

HIV hijacks a single host protein to thwart a powerful antiviral defense on two fronts.
Keywords: innate immunity, APOBEC3 restriction factors, Vif, HIV-1, CBF-β, RUNX, antiviral state, lentivirus, HIV-1 pathogenesis, Host-Pathogen interaction
Abstract
A diverse set of innate immune mechanisms protects cells from viral infections. The APOBEC3 family of DNA cytosine deaminases is an integral part of these defenses. For instance, APOBEC3D, APOBEC3F, APOBEC3G, and APOBEC3H would have the potential to destroy HIV-1 complementary DNA replication intermediates if not for neutralization by a proteasomal degradation mechanism directed by the viral protein Vif. At the core of this complex, Vif heterodimerizes with the transcription cofactor CBF-β, which results in fewer transcription complexes between CBF-β and its normal RUNX partners. Recent studies have shown that the Vif/CBF-β interaction is specific to the primate lentiviruses HIV-1 and SIV (simian immunodeficiency virus), although related nonprimate lentiviruses still require a Vif-dependent mechanism for protection from host species’ APOBEC3 enzymes. We provide a molecular explanation for this evolutionary conundrum by showing that CBF-β is required for expression of the aforementioned HIV-1–restrictive APOBEC3 gene repertoire. Knockdown and knockout studies demonstrate that CBF-β is required for APOBEC3 mRNA expression in the nonpermissive T cell line H9 and in primary CD4+ T lymphocytes. Complementation experiments using CBF-β separation-of-function alleles show that the interaction with RUNX transcription factors is required for APOBEC3 transcriptional regulation. Accordingly, the infectivity of Vif-deficient HIV-1 increases in cells lacking CBF-β, demonstrating the importance of CBF-β/RUNX–mediated transcription in establishing the APOBEC3 antiviral state. These findings demonstrate a major layer of APOBEC3 gene regulation in lymphocytes and suggest that primate lentiviruses evolved to hijack CBF-β in order to simultaneously suppress this potent antiviral defense system at both transcriptional and posttranslational levels.
INTRODUCTION
The overall innate immune response is composed of sensors, transducers, and effectors (1). Sensors bind to pathogen-associated molecular patterns and relay signals to effector proteins in order to mediate the clearance of sensed pathogens. The APOBEC3 family of DNA cytosine deaminases are vital innate immune effector proteins because they can be up-regulated by type I interferons and they afford protection against a wide range of DNA-based parasites including retroviruses, endogenous retrotransposons, and a variety of DNA viruses (2–5). Human cells can express up to seven distinct APOBEC3 enzymes (A/B/C/D/F/G/H), as well as more distantly related family members APOBEC1, AID, APOBEC2, and APOBEC4. The hallmark activity of most members of this family is single-stranded DNA cytosine-to-uracil deamination.
The AIDS virus HIV-1 is the most-studied APOBEC3-susceptible pathogen. A large number of studies using various model systems, including T cell lines and primary CD4+ T lymphocytes, have combined to show that APOBEC3D, APOBEC3F, APOBEC3G, and APOBEC3H have the potential to suppress HIV-1 replication [see recent publications (6–9) and references therein]. These enzymes block virus replication by interfering with reverse transcription and deaminating viral complementary DNA (cDNA) cytosines to uracils, which manifest as genomic strand guanine-to-adenine mutations. However, this potent antiviral mechanism is subverted by the viral infectivity factor (Vif), which nucleates the formation of an E3 ubiquitin ligase complex to promote their degradation [reviewed by (2–5)].
HIV-1 Vif forms an APOBEC3 ubiquitin ligase complex by hijacking the transcriptional cofactor CBF-β to form a scaffold for the recruitment of a cullin-RING ligase complex consisting of CUL5, ELOB, ELOC, and RBX2 (10, 11). Vif itself forms the substrate recognition component that directly binds cellular APOBEC3 enzymes and targets them for polyubiquitination and degradation. The requirement for CBF-β is conserved for Vif function among different HIV-1 subtypes and simian immunodeficiency virus (SIV) isolates, which are primate-specific pathogens, but CBF-β is dispensable for nonprimate lentiviral Vif proteins to degrade the APOBEC3 enzymes of their host species (that is, BIV, MVV, CAEV, and FIV Vif do not require CBF-β for degradation of bovine, ovine, caprine, and feline APOBEC3 proteins) (12–16). The essential nature of the primate lentiviral Vif/CBF-β interaction is highlighted by the extensive interface evident in a recent crystal structure (4800 Å2) (17). CBF-β functions canonically as a cofactor to the RUNX family of transcription factor proteins to regulate the expression of a battery of genes involved in hematopoietic and bone cell development (18, 19). The virus may therefore also exploit the Vif/CBF-β interaction to manipulate the T cell transcription program to somehow benefit virus replication. In support of this general idea, HIV-1 Vif overexpression in the APOBEC3-low Jurkat cell line has been shown to alter the expression of dozens of cellular genes that exhibit significant RUNX1 enrichment near their putative transcriptional start sites (20).
