

Abstract
2-Methyl-6-ethylaniline (MEA) is the main microbial degradation intermediate of the chloroacetanilide herbicides acetochlor and metolachlor. Sphingobium sp. strain MEA3-1 can utilize MEA and various alkyl-substituted aniline and phenol compounds as sole carbon and energy sources for growth. We isolated the mutant strain MEA3-1Mut, which converts MEA only to 2-methyl-6-ethyl-hydroquinone (MEHQ) and 2-methyl-6-ethyl-benzoquinone (MEBQ). MEA may be oxidized by the P450 monooxygenase system to 4-hydroxy-2-methyl-6-ethylaniline (4-OH-MEA), which can be hydrolytically spontaneously deaminated to MEBQ or MEHQ. The MEA microbial metabolic pathway was reconstituted based on the substrate spectra and identification of the intermediate metabolites in both the wild-type and mutant strains. Plasmidome sequencing indicated that both strains harbored 7 plasmids with sizes ranging from 6,108 bp to 287,745 bp. Among the 7 plasmids, 6 were identical, and pMEA02′ in strain MEA3-1Mut lost a 37,000-bp fragment compared to pMEA02 in strain MEA3-1. Two-dimensional electrophoresis (2-DE) and protein mass fingerprinting (PMF) showed that MEA3-1Mut lost the two-component flavin-dependent monooxygenase (TC-FDM) MeaBA, which was encoded by a gene in the lost fragment of pMEA02. MeaA shared 22% to 25% amino acid sequence identity with oxygenase components of some TC-FDMs, whereas MeaB showed no sequence identity with the reductase components of those TC-FDMs. Complementation with meaBA in MEA3-1Mut and heterologous expression in Pseudomonas putida strain KT2440 resulted in the production of an active MEHQ monooxygenase.
INTRODUCTION
Chloroacetanilide herbicides are widely used throughout the world (1) for the control of most annual grasses and certain broadleaf weeds (2). The majority of the commonly used chloroacetanilide herbicides, such as alachlor, acetochlor, butachlor, and metolachlor, are N-alkoxyalkyl-N-chloroacetyl-substituted aniline derivatives (3). Long-term application of these herbicides has caused negative impacts on both the aquatic environment and agricultural ecosystems (4, 5). Acetochlor has been classified by the U.S. Environmental Protection Agency (EPA) (4) as a B-2 carcinogen and a probable human carcinogen, and several chloroacetanilide herbicides have been proven to cause tumors in rats (6). Thus, there is great concern about the behavior and fate of chloroacetamide herbicides and their degradation metabolites in the environment.
Although chloroacetamide herbicides may be degraded through chemical and physical processes, microbial metabolism is the main mechanism responsible for herbicidal degradation in natural soils (7, 8). A variety of bacterial strains that are able to degrade butachlor, alachlor, acetochlor, and metolachlor have been characterized (9, 10). In the degradation pathway, these herbicides are N-dealkylated to 2-chloro-N-(2,6-diethylphenyl) acetamide (CDEPA) (for alachlor and butachlor) or 2-chloro-N-(2-methyl-6-ethylphenyl) acetamide (CMEPA) (for acetochlor and metolachlor), which are then converted to 2,6-diethylaniline (DEA) or 2-methyl-6-ethylaniline (MEA), respectively (2, 10). Oxygenase systems are involved in bacterial degradative N-dealkylation. A novel three-component Rieske non-heme iron oxygenase system was determined to catalyze the N-dealkylation of chloroacetamide herbicides in Sphingomonas sp. strains DC-6 and DC-2 (3). A cytochrome P450 system, EthBAD, was reported to be involved in the N-deethoxymethylation of acetochlor by Rhodococcus sp. strain T3-1 (11). Two genes, cmeH and damH, were cloned from Sphingobium quisquiliarum strain DC-2 and Delftia sp. strain T3-6, respectively, and were shown to encode amidases that catalyze the amide bond cleavage of CDEPA or CMEPA to DEA or MEA, respectively (10, 12).
Alkyl substituents of the aniline derivatives of MEA are located on both sides of the amine group. This special chemical structure makes MEA more difficult to be degraded in natural soils (13). Under conditions of both batch and continuous operations, an oxidation pathway for 2,6-dimethylaniline (DMA) via hydroxyl radicals (OH) was proposed to use Fenton's reactions, in which 2,6-dimethyl-phenol (DMP), 2,6-dimethyl-hydroquinone (DMHQ), 2,6-dimethyl-p-benzoquinone (DMBQ), and 3-hydroxy-2,6-dimethyl-p-benzoquinone (3-OH-DMBQ) were detected as aromatic by-products (13). Chao et al. (14) reported that DMA was transformed to 4-hydroxy-2,6-dimethylaniline (4-OH-DMA) by a P450 monooxygenase system in cultured mammalian cells. Sphingobium baderi strain DE-13 is capable of degrading MEA to the intermediate 4-OH-MEA, which is further transformed to 2-methyl-6-ethyl-benzoquinone imine (MEBQI) (10). Zhang et al. (9) proposed a DEA dealkylation process during the degradation of butachlor by Paracoccus sp. strain FLY-8. However, the molecular basis of and further metabolic pathways for DEA, MEA, DMA, and DMP in microorganisms remain unclear.
The biochemical pathway of acetochlor degradation by a three-bacterium consortium was proposed (acetochlor to CMEPA by Rhodococcus sp. strain T3-1, CMEPA to MEA by Delftia sp. strain T3-6, and MEA by Sphingobium sp. strain MEA3-1) based on the identified degradation intermediates (2). Recently, acetochlor and CMEPA metabolisms have been described in Rhodococcus sp. T3-1 and Delftia sp. T3-6 (11, 12). In this study, we clarified the partial metabolic pathway responsible for MEA degradation and cloned a novel flavin-dependent monooxygenase system, MeaBA, involved in MEA degradation.
MATERIALS AND METHODS
Chemicals and media.
MEA was purchased from Qingdao Vochem Co., Ltd. (Qingdao, China), and 4-OH-DMA and DMHQ (structural analogues of 4-OH-MEA and MEHQ, respectively) were purchased from Sigma-Aldrich (Shanghai, China). Other alkyl-substituted aniline or phenol compounds were purchased from Sinopharm Chemical Reagent Co., Ltd. (Beijing, China). All molecular reagents were purchased from TaKaRa Co., Ltd. (Dalian, China). All chemicals used in this study were of analytical grade or higher purity. Minimal salts medium (MSM), lysogeny broth (LB) medium, and 1/3 LB medium were used for the strains cultured in this study (15).
