxobut-2-enoic acid (58)
Compound 58 was prepared from compound 60 using the following method. In a 50 mL round-bottomed flask, ethanol (28.2 mL), 60 (0.500 g, 0.850 mmol) and 1M NaOH (3.22 mL, 3.22 mmol) were stirred to give a yellow solution. After four hours, 1M HCl (3.22 mL) was added and a yellow solid precipitated. The solid was filtered and rinsed with water. The resulting solid was dissolved in DCM, washed with brine and the organics were dried over magnesium sulfate in vacuo. The title compound was obtained from column chromatography (0–10% MeOH in DCM) as an off-white solid. Yield 0.320 g, 65.6%. 1H NMR (400 MHz, DMSO-d6) δ 12.98 (s, 1H), 12.28 (s, 1H), 7.71 – 7.51 (m, 3H), 7.48 – 7.19 (m, 6H), 7.12 (t, J = 7.6 Hz, 1H), 7.02 (d, J = 8.2 Hz, 1H), 6.85 (d, J = 8.2 Hz, 2H), 6.44 (d, J = 15.7 Hz, 1H), 5.45 (dd, J = 11.7, 4.4 Hz, 1H), 3.73 (dd, J = 18.7, 11.8 Hz, 1H), 2.89 (dd, J = 18.6, 4.4 Hz, 1H). 13C NMR (150 MHz, DMSO-d6) δ 168.08, 166.05, 160.24, 160.04, 154.75, 152.81, 152.77, 150.29, 140.25, 138.65, 134.49, 132.80, 131.95, 131.60, 131.36, 131.26, 130.74, 128.51, 127.55, 127.43, 122.92, 122.47, 121.84, 119.01, 115.61, 58.50, 45.19. HRMS (m/z): [M+H]+ calcd for C28H20ClBrN3O4, 576.03202; found, 576.03294. Anal. Calcd for C28H19ClBrN3O4·0.40H2O: C, 57.58; H, 3.42; N, 7.19. Found C, 57.46; H, 3.50; N, 7.23.
(Z)-4-{3-[4-(4-Bromophenyl)-2-oxo-1,2-dihydroquinolin-3-yl]-5-(4-chlorophenyl)-4,5-dihydro-1H-pyrazol-1-yl}-4-oxobut-2-enoic acid (59)
Compound 59 was prepared according to general procedure G using 89 (0.061 g, 0.63 mmol) and 22f (0.300 g, 0.630 mmol). The THF was removed under vacuum and the resultant residue was dissolved in hot EtOAc. A yellow solid was present upon cooling which was filtered and determined to be the desired product. Yield 0.171 g, 47.3%. 1H NMR (400 MHz, DMSO-d6) δ 12.74 (s, 1H), 12.30 (s, 1H), 7.75 (d, J = 8.3 Hz, 1H), 7.67 (d, J = 8.4 Hz, 1H), 7.58 (t, J = 7.9 Hz, 1H), 7.43 (d, J = 8.4 Hz, 1H), 7.32 (t, J = 8.6 Hz, 3H), 7.22 (d, J = 8.2 Hz, 1H), 7.15 (t, J = 7.8 Hz, 1H), 7.03 (d, J = 8.2 Hz, 1H), 6.87 (d, J = 8.1 Hz, 2H), 6.48 (d, J = 12.2 Hz, 1H), 6.20 (d, J = 12.1 Hz, 1H), 5.47 – 5.38 (m, 1H), 3.76 (dd, J = 18.8, 11.9 Hz, 1H), 2.84 – 2.74 (m, 1H). 13C NMR (100 MHz, DMSO-d6) δ 166.80, 161.77, 160.05, 153.90, 150.03, 140.50, 138.60, 134.27, 131.76, 131.69, 131.55, 131.28, 130.82, 129.68, 129.41, 128.36, 127.51, 127.32, 123.10, 122.47, 121.89, 119.04, 115.61, 93.88, 67.04, 58.28, 45.27, 25.15. HRMS (m/z): [M-H]− calcd for C28H18ClBrN3O4, 576.03202; found, 576.03371. Anal. Calcd for C28H19ClBrN3O4·0.60H2O: C, 57.23; H, 3.46; N, 7.15. Found C, 57.22; H, 3.54; N, 6.91.