Surprisingly, during the course of interrogating CBF-β function in T cells, we discovered that this protein is actually a positive regulator of transcription of the restrictive APOBEC3 genes themselves. Both knockdown and knockout experiments demonstrate that CBF-β is required for APOBEC3 gene expression. Complementation experiments with previously characterized CBF-β separation-of-function alleles show that the CBF-β interaction with RUNX proteins is required for APOBEC3 transcription. These results suggest that Vif may hijack CBF-β for two major reasons: to form the canonical APOBEC3 degradation complex and to down-regulate the expression of the APOBEC3 genes. In support of this idea, Vif-proficient HIV-1 replication is virtually unimpeded and Vif-deficient HIV-1 is partly recovered in CBF-β–depleted T cells, in both instances because the endogenous APOBEC3 genes are now poorly expressed. This molecular “double whammy” is the first example to our knowledge whereby a viral protein hijacks a single host protein to leverage two distinct mechanisms to overcome a potent and multifaceted antiviral defense system.
RESULTS
CBF-β depletion in T cells results in down-regulation of multiple APOBEC3 genes
To study the role of CBF-β in T cells, we transduced the H9 T cell line with a short hairpin RNA (shRNA) construct to knock down CBF-β or with a nonspecific shRNA as a negative control. As expected from our previous studies, this procedure reduced CBF-β mRNA levels by >90% and caused a corresponding reduction in CBF-β protein levels (Fig. 1, A and B) (10, 12, 21). However, unexpectedly, CBF-β depletion also resulted in a strong reduction in endogenous APOBEC3F and APOBEC3G protein levels (Fig. 1B and fig. S1A). The RT-qPCR analyses revealed a corresponding decline in APOBEC3G mRNA levels, as well as large decreases in APOBEC3C, APOBEC3D, APOBEC3F, and APOBEC3H mRNA levels (Fig. 1C). CBF-β knockdown in primary human CD4+ T cells also caused reduced APOBEC3D, APOBEC3F, and APOBEC3G mRNA levels, as well as those of a positive control (TBX21) (fig. S2) (20).
Fig. 1. CBF-β knockdown and deletion decreases expression of APOBEC3 mRNAs and proteins.

(A) CBF-β mRNA levels relative to TBP in H9 and knockdown derivatives by real-time quantitative polymerase chain reaction (RT-qPCR) (n = 3 with mean ± SD shown). (B) Representative immunoblots of CBF-β and APOBEC3G protein levels in the parental H9 T cell line and shRNA-transduced pools. Tubulin (TUB) is a loading control. (C) RT-qPCR of APOBEC3 mRNA levels relative to TBP in cells transduced with a control shRNA or a CBF-β–specific shRNA (n = 3 with mean ± SD shown; N.D., not detected). (D) Schematic of CRISPR/Cas9 disruption of CBF-β exon 2. (E) Representative immunoblots of CBF-β and APOBEC3G protein levels in H9 cells and a CBF-β knockout derivative. (F) RT-qPCR of APOBEC3 mRNA levels relative to TBP in H9 cells and a CBF-β knockout derivative (n = 3 with mean ± SD shown).
To rule out potential off-target effects of the shRNA-mediated knockdown approach, we used CRISPR/Cas9 (22) to disrupt exon 2 of the CBF-β gene (Fig. 1D). CBF-β disruption did not appear to adversely affect the H9 T cell line, evidenced by high targeting frequencies (>30%) and by parental and null lines showing similar growth kinetics. CBF-β–null H9 derivatives also showed large reductions in APOBEC3C, APOBEC3D, APOBEC3F, APOBEC3G, and APOBEC3H mRNA and APOBEC3F and APOBEC3G protein levels in comparison to the parental cell line (Fig. 1, E and F, and fig. S1B). Thus, both knockdown and knockout results combined to strongly implicate CBF-β as a positive transcriptional factor for APOBEC3C, APOBEC3D, APOBEC3F, APOBEC3G, and APOBEC3H expression in T cells.
The CBF-β/RUNX interaction is required for APOBEC3 transcriptional regulation
We next used CBF-β separation-of-function mutants (21, 23) and focused on APOBEC3G expression as a phenotypic readout to determine whether regulation of APOBEC3 transcription is mediated through the canonical CBF-β/RUNX pathway. Specifically, we used CBF-β N104K, which disrupts the interaction with RUNX proteins but not Vif, and CBF-β F68D, which disrupts the interaction with Vif but not RUNX proteins (Fig. 2, A to C). To confirm the monofunctionality of each of these mutants, we showed that N104K blocks RUNX1-dependent transcriptional activation of a FOXP3 luciferase reporter construct but is still fully competent for Vif-mediated degradation of APOBEC3G (Fig. 2, B and C) (21). In contrast, F68D still retains the full capacity to promote RUNX1-dependent transcription but has compromised Vif degradation function (Fig. 2, B and C).
Fig. 2. RUNX interaction is necessary to restore APOBEC3G expression in CBF-β–depleted cells.