Bacterial strains, plasmids, and culture conditions.
The strains, plasmids, and primers used in this study are listed in Tables 1 and 2. Escherichia coli strains were routinely grown aerobically at 37°C and 180 rpm in LB medium. Strains MEA3-1 and MEA3-1Mut were grown aerobically at 30°C and 180 rpm in 1/3 LB medium (16), unless otherwise indicated. Pseudomonas putida strain KT2440 was grown aerobically at 30°C and 180 rpm in LB medium.
TABLE 1.
Strains and plasmids used in this study
| Strain or plasmid | Characteristic(s)a | Source or reference |
|---|---|---|
| Strains | ||
| Sphingobium sp. MEA3-1 | Wild-type MEA degrader, pMEA01–pMEA07, Smr | 2 |
| Sphingobium sp. MEA3-1Mut | Derivative of MEA3-1, deletion within pMEA02 | This study |
| P. putida KT2440 | A model strain for aromatic catabolism, Cmr Ampr | 31 |
| E. coli DH5α | Host strain for cloning vectors | TaKaRa |
| E. coli HB101(pRK600) | Conjugation helper strain, Cmr | 33 |
| Plasmids | ||
| pBBR1MCS-2 | Broad-host-range cloning vector, Kmr | 30 |
| pBBR-meaA | pBBR1MCS-5 derivative carrying meaA, Kmr | This study |
| pBBR-meaBA | pBBR1MCS-5 derivative carrying meaBA, Kmr | This study |
| pBBR-meaBA-orf1-4 | pBBR1MCS-5 derivative carrying 7,479 bp | This study |
| pET29a-meaBAT7 | pET29a(+) derivative carrying meaBA, Kmr | This study |
| pMD19-T | T-A cloning vectors, Ampr | TaKaRa |
Smr, streptomycin resistant; Cmr, chloramphenicol resistant; Ampr, ampicillin resistant; Kmr, kanamycin resistant.
TABLE 2.
Primers used in this study
| Primer | Sequence (5′ to 3′)a | Target gene(s) |
|---|---|---|
| MeaBp-F | CTCGAGGATCGGCCATCCTATCGCTG | |
| MeaA-R | GAGCTCTCATCGCGCCTCCGTCAGCGC | meaBA |
| MeaAp-F | CTCGAGGATCGGCCATCCTATCGCTGAACAGCTTCGGCGGTATCTAGAGGAGGTACTTTGAATGGAAGAGGCGCATAACAA | meaA (fusion promoter sequence) |
| Orf4-R | GAGCTCCTAACAATCCACCCGGACCA | meaBA-orf1-4 |
| 16S-F | GCGTAGGATTAGCTAGTTGGT | Partial 16S rRNA sequence |
| 16S-R | AGCTAGTTATCATCGTTTACG | Partial 16S rRNA sequence |
Restriction sites are underlined.
Isolation of spontaneous mutant strains.
Sphingobium sp. MEA3-1 (China Center for Type Culture Collection [CCTCC] M 2012527) was cultured on 1/3 LB plates without selection, and isolated colonies were transferred to fresh 1/3 LB plates. After continuous transfers, colonies with different morphotypes were observed and purified. The ability of the resulting isolates to degrade MEA was determined, as described below. The mutant strain that lost the ability to mineralize MEA was designated MEA3-1Mut. The enterobacterial repetitive intergenic consensus PCR (ERIC-PCR) pattern (17) and 16S rRNA gene sequence of strain MEA3-1Mut were determined and compared with those of the wild-type strain MEA3-1.
Degradation experiment.
The wild-type and mutant strains were cultured in 1/3 LB medium, harvested by centrifugation (Allegra X-22R centrifuge, F0630 rotor; Beckman Coulter, USA) at 2,180 × g and 4°C for 10 min, washed twice with fresh MSM, and then resuspended in MSM to an approximate optical density at 600 nm (OD600) of 2.0 as the inoculum. The 1.0-ml inocula of wild-type and mutant strains were inoculated into a 100-ml Erlenmeyer flask containing 50 ml of MSM supplemented with 100 mg/liter different aromatic compounds as the sole carbon sources at 180 rpm and 30°C, respectively. These aromatic compounds included phenol and the aniline derivatives DEA, DMA, aniline, 2,3-dimethyl-aniline (2,3-DMA), 2,4-dimethyl-aniline (2,4-DMA), DMHQ, 4-OH-DMA, 2-methyl-aniline (2-MA), toluene, O-xylene, phenol, catechol, hydroxyquinol, 2-methyl-phenol (2-MP), 2,6-dimethyl-phenol (2,6-DMP), 3,5-dimethyl-phenol (3,5-DMP), and 3,4-dimethyl-phenol (3,4-DMP). The degradation of these aromatic compounds was measured using high-performance liquid chromatography (HPLC). All treatments were performed in triplicate, and control experiments without inoculation or without substrates were performed under the same conditions.
Identification of MEA-degrading metabolites.
The 2.0-ml inocula of strains MEA3-1 and MEA3-1Mut, as described above, were inoculated into a 250-ml Erlenmeyer flask containing 100 ml of MSM supplemented with 100 mg/liter MEA. When the concentration of MEA decreased to approximately 50 mg/liter, the supernatant was collected by centrifugation at 12,580 × g and 4°C for 10 min and lyophilized. The residual was dissolved in 1 ml of methanol, which was filtered through a 0.22-μm-pore-size Millipore membrane (Sangon Biotech, Shanghai, China) for HPLC-tandem mass spectrometry (HPLC-MS/MS) analysis. A 5-μm C18 separation column (internal diameter, 4.6 mm; length, 250 mm) filled with Kromasil 100 Å was used for HPLC analysis. The mobile phase was methanol-water (80:20 [vol/vol]), and the flow rate was 0.8 ml/min. The detection wavelength was 240 nm, and the injection volume was 20 μl. MS analysis was performed in electrospray ionization (ESI) mode with an Agilent G6410B triple-quad mass spectrometer. In the MS analysis, the metabolites were separated and ionized by electrospray ionization to obtain a positive polarity. Characteristic fragment ions were identified by second-order MS and compared to those generated with authentic or structural analogue standards.
Plasmid profiling.
