

Abstract
Four Rhodococcus spp. exhibited the ability to use 4,4′-dithiodibutyric acid (DTDB) as a sole carbon source for growth. The most important step for the production of a novel polythioester (PTE) using DTDB as a precursor substrate is the initial cleavage of DTDB. Thus, identification of the enzyme responsible for this step was mandatory. Because Rhodococcus erythropolis strain MI2 serves as a model organism for elucidation of the biodegradation of DTDB, it was used to identify the genes encoding the enzymes involved in DTDB utilization. To identify these genes, transposon mutagenesis of R. erythropolis MI2 was carried out using transposon pTNR-TA. Among 3,261 mutants screened, 8 showed no growth with DTDB as the sole carbon source. In five mutants, the insertion locus was mapped either within a gene coding for a polysaccharide deacetyltransferase, a putative ATPase, or an acetyl coenzyme A transferase, 1 bp upstream of a gene coding for a putative methylase, or 176 bp downstream of a gene coding for a putative kinase. In another mutant, the insertion was localized between genes encoding a putative transcriptional regulator of the TetR family (noxR) and an NADH:flavin oxidoreductase (nox). Moreover, in two other mutants, the insertion loci were mapped within a gene encoding a hypothetical protein in the vicinity of noxR and nox. The interruption mutant generated, R. erythropolis MI2 noxΩtsr, was unable to grow with DTDB as the sole carbon source. Subsequently, nox was overexpressed and purified, and its activity with DTDB was measured. The specific enzyme activity of Nox amounted to 1.2 ± 0.15 U/mg. Therefore, we propose that Nox is responsible for the initial cleavage of DTDB into 2 molecules of 4-mercaptobutyric acid (4MB).
INTRODUCTION
The genus Rhodococcus has attracted great interest in recent years due to the ability of many of its species to degrade and transform a wide range of xenobiotic substances (1). Rhodococci are described as aerobic, Gram-positive, mycolate-containing actinomycetes with high genomic G+C contents. They have a diverse set of genetic and catabolic features, because of the presence of large linear plasmids in addition to their large chromosomes (2). The number of publications and patents involving Rhodococcus spp. has increased significantly in recent years (3).
Rhodococcus spp. have been isolated from a variety of sources. They are ubiquitous in soils contaminated with crude oil and/or other xenobiotic compounds. The ability of Rhodococcus species to degrade substituted hydrocarbons and other chemicals allows them to play a role in the natural degradation and bioremediation of such compounds (4). Many Rhodococcus species are characterized by broad metabolic diversity and an array of unique enzymatic capabilities; therefore, they are also of interest for pharmaceutical, environmental, chemical, and energy studies. They play a significant role in the process of desulfurization of fossil fuels (5) and in the industrial production of acrylamide (6). Strains of Rhodococcus erythropolis have been identified as the most promising bacteria for biodesulfurization. For example, strain ATCC 4277 is most effective for the desulfurization of dibenzothiophene (DBT) (7). Moreover, R. erythropolis CCM2595 is studied as a model strain for the bioremediation of phenol and other aromatic compounds (8). The complexity of Rhodococcus biology is due to its large genome, which contains a multiplicity of catabolic genes, a sophisticated regulatory network, and a high genetic redundancy of biosynthetic pathways (3).
Rhodococcus species are highly accessible for investigations, both because they exhibit high growth rates and a simple developmental cycle (9) and because several transposon mutagenesis systems have been established for these bacteria (10–12). The transposon-based vector system recently established by Sallam et al. (9, 13) can efficiently generate random mutagenesis when transfected into various Rhodococcus species.
4,4′-Dithiodibutyric acid (DTDB), also known as 3-carboxypropyl disulfide, is a white, solid organic sulfur compound (OSC). This disulfide is used as an alternative monolayer for the manufacture of protein chips that are based on a gold surface (14), which are then used for the recognition of various sugars by surface-enhanced Raman spectroscopy and cyclic voltammetry (15). DTDB has also been employed in studies concerned with the improvement of polythioester (PTE) production (16). DTDB biodegradation is restricted to only a few Rhodococcus strains, although DTDB is a structural analogue of homocysteine, a common cell metabolite (17).
According to the pathway proposed previously for the microbial utilization of DTDB in R. erythropolis strain MI2 (17) (Fig. 1), the catabolism of DTDB is initiated by a symmetrical cleavage of DTDB into 2 molecules of 4-mercaptobutyric acid (4MB) by an unknown disulfide reductase. The toxicity of 4MB is due to the presence of a free sulfhydryl group that inhibits bacterial growth at concentrations as low as 0.01% (vol/vol). Therefore, to date, 4MB could not be successfully used directly as a PTE precursor (18).
FIG 1.

Proposed pathway for the degradation of DTDB in R. erythropolis strain MI2. DTDB is initially cleaved by the NADH:flavin oxidoreductase (Nox) into 2 molecules of 4-mercaptobutyric acid, which is then probably oxidized by a putative oxygenase, yielding 4-oxo-4-sulfanylbutyric acid. Afterwards, the sulfur moieties could be removed by a putative desulfhydrase as volatile hydrogen sulfide. The resulting succinic acid is then further metabolized.
The second proposed step in the catabolism of DTDB is the oxidation of 4MB into 4-oxo-4-sulfanylbutyric acid by a putative oxygenase enzyme. Finally, 4-oxo-4-sulfanylbutyric acid is presumably desulfurized into succinic acid by a putative desulfhydrase, thus releasing the sulfur moiety as volatile hydrogen sulfide (17).
Microbially synthesized PTEs are nonbiodegradable polymers containing sulfur in their backbones (19, 20). PTEs are accumulated in the cytoplasm of some bacterial cells, forming hydrophobic inclusions, such as the well-known polyhydroxyalkanoates (PHAs) (21, 22). These PTEs have many industrially and scientifically unique characteristics, such as their nonbiodegradability, microstructure, thermal behavior, and diverse mechanical properties, which allow them to be incorporated in various industrial applications (22, 23).
PTE heteropolymers were detected in Ralstonia eutropha (24, 25). Later, PTE homopolymers were synthesized in recombinant strains of Escherichia coli and Advenella mimigardefordensis (20, 26, 27). Although various precursors, different strains, and alternative fermentation strategies were applied, neither homopolymers nor heteropolymers of 4MB could be produced so far (18, 21).
Therefore, the current study provides the first detailed analysis of the catabolism of the promising PTE precursor DTDB by Rhodococcus spp. Moreover, we identified the gene coding for the enzyme that catalyzes the initial cleavage step of DTDB, which is considered to be the first important step for microbial production of poly(4MB). Because poly(4MB) has a backbone of 4 carbons, it is expected that poly(4MB) will show interesting new features, which may allow it to be used in various industrial applications.
MATERIALS AND METHODS
Sulfur-containing chemicals.
DTDB and 4MB were purchased from Sigma-Aldrich Chemie (Steinheim, Germany).
Bacterial strains, plasmids, and culture conditions.
The bacterial strains and plasmids used in this study are listed in Table 1. All Rhodococcus strains investigated for their abilities to utilize DTDB as the sole source of carbon and energy are shown in Table 2. Rhodococcus strains and transposon-induced mutants of Rhodococcus erythropolis strain MI2 were cultivated aerobically at 30°C in lysogeny broth (LB) medium (28) or in mineral salts medium (MSM) (29, 30) containing the required carbon source. All carbon sources were prepared as filter-sterilized 20% (wt/vol) stock solutions, and the pH was adjusted to 7.0. DTDB was added as a carbon source at concentrations between 20 and 40 mM. Solid medium contained 1.8% (wt/vol) purified agar agar. E. coli strains were cultivated aerobically in LB medium (28) at 37°C. Antibiotics at the following concentrations were added to the required growth medium to maintain plasmids: ampicillin, 75 μg/ml; chloramphenicol, 34 μg/ml; thiostrepton, 12.5 μg/ml; gentamicin, 50 μg/ml.
TABLE 1.