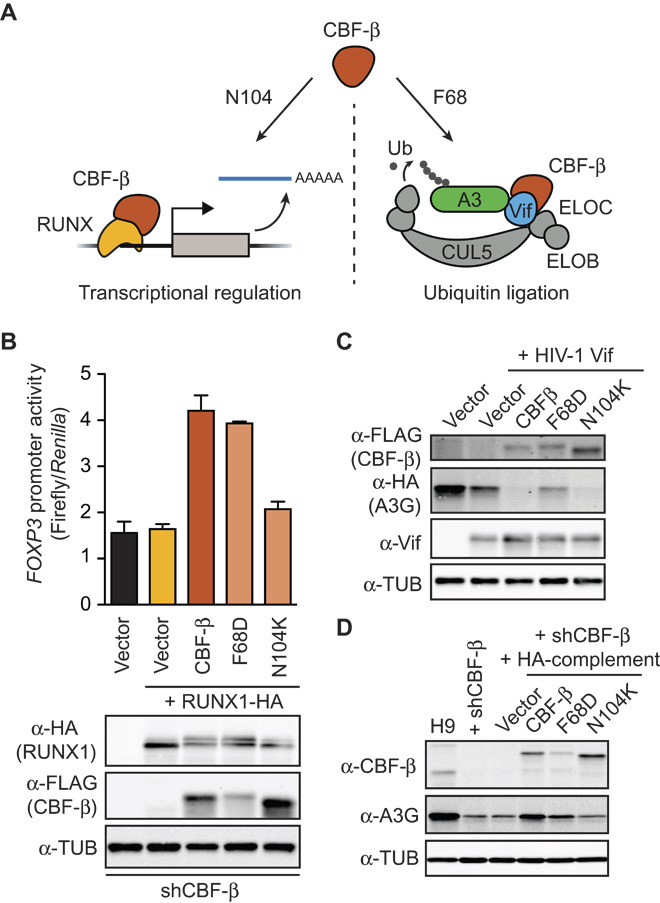
(A) Schematic depicting established phenotypes of CBF-β separation-of-function mutants. Residue N104 is required for CBF-β transcription function with RUNX proteins, whereas F68 is required for Vif-E3 ligase-mediated degradation of APOBEC3 enzymes. (B) Histogram reporting FOXP3 promoter activity as measured by firefly luciferase levels relative to a Renilla luciferase cotransfection control. The indicated CBF-β expression construct, RUNX1 expression construct, and appropriate empty vector controls were cotransfected with luciferase vectors into CBF-β knockdown 293T cells 48 hours before luciferase activity measurement (n = 3; mean ± SD shown). Representative immunoblots from a single experimental replicate are shown below. (C) Representative immunoblots showing Vif functionality [APOBEC3G-HA (hemagglutinin) degradation activity] in the presence of the indicated FLAG–CBF-β constructs 48 hours after transfection into CBF-β knockdown 293T cells. (D) Immunoblots showing the results of a representative complementation experiment using CBF-β knockdown H9 cells and the indicated HA–CBF-β expression constructs or controls. APOBEC3G levels are low in the absence of CBF-β or in the presence of CBF-β N104K (even with higher expression levels relative to the other constructs). In contrast, APOBEC3G levels are restored by expressing wild-type CBF-β or the F68D mutant.
Each CBF-β separation-of-function mutant was then used in a series of complementation experiments. Only CBF-β F68D, which still interacts with RUNX proteins, was able to complement the CBF-β knockdown and cause a restoration in cellular APOBEC3G protein levels (Fig. 2D). The CBF-β N104K mutant, which is defective for both physical and functional interactions with RUNX proteins, failed to rescue APOBEC3G expression levels (Fig. 2D). Together, these results demonstrate that CBF-β regulation of APOBEC3G expression requires a functional interaction with at least one RUNX transcription factor.
CBF-β depletion renders H9 cells permissive for HIV-1 replication independent of Vif function
All current models for HIV-1 Vif function predict that CBF-β depletion should lead to Vif instability and degradation, compromised Vif-E3 ubiquitin ligase function, and a diminished ability to counteract restriction by APOBEC3G and related APOBEC3 restriction factors (2–5). In contrast, HIV-1 particles produced in CBF-β–depleted cells were not restricted and showed slightly higher infectivity in spreading infections in comparison to the same virus replicating in parallel in the H9 parental cell line (fig. S3). The CBF-β knockdown was durable through the entire duration of two independent experiments as judged by immunoblotting. This counterintuitive phenotype was even clearer for Vif-deficient viral particles produced in CBF-β–depleted cells because these retained about 60% of Vif-proficient HIV-1 infectivity (Fig. 3, A and B). In contrast and as expected from many previous studies, Vif-deficient particles were poorly infectious after production in the multi-APOBEC3–expressing H9 cell line with endogenous CBF-β intact (Fig. 3B). These observations are supported by immunoblots of cellular and viral particles showing, as above, that CBF-β is required to maintain endogenous APOBEC3G expression and that Vif expression is diminished when CBF-β is depleted (Fig. 3C).
Fig. 3. CBF-β knockout protects HIV-1 from APOBEC3-mediated restriction.