Plasmids from strains MEA3-1 and MEA3-1Mut were isolated using a modified alkaline extraction method (18). After 12 h of incubation (logarithmic phase), approximately 5 × 109 cells were harvested by centrifugation at 12,580 × g and 4°C for 10 min and resuspended in 1.0 ml of buffer I (1 M NaCl and 50 mM glucose in Tris-EDTA [TE] buffer [pH 8.0]) after they were washed with 5 ml of TE buffer (10 mM Tris-HCl, 1 mM EDTA [pH 8.0]). The cells were incubated with 20 μg/ml RNase and 100 μg/ml lysozyme at 37°C for 30 min until the cells began to lyse. Next, 2.0 ml of freshly prepared buffer II (0.2 M NaOH, 10 g/liter SDS) was added and mixed gently. The samples were placed on ice for 15 min. Next, 1.5 ml of ice-cold buffer III (5 M potassium acetate, 11.5% acetic acid [vol/vol]) was added and mixed gently until a white precipitate formed. The supernatant was collected by centrifugation at 12,580 × g and 4°C for 15 min. Other procedures and manipulations (phenol-chloroform extraction and isopropanol precipitation) were operated as described by O'Sullivan and Klaenhammer (18).
The isolated plasmids were visualized using pulsed-field gel electrophoresis (PFGE) (Bio-Rad) (19). PFGE was performed in TBE buffer (20 mM Tris-HCl, 20 mM boric acid, 0.5 mM EDTA [pH 8.0]) at 12.5°C at 6 V/cm, with linearly increasing pulse times from 10 to 20 s for 24 h. HindIII-digested lambda phage DNA was used as a molecular size standard.
Plasmidome sequencing, assembly, and annotation.
The plasmids from strains MEA3-1 and MEA3-1Mut were isolated, as described above, and the linear DNA in the plasmidomes was digested using ATP-dependent DNase. The 16S-F/16S-R primer pair, which targets the 16S rRNA gene, was used to detect residual chromosomal DNA. The plasmidomes of strains MEA3-1 and MEA3-1Mut were shotgun sequenced (20) with a Roche 454 genome sequencer FLX Titanium platform (Han-Yu Biological Technology Co. Ltd.) (21). The sequencing depth and the fold coverage of the plasmid draft were 20× and 99-fold, respectively. Sequencing reads were assembled using the SOAPdenovo software (version 1.05; http://soap.genomics.org.cn/soapdenovo.html). De novo gene prediction was conducted using Glimmer (version 3.0; http://ccb.jhu.edu/software/glimmer/index.shtml). The BLAST program (22), combined with sequences from the KEGG, COG, Swiss-Prot, and nonredundant protein databases, was used to accomplish functional annotation, with an E value cutoff of 1E−5.
The gaps predicted by MUMmer and BLAST were closed using ContigScape (23), which interactively displayed the relationships between plasmidome contigs, thereby allowing a faster and more precise determination of linkages and greatly improving the efficiency of gap closure (23). Gap closure was verified by PCR and self-formed adaptor PCR (SEFA PCR) (24) amplification; all gap closure primers are shown in Table S1 in the supplemental material.
Protein extraction and 2-DE analysis.
Crude enzyme extracts of strains MEA3-1 and MEA3-1Mut were prepared using ultrasonic disruption (Sonicator 201 M; Kubota, Japan) for 10 min at 4°C in a lysis buffer {7 M urea, 2 M thiourea, 4% (wt/vol) 3-[(3-cholamidopropyl)-dimethylammonio]-1-propanesulfonate (CHAPS), 0.2% (wt/vol) Bio-Lyte (Bio-Rad) (pH 3 to 10), 65 mM dithiothreitol (DTT)} and centrifuged at 12,580 × g at 4°C for 20 min to remove cell debris. Protein concentrations were determined using a modified Bradford assay (25), with ovalbumin as the standard.
The proteins in the supernatant were analyzed using 2-DE. For each replicate, 100 μg of total protein extract was loaded onto 17-cm immobilized pH gradient dry strips (pH 4 to 7 linear gradient; Bio-Rad, USA) during the rehydration step (13 h), followed by focusing for a total of 60,000 V · h using a Protean isoelectric focusing (IEF) cell (Bio-Rad). After isoelectric focusing, the gel strips were equilibrated in 5 ml of equilibration buffer (0.375 M Tris-HCl [pH 8.8], 6 M urea, 20% [vol/vol] glycerol, 2% [wt/vol] SDS, and 2% [wt/vol] DTT) for 15 min and then reequilibrated for 15 min in the same buffer but without DTT, which was replaced with iodoacetamide (2.5% [wt/vol]) (26). SDS-PAGE in the second dimension was performed with 12% SDS-polyacrylamide gels (25 cm by 20 cm) and sealed with low-melting-point agarose (0.5% [wt/vol]). Electrophoresis was performed using a Protean Plus Dodeca cell apparatus (Bio-Rad) at 50 V for the first 30 min, followed by 150 V for 8 h. The protein spots were visualized using mass spectrometry-compatible silver staining (27).
Protein mass fingerprinting (PMF).
The gels were scanned using a Umax PowerLook III scanner (Umax Technologies, USA) at a resolution of 300 dots per in. (dpi), and the images of the gels were analyzed with PDQuest (version 8.0; Bio-Rad). Selected protein spots were manually excised from the gels for matrix-assisted laser desorption ionization–time of flight mass spectrometry (MALDI-TOF MS) analysis (Bo-Yuan Biological Technology Co. Ltd.). The resulting peptide fragments were analyzed by searching against the plasmidome databases of strains MEA3-1 and MEA3-1Mut, as described below.
Cloning of meaBA.
The lost genes in Sphingobium sp. strain MEA3-1 that may be involved in MEA degradation were analyzed in detail by searching against the NCBI database and the Protein Data Bank (PDB). Possible monooxygenase genes involved in MEA degradation, designated meaBA, were further studied by sequence analysis and functional verification, as described below.
For phylogenetic analysis, the amino acid sequences of MeaA and MeaB were first aligned using Clustal X (version 2.1) (28) and then imported into MEGA (version 5.0) (29) to construct a phylogenetic tree via the neighbor-joining method. Distances were calculated using the Kimura two-parameter distance model. Confidence values for the branches of the phylogenetic tree were determined using bootstrap analysis based on 1,000 resamplings.
Functional complementation of MEA3-1Mut and KT2440 with MeaBA.