Bacterial strains, plasmids, and oligonucleotides used in this study
| Strain, plasmid, or oligonucleotide | Relevant phenotype or genotypea | Source or reference |
|---|---|---|
| Strains | ||
| Rhodococcus erythropolis | ||
| MI2 | Wild type, DTDB degrading | 17 |
| HK1 | Transposon-induced mutant of R. erythropolis MI2; no growth with DTDB as the sole carbon source | This study |
| HK2 | Transposon-induced mutant of R. erythropolis MI2; no growth with DTDB as the sole carbon source | This study |
| HK3 | Transposon-induced mutant of R. erythropolis MI2; no growth with DTDB as the sole carbon source | This study |
| HK4 | Transposon-induced mutant of R. erythropolis MI2; no growth with DTDB as the sole carbon source | This study |
| HK5 | Transposon-induced mutant of R. erythropolis MI2; no growth with DTDB as the sole carbon source | This study |
| HK6 | Transposon-induced mutant of R. erythropolis MI2; no growth with DTDB as the sole carbon source | This study |
| HK7 | Transposon-induced mutant of R. erythropolis MI2; no growth with DTDB as the sole carbon source | This study |
| HK8 | Transposon-induced mutant of R. erythropolis MI2; no growth with DTDB as the sole carbon source | This study |
| MI2::noxΩtsr | No growth with DTDB as the sole carbon source | This study |
| PR4 | Wild type; degradation of polychlorinated biphenyls | NBRC100887 |
| IGTS8 | Wild type; desulfurization of fossil fuel and DBT | ATCC 53968 |
| Rhodococcus ruber | Wild type; PHA producer | NCIMB 20146 |
| Escherichia coli | ||
| TOP10 | F− mcrA Δ(mrr-hsdRMS-mcrBC) rpsL nupG ϕ80lacZΔM15 ΔlacX74 deoR recA1 araD139 Δ(ara-leu)7697 galU galK endA1 | Invitrogen, Carlsbad, CA |
| XL1-Blue | recA1 endA1 gyrA96 thi-1 hsdR17 (rK− mK+) supE44 relA1 λ− lac [F′ proAB lacIq lacZΔM15 Tn10 (Tcr)] | Stratagene, San Diego, CA |
| BL21(DE3)pLysS | F− ompT hsdSB(rB− mB−) gal dcm λ(DE3) pLysS(Cmr) | Novagen, Madison, WI |
| Plasmids | ||
| pTNR-TA | Apr Tsr | 9 |
| pJQ200mp18 | sacB oriV oriT traJ; Gmr | 77 |
| pJQ200mp18::noxΩtsr | pJQ200mp18 harboring the interrupted oxidoreductase gene | This study |
| pET23a(+) | pBR322 ori, f1 ori His6; Apr T7lac | Novagen, Madison, WI |
| pET23a(+)::nox | pET23a(+) harboring the oxidoreductase gene | This study |
| Primers | ||
| Walking IS1 | GGAAAAGGACTGCTGTCGCTGCC | MWG-Biotech AG |
| Sequencing IS1 | GCCGTAGACCTCGATGAACTCCAC | MWG-Biotech AG |
| Forward-nox-XbaI | AAAATCTAGAATGACCACAGCGCCCACAC | MWG-Biotech AG |
| Reverse-nox-XhoI | AAAACTCGAGTCACTGGCCGAGGCGC | MWG-Biotech AG |
| Forward-nox-NdeI | AAAACATATGACCACAGCGCCCACACC | MWG-Biotech AG |
| Forward-thio-BamHI | AAAAGGATCCATGAATGACTGAGTTGGAC | MWG-Biotech AG |
| Reverse-thio-BamHI | AAAAGGATCCTTATCGGTTGGCCGCGAG | MWG-Biotech AG |
Underlined letters in primer sequences indicate restriction sites.
TABLE 2.
Bacterial isolates from this study and previous studies capable of using the indicated OSC as the sole source of carbon and energy
| Strain designation | DTDB utilization | Susceptibilitya | Source or reference |
|---|---|---|---|
| Rhodococcus erythropolis MI2 | + | Cm, Ts | This study; reference 17 |
| Rhodococcus erythropolis PR4 | + | Cm, Ts | This study |
| Rhodococcus erythropolis IGTS8 | + | Cm, Ts | This study |
| Rhodococcus ruber | + | Cm, Ts, Km, Ap | This study |
| Rhodococcus erythropolis ATCC 4277T | − | Ap, Cm, Gm, Km, Tc, Ts | This study |
| Rhodococcus jostii RHA1 | − | Cm, Ts, Km, Ap | This study |
| Rhodococcus opacus R7 | − | Cm, Ts, Km, Ap | This study |
| Rhodococcus sp. strain BCP1 | − | Cm, Ts, Km, Ap | This study |
| Rhodococcus erythropolis SQ1 | − | Cm, Ts, Km, Ap | This study |
| Rhodococcus erythropolis BEA1 | − | Ap, Cm, Gm, Km, Pn, Sm, Tc, Ts | 17 |
| Rhodococcus erythropolis BEA2 | − | Ap, Cm, Gm, Km, Pn, Sm, Tc, Ts | 17 |
| Rhodococcus erythropolis BEA3 | − | Ap, Cm, Gm, Pn, Sm, Tc, Ts | 17 |
| Rhodococcus erythropolis BEA4 | − | Ap, Cm, Gm, Km, Pn, Sm, Tc, Ts | 17 |
Ap, ampicillin; Cm, chloramphenicol; Gm, gentamicin; Km, kanamycin; Pn, penicillin; Sm, streptomycin; Tc, tetracycline, Ts, thiostrepton.
Isolation, amplification, and transfer of DNA.
Chromosomal DNAs of the wild-type strain and the transposon-induced mutants were isolated with the DNeasy blood and tissue kit (Qiagen). Plasmid DNA was isolated using the PeqGOLD plasmid miniprep kit I (Peqlab, Erlangen, Germany). DNA fragments were purified from agarose gels by using the PeqGOLD gel extraction kit (Peqlab). DNA polymerases, ligases, and restriction enzymes were used according to the manufacturer's instructions. PCR was conducted in a peqSTAR 2× gradient thermal cycler (Peqlab, Erlangen, Germany) using Phusion high-fidelity DNA polymerase. Oligonucleotides are listed in Table 1.
Competent cells of Rhodococcus spp. were generated according to the work of Desomer et al. (31) and Kalscheuer et al. (32). Competent cells of E. coli were prepared and transformed by the CaCl2 procedure (33).
DNA sequencing.
Sequencing reactions were carried out according to the standard sequencing laboratory protocols of the Seqlab in Göttingen, Germany. Sequencing data were analyzed using the SeqMan, Chromas 1.45, and Bioedit programs, as well as the Basic Local Alignment Search Tool (BLAST) (http://blast.ncbi.nlm.nih.gov/Blast.cgi) (34).
Polymer production, extraction, and content analysis.
PTEs were produced in MSM supplemented with DTDB and with a reduced nitrogen concentration (0.05% [vol/vol]) in order to enhance polymer accumulation. Polymer contents and compositions were determined by gas chromatography (GC)-mass spectrometry (MS). Polymers were extracted from freeze-dried cells of Rhodococcus ruber using a Soxhlet apparatus and were then subjected to GC-MS after acid methanolysis (35, 36). Nuclear magnetic resonance (NMR) analysis was carried out in the NMR spectroscopy laboratory at the Organic Chemistry Institute in the Westfälische Wilhelms-Universität Münster. The NMR experiment was performed on a Varian Unity Plus system with a 600-MHz spectrum for 1H NMR and a 300-MHz spectrum for 13C NMR.
Utilization of DTDB as a sole sulfur source.
In order to study the abilities of Rhodococcus strains to use DTDB as a sole sulfur source, R. erythropolis MI2, R. erythropolis IGTS8, R. erythropolis PR4, and R. ruber cells were grown aerobically at 30°C in a modified MSM in which 2 mM magnesium sulfate was replaced with 1 mM DTDB. The optical density was monitored every 12 h.
Transposon-induced mutagenesis in R. erythropolis MI2 using IS1415.
Plasmid pTNR-TA (37) was transferred to R. erythropolis strain MI2 by electroporation. A 400-μl volume of chilled competent cells was mixed with 0.5 μg pTNR-TA and was preincubated at 40°C for 5 min (32). Electroporation was carried out using a Bio-Rad Gene Pulser Xcell system set at a charging voltage of 2.5 kV, a resistance of 400 Ω, and a capacitance of 25.0 μF in a 2-mm-gap electrocuvette (Peqlab, Erlangen, Germany). Afterwards, the electroporation mixture was suspended in 600 μl LB and was incubated for 3 to 4 h at 30°C. Subsequently, the mixture was plated onto LB agar containing 12.5 μg/ml thiostrepton and was incubated for 5 days to select thiostrepton-resistant cells. Transposon-induced mutants were transferred in a coordinated pattern to MSM agar plates with 12.5 μg/ml thiostrepton (MSMTs) and 0.5% (wt/vol) sodium gluconate as master plates and to MSMTs agar plates with 30 mM DTDB as selective plates.
Analysis of pTNR-TA insertion sites.
The genomic DNA of each transposon-induced mutant was extracted, and a straightforward two-step gene walking PCR method was applied (38). Insertions of IS1415 into the genomes of these mutants were confirmed by PCR using primers hybridizing to the thiostrepton resistance (Tsr) cassette (forward primer) and inverted repeat 2 (IR2) (reverse primer). Genomic DNA of the wild-type strain was used as a negative control. Homology searches of the interrupted DNA sequences from mutants were conducted by BLAST (http://blast.ncbi.nlm.nih.gov/Blast.cgi) (34).
Detection of volatile hydrogen sulfide.
Volatile hydrogen sulfide produced by Rhodococcus strains and transposon-induced mutants of R. erythropolis strain MI2 cultivated in the presence of DTDB was detected using lead acetate test strips (Fluka, Steinheim, Germany).
Determination of DTDB degradation intermediates by GC-MS analyses.
The degradation intermediates in the cell-free supernatants and freeze-dried cells of wild-type Rhodococcus cultures and transposon-induced mutants of R. erythropolis MI2 were determined by gas chromatographic analyses after acid methylation of the lyophilized samples (35, 36). The commercially purchased DTDB and 4MB were calibrated after being subjected to methylation and GC-MS analysis.
Construction of nox gene interruption mutants.
The target gene was interrupted by cloning the thiostrepton resistance cassette directly into the gene coding for the NADH:flavin oxidoreductase; the resulting noxΩtsr gene was cloned into the XbaI restriction site of suicide plasmid pJQ200mp18 (see Fig. S1 in the supplemental material). The resulting plasmid was transferred to E. coli TOP10, and selection was carried out on LB agar plates with gentamicin. Subsequently, this plasmid was transferred to R. erythropolis strain MI2 as a recipient strain by electroporation, and the mutant was identified on LBTs agar plates. The DTDB-negative mutant was cultivated on MSMTs agar plates supplemented with gluconate as master plates and on MSMTs agar plates supplemented with DTDB as selection plates to confirm the mutation.