(E)-Methyl 4-{3-[4-(4-bromophenyl)-2-oxo-1,2-dihydroquinolin-3-yl]-5-(4-chlorophenyl)-4,5-dihydro-1H-pyrazol-1-yl}-4-oxobut-2-enoate (60)
Compound 60 was prepared from 22f (0.750g, 1.60 mmol) and (E)-methyl 4-chloro-4-oxobut-2-enoate (0.280 g, 1.90 mmol) using standard procedure G. The THF was removed under vacuum, the residue dissolved in DCM, washed 3X with brine and the organics concentrated. The title compound was obtained as a yellow solid by flash chromatography using a 0–10% MeOH gradient in DCM. Yield 0.546 g, 59.0%. 1H NMR (600 MHz, DMSO-d6) δ 12.32 (s, 1H), 7.70 (dt, J = 8.3, 1.9 Hz, 1H), 7.67 (dt, J = 8.1, 2.0 Hz, 1H), 7.60 (t, J = 7.8 Hz, 1H), 7.44 (d, J = 8.3 Hz, 1H), 7.37 – 7.32 (m, 3H), 7.30 (dt, J = 8.2, 2.0 Hz, 1H), 7.27 – 7.22 (m, 1H), 7.16 (t, J = 7.6 Hz, 1H), 7.08 (d, J = 8.2 Hz, 1H), 6.95 (d, J = 7.6 Hz, 2H), 6.53 (dd, J = 15.5, 1.0 Hz, 1H), 5.49 (dd, J = 11.6, 4.3 Hz, 1H), 3.86 – 3.73 (m, 4H), 3.03 (dd, J = 18.6, 4.2 Hz, 1H).13C NMR (100 MHz, DMSO-d6) δ 165.13, 160.11, 159.92, 154.93, 150.40, 140.16, 138.71, 134.71, 133.36, 132.02, 131.69, 131.37, 131.30, 131.11, 130.80, 129.93, 128.65, 128.56, 127.57, 127.46, 122.77, 122.50, 121.77, 119.01, 115.63, 58.56, 52.16, 45.15. HRMS (m/z): [M+H]+ calcd for C29H22ClBrN3O4, 590.04767; found, 590.04887. HPLC 85% MeOH:H2O (0.1% Formic Acid) Rt 1.56 min; > 90% purity; 75% ACN: H2O (0.1% Formic Acid) Rt 1.39 min; > 90% purity.
5-{3-[4-(4-Bromophenyl)-2-oxo-1,2-dihydroquinolin-3-yl]-5-(4-chlorophenyl)-4,5-dihydro-1H-pyrazol-1-yl}-5-oxopentanoic acid (61)
Compound 61 was prepared according to general procedure G using 88 (0.071 g, 0.63 mmol) and 22f (0.30 g, 0.63 mmol). Upon completion, the THF was removed under vacuum and the residue was dissolved in hot EtOAc. Upon cooling, a yellow solid formed which was filtered, dried under vacuum and determined to be the desired product. Yield 0.204 g, 54.9%. 1H NMR (400 MHz, DMSO-d6) δ 12.29 (s, 1H), 12.09 (s, 1H), 7.77 (dd, J = 8.2, 2.1 Hz, 1H), 7.70 (dd, J = 8.2, 2.1 Hz, 1H), 7.58 (t, J = 7.7 Hz, 1H), 7.43 (d, J = 8.3 Hz, 1H), 7.36 (dd, J = 8.2, 2.2 Hz, 1H), 7.34 – 7.28 (m, 2H), 7.23 (dd, J = 8.1, 2.3 Hz, 1H), 7.15 (t, J = 7.6 Hz, 1H), 7.04 (d, J = 8.2 Hz, 1H), 6.86 – 6.76 (m, 2H), 5.36 (dd, J = 12.0, 4.4 Hz, 1H), 3.81 – 3.68 (m, 1H), 2.80 (dd, J = 18.7, 4.3 Hz, 1H), 2.40 (dt, J = 15.3, 7.4 Hz, 1H), 2.29 – 2.06 (m, 3H), 1.68 – 1.51 (m, 2H). 13C NMR (100 MHz, DMSO-d6) δ 174.11, 169.29, 160.18, 152.73, 149.89, 141.29, 138.57, 134.59, 131.73, 131.64, 131.47, 131.30, 131.25, 130.88, 128.40, 127.31, 127.27, 123.41, 122.44, 121.77, 119.09, 115.60, 58.13, 45.10, 32.96, 32.58, 19.82. HRMS (m/z): [M+H]+ calcd for C29H24ClBrN3O4, 592.06332; found, 592.06461. Anal. Calcd for C29H23ClBrN3O4: C, 58.75; H, 3.91; N, 7.09. Found C, 58.47; H, 3.96; N, 6.91.
5-[3-(6-Chloro-2-oxo-4-phenyl-1,2-dihydroquinolin-3-yl)-5-(4-nitrophenyl)-4,5-dihydro-1H-pyrazol-1-yl]-5-oxopentanoic acid (63)
Compound 63 was prepared according to general procedure G using glutaric anhydride (89) (0.077 g, 0.67 mmol) and 63f (0.30 g, 0.67 mmol). The title compound was obtained after being dissolved in hot EtOAc followed by slow addition of hot hexanes to yield the title compound as a yellow solid. Yield 0.220 g, 58.4%. 1H NMR (400 MHz, DMSO-d6) δ 12.44 (s, 1H), 12.12 (s, 1H), 8.12 (d, J = 8.3 Hz, 2H), 7.70 – 7.51 (m, 4H), 7.51 – 7.38 (m, 2H), 7.28 (d, J = 6.6 Hz, 1H), 7.07 (d, J = 8.5 Hz, 2H), 6.95 (d, J = 2.5 Hz, 1H), 5.50 (dd, J = 12.3, 4.8 Hz, 1H), 3.83 (dd, J = 18.6, 12.2 Hz, 1H), 2.86 (dd, J = 18.6, 4.9 Hz, 1H), 2.45 – 2.30 (m, 1H), 2.29 – 2.17 (m, 1H), 2.13 (t, J = 7.4 Hz, 2H), 1.65 – 1.45 (m, 2H). 13C NMR (100 MHz, DMSO-d6) δ 174.07, 169.49, 160.10, 152.49, 149.59, 146.59, 137.35, 131.30, 129.39, 128.47, 126.68, 126.08, 124.34, 123.82, 120.63, 117.67, 58.32, 44.90, 32.94, 32.53, 19.70. HRMS (m/z): [M+H]+ calcd for C29H24ClN4O6, 559.13789; found, 559.13826. Anal. Calcd for C29H23ClN4O6: C, 62.31; H, 4.15; N, 10.02. Found C, 62.42; H, 4.39; N, 9.72.