Genomic DNA was extracted and purified from strain MEA3-1 via high-salt precipitation (11). Three PCR fragments (1,224 bp, 2,069 bp, and 6,247 bp) containing the 60-bp potential promoter region of meaB were amplified using the primer pairs MeaAp-F/MeaA-R, MeaBp-F/MeaA-R, and MeaBp-F/Orf4-R (Table 2), designated meaA, meaBA, and meaBA-orf1-4 (i.e., meaBA with orf1 to orf4), respectively. The three fragments were digested with SacI and XhoI and inserted into the corresponding sites of the broad-host-range plasmid pBBR1MCS-2 (30) to generate pBBR-meaA, pBBR-meaBA, and pBBR-meaBA-orf1-4, respectively. The inserted fragments in the three plasmids were verified by sequencing. The resulting three plasmids were transformed into E. coli DH5α. Next, three plasmids were introduced into strains MEA3-1Mut and KT2440 (31) via triparental mating (32), using pRK600 (33) as the helper plasmid. The abilities of the strains harboring different plasmids to degrade MEA and MEHQ were determined using a whole-cell biotransformation assay, as described by Liu et al. (34). Samples were taken at regular intervals, the concentrations of the substrates were determined rapidly using a UV-visible spectrophotometer, and bacterial growth was monitored by measuring the CFU (CFU per milliliter).
Effect of metyrapone on the degradation of MEA.
Metyrapone is a specific inhibitor of cytochrome P450 monooxygenase systems (35). Therefore, 200 μl of MSM containing 0.5 mM MEA and different concentrations of metyrapone (100 mg/liter, 200 mg/liter, and 300 mg/liter) were added into 96-well plates inoculated with strain MEA3-1. The concentration of MEA was measured by color development with 4-aminoantipyrene and potassium hexacyanoferrate (12). Reaction mixtures without inoculation and metyrapone were used as negative and positive controls, respectively.
Simulation of 4-OH-MEA hydrolytic deamination.
4-OH-DMA, a structural analogue of 4-OH-MEA, was used to simulate the spontaneous hydrolytic deamination process. Briefly, 200 μl of pure water containing 0.5 mM 4-OH-DMA was added to 96-well plates, and Nessler's reagent was added at intervals to determine the release of ammonia (36).
Nucleotide sequence accession numbers.
The GenBank accession number of the 12,052-bp DNA fragment containing the meaBA gene cluster and orf1-9 is KP752077. The sequence of the plasmidome from Sphingobium sp. strain MEA3-1 has been deposited in GenBank under the accession numbers CM003352 to CM003358. The accession numbers for the oxygenase components are in the legend to Fig. 4.
FIG 4.

Neighbor-joining tree constructed using the alignment of MeaA with the amino acid sequences of the oxidation components of some characterized FDMs. The branches corresponding to partitions reproduced in <50% of the bootstrap replicates are collapsed. The names of the strains and proteins are displayed in the phylogenetic tree. The oxygenase components and their accession numbers are as follows: MeaA, KP752077; C2-hpaH, AY566612; HsaA, Q0S811; NcnH, AAG44124; TcpA, AAM55214; HpaB, YP003001901; PheA1, ABS30825; and CndA, KJ461679. CndA belongs to the oxygenase component of a Rieske non-heme iron monooxygenase and was used to provide a frame of reference for the phylogeny. R. erythropolis, Rhodococcus erythropolis.
RESULTS
Isolation of MEA degradation-deficient mutants and metabolite identification.
The MEA degradation phenotype of Sphingobium sp. strain MEA3-1 was quite unstable. Large and small colonies appeared when the inoculum was freshly streaked from samples frozen at −80°C or after continuous transfers on 1/3 LB medium without selective pressure. The 16S rRNA gene sequence of the mutant strain exhibited 100% similarity with that of strain MEA3-1. The ERIC-PCR (17, 37) patterns of the two strains exhibited identical fragment distributions (see Fig. S1 in the supplemental material). These results indicated that the larger-size colony was indeed an MEA degradation-deficient mutant of strain MEA3-1 and was designated Sphingobium sp. strain MEA3-1Mut.
Strain MEA3-1 was able to completely degrade MEA, as detected by HPLC analysis, and to utilize MEA as the sole carbon source for growth (2). However, the whole-cell transformation experiments indicated that strain MEA3-1Mut could transform MEA only into an unidentified metabolite (Fig. 1). The MS/MS results indicated that the MEA substrate, with a retention time (tR) of 5.68 min, has a prominent protonated molecular ion at m/z 136 and fragment ions of m/z 120, 108, and 91 (Fig. 1A). Product A, with a tR of 5.08 min, has a molecular ion at m/z 152 and fragment ions of m/z 135, 123, and 107, which correspond to a hydroxylated form of MEA (Fig. 1C). Product A was proposed to be 4-hydroxy-2-methyl-6-ethylaniline (4-OH-MEA). Product B, with a tR of 4.61 min, has a molecular ion at m/z 153 and fragment ions of m/z 136, 121, and 108, which correspond to the hydrolytic deamination of product A (Fig. 1B). Product B was proposed to be 2-methyl-6-ethyl-hydroquinone (MEHQ). The possible spontaneous oxidation products of 4-OH-MEA and MEHQ, 2-methyl-6-ethyl-benzoquinone imine (MEBQI) at m/z 150 and 2-methyl-6-ethyl-benzoquinone (MEBQ) at m/z 151, respectively, were also detected using HPLC-MS (data not shown). The MS/MS fragment results were analyzed based on the compound structures (Fig. 1).
FIG 1.

HPLC and mass spectrum detection of the intermediate metabolites. (A to D) HPLC and positive-ion mass spectra of the substrate MEA (A), of the metabolite product (MEHQ) by strain MEA3-1Mut (B), of the metabolite product (4-OH-MEA) by strain MEA3-1Mut (C), and of the metabolite product (3-OH-MEHQ) by strain MEA3-1 (D).
Identification of MEA-degrading metabolites from strain MEA3-1.
Sphingobium sp. strain MEA3-1 can utilize MEA as a sole carbon and energy source but cannot utilize aniline for growth (38), indicating that strain MEA3-1 employs different pathways to degrade alkyl-substituted aniline compounds. To clarify the MEA degradation pathway in this strain, the degradation intermediates were analyzed. Using HPLC-MS, the same degradation products, 4-OH-MEA and MEHQ, were detected in the culture medium extracts of strain MEA3-1 during the biodegradation of MEA (data not shown). Another metabolite, product C (tR, 2.70 min), exhibited a molecular ion at m/z 169 and fragment ions of m/z 152, 134 and 115.7, which correspond to the hydroxylated form of MEHQ. Product C was proposed to be 3-hydroxy-2-methyl-6-ethyl-hydroquinone (3-OH-MEHQ) (Fig. 1D), and the possible spontaneous oxidation product 3-hydroxy-2-methyl-6-ethyl-benzoquinone (3-OH-MEBQ), with a molecular ion of m/z 167 and the same MS/MS fragment ions, was also detected.