The genotype of R. erythropolis MI2 noxΩtsr was then verified by PCR using primers binding outside the target gene, yielding an amplified product covering the complete interrupted target gene, while another primer pair binding inside the target gene was suitable for amplifying the fragment of the thiostrepton resistance cassette. All PCR fragments were then confirmed by sequencing.
Preparation of crude extracts.
R. erythropolis MI2 cells were first cultivated for 48 h in liquid MSM containing 0.5% (wt/vol) sodium gluconate and then washed, transferred to fresh MSM supplemented with 30 mM DTDB, and cultivated for another 48 h before harvesting. For cell disruption, cells were first suspended in an appropriate amount of buffer (50 mM Tris-HCl buffer [pH 8.0] containing 10% glycerol) and then disrupted by sonication (30 Hz, 50% amplitude, 1 min/ml) (Sonopuls HD2200 MS72 system; Bandelin Electronic GmbH & Co. KG, Berlin, Germany). Protein concentrations were determined as described by Bradford (39).
Cloning, expression, and purification of nox.
A PCR for the amplification of noxMI2 was carried out with Phusion high-fidelity DNA polymerase (Thermo Scientific, Schwerte, Germany) using total genomic DNA of R. erythropolis strain MI2 and the oligonucleotides forward-nox-NdeI and reverse-nox-XhoI (Table 1). The PCR product was isolated from an agarose gel by using the peqGOLD Gel Extraction kit (Peqlab, Erlangen, Germany) according to the manufacturer's instructions. Then nox and the pET23a(+) vector were digested with FastDigest NdeI and XhoI. Ligation was achieved with T4 DNA ligase (Thermo Scientific, Schwerte, Germany). The pET23a(+)::nox expression vector was used for the transformation of E. coli TOP10 cells. Transformants were selected on LB medium containing ampicillin (LBAp), and hybrid plasmids were isolated, analyzed by sequencing, and used for the transformation of E. coli BL21(DE3)pLysS cells (Novagen). For expression, BL21 harboring pET23a(+)::nox was cultivated in 50 ml LBAp overnight at 37°C and 120 rpm. Then 2 ml of the preculture was used to inoculate the main culture consisting of 100 ml autoinduction medium (ZYP) (40), and the mixture was incubated at 20°C and 120 rpm for approximately 18 h.
Cells were harvested by centrifugation (20 min, 4°C, 4,000 × g), washed twice, and resuspended in disruption buffer (50 mM potassium phosphate buffer [pH 6.5] containing 1 mM dithiothreitol [DTT], 1 mM EDTA, and 0.5 mM phenylmethylsulfonyl fluoride [PMSF]). Cells were disrupted by a 3-fold passage through a French press (100 times at 106 Pa). Lysates with the soluble protein fractions were obtained after centrifugation (1 h, 4°C, 16,000 × g) of the crude extracts and were used for enzyme purification. For purification, the cell-free supernatant obtained was subjected to nickel-nitriloacetate (Ni-NTA) affinity chromatography via HisTrap HP affinity columns (GE Healthcare, Uppsala, Sweden) according to the manufacturer's instructions. Soluble Nox protein was eluted by a buffer containing 300 mM imidazole. Protein concentrations were determined as described by Bradford (39). The purified protein was used for the enzyme activity assays.
Mass spectrometry and data analysis.
Prior to enzyme assay measurements, protein bands, which were bound to HisTrap columns with the overexpressed Nox, were cut from the SDS-PAGE gel and were transferred to 1.5-ml reaction tubes containing 500 μl of 10% (vol/vol) acetic acid. Those bands were subjected to matrix-assisted laser desorption ionization-time of flight tandem-MS (MALDI-TOF MS-MS) analysis. The MALDI-TOF MS-MS analysis was performed at the Institute for Microbiology of the Ernst-August-Arndt University Greifswald (Greifswald, Germany) as described previously (41) by using a 5800 proteomics analyzer (AB Sciex, Framingham, MA, USA). Mass spectrometry data were searched using the Mascot engine (version 2.1.0.4) and were compared with the amino acid sequence of Nox from R. erythropolis strain MI2 and the proteomes of E. coli O157:H7, avian pathogenic E. coli (APEC) O1:K1, and E. coli O6.
Qualitative enzyme activity assay using Ellman's reagent.
According to the method of Ellman (42), an enzyme reaction mixture with a final volume of 1 ml consisting of 50 mM potassium phosphate buffer (pH 7.0), 0.1 mM 5,5′-dithiobis(2-nitrobenzoic acid) (DTNB), 0.1 mM NADH, 5 mM DTDB, and either 4 μg/ml MI2 crude extract or 10 μg Nox was incubated in a cuvette for 3 min at room temperature. Finally, the yellow color (ε412 = 14,150 M−1 cm−1) (42, 43) could be measured spectrophotometrically at 412 nm. Controls were run without a substrate, without a cofactor (NADH), without an enzyme, or without a crude cell extract from R. erythropolis MI2. The reaction mixtures were subjected to high-performance liquid chromatography (HPLC) analysis in order to analyze the product of the enzyme reaction. As a control reference, a reaction sample with a final volume of 1 ml containing 50 mM Tris-HCl, 0.1 mM DTNB, and 10 mM 4MB was injected into the HPLC system.
Qualitative enzyme activity assay using NADH.
The enzyme activity was determined at 30°C by monitoring the decrease in the absorbance at 340 nm due to the oxidation of NADH. The activity was evaluated using the extinction coefficient for NADH of 6.22 mM−1 cm−1. Controls were run without a substrate and without an enzyme. The specific enzyme activity was calculated in units (micromoles per minute) per milligram of protein.
Nucleotide sequence accession numbers.
The complete DNA sequences and deduced amino acid sequences for the genes coding for (i) Nox (accession number KT600797), (ii) HipAC (accession number KT600798), (iii) acetyl coenzyme A (CoA) acyltransferase (Act) (accession number KT600799), (iv) adenosine methyltransferase/aminoglycoside phosphotransferase (accession number KT600800), (v) polysaccharide deacetyltransferase (accession number KT600801), and (vi) FtsK and an adjacent hypothetical protein (FtsK/SpoIIIE) (accession number KT600802) of Rhodococcus erythropolis strain MI2 have been deposited in the GenBank database.
RESULTS
Biodegradation of DTDB by available Rhodococcus spp.
To examine the abilities of Rhodococcus strains (Table 2) to utilize DTDB as the sole carbon and energy source, growth experiments were conducted on solid MSM containing different concentrations of DTDB ranging from 10 to 40 mM. According to Wübbeler et al. (17), R. erythropolis MI2 was the best-investigated strain that grew well in the presence of DTDB. In this study, nine Rhodococcus strains from our strain collection were screened for their abilities to catabolize DTDB. R. erythropolis strain MI2 was able to utilize DTDB for growth, while all eight other strains showed no growth with DTDB as the sole carbon source. These eight strains (Table 2) were subjected to an adaptation phase in liquid MSM containing 10 mM DTDB and 0.5% (wt/vol) sodium gluconate as a second carbon source. Subsequently, R. erythropolis PR4, R. erythropolis IGTS8, and R. ruber were capable of utilizing DTDB as a sole carbon and energy source, but only after an adaptation phase of 10 days in liquid MSM containing the compounds mentioned above (see Fig. S2 in the supplemental material). However, R. erythropolis strain IGTS8 and strain PR4 lost the ability to grow with DTDB after several subcultivations using DTDB as a sole carbon source, though the cells could be readapted again. These results were confirmed in liquid MSM, where an increase in turbidity was accompanied by a concomitant decrease in the concentration of DTDB, while controls (cultures without an inoculum) showed no decrease in the DTDB concentration.
The metabolic intermediates resulting from the biodegradation of DTDB were determined by GC-MS analysis in lyophilized cell-free supernatants of the four Rhodococcus species that grew with DTDB as the sole carbon source. After 72 h of cultivation, R. erythropolis MI2 had consumed 16.2 ± 0.3 mM DTDB from the 30 mM DTDB initially present. DTDB consumption was slightly delayed in cultures of R. ruber relative to that in R. erythropolis MI2, and it stopped after 36 h of cultivation at a concentration of 16.5 ± 0.6 mM. For R. erythropolis IGTS8 and R. erythropolis PR4, DTDB consumption started after 48 h of cultivation. 4MB was detected as an abundant intermediate in the cell-free supernatant of strain MI2, with a concentration as high as 1.1 mM (see Fig. S3 in the supplemental material).
4-Oxo-4-sulfanylbutyric acid and succinic acid were also detected in the cell-free culture supernatants of the four Rhodococcus strains, with the highest concentrations in the supernatant of strain MI2 (data not shown). Furthermore, no increase in the concentration of sulfite was detected in any of the cultures. Instead, the release of volatile hydrogen sulfide was detected.
Utilization of DTDB as a sulfur source.
To determine the abilities of R. erythropolis MI2, R. erythropolis PR4, R. erythropolis IGTS8, and R. ruber to utilize DTDB as a sole sulfur source, growth experiments were conducted in liquid MSM containing 1 mM DTDB instead of 2 mM magnesium sulfate. The results showed increases in turbidity (optical density) in the cultures of all four Rhodococcus strains investigated, while controls (cultures without any sulfur source) did not show growth (see Fig. S4 in the supplemental material).
Polyester accumulation.