4-(4-Chlorophenyl)-3-[5-(4-chlorophenyl)-1-(4-hydroxybutanoyl)-4,5-dihydro-1H-pyrazol-3-yl]quinolin-2(1H)-one (64)
In a flame dried 25 mL round bottomed flask, 26 (0.300 g, 0.560 mmol) was dissolved in THF (10 mL) and cooled on an ice bath to 0 °C under nitrogen with stirring. BH3-Me2S (2.0M in Hexanes, 0.561 ml, 2 eq.) was added drop-wise. The reaction was stirred for thirty minutes, quenched with MeOH and the solvent was removed under vacuum. The resultant residue was dissolved in DCM, washed three times with brine and the organics combined, dried over magnesium sulfate, concentrated in vacuo and column chromatographed using a 0–8% gradient of MeOH in DCM to give the title compound as a yellow solid. Yield 0.063 g, 21.6%. 1H NMR (400 MHz, DMSO-d6) δ 12.29 (s, 1H), 7.65 – 7.49 (m, 3H), 7.46 – 7.24 (m, 5H), 7.20 – 7.00 (m, 2H), 6.89 – 6.78 (m, 2H), 5.35 (dd, J = 12.0, 4.4 Hz, 1H), 4.45 (t, J = 5.2 Hz, 1H), 3.74 (dd, J = 18.5, 12.1 Hz, 1H), 3.41 – 3.27 (m, 2H), 2.80 (dd, J = 18.5, 4.5 Hz, 1H), 2.44 – 2.19 (m, 2H), 1.59 – 1.39 (m, 2H). 13C NMR (150 MHz, DMSO-d6) δ 169.82, 160.17, 152.52, 149.83, 141.31, 138.53, 134.18, 133.14, 131.58, 131.43, 131.39, 130.57, 129.11, 128.33, 127.28, 123.50, 122.41, 119.14, 115.57, 60.19, 58.10, 45.06, 30.14, 27.71. HRMS (m/z): [M+H]+ calcd for C28H24Cl2N3O3, 520.11892; found, 520.11993. Anal. Calcd for C28H23Cl2N3O3·0.40H2O: C, 63.74; H, 4.55; N, 7.96. Found C, 63.75; H, 4.33; N, 7.89.
4-{5-(4-Chlorophenyl)-3-[4-(4-chlorophenyl)-2-oxo-1,2-dihydroquinolin-3-yl]-4,5-dihydro-1H-pyrazol-1-yl}-4-oxobutanamide (65)
In a flame dried 25 mL round bottomed flask, 26 (0.300 g, 0.560 mmol), 4-dimethylaminopyridine (0.069 g, 0.560 mmol) and N1-((ethylimino)methylene)-N3,N3-dimethylpropane-1,3-diamine (0.096 g, 0.618 mmol) were added to THF (11.23 mL) at 0 °C and stirred for 45 minutes. Ammonia (0.5 M in dioxane, 1.0 eq, 1.1 mL) was added to the flask and the reaction mixture was stirred overnight while being allowed to warm to room temperature. The reaction was quenched with dilute HCl (0.1 M) and the organics were removed under vacuum. The resultant residue was dissolved in DCM, washed 3X with brine and the organics dried over magnesium sulfate and concentrated in vacuo prior to column chromatography using a 0–8% MeOH gradient in DCM (0.1 % Et3N). The title compound was obtained as a white solid. Yield 0.019 g, 6.35%. 1H NMR (400 MHz, DMSO-d6) δ 12.29 (s, 1H), 7.66 – 7.56 (m, 2H), 7.55 – 7.49 (m, 1H), 7.43 (d, J = 8.4 Hz, 2H), 7.28 (dd, J = 8.2, 4.4 Hz, 3H), 7.24 (s, 1H), 7.15 (t, J = 7.6 Hz, 1H), 7.03 (d, J = 8.2 Hz, 1H), 6.79 (d, J = 8.5 Hz, 2H), 6.73 (s, 1H), 5.34 (dd, J = 12.1, 4.5 Hz, 1H), 3.71 (dd, J = 18.4, 12.2 Hz, 1H), 2.72 (dd, J = 18.4, 4.5 Hz, 1H), 2.59 – 2.50 (m, 2H), 2.20 (t, J = 7.2 Hz, 2H). 13C NMR (150 MHz, DMSO-d6) δ 173.69, 169.78, 160.79, 153.18, 150.49, 141.85, 139.21, 134.63, 133.87, 132.22, 132.19, 132.09, 131.23, 128.99, 128.95, 127.94, 124.23, 123.07, 119.80, 116.24, 58.83, 46.27, 45.80, 29.94, 29.44. HRMS (m/z): [M+H]+ calcd for C28H23Cl2N4O3, 533.11417; found, 533.11517. HPLC 85% MeOH:H2O (0.1% Formic Acid) Rt 0.89 min; > 90% purity; 75% ACN: H2O (0.1% Formic Acid) Rt 1.00 min; > 95% purity.