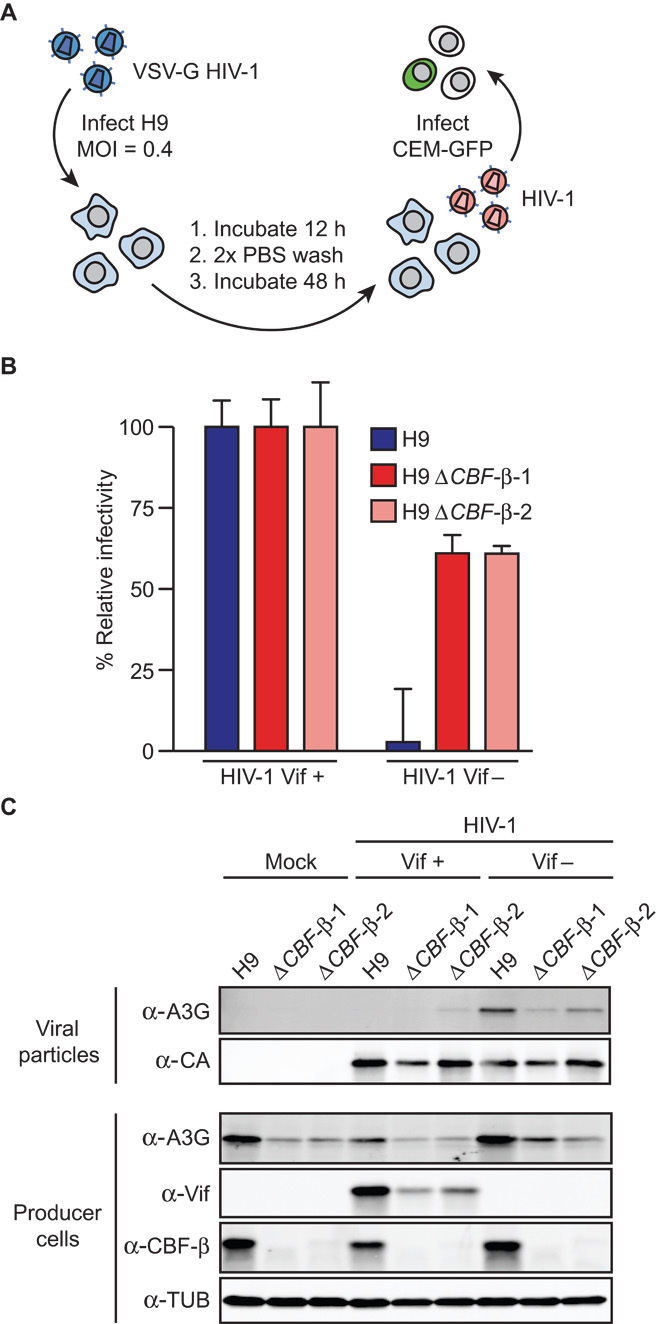
(A) Schematic of HIV-1 single-cycle infectivity assay. (B) Relative infectivity of Vif-proficient and Vif-deficient viruses produced in H9 cells or CBF-β knockout clones (n = 2; mean ± SD shown). Vif-proficient viral infectivity for each cell line is set to 100% to facilitate comparisons. (C) Representative immunoblots of relevant cellular and viral proteins from the experiment depicted in (B), as well as additional data from mock-treated cells.
DISCUSSION
The mechanisms regulating the APOBEC3 antiviral state in T lymphocytes have remained poorly defined, despite extensive literature documenting the potent restriction activities of these enzymes [reviewed by (2–5)]. At the transcriptional level, it is clear that type I interferons can stimulate APOBEC3 transcription, but this is strongly cell type–dependent. For instance, APOBEC3A is strongly up-regulated by interferon treatment of myeloid lineage cells but not T cells, and APOBEC3G has a strong interferon response in primary hepatocytes but minimal responsiveness in T lymphocytes (24–29). Additionally, overexpression of the transcription factors NFAT and IRF4 can induce APOBEC3G mRNA and protein expression in cell culture experiments, though the importance of these transcription factors in endogenous APOBEC3G regulation or in regulating other family members has not been addressed (30). Recent work has also demonstrated specific up-regulation of APOBEC3B in multiple distinct human cancers, and the underlying mechanisms have yet to be defined except for human papillomavirus (HPV)–positive cancers where the virus itself promotes APOBEC3B transcriptional up-regulation and induces genomic mutagenesis as collateral damage (31–35). Despite obvious complexities with both specialized and general APOBEC3 gene expression programs, a full understanding of these processes is likely to be important for future antiviral and anticancer strategies.
Here, we show that the transcription cofactor CBF-β is a strong positive regulator of multiple APOBEC3 genes in CD4+ T cells and that this regulation and the overall antiviral state are dependent on a functional interaction between CBF-β and the RUNX proteins (Fig. 4A). CBF-β depletion renders H9 cells permissive to Vif-deficient HIV-1 infection, demonstrating that CBF-β is critical for maintaining the overall APOBEC3 antiviral state in T lymphocytes. Our results suggest a model for virus-infected cells in which Vif hijacks CBF-β to counteract the APOBEC3 antiviral defense through a polyubiquitination mechanism while simultaneously down-regulating transcription of the APOBEC3 genes themselves by precluding CBF-β/RUNX complex formation (Fig. 4B). This model provides a parsimonious and evolutionarily attractive explanation for why the earliest primate lentivirus, likely a simian tropic ancestor to modern HIV-1, adapted Vif to bind CBF-β from among a large pool of other cellular scaffolding possibilities. One can only imagine that the expanded APOBEC3 protein repertoire of higher primates posed too great of a restrictive barrier for an ancient lentiviral zoonosis, until the Vif protein adapted to use CBF-β and down-regulate this restriction mechanism at both the transcriptional and posttranslational levels. Because the APOBEC3 restriction factors are at least as ancient as placental mammals, all modern lentiviruses have a Vif protein to counteract restriction by cellular APOBEC3 enzymes (36). It is likely that the ancestral APOBEC3 counteraction mechanism was CBF-β–independent and that adaption to bind CBF-β was necessary for lentiviral invasion of the primate branches of the mammalian phylogenetic tree. This scenario is concordant with recent data demonstrating that the Vif proteins of nonprimate lentiviruses—FIV, BIV, MVV, and CAEV—function independently of CBF-β (13–16) and with the fact that the mammalian hosts of these viruses have simpler APOBEC3 repertoires (4). It is supported by the existence of many canonical RUNX-binding sites throughout the human APOBEC3 locus (fig. S4) and also by a previous study showing that Vif overexpression in the Jurkat T cell line could affect the transcriptional activity of dozens of RUNX-regulated genes (although APOBEC3 expression was not altered possibly because basal mRNA levels are already too low in Jurkat) (20).