R. erythropolis strains MI2, PR4, and IGTS8 and R. ruber were investigated for their abilities to accumulate polyhydroxyalkanoates (PHAs). The cells were incubated in nitrogen-limited MSM containing DTDB either as the sole carbon source or together with sodium succinate or sodium lactate as a second carbon source. GC-MS analysis of the freeze-dried cells of all Rhodococcus strains investigated showed that only R. ruber accumulated PHAs when growing in a medium containing DTDB plus sodium succinate or sodium lactate. No PHAs were accumulated when R. ruber cells grew on DTDB as the sole carbon source. GC-MS analysis of the purified PHA showed that the polymer consisted of 3-hydroxybutyric acid (3HB) and 3-hydroxyvaleric acid (3HV), as shown in Fig. S5A and B in the supplemental material. The structure of the polymer was confirmed by NMR analysis (data not shown). Mercaptoalkanoic acids, such as 4-mercaptobutyric acid, were not incorporated by any of the strains investigated.
Isolation and phenotypic characterization of transposon-induced mutants.
R. erythropolis strain MI2 was chosen for further studies due to its high and stable ability to degrade DTDB and its susceptibility to thiostrepton. Strain MI2 was subjected to transposon mutagenesis employing the pTNR-TA vector (13, 37), which confers thiostrepton resistance on the recipient. This mutagenesis was aimed at generating mutants with defective growth on DTDB in order to elucidate the catabolism of DTDB.
Among about 3,261 mutants screened, 8 showed no growth with DTDB as the sole carbon source when cultivated on MSM agar plates; these were referred to as DTDB-negative mutants. The abilities of the wild type and the transposon-induced mutants to utilize 49 carbon sources were also tested and compared. Both the wild type and the mutants showed largely similar growth profiles on all sugars tested (see Table S1 in the supplemental material).
Analysis of pTNR insertion sites.
The mutants were analyzed at a molecular level. The results of sequence analyses and homologies obtained from the GenBank database are summarized and graphically illustrated in Fig. 2. This figure also provides the array of the coding DNA sequence (CDS) including the transposon insertion loci. The genotypic characterization of IS1415-induced mutants of R. erythropolis MI2 defective in the utilization of DTDB is shown in Table S2 in the supplemental material.
FIG 2.
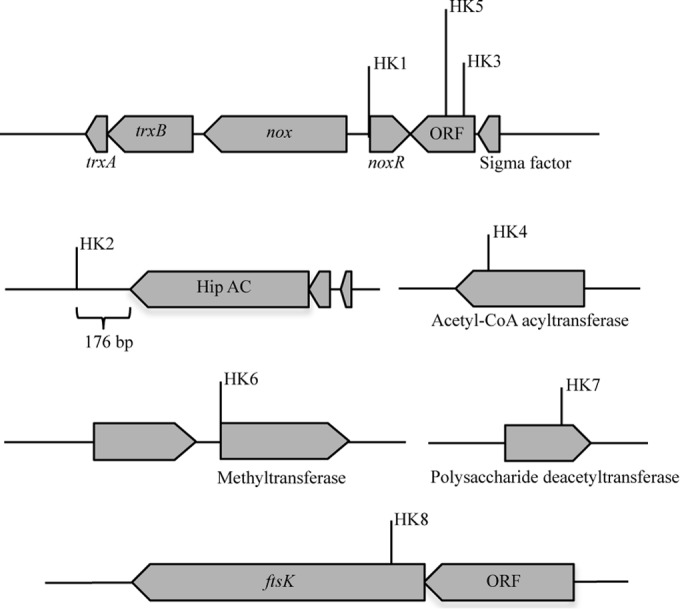
IS1415 insertions in the genome of R. erythropolis strain MI2 and identification of genes adjacent to the insertion loci. Insertion of the IS1415 element into the genome of R. erythropolis strain MI2 fully impaired the growth of the cells on DTDB. The insertion loci in all the transposon-induced mutants are indicated. The lengths and directions of arrows representing the genes affected indicate the proportional lengths and directions of transcription of those genes. Putative identities of the gene products are suggested from the identities of amino acid sequences to those of proteins in the GenBank database. ORF, open reading frame. The genes shown encode the following proteins: noxR, transcriptional regulator of the TetR family; nox, NADH:flavin oxidoreductase; trx, thioredoxin reductase; the Hip A-C gene, homeodomain-interacting protein kinase; ftsK, bacterial cell division protein.
The amino acid sequence of a putative translational product deduced from the pTNR-TA insertion locus in one of the DTDB-negative mutants (HK1) exhibited 98.9% identity to the transcriptional regulator encoded by tetR (locus tag O5Y-28895) in R. erythropolis CCM2595, which regulates the expression of a cluster of genes coding for an NADH:flavin oxidoreductase and a thioredoxin reductase. Amplification and sequence analyses of the region upstream of the transcriptional regulator showed two genes with 99.0% amino acid identity to an NADH:flavin oxidoreductase (locus tag O5Y-28900) and 98.8% sequence identity to a thioredoxin reductase (locus tag O5Y-28905) from R. erythropolis CCM2595, respectively (Fig. 2).
The first flavin-dependent enzyme characterized was the old yellow enzyme (OYE)-like FMN (flavin mononucleotide) binding domain. Its physiological function is still unknown. Each monomer of OYE contains FMN as a noncovalently bound cofactor. NAD(P)H is used as a reducing agent in the presence of oxygen, quinones, and α,β-unsaturated aldehydes and ketones (44). In two mutants (HK3 and HK5), the pTNR-TA insertion loci were mapped twice in a gene coding for a hypothetical protein in the vicinity of nox (Fig. 2).
In the genome of mutant HK2, the transposon was localized 176 bp downstream of a gene coding for a protein kinase family (Hip A-C) that is a member of a group of enzymes involved in cellular functions such as cell growth, proliferation, differentiation, motility, survival, and intracellular trafficking (45). In mutant HK4, the transposon locus was mapped in a gene encoding a putative acetyl-CoA acetyltransferase (EC 2.3.1.16). This acetyl-CoA acyltransferase belongs to the thiolase family, members of which catalyze the final step in fatty acid beta oxidation (46). In mutant HK6, the transposon insertion was localized 1 bp upstream of a gene coding for a tRNA G18 (ribose-2-O)-methyltransferase, whereas in mutant HK7, it was detected in a gene coding for a polysaccharide deacetyltransferase. The latter is an enzyme belonging to the family of hydrolases, specifically to those acting on carboxylic ester bonds (47) (Fig. 2). Sequence analyses of the pTNR-TA insertion locus in the genome of mutant HK8 showed similarity to a gene encoding an FtsK/SpoIII ATPase superfamily protein with 76% amino acid identity to the corresponding protein in R. erythropolis PR4 (Fig. 2). The FtsK domain, which is also called the bacterial cell division protein, contains a highly conserved putative ATP-binding P-loop cytoplasmic motif (48).
DTDB degradation intermediates.
To compare the intermediates that eventually accumulate during the catabolism of DTDB, all eight mutants and the wild-type R. erythropolis strain MI2 were cultivated in MSM containing 0.5% (wt/vol) sodium gluconate as a utilizable carbon source (preculture). The cells were then harvested by centrifugation after 48 h, washed, and transferred to MSM containing 0.5% (wt/vol) sodium gluconate plus 30 mM DTDB (main culture). Samples were taken every 12 h, and the cell-free supernatants obtained were subjected to GC-MS analyses. After 72 h of cultivation, the wild type had consumed all the DTDB. In all mutants, DTDB consumption occurred with some delay, and no further degradation was noticed after 36 h of cultivation. However, between 24 and 36 h of cultivation, a significant decrease in the DTDB concentration in the mutants was observed (data not shown).
4MB, which is the product of symmetrical cleavage of DTDB, was detected in the supernatants of both the wild type and the transposon-induced mutants (Fig. 3A). As also described by Wübbeler et al. (17), a considerable amount of 4-oxo-4-sulfanylbutyric acid was detected in the culture supernatants of the wild type and the transposon-induced mutants (Fig. 3B). In HK8 only, 4-oxo-4-sulfanylbutyric acid was detected at a very low concentration, which may indicate that the mutant cell could not oxidize 4MB that had accumulated in the culture supernatant, resulting in very slow growth of HK8. Moreover, succinic acid was detected inside the wild-type cells at a very low concentration (0.1 mM). However, among the transposon-induced mutants, succinic acid was detected only inside the cells of HK2, HK4, HK6, and HK7, at concentrations ranging from 0.04 to 0.06 mM. Moreover, it was detected at very low concentrations in the cell-free supernatants of the same mutants after 12 h of cultivation (data not shown).
FIG 3.

Analyses of cell-free supernatants of R. erythropolis strain MI2 and transposon-induced mutants in MSM with 30 mM DTDB and 0.5% (wt/vol) sodium gluconate. (A) Detection of the degradation intermediates of 4MB. (B) Because 4-oxo-4-sulfanylbutyric acid is not commercially available, it could not be used as a control, as a standard, or for calibration. Therefore, detection of this compound is shown as the “peak area” depending on the respective signal intensity.
Furthermore, volatile hydrogen sulfide was detected in cultures of the transposon-induced mutants and the wild type, as evidenced by the occurrence of a black precipitate on the test strips due to the conversion of lead(II) acetate into lead(II) sulfide. No hydrogen sulfide was detected in cells cultivated in a medium supplemented only with sodium gluconate.
Interruption of nox in R. erythropolis strain MI2.