In addition to 4-OH-MEA, MEHQ was detected in the MEA metabolites. We speculate that MEBQI, the 4-OH-MEA oxidation product, can spontaneously hydrolytically deaminate to MEBQ or MEHQ. 4-OH-DMA, a structural analogue of 4-OH-MEA, was used to simulate the spontaneous hydrolytic deamination process using Nessler's reagent colorimetry. As shown in Fig. S2 in the supplemental material, 4-OH-DMA was rapidly spontaneously deaminated by hydrolysis in pure water. Considering the similar structures of 4-OH-MEA and 4-OH-DMA, a similar reaction may also occur with 4-OH-MEA.
The substrate degradation spectra of the wild-type and mutant strains.
Using HPLC analysis, the degradation of alkyl-substituted aniline or phenol compounds by wild-type and mutant strains was monitored. The degradation results are shown in Table 3. Wild-type strain MEA3-1 degraded most alkyl-substituted aniline or phenol compounds, except for toluene, xylene, aniline, 2,3-DMA, 2,4-DMA, and 3,4-DMP. These results indicated that strain MEA3-1 employs different pathways to degrade alkyl-substituted aniline compounds, and this metabolic pathway is similar to that of alkyl-substituted phenol compounds. However, the mutant strain MEA3-1Mut was able to transform only alkyl-substituted aniline and phenol compounds; it is worth noting that the mutant strain did not degrade 4-OH-DMA and DMHQ, which are structural analogues of 4-OH-MEA and MEHQ, respectively. These results indicate that the degradation of MEA by strain MEA3-1 is subject to para-hydroxylation of the ring. Given the MEA-degrading metabolites of strains MEA3-1 and MEA3-1Mut, a possible pathway for MEA degradation by Sphingobium sp. MEA3-1 is illustrated in Fig. 2, and MEA3-1Mut has likely lost a key gene or gene cluster for MEBQ degradation.
TABLE 3.
Substrate spectra of strains MEA3-1 and MEA3-1Mut
| Substrate | Result witha: |
|
|---|---|---|
| MEA3-1 | MEA3-1Mut | |
| MEA | + | * |
| DEA | + | * |
| DMA | + | * |
| Aniline | − | − |
| 2,3-DMA | − | − |
| 2,4-DMA | − | − |
| 2-MA | + | * |
| 4-OH-DMA | + | − |
| DMHQ | + | − |
| Toluene | − | − |
| O-Xylene | − | − |
| Phenol | + | + |
| Catechol | + | + |
| Hydroxyquinol | + | + |
| Hydroquinone | + | + |
| 2-MP | + | * |
| 2,6-DMP | + | * |
| 3,5-DMP | + | * |
| 3,4-DMP | − | − |
+, positive for strain growth and substrates were degraded; −, negative for strain growth and substrates were not degraded; *, negative for strain growth and substrates were transformed to other compounds. The biomass was measured at 600 nm (OD600) using a Shimadzu UV-visible spectrophotometer. The mineralization or cometabolic transformation of these aromatic compounds was measured using HPLC, all treatments were performed in triplicate, and control experiments without inoculation and without substrate were performed under the same conditions.
FIG 2.

Partial metabolic pathway of MEA mineralization by Sphingobium sp. strain MEA3-1.
Plasmidome sequencing.
Considering the unstable degradative phenotype, we deduced that the genes encoding the MEA-degrading enzymes were located on plasmids. The plasmid profiles of MEA3-1 and MEA3-1Mut were analyzed using PFGE. Seven plasmids, designated pMEA01, pMEA02 (pMEA02′ for strain MEA3-1Mut), pMEA03, pMEA04, pMEA05, pMEA06, and pMEA07, were detected (Fig. 3). By comparing the plasmid profiles of strains MEA3-1 and MEA3-1Mut, six of the seven plasmids were found to shift identically during the PFGE of both strains. Compared with its counterpart pMEA02 in strain MEA3-1, a smaller plasmid, pMEA02′, was observed in the mutant (Fig. 3). We deduced that pMEA02′ is a derivative of pMEA02, with some portions deleted.
FIG 3.
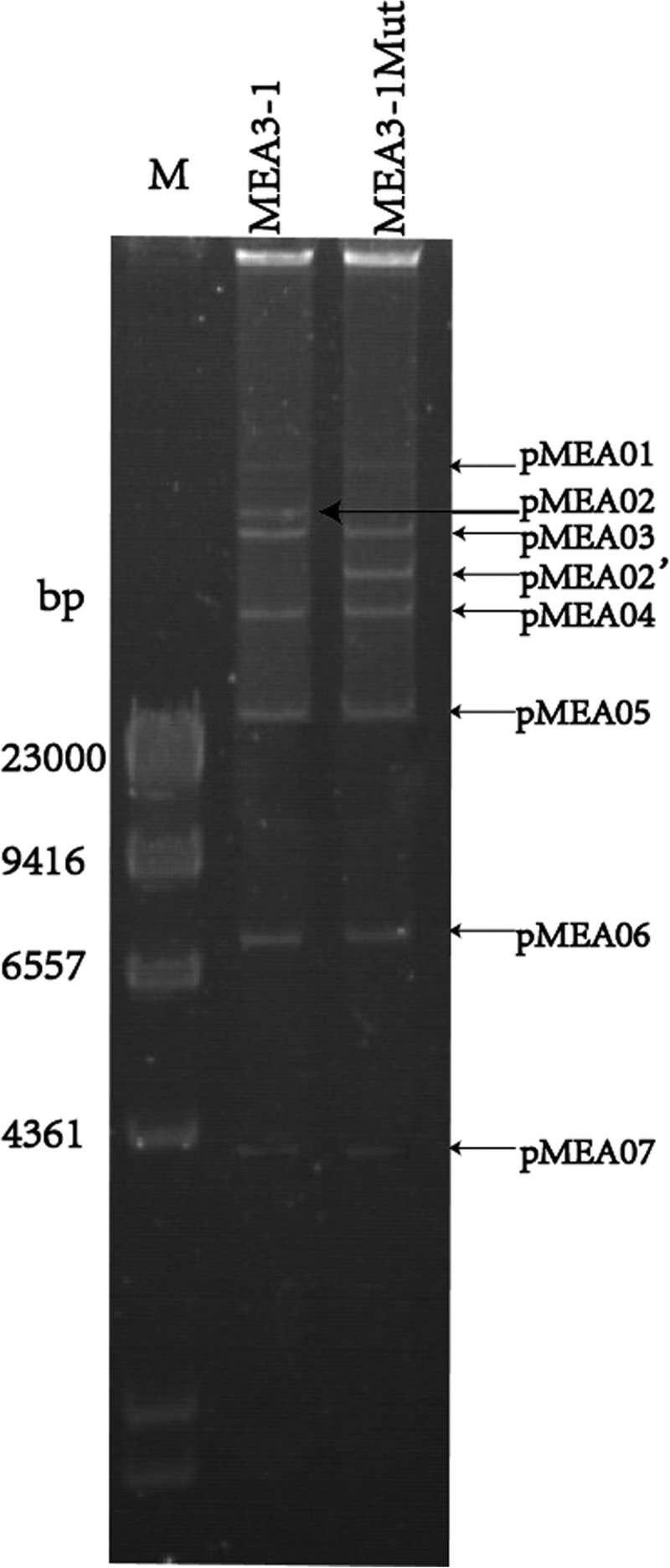
Plasmid profiles of strains MEA3-1 and MEA3-1Mut using PFGE. Lane M, HindIII digest of lambda phage DNA.