Fig. 4. New models for APOBEC3-mediated antiviral state and Vif function.

(A) CBF-β/RUNX drives transcription of APOBEC3 genes and maintains a robust antiviral state in the absence of HIV-1 infection in CD4+ T cells. (B) In HIV-1– or SIV-infected cells, Vif prevents CBF-β from binding RUNX transcription complexes to down-regulate APOBEC3 gene transcription and simultaneously promote APOBEC3 protein polyubiquitination and proteasomal degradation.
The results presented here are also encouraging from a drug development perspective because primate lentiviruses may be fully addicted to cellular CBF-β. In support of this idea, primate lentiviruses are largely confined to cell types of the hematopoietic compartment such as CD4+ T lymphocytes that express this cellular protein. Moreover, an obligate relationship between Vif and CBF-β is evidenced by the extensive heterodimer interface (4800 Å2), which would constrain Vif physically and evolutionarily and block it from interacting with many other cellular proteins. This is an attractive drug target because small molecules that disrupt the Vif/CBF-β interaction may preserve high levels of APOBEC3 gene expression and correspondingly high levels of APOBEC3-mediated antiviral immunity. Because the APOBEC3 enzymes directly mutate viral cDNA and can do so at lethal levels, reactivating this potent antiviral response may be able to suppress virus replication indefinitely.
MATERIALS AND METHODS
Human cell lines
H9 and derivative cell lines were maintained in RPMI (HyClone) supplemented with 10% fetal bovine serum (FBS) (Gibco) and 1% penicillin/streptomycin. 293T and derivative cell lines were maintained in Dulbecco’s modified Eagle’s medium (HyClone) supplemented with 10% FBS (Gibco) and 1% penicillin/streptomycin.
Human primary CD4+ T cells
Human primary CD4+ T cells were isolated from same-day-draw peripheral whole blood (Memorial Blood Center, Saint Paul, MN). Peripheral blood mononuclear cells were enriched by whole-blood fractionation over a Ficoll-Paque (GE Healthcare) gradient, and naïve CD4+ T cells were subsequently purified by negative selection (CD4+ T Cell Isolation Kit II, Miltenyi Biotec). CD4+ T cells were stimulated with phytohemagglutinin (10 μg/ml) and interleukin-2 (IL-2) (20 U/ml) and maintained in RPMI medium supplemented with 10% FBS, 1% penicillin/streptomycin, and IL-2 (20 U/ml).
Plasmid DNA transfection experiments
293T cells were transfected using Transit LTI (Mirus) according to the manufacturer’s protocol. All downstream analyses were performed 48 hours after transfection.
Knockdown experiments
The CBF-β shRNA was derived by subcloning the short hairpin sequence from the pGIPZ-based Open Biosystems catalog no. RHS4430-99161432 into the pLKO.1 puro vector (Addgene 8453) by direct ligation using Age I and Eco RI restriction sites (pGIPZ-based vectors showed poor infectivity in T cell lines). The shControl construct was obtained from Open Biosystems (catalog no. RHS4346). shRNA-expressing lentiviruses were prepared as described by Jager et al. (10).
CRISPR/Cas9-mediated knockouts
Guide RNA sequences identical to CBF-β exon 2 were ligated into the lentiCRISPRv1 vector (Addgene 49535) according to accompanying Zhang laboratory protocols (37). Knockout derivative cell lines were generated by targeting the 5′-GGCCGTCGCGGCAGGCGTTC-3′ site in exon 2 of the CBF-β gene. This targeting construct was generated by annealing oligos 5′-CACCGGCCGTCGCGGCAGGCGTTCG-3′ and 5′-AAACGAACGCCTGCCGCGACGGCC-3′ and ligating the resulting double-stranded DNA into Bsm BI–digested lentiCRISPRv1. The transducing vector was produced by transfecting 293 cells with the lentiCRISPRv1-targeting construct along with pΔ-NRF (HIV-1 gag, pol, rev, tat genes) and pMDG (VSV-G) expression vectors. Virus-containing culture supernatants were filtered (0.45 μm) and concentrated by centrifugation (22,000g, 2 hours, 10°C), and viral pellets were resuspended in complete RPMI. H9 cells were incubated with Cas9/CRISPR-encoding lentiviruses for 48 hours and then placed under drug selection (puromycin, 1 μg/ml). Clones were generated by limiting dilution in 96-well U-bottom plates, followed by outgrowth and screening by immunoblotting and target DNA sequencing.