In mutant HK1, the pTNR-TA insertion locus was localized 6 bp upstream of the gene encoding the putative transcriptional regulator (NoxR) of a gene cluster encoding the NADH:flavin oxidoreductase. This enzyme was a reasonable candidate for the initial step of DTDB catabolism. Therefore, a defined gene interruption mutant, R. erythropolis MI2 noxΩtsr, was generated. In contrast to the wild type, this mutant did not grow if DTDB was offered as the sole carbon source. Growth experiments were carried out in parallel with the wild type and R. erythropolis noxΩtsr in MSM containing 0.5% (wt/vol) sodium gluconate plus 30 mM DTDB. Both the wild type and the noxΩtsr mutant exhibited growth. Interestingly, no DTDB was utilized, and no intermediates were detected, by R. erythropolis noxΩtsr, in contrast to the wild type, which consumed all available DTDB within 72 h (Fig. 4). These results suggested that Nox is probably responsible for the cleavage of DTDB into two 4MB molecules.
FIG 4.

Utilization of DTDB and increase in the turbidity of cultures of R. erythropolis strain MI2 and R. erythropolis MI2 noxΩtsr. Cells were cultivated in 250-ml Klett flasks with baffles on a rotary shaker at 30°C and 120 rpm. The flasks contained 100 ml liquid MSM with 30 mM DTDB and 0.5% (wt/vol) sodium gluconate. ●, increase in the optical density of a culture of R. erythropolis strain MI2; ■, increase in the optical density of a culture of R. erythropolis MI2 noxΩtsr; ○, consumption of DTDB as the sole carbon source by R. erythropolis strain MI2; □, concentration of DTDB in a culture of R. erythropolis MI2 noxΩtsr.
Heterologous expression and purification of soluble Nox.
For the production of Nox protein, several E. coli strains and expression techniques were tested. To yield soluble Nox, E. coli strain BL21(DE3)pLysS was used, but most of the expressed protein was still insoluble (see Fig. S6 in the supplemental material). The overexpressed His-tagged Nox (Nox-His6) was purified using several HisTrap columns and several washing and elution protocols and was observed on an SDS-PAGE gel. In addition to Nox, three other protein bands were present in the elution fraction (see Fig. S6, lane 5). These three protein bands were subjected to MALDI-TOF MS-MS analysis and were identified as periplasmic proteins from E. coli: (i) a bifunctional polymyxin resistance protein with a molecular mass of 74 kDa; (ii) OmpF (molecular mass, 39 kDa), an outer membrane protein; and (iii) a catabolic gene activator protein (CAP) with a molecular mass of 23 kDa. The apparent molecular mass of Nox (∼44 kDa) agreed with its expected molecular mass (see Fig. S6).
Enzyme activity assays with a crude extract of strain MI2 and Nox.
After overexpression of Nox as a His6-tagged fusion protein in E. coli, an enzyme activity assay was carried out using DTDB as the substrate in the presence of the cofactor NADH and Ellman's reagent. DTNB reacts with the free thiol group of 4MB, which results from the cleavage of DTDB in the presence of active enzyme. This reaction gave a yellow solution consisting of 2-nitro-5-thiobenzoic acid (TNB−) and 4MB, indicating the following reaction:

The reaction between DTNB and 4MB was confirmed by HPLC analysis. There was an increase in absorbance at 412 nm when the crude cell extract of MI2 containing Nox or the purified Nox-His6 fusion protein was added to the reaction mixture. However, when either the substrate DTDB, the enzyme Nox, or the cofactor NADH was omitted, no increase in the absorbance at 412 nm or at 340 nm was observed. Moreover, HPLC analysis of the reaction samples at the end of the enzyme assay confirmed the presence of TNB-4MB (as in the reaction presented above) only if the active enzyme was present (Fig. 5A and B). The specific enzyme activities were determined using the crude cell extract of strain MI2 and Nox-His6. Nox was highly specific for NADH, and no reaction was observed if NADPH was added as a cofactor. The specific activity of the elution fraction containing Nox was 1.2 ± 0.15 U/mg protein, and the specific activity in the crude extract of strain MI2 was 0.1902 ± 0.12 U/mg.
FIG 5.

Chromatograms and mass spectra obtained after HPLC analysis of samples withdrawn at the end of the enzyme assay. RT, retention time. (A) TNB-4MB (arrows) was identified in the sample with active Nox. (B) Sample without Nox.
DISCUSSION
R. erythropolis strains MI2, PR4, and IGTS8 and R. ruber were all able to catabolize DTDB and to use it as the sole carbon and energy source. Thus, these Rhodococcus species possess the enzymatic machinery for the utilization of DTDB. Many strains of R. erythropolis are metabolically flexible and able to perform reactions such as oxidations, dehydrogenations, epoxidations, dehalogenations, hydrolysis, and hydroxylations due to a diverse set of enzymes (49). In addition, all members of the genus Rhodococcus have the capacity to cleave sulfur-sulfur bonds. R. erythropolis IGTS8 is used to study the microbial desulfurization of fossil fuels (50); however, in order to catabolize DTDB, it requires an adaptation phase. Moreover, the efficiency of DTDB degradation was lower in R. erythropolis IGTS8, R. erythropolis PR4, and R. ruber than in R. erythropolis MI2 (see Fig. S3 in the supplemental material).
R. erythropolis CCM2595 is used for the bioremediation of phenol and other aromatic compounds, and the enzymes involved in the phenol degradation pathway are induced only when phenol is present in the culture medium (8). Necessary genes encoding enzymes for the metabolization of most carbon sources are expressed via the induction of specific operons, which are mostly induced by the substrate itself, when bacterial cells come into contact with this carbon source (51). Based on these considerations, the enzyme involved in DTDB degradation is assumed to be induced in the presence of DTDB.
R. erythropolis strains IGTS8 and PR4 showed a long adaptation phase and degraded DTDB at a low rate. Additionally, they lost the ability to degrade the xenobiotic compound after several subcultivations on DTDB as the sole carbon source. In R. erythropolis IGTS8, the expression of the desulfurization genes (dsz), which catalyze the degradation of benzothiophene (BT) or dibenzothiophene (DBT), is repressed by the presence or accumulation of some metabolites, such as sulfate, methionine, and cysteine (52). It is possible that in R. erythropolis strains IGTS8 and PR4 also, the presence or accumulation of the DTDB degradation intermediates could repress the catabolic pathway and eventually cause the loss of the ability to degrade DTDB in these strains.
Moreover, R. erythropolis strains MI2, PR4, and IGTS8 and R. ruber were all able to utilize DTDB as a sole sulfur source (see Fig. S4 in the supplemental material). Many Rhodococcus spp. have the ability to utilize DBT as a sole sulfur source without cleaving its carbon-carbon backbone. This sulfur-specific pathway has been extensively studied in two R. erythropolis strains, IGTS8 and D-1 (53, 54). Also, the genes coding for the enzymes involved in the DBT desulfurization process were determined and sequenced in strain IGTS8 (55–57).
As mentioned above, DTDB is considered to be a promising PTE precursor substrate for the synthesis of poly(4MB). The three strains of R. erythropolis with the ability to utilize DTDB as the sole carbon source were unable to produce PHAs or PTEs. The PHA synthase structural gene of R. ruber (phbCRr) was the first identified PHA synthase gene of a Gram-positive bacterium (58). R. ruber was able to synthesize the copolyester poly(3HB-co-3HV) and the terpolyester poly(3HB-co-3HV-co-3HPi) when the cells were cultivated in the presence of 3-hydroxypivalic acid (3HPi) as the sole carbon source or with a second carbon source, such as glucose (59). Although R. ruber utilizes DTDB as a sole carbon source, it was unable to produce homopolymers or copolymers containing 4MB as a building block. When R. ruber was cultivated in the presence of DTDB plus sodium lactate as a second carbon source, it produced the copolymer poly(3HB-co-3HV), as shown in Fig. S5A in the supplemental material.
According to the DTDB degradation pathway proposed previously (17) (Fig. 1), the initial step is the reduction of the disulfide bond, resulting in the formation of two 4MB molecules. The formation of 4MB in cell-free supernatants was confirmed by GC-MS analyses (see Fig. S3 in the supplemental material). Sequence analysis of the genomic region in the vicinity of the transposon insertion in HK1 showed a gene (nox) coding for an NADH:flavin oxidoreductase, which is most likely the enzyme catalyzing the initial cleavage of DTDB. The noxΩtsr interruption mutant confirmed that the enzyme encoded by this gene is mandatory for DTDB catabolism. Whereas R. erythropolis MI2 noxΩtsr completely lost the ability to catalyze DTDB (Fig. 4), HK1 (Fig. 2) exhibited only a delay in DTDB consumption. These results showed very clearly that the disruption of nox resulted in a complete loss of DTDB degradation.
Nox is a member of the OYE family, which is a member of the flavin mononucleotide (FMN) binding domain family. Members of the OYE family are characterized by their ability to catalyze a diverse range of transformations. Despite this, the true physiological role of OYE members remains a mystery (60). All OYEs can act as electron acceptors in a catalytic reaction (61). Many compounds structurally related to DTDB and linked by disulfide bonds are catalyzed by flavoprotein disulfide reductases (62), such as the dihydrolipoamide dehydrogenases (44) from Advenella mimigardefordensis strain DPN7T (LpdAAm) and Ralstonia eutropha H16 (PdhLRe). Both catalyze the cleavage of the disulfide 3,3′-dithiodipropionic acid (DTDP) into 2 molecules of 3-mercaptopropionic acid (3MP) (63).