The plasmidomes of strains MEA3-1 and MEA3-1Mut were sequenced and subjected to gap closure. The total number of contigs of strain MEA3-1 was 213, with a total length of 785,046 bp. Gaps between the contigs were closed via bioinformatic analysis and primer walking with PCR products. Seven complete plasmid sequences (pMEA01 to pMEA07) were obtained, and a circle graph of pMEA02 from strain MEA3-1 is shown in Fig. S3 in the supplemental material.
The largest and smallest plasmids were 287,745 bp and 6,108 bp, with 413 and 10 open reading frames (ORFs), respectively. The seven plasmids were not homologous to other large plasmids from Sphingomonas spp., and most of the ORFs were annotated as hypothetical proteins, plasmid-relevant proteins, or transposases. The 134,691-bp pMEA02 plasmid contained 164 ORFs with six IS6100 transposase regions and many other transposase family regions. A 24,948-bp fragment (from bp 132840 to 23097 in pMEA02) and a 12,052-bp fragment (from bp 43824 to 55875 in pMEA02) were lost to generate the plasmid pMEA02′ in strain MEA3-1Mut (see Fig. S3). Therefore, MEA degradation-related genes may be located in these contigs in pMEA02 from strain MEA3-1.
2-DE and PMF analyses.
Sphingobium sp. strains MEA3-1 and MEA3-1Mut total proteins were analyzed using 2-DE to determine the differences associated with pMEA02 in the wild-type and mutant strains. As revealed by PDQuest software analysis, the protein profiles produced from the 2-DE gels were highly reproducible among the three independent extractions. Figure S4 in the supplemental material shows representative gels of the soluble proteins extracted from strains MEA3-1 and MEA3-1Mut. Compared with MEA3-1Mut, strain MEA3-1 had three significantly distinct protein spots, which were designated A, B, and C.
PMF analysis identified several peptide fragments from the three protein spots (see Table S2 in the supplemental material). Genes encoding the three proteins were located at bp 9382 to 10158, bp 12713 to 13816, and bp 45926 to 47104 in pMEA02, which were lost in MEA3-1Mut. The three proteins exhibited 100%, 99%, and 99% amino acid sequence similarity with 3-hydroxy-2-methylbutyryl-coenzyme A (CoA) dehydrogenase (spot A), acyl-CoA dehydrogenase (ACAD) (spot B), and hypothetical protein (spot C) from Sphingomonas sp. DC-6, respectively, as determined by searching against the NCBI protein database. It is worth noting that the strain DC-6 was reported to completely mineralize acetochlor (3, 10).
ORF analysis.
Spots A and B are located in the lost 24,948-bp fragment, and spot C is located in the lost 12,052-bp fragment. The two fragments are surrounded by the same transposable element, Tnp1, in pMEA02. Tnp1 exhibited high levels of amino acid sequence identity (99% and 100%) with the IS6100 transposase-like protein from the carbazole-degrading strain Sphingomonas sp. strain XLDN2-5 and E. coli, respectively (39).
There were no relevant hydroxylase genes in the 24,948-bp fragment encoding the proteins in spots A and B (data not shown); therefore, it was not analyzed in detail. A detailed search for ORFs in the 12,052-bp fragment revealed that orf5, encoding the hypothetical protein (HP1) in spot C, was an oxygenase gene (Table 4). Phylogenetic analysis showed that orf5 encodes an amino acid sequence that shares significant identity with the oxygenase components of some two-component flavin-dependent monooxygenases (TC-FDMs) (Fig. 4); for example, HP1 exhibits 25% identity with C2-hpaH (p-hydroxyphenylacetate 3-hydroxylase) from Acinetobacter baumannii (40), 24% identity with HsaA [3-hydroxy-9,10-secoandrosta-1,3,5(10)-triene-9,17-dione monooxygenase] from Rhodococcus sp. strain RHA1 (41), and 22% identity with NcnH (naphthocyclinone hydroxylase) from Streptomyces arenae (42).
TABLE 4.