Methyl 4-{5-(4-chlorophenyl)-3-[4-(4-chlorophenyl)-2-oxo-1,2-dihydroquinolin-3-yl]-4,5-dihydro-1H-pyrazol-1-yl}butanoate (66)
In a 20 mL round-bottomed flask, 26f (0.500 g, 1.151 mmol) and methyl 4-oxobutanoate (0.121 ml, 1.151 mmol) were dissolved in DCE (11.51 ml). The mixture was allowed to stir at room temperature for four hours and sodium triacetoxyborohydride (0.293 g, 1.381 mmol) was added in one portion. The reaction was monitored by TLC and HPLC-MS. Upon completion, the DCE was removed under vacuum; the residue diluted with DCM and washed 3X with brine. The organics were concentrated and the title compound was obtained from flash column chromatography using 0–10% MeOH in DCM and trituration of the compound from ether as a yellow solid. Yield 0.150 g, 24.4%. 1H NMR (400 MHz, CDCl3) δ 12.22 (s, 1H), 7.55 – 7.45 (m, 3H), 7.40 (s, 1H), 7.34 – 7.26 (m, 5H), 7.22 (s, 2H), 7.13 (t, J = 7.6 Hz, 1H), 4.11 (dd, J = 13.6, 10.1 Hz, 1H), 3.62 (s, 3H), 3.41 (dd, J = 16.4, 10.1 Hz, 1H), 2.91 – 2.78 (m, 1H), 2.70 – 2.53 (m, 2H), 2.30 – 2.08 (m, 2H), 1.84 – 1.66 (m, 2H). 13C NMR (100 MHz, CDCl3) δ 174.18, 162.94, 150.63, 146.34, 139.59, 138.08, 135.07, 134.23, 133.49, 131.12, 130.90, 129.08, 128.89, 128.51, 128.33, 127.82, 124.96, 122.96, 120.65, 116.18, 70.81, 52.20, 51.66, 46.60, 31.30, 23.04. HRMS (m/z): [M+H]+ calcd for C29H26Cl2N3O3, 534.13457; found, 534.13624. HPLC 85% MeOH:H2O (0.1% Formic Acid) Rt 2.35 min; > 95% purity; 75% ACN: H2O (0.1% Formic Acid) Rt 2.09 min; > 95% purity.
4-{5-(4-Chlorophenyl)-3-[4-(4-chlorophenyl)-2-oxo-1,2-dihydroquinolin-3-yl]-4,5-dihydro-1H-pyrazol-1-yl}butanoic acid (67)
In a 10 mL round-bottomed flask, compound 66 (0.100 g, 0.187 mmol) was dissolved in NaOH (0.711 ml, 0.711 mmol), H2O (10.00 ml) and Ethanol (6.24 ml). The mixture was stirred at room temperature for four hours and monitored by TLC/LC-MS. Upon completion, HCl (0.711 ml, 0.711 mmol) was added, giving a bright yellow solid which was filtered, dissolved in DCM and washed 3X with acidified (pH 2, HCl) brine. The organics were combined, dried over magnesium sulfate and concentrated in vacuo. The title compound was obtained by flash chromatography using 0–8% MeOH in DCM as a yellow solid. Yield 0.050 g, 51.3%. 1H NMR (600 MHz, DMSO-d6) δ 12.16 (s, 1H), 11.92 (s, 1H), 7.59 – 7.49 (m, 3H), 7.42 – 7.37 (m, 3H), 7.35 – 7.29 (m, 4H), 7.13 (t, J = 7.6 Hz, 1H), 7.07 (d, J = 8.2 Hz, 1H), 4.03 (dd, J = 13.8, 10.2 Hz, 1H), 3.33 (dd, J = 16.4, 10.2 Hz, 1H), 2.73 (dd, J = 16.5, 13.8 Hz, 1H), 2.54 – 2.46 (m, 1H), 2.40 – 2.31 (m, 1H), 2.12 – 1.96 (m, 2H), 1.55 – 1.39 (m, 2H). 13C NMR (150 MHz, DMSO-d6) δ 174.25, 160.57, 148.75, 146.65, 140.21, 138.31, 135.02, 132.63, 131.86, 131.23, 131.19, 130.84, 130.75, 129.09, 128.44, 128.08, 127.81, 127.04, 125.16, 122.15, 119.31, 115.43, 69.48, 52.16, 46.06, 30.83, 22.35. HRMS (m/z): [M+H]+ calcd for C28H24Cl2N3O3, 520.11892; found, 520.11970. HPLC 85% MeOH:H2O (0.1% Formic Acid) Rt 1.33 min; > 95% purity; 75% ACN: H2O (0.1% Formic Acid) Rt 0.98 min; > 95% purity.