Immunoblotting experiments
Cell lysates prepared in 2.5× Laemmli sample buffer were separated by SDS–polyacrylamide gel electrophoresis and electrophoretically transferred to PVDF-FL membranes (Millipore). Membranes were blocked in 4% milk in phosphate-buffered saline (PBS) and incubated with primary antibodies diluted in 4% milk in PBS + 0.1% Tween 20. Secondary antibodies were diluted in 4% milk in PBS + 0.1% Tween 20 + 0.01% SDS. Membranes were imaged on a LI-COR Odyssey instrument. Primary antibodies used in these studies were rabbit anti-APOBEC3G [National Institutes of Health (NIH) ARRRP 10201 courtesy of J. Lingappa], rabbit anti-APOBEC3F (NIH ARRRP 11474 courtesy of M. Malim), rabbit anti–CBF-β (Epitomics), rabbit anti-HA (Cell Signaling), rabbit anti-FLAG (Sigma F7425), mouse anti–HIV-1 p24/CA (NIH ARRRP 3537 courtesy of B. Chesebro and K. Wehrly), mouse anti–HIV-1 Vif (NIH ARRRP 6459 courtesy of M. Malim), and mouse anti-tubulin (Covance). Secondary antibodies used were IRDye 800CW goat anti-rabbit (LI-COR 827-08365) and Alexa Fluor 680 goat anti-mouse (Molecular Probes A-21057).
RNA isolation, cDNA synthesis, and RT-qPCR
RNA was extracted from cell lines and primary cells (RNeasy, Qiagen) and used as a template for random hexamer-primed cDNA synthesis (Transcriptor, Roche). Gene expression was analyzed on a Roche LightCycler 480 instrument as described by Refsland et al. (24). Primer and probe sequences are listed in table S1.
CBF-β complementation experiments
HA-tagged CBF-β variants were subcloned into the CSU6-IDR2 IRES-dsRed lentiviral packaging vector (courtesy of W. Sundquist) and cotransfected into 293 cells along with pΔ-NRF (HIV-1 gag-pol-rev-tat) and pMDG (VSV-G) expression vectors. Forty-eight hours after transfection, culture supernatants were filtered (0.45 μm) and concentrated by centrifugation (22,000g, 2 hours, 10°C). Viral pellets were resuspended in complete RPMI and used to directly infect H9 cells. Transduced cell populations were assayed for dsRed expression by flow cytometry and analyzed by immunoblotting 72 hours after transduction.
Dual luciferase transcription reporter assays
Transcriptional reporter assays were performed in CBF-β knockdown 293 cells using a FOXP3 promoter luciferase reporter construct as described by Hultquist et al. (21).
HIV-1 single-cycle and spreading infectivity experiments
VSV-G pseudotyped HIV-1 was produced by cotransfecting 293 cells with HIV-1IIIB A200C molecular clone derivatives (pIIIB or pIIIB Δvif) and pMDG (VSV-G). Forty-eight hours after transfection, culture supernatants containing viruses were filtered (0.45 μm) and concentrated by centrifugation (22,000g, 2 hours, 10°C). Virus pellets were resuspended in complete RPMI, and viral titer was determined by serial dilution on CEM-GFP indicator cells. Viruses were added to H9 cultures (multiplicity of infection, 0.4), and cells were washed twice in PBS 12 hours after infection to remove any residual pseudotyped virus. Forty-eight hours later, cell-free supernatants containing progeny viruses were used to infect CEM-GFP reporter cells. HIV-1–spreading infections were performed as described by Hultquist et al. (38).
Acknowledgments
We thank A. Haase, T. Ikeda, A. Land, C. Richards, N. Shaban, and P. Southern for helpful comments. LentiCRISPRv1 was a gift from F. Zhang. RUNX3 ChIP-seq data were obtained from the ENCODE consortium (ENCSR000BRI) and originally provided by R. Myers. Funding: Salary support for B.D.A. was provided in part by NIH T32 AI83196, T32 AI007313, and F31 AI116305. This work was supported by grants to R.S.H. from the NIH (R01 AI064046 and P01 GM091743). Author contributions: B.D.A. performed all of the experiments. B.D.A. and R.S.H. designed the experiments, interpreted the results, and wrote the manuscript. Competing interests: R.S.H. is a co-founder of ApoGen Biotechnologies Inc. B.D.A. has no competing interests. Data and materials availability: All of the reagents and primary data sets reported here are available upon written request.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/1/8/e1500296/DC1
Fig. S1. CBF-β knockdown or knockout causes decreased A3F protein levels.
Fig. S2. CBF-β knockdown in primary human CD4+ T cells results in a concomitant reduction in the mRNA levels of APOBEC3D, APOBEC3F, and APOBEC3G.
Fig. S3. CBF-β knockdown renders H9 cells more permissive to Vif-deficient HIV-1 replication.
Fig. S4. Schematic of predicted and ChIP-validated RUNX-binding sites within the human APOBEC3 locus.
Table S1. qPCR primer and probe information.