Deduced functions of ORFs within the missing 12,052-bp fragment sequence
| Gene | Product size (no. of amino acids) | Positions | Homologous protein | Source | GenBank accession no. | % amino acid identity |
|---|---|---|---|---|---|---|
| tnp1 | 264 | 1–795 | Transposase IS6100 | E. coli | YP_003108355.1 | 100 |
| orf7 | 138 | 835–1251 | Aminoglycoside phosphotransferase | Sphingobium chlorophenolicum L-1 | YP_004554070.1 | 68 |
| orf6 (meaB) | 168 | 1251–1757 | Hypothetical protein | S. chlorophenolicum | WP_013847811 | 31 |
| orf5 (meaA) | 392 | 2088–3266 | Pigment production hydroxylase | Rhodococcus opacus | WP_012691790 | 39 |
| orf1 | 480 | 3479–4921 | Isopropylmalate isomerase large subunit | Sphingomonas-like bacterium B12 | WP_022690399 | 81 |
| orf2 | 199 | 4918–5517 | Isopropylmalate isomerase small subunit | S. chlorophenolicum | WP_013849123 | 61 |
| orf3 | 441 | 5727–7052 | Homogentisate 1,2-dioxygenase | Sphingomonas sp. YL-JM2C | WP_037520129 | 52 |
| orf4 | 123 | 7073–7444 | Hypothetical protein Veis_4652 | Verminephrobacter eiseniae EF01-2 | YP_999367.1 | 58 |
| orf8 | 284 | 6625–7479 | Integrase catalytic subunit | S. wittichii RW1 | YP_001260297.1 | 88 |
| orf9 | 267 | 10253–11056 | Putative transposase | Magnetospirillum sp. SO-1 | WP_008622716.1 | 84 |
| tnp1 | 264 | 12052–11258 | Transposase IS6100 | E. coli | YP_003108355.1 | 100 |
HsaA, NcnH, and C2-hpaH were previously classified as enzymes in the TC-FDM family, and the consensus sequences of this family were determined (43). HP1 has partially conserved residues (W86, W183, S145, W143, R264, H367, and M363) identical to residues in the consensus amino acid sequences interacting with flavin mononucleotide (FMN) in TC-FDM (see Fig. S5 in the supplemental material) (43). HP1 also contained the conserved domains COG1960 and KOG0139 belonging to the ACAD superfamily (44). These results indicate that HP1 is a novel oxygenase component of the TC-FDM family, and its absence in mutant MEA3-1Mut suggests that HP1 is responsible for the hydroxylation of MEHQ. orf5 was designated meaA and encodes the oxygenase component MeaA of the MEHQ monooxygenase.
A BLAST analysis indicated that another hypothetical protein (HP2, encoded by orf6), located upstream of the meaA gene shared significant identity with cytochrome C (30% amino acid sequence identity with 40% coverage) from Bacillus altitudinis. We aligned HP2 with the reductase components of the TC-FDMs described above and found that the sequence identities with some hypothetical proteins ranged from 6 to 14% (40–42). We deduced that HP2 was the reduction component of a TC-FDM. The HP2 was assumed to be the reductase component of MeaA and was designated MeaB. The reductase component of the MEHQ hydroxylase (MeaB) may perform the NADH-dependent reduction of free FMN, which may be subsequently transferred to the larger monooxygenase component (MeaA) and used for MEHQ monooxygenation (Fig. 5C).
FIG 5.

Physical map of the 12,052-bp putative transposable element containing meaBA in Sphingobium sp. strain MEA3-1. (A) Arrows indicate the sizes, locations, and directions of transcription of the ORFs. Complementation of the meaBA-disrupted mutants with different regions is illustrated below the physical map. (B) Degradation curve of MEHQ and the growth curve of MEA3-1Mut(pBBR-meaBA-orf1-4, pBBR-meaBA, and pBBR-meaA). (C) Probable scheme of the catalytic cycle of MEHQ hydroxylase MeaA. Error bars represent the standard deviations for three replicates.
Functional expression of MeaBA.
To further confirm the functions of MeaA, MeaB, and the products of the gene completely lacking the fragment (meaBA-orf1-4, excluding the transposable element), plasmids pBBR1MSC-2, pBBR-meaA, pBBR-meaBA, and pBBR-meaBA-orf1-4 were introduced into strains MEA3-1Mut, DH5α, and KT2440. The whole-cell transformation experiments revealed that strains MEA3-1Mut and KT2440 harboring pBBR-meaBA or pBBR-meaBA-orf1-4, but not pBBR-meaA, acquired the ability to degrade or transform MEHQ (Fig. 5A). The initial and final cell densities of strain MEA3-1Mut harboring pBBR-meaBA or pBBR-meaBA-orf1-4 were approximately 2 × 106 and 7.2 × 106 CFU/ml, respectively (Fig. 5B). In addition, strain KT2440 harboring pBBR-meaBA or pBBR-meaBA-orf1-4 acquired the ability to transform MEHQ but could not utilize MEHQ as a sole carbon source for growth (data not shown). Therefore, we confirmed that MeaBA contains the two components of the MEHQ monooxygenase, and orf1 to orf4 in the 12,052-bp fragment likely do not participate in the degradation of MEA. However, E. coli DH5α harboring either pBBR-meaBA or pBBR-meaBA-orf1-4 did not degrade MEHQ (Fig. 5A). This failure might be caused by codon usage, protein folding, or the low efficiency of the native meaBA promoter in E. coli DH5α (45). To exclude the third possibility, meaBA was placed under the control of a T7 promoter in the vector pET-29a(+) and introduced into E. coli BL21(DE3) (45). The results of the whole-cell transformation assay showed that isopropyl-β-d-thiogalactopyranoside (IPTG)-induced E. coli BL21(DE3) harboring pET29a-meaBAT7 was still unable to degrade MEHQ.
Possible enzyme involved in the upstream MEA metabolic pathway.
We deduced that a P450 monooxygenase system initiates the degradation of MEA via aromatic ring hydroxylation at the para position. Metyrapone is a P450 inhibitor, and its effect on MEA degradation by strain MEA3-1 was tested (see Fig. S6 in the supplemental material). MEA degradation was visually observed, because MEA reacts with 4-aminoantipyrene to form a purple compound, and the color gradually deepens as MEA concentration increases. MEA degradation was inhibited when the metyrapone concentration was >200 mg/liter. These results show that a P450 monooxygenase system possibly initiates MEA degradation to transform MEA to 4-OH-MEA.
DISCUSSION
MEA and DEA are important degradation intermediates of several chloroacetanilide herbicides. Located on both sides of the amine group, the alkyl substituents of these aniline derivatives make their degradation processes different from that of anilines in natural soils (3, 10). Microbial degradation is an important process in the biogeochemical cycling and detoxification of organic pollutants in the environment. Several strains have been reported to degrade MEA, but the detailed degradation process is unclear (2, 10). It was proposed that DEA, a metabolite of butachlor, was degraded through dealkylation to an aniline, oxidation to a catechol, and ring cleavage through an ortho-cleavage pathway in Paracoccus sp. FLY-8 in vitro (9). In the present study, a novel flavin-dependent monooxygenase system for MEHQ hydroxylation, MeaBA, was cloned and characterized, and the possible metabolic pathway responsible for MEA degradation was proposed.