Flavoprotein oxidases catalyze 2-electron oxidations of a wide variety of substrates, including amines and alcohols. For example, the flavoprotein putrescine oxidase from R. erythropolis PuO catalyzes an oxidative deamination of the aliphatic diamine (64). The completely sequenced genome of Rhodococcus jostii RHA1 (65) encodes about 1,085 oxidoreductases. This large number reflects the abundance of members of this enzyme class in all sequenced genomes of Rhodococcus species. To summarize all these findings, Nox is most likely the enzyme cleaving the sulfur-sulfur bond of the DTDB in R. erythropolis MI2, as confirmed by the enzymatic in vitro analysis employing the purified enzyme.
4MB is a highly toxic mercaptoalkanoic acid, more toxic than other mercaptoalkanoates, as documented during studies on the synthesis of PTEs (16, 18). In the present study, low concentrations of 4MB were detected in the culture supernatants of the HK1, HK5, and HK3 mutants (Fig. 3A), in which the transposon insertion loci were mapped in three different locations in the vicinity of nox. The oxidation of 4MB to 4-oxo-4-sulfanylbutyric acid is the second proposed step in DTDB catabolism. This reaction is performed by a putative oxygenase. The high toxicity of 4MB is due to the sulfhydryl group, since a concentration of only 0.01% (vol/vol) 4MB is sufficient to inhibit bacterial growth (18); hence, its oxidation is probably a very important step for the survival of the bacterial cells.
4-Oxo-4-sulfanylbutyric acid was detected in the culture supernatants of both the wild-type strain and the transposon-induced mutants. On the other hand, no intermediates were detected in R. erythropolis MI2 noxΩtsr.
In conclusion, R. erythropolis MI2 noxΩtsr cannot cleave DTDB, whether DTDB is used as the sole carbon source or with a second carbon source. The eight transposon-induced mutants showed a DTDB-negative phenotype with DTDB as the sole carbon source. However, these mutants can utilize DTDB partially if another carbon source is also available. This finding may indicate the presence of residual expression of the genes involved in DTDB degradation in the transposon-induced mutants. The final step in DTDB degradation is the conversion of 4-oxo-4-sulfanylbutyric acid into succinic acid, which is then further metabolized in the central citric acid cycle (17).
Since NoxMI2 possibly represents the key enzyme in the degradation of DTDB, it was cloned, overexpressed, and characterized. NoxMI2 consists of 403 amino acids and has a calculated molecular mass of 43.573 kDa (isotopically averaged) and a calculated pI of 8.76. The instability index (II) is 44.07, and the grand average of hydropathicity (GRAVY) is 0.044, which classifies this protein as unstably hydrophobic. The contaminating proteins identified in the elution fraction should not be able to cleave DTDB. The first protein was identified as a bifunctional polymyxin resistance protein. This protein is found in most Gram-negative bacteria and catalyzes the decarboxylation of UDP-glucuronic acid to UDP-4-keto-arabinose by adding a formyl group. The modified arabinose, which is then attached to lipid A, is essential for resistance to cationic antimicrobial peptides such as polymyxin (66–68). The second protein is an outer membrane protein (OmpF), and its function is limited to the passive diffusion of small molecules across the outer membrane pores (69). Finally, the third protein was identified as a catabolic gene activator protein (CAP) that acts as a global transcription regulator. The CAP plays a major role in carbon catabolite repression (CCR) (70). Taking into account the modes of action of the E. coli proteins identified, they are probably not involved in the cleavage of DTDB.
The key enzyme for microbial desulfurization of dibenzothiophene in R. erythropolis IGTS8 and R. erythropolis D-1 is an NADH-dependent FMN oxidoreductase (DszD). This enzyme was overexpressed, purified, and characterized in previous studies (71, 72), and the specific enzyme activity of the purified flavin reductase from strain D-1 was measured (122 U/mg) (71). On the other hand, the soluble NADH:flavin oxidoreductase (StyB) from Pseudomonas putida strain SN1 catalyzes styrene with a maximum specific activity (Vmax) of 1,867 U/mg (73). Moreover, the disulfide reductase from Achromobacter starkeyi has a specific activity of 1.18 U/mg when catalyzing DTNB or glutathione and was observed to have a higher affinity for DTNB than for glutathione (74). In agreement with observations in other studies, flavin oxidoreductases include many enzymes, which catalyze the initial degradation steps of a wide range of natural and xenobiotic substrates. Oxidoreductases that catalyze natural substrates, such as styrene, perform such reactions with relatively high enzymatic activities. In contrast, for oxidoreductases that catalyze the degradation of xenobiotic compounds, such as DTNB and DTDB, the specific activity is usually much lower.
To substantiate the involvement of Nox in the proposed initial step of DTDB degradation, further studies on NoxMI2 are in progress. Because R. erythropolis strain MI2 shows a high and stable capacity for DTDB degradation, successful heterologous expression of the BPEC (butyrate kinase [Buk], phosphotransbutyrylase [Ptb], PHA synthase [PhaE and PhaC]) pathway (75) or the PHA synthase genes of R. ruber (58) in a genetically engineered R. erythropolis strain could yield poly(4MB). The accumulation of 4MB inside R. erythropolis MI2 cells through deletion of the gene encoding the oxygenase, enhancement of Nox activity, and overexpression of PHA synthases from R. ruber by using the pTip QC2 expression vector (76) for expressing recombinant proteins in R. erythropolis could be a promising step toward the production of poly(4MB).
Supplementary Material
ACKNOWLEDGMENTS
We thank Klaus Bergander and his colleagues at the NMR spectroscopy laboratory (Institut für Organische Chemie, Westfälische Wilhelms-Universität Münster) for their cooperation. Moreover, we thank Birgit Voigt (Institut für Mikrobiologie, Ernst-Moritz-Arndt Universität, Greifswald, Germany) for MALDI-TOF analysis. We are grateful to Tomohiro Tamura (Director of the National Research Planning Office for Life Science and Biotechnology, Japan) for helpful discussions regarding the pTNR-TA vector.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.02059-15.
REFERENCES
- 1.Larkin MJ, Kulakov LA, Allen CCR. 2006. Biodegradation by members of the genus Rhodococcus: biochemistry, physiology and genetic adaptation. Adv Appl Microbiol 59:1–29. doi: 10.1016/S0065-2164(06)59001-X. [DOI] [PubMed] [Google Scholar]
- 2.van der Geize R, Dijkhuizen L. 2004. Harnessing the catabolic diversity of rhodococci for environmental and biotechnological applications. Curr Opin Microbiol 7:255–261. doi: 10.1016/j.mib.2004.04.001. [DOI] [PubMed] [Google Scholar]
- 3.Alvarez HM. (ed). 2010. Microbiology monographs, vol 16. Biology of Rhodococcus. Springer, Berlin, Germany. [Google Scholar]
- 4.Bell KS, Philp JC, Aw DWJ, Christofi N. 1998. The genus Rhodococcus. J Appl Microbiol 85:195–210. doi: 10.1046/j.1365-2672.1998.00525.x. [DOI] [PubMed] [Google Scholar]
- 5.Matsui T, Noda K, Tanaka Y, Maruhashi K, Kurane R. 2002. Recombinant Rhodococcus sp. strain T09 can desulfurize DBT in the presence of inorganic sulfate. Curr Microbiol 45:240–244. doi: 10.1007/s00284-002-3739-0. [DOI] [PubMed] [Google Scholar]
- 6.Komeda H, Hori Y, Kobayashi M, Shimizu S. 1996. Transcriptional regulation of the Rhodococcus rhodochrous J1 nitA gene encoding a nitrilase. Proc Natl Acad Sci U S A 93:10572–10577. doi: 10.1073/pnas.93.20.10572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Maass D, de Oliveira D, de Souza AAU, Souza SMAGU. 2014. Biodesulfurization of a system containing synthetic fuel using Rhodococcus erythropolis ATCC 4277. Appl Biochem Biotechnol 174:2079–2085. doi: 10.1007/s12010-014-1189-3. [DOI] [PubMed] [Google Scholar]
- 8.Szőköl J, Rucká L, Šimčíková M, Halada P, Nešvera J, Pátek M. 2014. Induction and carbon catabolite repression of phenol degradation genes in Rhodococcus erythropolis and Rhodococcus jostii. Appl Microbiol Biotechnol 98:8267–8279. doi: 10.1007/s00253-014-5881-6. [DOI] [PubMed] [Google Scholar]