REFERENCES AND NOTES
- 1.Beutler B., Eidenschenk C., Crozat K., Imler J.-L., Takeuchi O., Hoffmann J. A., Akira S., Genetic analysis of resistance to viral infection. Nat. Rev. Immunol. 7, 753–766 (2007). [DOI] [PubMed] [Google Scholar]
- 2.Malim M. H., Bieniasz P. D., HIV restriction factors and mechanisms of evasion. Cold Spring Harb. Perspect. Med. 2, a006940 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Desimmie B. A., Delviks-Frankenberrry K. A., Burdick R. C., Qi D., Izumi T., Pathak V. K., Multiple APOBEC3 restriction factors for HIV-1 and one Vif to rule them all. J. Mol. Biol. 426, 1220–1245 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Refsland E. W., Harris R. S., The APOBEC3 family of retroelement restriction factors. Curr. Top. Microbiol. Immunol. 371, 1–27 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Strebel K., HIV accessory proteins versus host restriction factors. Curr. Opin. Virol. 3, 692–699 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Refsland E. W., Hultquist J. F., Harris R. S., Endogenous origins of HIV-1 G-to-A hypermutation and restriction in the nonpermissive T cell line CEM2n. PLOS Pathog. 8, e1002800 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ooms M., Brayton B., Letko M., Maio S. M., Pilcher C. D., Hecht F. M., Barbour J. D., Simon V., HIV-1 Vif adaptation to human APOBEC3H haplotypes. Cell Host Microbe 14, 411–421 (2013). [DOI] [PubMed] [Google Scholar]
- 8.Refsland E. W., Hultquist J. F., Luengas E. M., Ikeda T., Shaban N. M., Law E. K., Brown W. L., Reilly C., Emerman M., Harris R. S., Natural polymorphisms in human APOBEC3H and HIV-1 Vif combine in primary T lymphocytes to affect viral G-to-A mutation levels and infectivity. PLOS Genet. 10, e1004761 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sato K., Izumi T., Misawa N., Kobayashi T., Yamashita Y., Ohmichi M., Ito M., Takaori-Kondo A., Koyanagi Y., Remarkable lethal G-to-A mutations in vif-proficient HIV-1 provirus by individual APOBEC3 proteins in humanized mice. J. Virol. 84, 9546–9556 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jäger S., Kim D. Y., Hultquist J. F., Shindo K., LaRue R. S., Kwon E., Li M., Anderson B. D., Yen L., Stanley D., Mahon C., Kane J., Franks-Skiba K., Cimermancic P., Burlingame A., Sali A., Craik C. S., Harris R. S., Gross J. D., Krogan N. J., Vif hijacks CBF-β to degrade APOBEC3G and promote HIV-1 infection. Nature 481, 371–375 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang W., Du J., Evans S. L., Yu Y., Yu X.-F., T-cell differentiation factor CBF-β regulates HIV-1 Vif-mediated evasion of host restriction. Nature 481, 376–379 (2011). [DOI] [PubMed] [Google Scholar]
- 12.Hultquist J. F., Binka M., LaRue R. S., Simon V., Harris R. S., Vif proteins of human and simian immunodeficiency viruses require cellular CBFβ to degrade APOBEC3 restriction factors. J. Virol. 86, 2874–2877 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang W., Wang H., Li Z., Liu X., Liu G., Harris R. S., Yu X. F., Cellular requirements for bovine immunodeficiency virus Vif-mediated inactivation of bovine APOBEC3 proteins. J. Virol. 88, 12528–12540 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Han X., Liang W., Hua D., Zhou X., Du J., Evans S. L., Gao Q., Wang H., Viqueira R., Wei W., Zhang W., Yu X.-F., Evolutionarily conserved requirement for core binding factor beta in the assembly of the human immunodeficiency virus/simian immunodeficiency virus Vif-cullin 5-RING E3 ubiquitin ligase. J. Virol. 88, 3320–3328 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ai Y., Zhu D., Wang C., Su C., Ma J., Ma J., Wang X., Core-binding factor subunit beta is not required for non-primate lentiviral Vif-mediated APOBEC3 degradation. J. Virol. 88, 12112–12122 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang J., Wu J., Wang W., Wu H., Yu B., Wang J., Lv M., Wang X., Zhang H., Kong W., Yu X., Role of cullin-elonginB-elonginC E3 complex in bovine immunodeficiency virus and maedi-visna virus Vif-mediated degradation of host A3Z2-Z3 proteins. Retrovirology 11, 77 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guo Y., Dong L., Qiu X., Wang Y., Zhang B., Liu H., Yu Y., Zang Y., Yang M., Huang Z., Structural basis for hijacking CBF-β and CUL5 E3 ligase complex by HIV-1 Vif. Nature 505, 229–233 (2014). [DOI] [PubMed] [Google Scholar]
- 18.Wong W. F., Kohu K., Chiba T., Sato T., Satake M., Interplay of transcription factors in T-cell differentiation and function: The role of Runx. Immunology 132, 157–164 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ito Y., Oncogenic potential of the RUNX gene family: ‘Overview’. Oncogene 23, 4198–4208 (2004). [DOI] [PubMed] [Google Scholar]
- 20.Kim D. Y., Kwon E., Hartley P. D., Crosby D. C., Mann S., Krogan N. J., Gross J. D., CBFβ stabilizes HIV Vif to counteract APOBEC3 at the expense of RUNX1 target gene expression. Mol. Cell 49, 632–644 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hultquist J. F., McDougle R. M., Anderson B. D., Harris R., HIV type 1 viral infectivity factor and the RUNX transcription factors interact with core binding factor β on genetically distinct surfaces. AIDS Res. Hum. Retroviruses 28, 1543–1551 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hsu P. D., Lander E. S., Zhang F., Development and applications of CRISPR-Cas9 for genome engineering. Cell 157, 1262–1278 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nagata T., Werner M. H., Functional mutagenesis of AML1/RUNX1 and PEBP2β/CBFβ define distinct, non-overlapping sites for DNA recognition and heterodimerization by the Runt domain. J. Mol. Biol. 308, 191–203 (2001). [DOI] [PubMed] [Google Scholar]