4-(4-Chlorophenyl)-3-[5-(4-chlorophenyl)-1-(4-fluorobutanoyl)-4,5-dihydro-1H-pyrazol-3-yl]quinolin-2(1H)-one (68)
In a 10 mL round-bottomed flask, 4-fluorobutanoic acid 68g (0.100 g, 0.943 mmol), DMAP (0.127 g, 1.037 mmol) and EDCI (0.199 g, 1.037 mmol) were dissolved in DCM (9.43 ml) which had been pre-cooled to 0°C. The mixture was stirred for 45 minutes prior to the addition of the 26f (0.409 g, 0.943 mmol). The reaction was allowed to warm to room temperature, and monitored by TLC. The reaction was quenched with 0.2 N HCl and extracted into DCM. The organics were washed 3X with brine, dried over magnesium sulfate and concentrated. The title compound was obtained as a yellow solid after column chromatography using a gradient of 0–50% EtOAc in DCM as a yellow solid. Yield 0.050 g, 21.6%. 1H NMR (400 MHz, CDCl3) δ 13.16 (s, 1H), 7.62 – 7.43 (m, 4H), 7.38 (d, J = 8.2 Hz, 1H), 7.34 – 7.16 (m, 5H), 7.02 (d, J = 8.3 Hz, 2H), 5.42 (dd, J = 11.8, 4.0 Hz, 1H), 4.44 (dtd, J = 47.3, 5.8, 1.7 Hz, 2H), 3.70 (dd, J = 18.2, 11.8 Hz, 1H), 3.15 (dd, J = 18.3, 4.1 Hz, 1H), 2.68 – 2.45 (m, 2H), 2.18 – 1.82 (m, 2H). 13C NMR (150 MHz, CDCl3) δ 170.45, 162.91, 152.52, 151.98, 151.92, 140.41, 138.42, 135.43, 134.99, 133.97, 133.56, 132.00, 130.81, 130.72, 130.68, 130.36, 129.05, 128.96, 128.85, 128.73, 128.70, 128.04, 127.60, 127.52, 126.86, 123.47, 123.09, 120.36, 116.41, 84.06, 82.97, 59.35, 45.44, 29.91, 29.88, 25.69, 25.62, 25.56. 19F NMR (282 MHz, CDCl3) δ −220.53 (tt, J = 47.3, 26.3 Hz). HRMS (m/z): [M+H]+ calcd for C28H23Cl2N3O2F, 522.11459; found, 522.11462. HPLC 85% MeOH:H2O (0.1% Formic Acid) Rt 1.46 min; > 95% purity; 75% ACN: H2O (0.1% Formic Acid) Rt 1.32 min; > 95% purity.
Supplementary Material
Acknowledgments
The authors thank the Emory Institute for Drug Development for performing the plasma stability assays, Dr. Terry W. Moore, Dr. Alexandra Orchard and Eric Miller for helpful discussion and comments on the manuscript, Drs. Shaoxiong Wu, Fred Strobel for excellent core facility support, and Phuong Le and Jing Zhang for excellent technical assistance. This work was support by the National Institutes of Health (NIH-NINDS NS071802 TMA and NS065371, NS036654 SFT), an NIH Graduate training in the Pharmacological Sciences (GM008602 TMA), an NIH Translational Research in Neurology (5T32-NS007480-07 KMV), as well as the University of KwaZulu-Natal and the NRF (CS ), and Lundbeck A/S and Pfizer Inc. research grants to Emory University.