- 9.Sallam KI, Tamura N, Imoto N, Tamura T. 2010. New vector system for random, single-step integration of multiple copies of DNA into the Rhodococcus genome. Appl Environ Microbiol 76:2531–2539. doi: 10.1128/AEM.02131-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ashour J, Hondalus MK. 2003. Phenotypic mutants of the intracellular actinomycete Rhodococcus equi created by in vivo Himar1 transposon mutagenesis. J Bacteriol 185:2644–2652. doi: 10.1128/JB.185.8.2644-2652.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fernandes PJ, Powell JA, Archer JA. 2001. Construction of Rhodococcus random mutagenesis libraries using Tn5 transposition complexes. Microbiology 147:2529–2536. doi: 10.1099/00221287-147-9-2529. [DOI] [PubMed] [Google Scholar]
- 12.Mangan MW, Meijer WG. 2001. Random insertion mutagenesis of the intracellular pathogen Rhodococcus equi using transposomes. FEMS Microbiol Lett 205:243–246. doi: 10.1111/j.1574-6968.2001.tb10955.x. [DOI] [PubMed] [Google Scholar]
- 13.Sallam KI, Tamura N, Tamura T. 2007. A multipurpose transposon-based vector system mediates protein expression in Rhodococcus erythropolis. Gene 386:173–182. doi: 10.1016/j.gene.2006.09.006. [DOI] [PubMed] [Google Scholar]
- 14.Jang LS, Keng HK. 2006. Development and characterization of 4,4′-dithiodibutyric acid as a monolayer for protein chips. Sensors Mater 18:367–380. [Google Scholar]
- 15.Kanayama N, Kitano H. 2000. Interfacial recognition of sugars by boronic acid-carrying self-assembled monolayers. Langmuir 16:577–583. doi: 10.1021/la990182e. [DOI] [Google Scholar]
- 16.Lütke-Eversloh T, Steinbüchel A. 2003. Polythioesters. Biopolymers 9:63–80. [Google Scholar]
- 17.Wübbeler JH, Bruland N, Wozniczka M, Steinbüchel A. 2010. Biodegradation of the xenobiotic organic disulphide 4,4′-dithiodibutyric acid by Rhodococcus erythropolis strain MI2 and comparison to the microbial utilisation of 3,3′-dithiodipropionic acid and 3,3′-thiodipropionic acid. Microbiology 156:1221–1233. doi: 10.1099/mic.0.036178-0. [DOI] [PubMed] [Google Scholar]
- 18.Lütke-Eversloh T, Steinbüchel A. 2003. Novel precursor substrates for polythioesters (PTE) and limits of PTE biosynthesis in Ralstonia eutropha. FEMS Microbiol Lett 221:191–196. doi: 10.1016/S0378-1097(03)00185-X. [DOI] [PubMed] [Google Scholar]
- 19.Kim DY, Elbanna K, Thakor N, Lütke-Eversloh T, Steinbüchel A. 2005. Poly(3-mercaptopropionate): a non-biodegradable biopolymer? Biomacromolecules 6:897–901. doi: 10.1021/bm049334x. [DOI] [PubMed] [Google Scholar]
- 20.Lütke-Eversloh T, Kawada J, Marchessault RH, Steinbüchel A. 2002. Characterization of biological polythioesters: physical properties of novel copolymers synthesized by Ralstonia eutropha. Biomacromolecules 3:159–166. doi: 10.1021/bm015603x. [DOI] [PubMed] [Google Scholar]
- 21.Lütke-Eversloh T, Steinbüchel A. 2004. Microbial polythioesters. Macromol Biosci 4:165–174. doi: 10.1002/mabi.200300084. [DOI] [PubMed] [Google Scholar]
- 22.Wübbeler JH, Steinbüchel A. 2014. New pathways for bacterial polythioesters. Curr Opin Biotechnol 29:85–92. doi: 10.1016/j.copbio.2014.02.017. [DOI] [PubMed] [Google Scholar]
- 23.Steinbüchel A. 2001. Perspectives for biotechnological production and utilization of biopolymers: metabolic engineering of polyhydroxyalkanoate biosynthesis pathways as a successful example. Macromol Biosci 1:1–24. doi:. [DOI] [Google Scholar]
- 24.Lütke-Eversloh T, Bergander K, Luftmann H, Steinbüchel A. 2001. Identification of a new class of biopolymer: bacterial synthesis of a sulfur-containing polymer with thioester linkages. Microbiology 147:11–19. doi: 10.1099/00221287-147-1-11. [DOI] [PubMed] [Google Scholar]
- 25.Lütke-Eversloh T, Bergander K, Luftmann H, Steinbüchel A. 2001. Biosynthesis of poly(3-hydroxybutyrate-co-3-mercaptobutyrate) as a sulfur analogue to poly(3-hydroxybutyrate) (PHB). Biomacromolecules 2:1061–1065. doi: 10.1021/bm015564p. [DOI] [PubMed] [Google Scholar]
- 26.Thakor N, Lütke-Eversloh T, Steinbüchel A. 2005. Application of the BPEC pathway for large scale biotechnological production of poly(3-mercaptopropionate) by recombinant Escherichia coli, including a novel in situ isolation method. Appl Environ Microbiol 71:835–841. doi: 10.1128/AEM.71.2.835-841.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xia Y, Wübbeler JH, Qi Q, Steinbüchel A. 2012. Employing a recombinant strain of Advenella mimigardefordensis for biotechnical production of homopolythioesters from 3,3′-dithiodipropionic acid. Appl Environ Microbiol 78:3286–3297. doi: 10.1128/AEM.00007-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sambrook J, Fritsch EF, Maniatis T. 1989. Molecular cloning: a laboratory manual, 2nd ed Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 29.Schlegel HG, Gottschalk G, Bartha RV. 1961. Formation and utilization of poly-β-hydroxybutyric acid by Knallgas bacteria (Hydrogenomonas). Nature 191:463–465. doi: 10.1038/191463a0. [DOI] [PubMed] [Google Scholar]
- 30.Schlegel HG, Kaltwasser H, Gottschalk G. 1961. Ein Submersverfahren zur Kultur wasserstoffoxidierender Bakterien: Wachstumsphysiologische Untersuchungen. Arch Mikrobiol 38:209–222. doi: 10.1007/BF00422356. [DOI] [PubMed] [Google Scholar]
- 31.Desomer J, Dhaese P, Montagu MV. 1990. Transformation of Rhodococcus fascians by high-voltage electroporation and development of R. fascians cloning vectors. Appl Environ Microbiol 56:2818–2825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kalscheuer R, Arenskötter M, Steinbüchel A. 1999. Establishment of a gene transfer system for Rhodococcus opacus PD630 based on electroporation and its application for recombinant biosynthesis of poly(3-hydroxyalkanoic acids). Appl Microbiol Biotechnol 52:508–515. doi: 10.1007/s002530051553. [DOI] [PubMed] [Google Scholar]
- 33.Sambrook J, Russell DW. 2001. Molecular cloning: a laboratory manual, 3rd ed Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 34.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. 1990. Basic local alignment search tool. J Mol Biol 215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 35.Brandl H, Gross A, Lenz RW, Fuller RC. 1988. Pseudomonas oleovorans as a source of poly(β-hydroxyalkanoates) for potential applications as biodegradable polyesters. Appl Environ Microbiol 54:1977–1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Timm A, Byrom D, Steinbüchel A. 1990. Formation of blends of various poly(3-hydroxyalkanoic acids) by a recombinant strain of Pseudomonas oleovorans. Appl Microbiol Biotechnol 33:296–301. doi: 10.1007/BF00164525. [DOI] [Google Scholar]
- 37.Sallam KI, Mitani Y, Tamura T. 2006. Construction of random transposition mutagenesis system in Rhodococcus erythropolis using IS1415. J Biotechnol 121:13–22. doi: 10.1016/j.jbiotec.2005.07.007. [DOI] [PubMed] [Google Scholar]
- 38.Pilhofer M, Bauer AP, Schrallhammer M, Richter L, Ludwig W, Schleifer KH, Petroni G. 2007. Characterization of bacterial operons consisting of two tubulins and a kinesin-like gene by the novel two-step gene walking method. Nucleic Acids Res 35:e135. doi: 10.1093/nar/gkm836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bradford MM. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 40.Studier FW. 2005. Protein production by auto-induction in high density shaking cultures. Protein Expr Purif 41:207–234. doi: 10.1016/j.pep.2005.01.016. [DOI] [PubMed] [Google Scholar]
- 41.Wolf C, Hochgräfe F, Kusch H, Albrecht D, Hecker M, Engelmann S. 2008. Proteomic analysis of antioxidant strategies of Staphylococcus aureus: diverse responses to different oxidants. Proteomics 8:3139–3153. doi: 10.1002/pmic.200701062. [DOI] [PubMed] [Google Scholar]
- 42.Ellman GL. 1959. Tissue sulfhydryl groups. Arch Biochem Biophys 82:70–77. doi: 10.1016/0003-9861(59)90090-6. [DOI] [PubMed] [Google Scholar]
- 43.Riddles PW, Blakeley RL, Zerner B. 1983. Reassessment of Ellman's reagent. Methods Enzymol 91:49–60. doi: 10.1016/S0076-6879(83)91010-8. [DOI] [PubMed] [Google Scholar]
- 44.Warburg O, Christian W. 1932. Ein zweites Sauerstoffübertragendes Ferment und sein Absorptionsspektrum. Naturwissenschaften 20:688. doi: 10.1007/BF01494406. [DOI] [Google Scholar]
- 45.Knight ZA, Gonzalez B, Feldman ME, Zunder ER, Goldenberg DD, Williams O, Loewith R, Stokoe D, Balla A, Toth B, Balla T, Weiss WA, Williams RL, Shokat KM. 2006. A pharmacological map of the PI3-K family defines a role for p110α in insulin signaling. Cell 125:733–747. doi: 10.1016/j.cell.2006.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Beinert H, Bock RM, Goldman DS, Green DE, Mahler HR, Sanae M, Stansly PG, Wakil SJ. 1953. The reconstruction of the fatty acid oxidizing system of animal tissues. J Am Chem Soc 75:4111–4112. doi: 10.1021/ja01112a536. [DOI] [Google Scholar]