- 24.Refsland E. W., Stenglein M. D., Shindo K., Albin J. S., Brown W. L., Harris R. S., Quantitative profiling of the full APOBEC3 mRNA repertoire in lymphocytes and tissues: Implications for HIV-1 restriction. Nucleic Acids Res. 38, 4274–4284 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Koning F. A., Newman E. N., Kim E.-Y., Kunstman K. J., Wolinsky S. M., Malim M. H., Defining APOBEC3 expression patterns in human tissues and hematopoietic cell subsets. J. Virol. 83, 9474–9485 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Thielen B. K., McNevin J. P., McElrath M. J., Hunt B. V., Klein K. C., Lingappa J. R., Innate immune signaling induces high levels of TC-specific deaminase activity in primary monocyte-derived cells through expression of APOBEC3A isoforms. J. Biol. Chem. 285, 27753–27766 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stenglein M. D., Burns M. B., Li M., Lengyel J., Harris R. S., APOBEC3 proteins mediate the clearance of foreign DNA from human cells. Nat. Struct. Mol. Biol. 17, 222–229 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bonvin M., Achermann F., Greeve I., Stroka D., Keogh A., Inderbitzin D., Candinas D., Sommer P., Wain-Hobson S., Vartanian J. P., Greeve J., Interferon-inducible expression of APOBEC3 editing enzymes in human hepatocytes and inhibition of hepatitis B virus replication. Hepatology 43, 1364–1374 (2006). [DOI] [PubMed] [Google Scholar]
- 29.Tanaka Y., Marusawa H., Seno H., Matsumoto Y., Ueda Y., Kodama Y., Endo Y., Yamauchi J., Matsumoto T., Takaori-Kondo A., Ikai I., Chiba T., Anti-viral protein APOBEC3G is induced by interferon-α stimulation in human hepatocytes. Biochem. Biophys. Res. Commun. 341, 314–319 (2006). [DOI] [PubMed] [Google Scholar]
- 30.Farrow M. A., Kim E.-Y., Wolinsky S. M., Sheehy A. M., NFAT and IRF proteins regulate transcription of the anti-HIV gene, APOBEC3G. J. Biol. Chem. 286, 2567–2577 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Burns M. B., Lackey L., Carpenter M. A., Rathore A., Land A. M., Leonard B., Refsland E. W., Kotandeniya D., Tretyakova N., Nikas J. B., Yee D., Temiz N. A., Donohue D. E., McDougle R. M., Brown W. L., Law E. K., Harris R. S., APOBEC3B is an enzymatic source of mutation in breast cancer. Nature 494, 366–370 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Burns M. B., Temiz N. A., Harris R. S., Evidence for APOBEC3B mutagenesis in multiple human cancers. Nat. Genet. 45, 977–983 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Leonard B., Hart S. N., Burns M. B., Carpenter M. A., Temiz N. A., Rathore A., Vogel R. I., Nikas J. B., Law E. K., Brown W. L., Li Y., Zhang Y., Maurer M. J., Oberg A. L., Cunningham J. M., Shridhar V., Bell D. A., April C., Bentley D., Bibikova M., Cheetham R. K., Fan J.-B., Grocock R., Humphray S., Kingsbury Z., Peden J., Chien J., Swisher E. M., Hartmann L. C., Kalli K. R., Goode E. L., Sicotte H., Kaufmann S. H., Harris R. S., APOBEC3B upregulation and genomic mutation patterns in serous ovarian carcinoma. Cancer Res. 73, 7222–7231 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Warren C. J., Xu T., Guo K., Griffin L. M., Westrich J. A., Lee D., Lambert P. F., Santiago M. L., Pyeon D., APOBEC3A functions as a restriction factor of human papillomavirus. J. Virol. 89, 688–702 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vieira V. C., Leonard B., White E. A., Starrett G. J., Temiz N. A., Lorenz L. D., Lee D., Soares M. A., Lambert P. F., Howley P. M., Harris R. S., Human papillomavirus E6 triggers upregulation of the antiviral and cancer genomic DNA deaminase APOBEC3B. MBio 5, e02234-14 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.LaRue R. S., Jónsson S. R., Silverstein K. A. T., Lajoie M., Bertrand D., El-Mabrouk N., Hötzel I., Andrésdóttir V., Smith T. P. L., Harris R. S., The artiodactyl APOBEC3 innate immune repertoire shows evidence for a multi-functional domain organization that existed in the ancestor of placental mammals. BMC Mol. Biol. 9, 104 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sanjana N. E., Shalem O., Zhang F., Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 11, 783–784 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hultquist J. F., Lengyel J. A., Refsland E. W., LaRue R. S., Lackey L., Brown W. L., Harris R. S., Human and rhesus APOBEC3D, APOBEC3F, APOBEC3G, and APOBEC3H demonstrate a conserved capacity to restrict Vif-deficient HIV-1. J. Virol. 85, 11220–11234 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.ENCODE Project Consortium , An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/1/8/e1500296/DC1
Fig. S1. CBF-β knockdown or knockout causes decreased A3F protein levels.
Fig. S2. CBF-β knockdown in primary human CD4+ T cells results in a concomitant reduction in the mRNA levels of APOBEC3D, APOBEC3F, and APOBEC3G.
Fig. S3. CBF-β knockdown renders H9 cells more permissive to Vif-deficient HIV-1 replication.
Fig. S4. Schematic of predicted and ChIP-validated RUNX-binding sites within the human APOBEC3 locus.
Table S1. qPCR primer and probe information.