Abbreviations Used
- Ca2+
calcium ion
- Mg2+
magnesium ion
- ATD
amino-terminal domain
- LBD
ligand-binding domain
- DQP
dihydroquinolone-pyrazoline
- BH3-DMS
borane dimethylsulfide
- NH3
ammonia
- µM
micromolar
- mV
millivolt
- QNZ
quinazoline-4-one
- S2
segment two
- TPSA
topological polar surface area
- Papp
permeability coefficient
- MDR1-MDCK
multidrug resistance gene 1-Madin-Darby canine kidney cells
- cRNA
complementary RNA
- NaCl
sodium chloride
- KCl
potassium chloride
- BaCl2
barium chloride
- NaOH
sodium hydroxide
- MΩ
megaohm
- NIMH PDSP
National Institute of Mental Health Psychoactive Drug Screening Program
- ACN
acetonitrile
- EtOAc
ethyl acetate
Footnotes
Supporting Information Available: Supporting information including the experimental detail for the synthesis of all intermediates, solubility determination methods and data analysis, table S1 with A-ring modifications, table S2 with hetero-aromatic C–ring substitutions, table S3 with C-ring modifications, table S4 with off-target data from NIMH-PDSP, figure S1 showing mutant receptor responses, figure S2 showing plasma stability, compound solubility and Scheme S1 showing the synthesis of the mono-fluoro butyrate is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Mayer ML. Glutamate receptor ion channels. Curr. Opin. Neurobiol. 2005;15:282–288. doi: 10.1016/j.conb.2005.05.004. [DOI] [PubMed] [Google Scholar]
- 2.Traynelis SF, Wollmuth LP, McBain CJ, Menniti FS, Vance KM, Ogden KK, Hansen KB, Yuan H, Myers SJ, Dingledine R, Sibley D. Glutamate receptor ion channels: structure, regulation, and function. Pharmacol. Rev. 2010;62:405–496. doi: 10.1124/pr.109.002451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang PY, Petralia RS, Wang Y-X, Wenthold RJ, Brenowitz SD. Functional NMDA Receptors at Axonal Growth Cones of Young Hippocampal Neurons. J. Neurosci. 2011;31:9289–9297. doi: 10.1523/JNEUROSCI.5639-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cull-Candy SG, Leszkiewicz DN. Role of Distinct NMDA Receptor Subtypes at Central Synapses. Sci. STKE. 2004;2004:re16. doi: 10.1126/stke.2552004re16. [DOI] [PubMed] [Google Scholar]
- 5.Pérez-Otaño I, Ehlers MD. Homeostatic plasticity and NMDA receptor trafficking. Trends. Neurosci. 2005;28:229–238. doi: 10.1016/j.tins.2005.03.004. [DOI] [PubMed] [Google Scholar]
- 6.Mony L, Kew JNC, Gunthorpe MJ, Paoletti P. Allosteric modulators of NR2B–containing NMDA receptors: molecular mechanisms and therapeutic potential. Br. J. Pharmacol. 2009;157:1301–1317. doi: 10.1111/j.1476-5381.2009.00304.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen H-SV, Lipton AS. The chemical biology of clinically tolerated NMDA receptor antagonists. J. Neurochem. 2006;97:1611–1626. doi: 10.1111/j.1471-4159.2006.03991.x. [DOI] [PubMed] [Google Scholar]
- 8.Hallett PJ, Standaert DG. Rationale for and use of NMDA receptor antagonists in Parkinson's disease. Pharmacol. Ther. 2004;102:155–174. doi: 10.1016/j.pharmthera.2004.04.001. [DOI] [PubMed] [Google Scholar]
- 9.Hollmann M, Heinemann S. Cloned glutamate receptors. Annu. Rev. Neurosci. 1994;17:31–108. doi: 10.1146/annurev.ne.17.030194.000335. [DOI] [PubMed] [Google Scholar]
- 10.Durand GM, Gregor P, Zheng X, Bennett MV, Uhl GR, Zukin RS. Cloning of an apparent splice variant of the rat N-methyl-D-aspartate receptor NMDAR1 with altered sensitivity to polyamines and activators of protein kinase C. Proc. Natl. Acad. Sci. USA. 1992;89:9359–9363. doi: 10.1073/pnas.89.19.9359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Furukawa H, Gouaux E. Mechanisms of activation, inhibition and specificity: crystal structures of the NMDA receptor NR1 ligand-binding core. EMBO. J. 2003;22:2873–2885. doi: 10.1093/emboj/cdg303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Furukawa H, Singh SK, Mancusso R, Gouaux E. Subunit arrangement and function in NMDA receptors. Nature. 2005;438:185–192. doi: 10.1038/nature04089. [DOI] [PubMed] [Google Scholar]
- 13.Chen PE, Wyllie DJ. Pharmacological insights obtained from structure-function studies of ionotropic glutamate receptors. Br. J. Pharmacol. 2006;147:839–853. doi: 10.1038/sj.bjp.0706689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Paoletti P, Neyton J. NMDA receptor subunits: function and pharmacology. Curr. Opin. Pharmacol. 2007;7:39–47. doi: 10.1016/j.coph.2006.08.011. [DOI] [PubMed] [Google Scholar]