- 47.Jorge JA, Kinney SG, Reissig JL. 1982. Purification and characterization of Neurospora crassa N-acetyl galactosaminoglycan deacetylase. Braz J Med Biol Res 151:29–34. [PubMed] [Google Scholar]
- 48.Bi E, Lutkenhaus J. 1991. FtsZ structure associated with cell division in Escherichia coli. Nature 354:161–164. doi: 10.1038/354161a0. [DOI] [PubMed] [Google Scholar]
- 49.de Carvalho CC, da Fonseca MM. 2005. The remarkable Rhodococcus erythropolis. Appl Microbiol Biotechnol 67:715–726. doi: 10.1007/s00253-005-1932-3. [DOI] [PubMed] [Google Scholar]
- 50.Grossman MJ, Lee MK, Prince RC, Minak-Bernero V, George GN, Pickering IJ. 2001. Deep desulfurization of extensively hydrodesulfurized middle distillate oil by Rhodococcus sp. strain ECRD-1. Appl Environ Microbiol 67:1949–1952. doi: 10.1128/AEM.67.4.1949-1952.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Brückner R, Titgemeyer F. 2002. Carbon catabolite repression in bacteria: choice of the carbon source and autoregulatory limitation of sugar utilization. FEMS Microbiol Lett 209:141–148. doi: 10.1111/j.1574-6968.2002.tb11123.x. [DOI] [PubMed] [Google Scholar]
- 52.Li MZ, Squires CH, Monticello DJ, Childs JD. 1996. Genetic analysis of the dsz promoter and associated regulatory regions of Rhodococcus erythropolis IGTS8. J Bacteriol 178:6409–6418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gallagher JR, Olson SE, Stanley CD. 1993. Microbial desulfurization of dibenzothiophene: a sulfur-specific pathway. FEMS Microbiol Lett 107:31–36. doi: 10.1111/j.1574-6968.1993.tb05999.x. [DOI] [PubMed] [Google Scholar]
- 54.Izumi Y, Ohshiro T, Ogino H, Hine Y, Shimao M. 1994. Selective desulfurization of dibenzothiophene by Rhodococcus erythropolis D-1. Appl Environ Microbiol 60:223–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Denome AS, Olson ES, Young KD. 1993. Identification and cloning of genes involved in specific desulfurization of dibenzothiophene by Rhodococcus sp. strain IGTS8. Appl Environ Microbiol 59:2837–2843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Denome AS, Oldfield C, Nash LJ, Young KD. 1994. Characterization of the desulfurization genes from Rhodococcus sp. strain IGTS8. J Bacteriol 176:6707–6716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Piddington SA, Kovacevich BR, Rambosek J. 1995. Sequence and molecular characterization of a DNA region encoding the dibenzothiophene desulfurization operon of Rhodococcus sp. strain IGTS8. Appl Environ Microbiol 61:468–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pieper U, Steinbüchel A. 1992. Identification, cloning and sequence analysis of the poly(3-hydroxyalkanoic acid) synthase gene of the Gram-positive bacterium Rhodococcus ruber. FEMS Microbiol Lett 96:73–79. doi: 10.1111/j.1574-6968.1992.tb05396.x. [DOI] [PubMed] [Google Scholar]
- 59.Füchtenbusch B, Fabritius D, Wältermann M, Steinbüchel A. 1998. Biosynthesis of novel copolyesters containing 3-hydroxypivalic acid by Rhodococcus ruber NCIMB 40126 and related bacteria. FEMS Microbiol Lett 159:85–92. doi: 10.1111/j.1574-6968.1998.tb12845.x. [DOI] [Google Scholar]
- 60.Williams RE, Burce NC. 2002. ‘New uses for an Old Enzyme’—the Old Yellow Enzyme family of flavoenzymes. Microbiology 148:1607–1614. doi: 10.1099/00221287-148-6-1607. [DOI] [PubMed] [Google Scholar]
- 61.Fox KM, Karplus PA. 1994. Old yellow enzyme at 2 Å resolution: overall structure, ligand binding, and comparison with related flavoproteins. Structure 2:1089–1105. doi: 10.1016/S0969-2126(94)00111-1. [DOI] [PubMed] [Google Scholar]
- 62.Williams CH., Jr 1992. Lipoamide dehydrogenase, glutathione reductase, thioredoxin reductase, and mercuric ion reductase—a family of flavoenzyme transhydrogenases, p 121–211. In M̈uller F (ed), Chemistry and biochemistry of flavoenzymes, vol III. CRC Press, Boca Raton, FL. [Google Scholar]
- 63.Wübbeler JH, Raberg M, Brandt U, Steinbüchel A. 2010. Dihydrolipoamide dehydrogenases of Advenella mimigardefordensis and Ralstonia eutropha catalyze cleavage of 3,3′-dithiodipropionic acid into 3-mercaptopropionic acid. Appl Environ Microbiol 76:7023–7028. doi: 10.1128/AEM.01706-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kopacz MM, Heuts DPHM, Fraaije MW. 2014. Kinetic mechanism of putrescine oxidase from Rhodococcus erythropolis. FEBS J 281:4384–4393. doi: 10.1111/febs.12945. [DOI] [PubMed] [Google Scholar]
- 65.McLeod MP, Warren RL, Hsiao WW, Araki N, Myhre M, Fernandes C, Miyazawa D, Wong W, Lillquist AL, Wang D, Dosanjh M, Hara H, Petrescu A, Morin RD, Zang G, Stott JM, Schein JE, Shin H, Smailus D, Siddiqui AS, Marra MA, Jones SJM, Holt R, Brinkman FSL, Mizauchi K, Fukuda M, Davies JE, Mohn WW, Eltis LD. 2006. The complete genome of Rhodococcus sp. RHA1 provides insights into a catabolic powerhouse. Proc Natl Acad Sci U S A 103:15582–15587. doi: 10.1073/pnas.0607048103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Thoden JB, Hegeman AD, Wesenberg G, Chapeau MC, Frey PA, Holden HM. 1997. Structural analysis of UDP-sugar binding to UDP-galactose 4-epimerase from Escherichia coli. Biochemistry 36:6294–6304. doi: 10.1021/bi970025j. [DOI] [PubMed] [Google Scholar]
- 67.Gatzeva-Topalova PZ, May AP, Sousa MC. 2004. Crystal structure of Escherichia coli ArnA (PmrI) decarboxylase domain. A key enzyme for lipid A modification with 4-amino-4-deoxy-l-arabinose and polymyxin resistance. Biochemistry 43:13370–13379. doi: 10.1021/bi048551f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Jo H, Jeong EY, Jeon J, Ban C. 2014. Structural insights into Escherichia coli polymyxin B resistance protein D with X-ray scattering. BMC Struct Biol 14:24. doi: 10.1186/s12900-014-0024-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Duval V, Nicoloff H, Levy SB. 2009. Combined inactivation of lon and ycgE decreases multidrug susceptibility by reducing the amount of OmpF porin in Escherichia coli. Antimicrob Agents Chemother 53:4944–4948. doi: 10.1128/AAC.00787-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Welch RA, Burland V, Plunkett G III, Redford P, Roesch P, Rasko D, Buckles EL, Liou SR, Boutin A, Hackett J, Stroud D, Mayhew GF, Rose DJ, Zhou S, Schwartz DC, Perna NT, Mobley HLT, Donnenberg MS, Blattner FR. 2002. Extensive mosaic structure revealed by the complete genome sequence of uropathogenic Escherichia coli. Proc Natl Acad Sci U S A 99:17020–17024. doi: 10.1073/pnas.252529799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Matsubara T, Ohshiro T, Nishina Y, Izumi Y. 2001. Purification, characterization and overexpression of flavin reductase involved in dibenzothiophene desulfurization by Rhodococcus erythropolis D-1. Appl Environ Microbiol 67:1179–1184. doi: 10.1128/AEM.67.3.1179-1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ohshiro T, Kojima T, Torii K, Kawasoe H, Izumi Y. 1999. Purification and characterization of dibenzothiophene (DBT) sulfone monooxygenase, an enzyme involved in DBT desulfurization, from Rhodococcus erythropolis D-1. J Biosci Bioeng 88:610–616. doi: 10.1016/S1389-1723(00)87088-7. [DOI] [PubMed] [Google Scholar]
- 73.Yeo YJ, Shin S, Lee SG, Park S, Jeong YJ. 2009. Production, purification, and characterization of soluble NADH-flavin oxidoreductase (StyB) from Pseudomonas putida SN1. J Microbiol Biotechnol 19:362–367. doi: 10.4014/jmb.0806.382. [DOI] [PubMed] [Google Scholar]
- 74.Ruiz-Herrera J, Amezcua-Ortega R, Trujillo A. 1968. Purification and properties of a disulfide reductase obtained from Achromobacter starkeyi. J Biol Chem 243:4083–4088. [PubMed] [Google Scholar]
- 75.Liu SJ, Steinbüchel A. 2000. A novel genetically engineered pathway for synthesis of poly(hydroxyalkanoic acid) in Escherichia coli. Appl Environ Microbiol 66:739–743. doi: 10.1128/AEM.66.2.739-743.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Nakashima N, Tamura T. 2004. Isolation and characterization of a rolling-circle-type plasmid from Rhodococcus erythropolis and application of the plasmid to multiple-recombinant-protein expression. Appl Environ Microbiol 70:5557–5568. doi: 10.1128/AEM.70.9.5557-5568.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Quandt J, Hynes MF. 1993. Versatile suicide vectors which allow direct selection for gene replacement in gram-negative bacteria. Gene 127:15–21. doi: 10.1016/0378-1119(93)90611-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.