- 15.Vance KM SN, Traynelis SF, Furukawa H. Ligand-specific deactivation time course of GluN1/GluN2D NMDA receptors. Nat. Commun. 2011;2:294. doi: 10.1038/ncomms1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gielen M, Retchless BS, Mony L, Johnson JW, Paoletti P. Mechanism of differential control of NMDA receptor activity by NR2 subunits. Nature. 2009;459:703–707. doi: 10.1038/nature07993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yuan H, Hansen KB, Vance KM, Ogden KK, Traynelis SF. Control of NMDA Receptor Function by the NR2 Subunit Amino-Terminal Domain. J. Neurosci. 2009;29:12045–12058. doi: 10.1523/JNEUROSCI.1365-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lester RAJ, Clements JD, Westbrook GL, Jahr CE. Channel kinetics determine the time course of NMDA receptor-mediated synaptic currents. Nature. 1990;346:565–567. doi: 10.1038/346565a0. [DOI] [PubMed] [Google Scholar]
- 19.Nowak L, Bregestovski P, Ascher P, Herbet A, Prochiantz A. Magnesium gates glutamate-activated channels in mouse central neurons. Nature. 1984;307:462–465. doi: 10.1038/307462a0. [DOI] [PubMed] [Google Scholar]
- 20.Mayer ML, Westbrook GL, Guthrie PB. Voltage-dependent block by Mg2+ of NMDA responses in spinal-cord neurons. Nature. 1984;309:261–263. doi: 10.1038/309261a0. [DOI] [PubMed] [Google Scholar]
- 21.Clarke RJ, Johnson JW. NMDA Receptor NR2 Subunit Dependence of the Slow Component of Magnesium Unblock. J. Neurosci. 2006;26:5825–5834. doi: 10.1523/JNEUROSCI.0577-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Monyer H, Burnashev N, Laurie DJ, Sakmann B, Seeburg PH. Developmental and regional expression in the rat brain and functional properties of four NMDA receptors. Neuron. 1994;12:529–540. doi: 10.1016/0896-6273(94)90210-0. [DOI] [PubMed] [Google Scholar]
- 23.Akazawa C, Shigemoto R, Bessho Y, Nakanishi S, Mizuno N. Differential expression of five N-methyl-D-aspartate receptor subunit mRNAs in the cerebellum of developing and adult rats. J. Comp. Neurol. 1994;347:150–160. doi: 10.1002/cne.903470112. [DOI] [PubMed] [Google Scholar]
- 24.Galvan A, Wichmann T. Pathophysiology of parkinsonism. Clin. Neurophysiol. 2008;119:1459–1474. doi: 10.1016/j.clinph.2008.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bräuner-Osborne H, Egebjerg J, Nielsen EØ, Madsen U, Krogsgaard-Larsen P. Ligands for Glutamate Receptors:□ Design and Therapeutic Prospects. J. Med. Chem. 2000;43:2609–2645. doi: 10.1021/jm000007r. [DOI] [PubMed] [Google Scholar]
- 26.Standaert DG, Testa CM, Penney JB, Jr., Young AB. Alternatively spliced isoforms of the NMDAR1 glutamate receptor subunit: Differential expression in the basal ganglia of the rat. Neurosci. Lett. 1993;152:161–164. doi: 10.1016/0304-3940(93)90508-i. [DOI] [PubMed] [Google Scholar]
- 27.Laurie DJ, Seeburg PH. Regional and developmental heterogeneity in splicing of the rat brain NMDAR1 mRNA. J. Neurosci. 1994;14:3180–3194. doi: 10.1523/JNEUROSCI.14-05-03180.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Standaert DG, Testa CM, Young A, Penney JB., Jr. Organization of N-methyl-D-aspartate glutamate receptor gene expression in the basal ganglia of the rat. J. Comp. Neurol. 1994;343:1–16. doi: 10.1002/cne.903430102. [DOI] [PubMed] [Google Scholar]
- 29.Goff DC, Cather C, Gottlieb JD, Evins AE, Walsh J, Raeke L, Otto MW, Schoenfeld D, Green MF. Once-weekly D-cycloserine effects on negative symptoms and cognition in schizophrenia: An exploratory study. Schizophr. Res. 2008;106:320–327. doi: 10.1016/j.schres.2008.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Acker TM, Yuan H, Hansen KB, Vance KM, Ogden KK, Jensen HS, Burger PB, Mullasseril P, Snyder JP, Liotta DC, Traynelis SF. Mechanism for Noncompetitive Inhibition by Novel GluN2C/D N-Methyl-d-aspartate Receptor Subunit-Selective Modulators. Mol. Pharmacol. 2011;80:782–795. doi: 10.1124/mol.111.073239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Frye SV, Johnson MC, Valvano NL. Synthesis of 2-aminobenzophenones via rapid halogen-lithium exchange in the presence of a 2-amino-N-methoxy-N-methylbenzamide. J. Org. Chem. 1991;56:3750–3752. [Google Scholar]
- 32.Kim DW, Jeong; Lim ST, Sohn M-H, Katzenellenbogen JA, Chi DY. Facile Nucleophilic Fluorination Reactions Using tert-Alcohols as a Reaction Medium: Significantly Enhanced Reactivity of Alkali Metal Fluorides and Improved Selectivity. J. Org. Chem. 2008;73:957–962. doi: 10.1021/jo7021229. [DOI] [PubMed] [Google Scholar]
- 33.O'Hagan D. Preparation of monofluorocarboxylic acids using N,N-diethyl-1,1.2,3,3,3-hexafluoropropylamine. J. Fluorine. Chem. 1989;43:371–377. [Google Scholar]
- 34.Topliss JG. A manual method for applying the Hansch approach to drug design. J. Med. Chem. 1977;20:463–469. doi: 10.1021/jm00214a001. [DOI] [PubMed] [Google Scholar]
- 35.Hansch C, Leo AJ. Substituent Constants for Correlation Analysis in Chemistry and Biology. New York: John Wiley and Sons; 1979. pp. 67–161. [Google Scholar]
- 36.Hansen KB, Traynelis SF. Structural and Mechanistic Determinants of a Novel Site for Noncompetitive Inhibition of GluN2D-Containing NMDA Receptors. J. Neurosci. 2011;31:3650–3661. doi: 10.1523/JNEUROSCI.5565-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hitchcock SA, Pennington LD. Structure-Brain Exposure Relationships. J. Med. Chem. 2006;49:7559–7583. doi: 10.1021/jm060642i. [DOI] [PubMed] [Google Scholar]
- 38.Wang Q, Rager JD, Weinstein K, Kardos PS, Dobson GL, Li J, Hidalgo IJ. Evaluation of the MDR-MDCK cell line as a permeability screen for the blood–brain barrier. Int. J. Pharm. 2005;288:349–359. doi: 10.1016/j.ijpharm.2004.10.007. [DOI] [PubMed] [Google Scholar]
- 39.Hardegger LA, Kuhn B, Spinnler B, Anselm L, Ecabert R, Stihle M, Gsell B, Thoma R, Diez J, Benz J, Plancher J-M, Hartmann G, Banner DW, Haap W, Diederich F. Systematic Investigation of Halogen Bonding in Protein–Ligand Interactions. Angew. Chem. Int. Ed. 2011;50:314–318. doi: 10.1002/anie.201006781. [DOI] [PubMed] [Google Scholar]
- 40.Parisini E, Metrangolo P, Pilati T, Resnati G, Terraneo G. Halogen bonding in halocarbon-protein complexes: a structural survey. Chem. Soc. Rev. 2011;40:2267–2278. doi: 10.1039/c0cs00177e. [DOI] [PubMed] [Google Scholar]
- 41.Wilson CJ, Bevan MD. Intrinsic dynamics and synaptic inputs control the activity patterns of subthalamic nucleus neurons in health and in Parkinson's disease. Neuroscience. 2011;198:54–68. doi: 10.1016/j.neuroscience.2011.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wichmann T, DeLong MR. Models of basal ganglia function and pathophysiology of movement disorders. Neurosurg. Clin. N. Am. 1998;9:223–226. [PubMed] [Google Scholar]
- 43.Bolam JP, Hanley JJ, Booth PAC, Bevan MD. Synaptic organisation of the basal ganglia. J. Anat. 2000;196:527–542. doi: 10.1046/j.1469-7580.2000.19640527.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mullasseril P, Hansen KB, Vance KM, Ogden KK, Yuan H, Kurtkaya NL, Santangelo R, Orr AG, Le P, Vellano KM, Liotta DC, Traynelis SF. A subunit-selective potentiator of NR2C- and NR2D-containing NMDA receptors. Nat. Commun. 2010;1:90. doi: 10.1038/ncomms1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Surmeier DJ, Mercer JN, Chan CS. Autonomous pacemakers in the basal ganglia: who needs excitatory synapses anyway? Curr. Opin. Neurobio. 2005;15:312–318. doi: 10.1016/j.conb.2005.05.007. [DOI] [PubMed] [Google Scholar]
- 46.Dravid SM, Burger PB, Prakash A, Geballe MT, Yadav R, Le P, Vellano K, Snyder JP, Traynelis SF. Structural Determinants of D-Cycloserine Efficacy at the NR1/NR2C NMDA Receptors. J. Neurosci. 2010;30:2741–2754. doi: 10.1523/JNEUROSCI.5390-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lisman JE, Coyle JT, Green RW, Javitt DC, Benes FM, Heckers S, Grace AA. Circuit-based framework for understanding neurotransmitter and risk gene interactions in schizophrenia. Trends. Neurosci. 2008;31:234–242. doi: 10.1016/j.tins.2008.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hillman BG, Gupta SC, Stairs DJ, Buonanno A, Dravid SM. Behavioral analysis of NR2C knockout mouse reveals deficit in acquisition of conditioned fear and working memory. Neurobiol. Learn. Mem. 2011;95:404–414. doi: 10.1016/j.nlm.2011.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mosley CA, Acker TM, Hansen KB, Mullasseril P, Andersen KT, Le P, Vellano KM, Bräuner-Osborne H, Liotta DC, Traynelis SF. Quinazolin-4-one derivatives: A novel class of noncompetitive NR2C/D subunit-selective N-methyl-D-aspartate receptor antagonists. J. Med. Chem. 2010;53:5476–5490. doi: 10.1021/jm100027p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Santangelo Freel RM, Ogden KK, Strong KL, Khatri A, Chepiga KM, Jensen HS, Traynelis SF, Liotta DC. Synthesis and Structure Activity Relationship of Tetrahydroisoquinoline-based Potentiators of GluN2C and GluN2D Containing N-Methyl-D-Aspartate Receptors. J. Med. Chem. 2013 doi: 10.1021/jm400177t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dolomanov OV, Bourhis LJ, Gildea RJ, Howard JAK, Puschmann H. OLEX2: a complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009;42:339–341. [Google Scholar]
- 52.Sheldrick G. A short history of SHELX. Acta. Crystallogr. Sec. A: Found. Crystallogr. 2008;64:112–122. doi: 10.1107/S0108767307043930. [DOI] [PubMed] [Google Scholar]
- 53.Petříček V, Dušek M, Palatinus L JANA2006. The crystallographic computing system. Praha, Czech Republic: Institute of Physics; 2006. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.