MEA was first transformed into the transitory intermediate 4-OH-MEA by a potential P450 monooxygenase. The degradation of DMA, a structural analogue of MEA, has been extensively studied in mammals (6, 14). It was reported that DMA was transformed to 4-OH-DMA in cultured mammalian cells by a P450 monooxygenase system (14). It has also been shown that the human cytochrome P450 isoforms responsible for the metabolism of alkylated aniline derivatives were CYP3A4 and CYP2B6 (6). Cytochrome P450 was proposed to be involved in the first degradation step of MEA by Sphingobium sp. MEA3-1. The cytochrome P450 inhibitor metyrapone (35) was found to inhibit the degradation of MEA by strain MEA3-1 (see Fig. S6 in the supplemental material). Therefore, we hypothesize that MEA is transformed into 4-OH-MEA in the presence of a P450 monooxygenase system. We found five cytochrome P450 oxygenases annotated in the genome of strain MEA3-1. A BLASTp analysis indicates that these cytochrome P450 oxygenases may be involved in the metabolism or synthesis of N-(1-pyrene)-acetamide, N-butyl-isocyanide, and cholesterol. However, whether they are involved in MEA degradation still needs to be clarified.
4-OH-MEA is an unstable intermediate that is oxidized rapidly to MEBQI in animals and microbes (10, 14). In this study, using Nessler's reagent to determine the release of ammonia from 4-OH-DMA (36), we found that MEBQI was spontaneously deaminated to MEHQ (see Fig. S2 in the supplemental material). Levenberg and Hayaishi (46) reported the hydrolytic deamination of the naturally occurring 2-amino-4-hydroxy-pteridines (pterins), which yielded the corresponding 2,4-dihydroxy compounds (lumazines). 4-OH-DMA, the degradation product of sulfonated azo dyes, was not observed in the reaction mixtures because it spontaneously and rapidly hydrolyzed to DMBQ (47).
Based on metabolite identification, MEHQ is further hydroxylated to form 3-OH-MEHQ (Fig. 1). Similar transformations have been reported in the degradation of xylenol compounds. Cell extracts of Mycobacterium sp. strain DM1 catalyzed the reduction of 2,6-dimethyl-3-hydroxyquinone (DMHQ) to 3-hydroxy-2,6-dimethyl-hydroquinone (3-OH-DMHQ) (48). Hofrichter et al. (49) also reported that the metabolism of DMHQ occurred via DMHQ, DMBQ, 3-OH-DMHQ, and 3-OH-DMBQ in Penicillium frequentans Bi7/2. However, the biotransformation mechanism of DMHQ and MEHQ to 3-OH-DMHQ and 3-OH-MEHQ, respectively, is still unclear.
In this study, using heterologous expression and complementation experiments, we identified two genes, meaA and meaB, that encode enzymes responsible for MEHQ hydroxylation. The oxygenase MeaA and reductase MeaB were obviously different from previously reported TC-FDMs at the amino acid sequence level, such as C2-hpaH (41), HsaAB (43), and NcnH (42). MeaB shared significant sequence identity with cytochrome c from B. altitudinis. The hydroxylation of 4-ethylphenol by the bacterial flavocytochrome p-cresol methylhydroxylase (PCMH) from P. putida has been proposed to be involved in the initial formation of an enzyme-bound p-quinone methide product intermediate (50, 51). The reductase component of MEHQ hydroxylase (MeaB) performs the NADH-dependent reduction of free FMN, which is subsequently transferred to the larger monooxygenase component (MeaA) and used for the reaction of MEHQ monooxygenation (52).
Degradative plasmids are frequently isolated from Sphingobium species. Seven plasmid sequences (pMEA01 to pMEA07) were obtained from strain MEA3-1 (Fig. 3). Several instances of the degradation of aromatic compounds by genes carried on plasmids have been reported, such as those with pLB1 (a γ-hexachlorocyclohexane-degrading plasmid) from Sphingobium japonicum strain UT26 (53), pCF01-05 (a carbofuran-degrading plasmid) from Sphingomonas sp. strain CF06 (54), and pCAR3 (a carbazole-degrading plasmid) from Sphingomonas sp. strain KA1 (55). Until this study, Stolz (56) reported that, at most, five plasmids have been isolated from sphingomonads, namely, those found among two plasmids from Sphingomonas aromaticivorans strain F199, Sphingomonas wittichii strain RW1, and Novosphingobium pentaromativorans strain US6-1, three plasmids from S. japonicum strain UT26 and Novosphingobium sp. strain PP1Y, four plasmids from Sphingomonas xenophagum strain BN6 and Sphingobium fuliginis strain ATCC 27551, and five plasmids from Sphingomonas sp. strain MM-1 (56). The meaBA genes are located on plasmid pMEA02 and are surrounded by a transposable element from the IS6100 family (Fig. 5A). This location might be the reason that MEA degradation was prone to loss.
The horizontal transfer of genes plays a key role in the evolution of catabolic pathways, thereby facilitating bacterial adaptation to pollutant-contaminated sites (57). Notably, many monooxygenase genes are associated with mobile genetic elements, such as IS6100, an insertion sequence classified in the IS6 family. Wang et al. (58) reported that a novel 3-phenoxybenzoate 1,2-dioxygenase gene in Sphingobium wenxiniae strain JZ-1 was located between two IS6100 transposase genes, tnp1 and tnp2. Two identical nonylphenol monooxygenase genes were also surrounded by an IS21-type insertion sequence (IS) and IS6100 in Sphingomonas sp. strain NP5 (59). Dogra et al. (60) revealed that most of the lin genes in Sphingomonas paucimobilis strains B90A, Sp+, and UT26 are associated with IS6100. Further research suggests that the IS6100 elements have a very broad host range, and their presence on plasmids (even in strains in which their locations have not been ascertained) cannot be ruled out (60).
A downstream ring cleavage pathway and an initializing P450 monooxygenase still need to be identified in future research.
Supplementary Material
ACKNOWLEDGMENTS
This work was financially supported by the Natural Science Foundation of China (grants 31400098, 31270095, and 31560031), the National Basic Research Program (grant 2015CB1505002), the Natural Science Foundation of Jiangsu Province (grant BK2012029), the National Science and Technology Support Program (grants 2012BAD14B02 and 2014ZX08011-003), the 863 Project (grant 2013AA102804), and the Graduate Culture and Innovation Project of Jiangsu Province (grant KYLX_0514).
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.01883-15.
REFERENCES
- 1.Foley ME, Sigler V, Gruden CL. 2008. A multiphasic characterization of the impact of the herbicide acetochlor on freshwater bacterial communities. ISME J 2:56–66. doi: 10.1038/ismej.2007.99. [DOI] [PubMed] [Google Scholar]
- 2.Hou Y, Dong W, Wang F, Li J, Shen W, Li Y, Cui Z. 2014. Degradation of acetochlor by a bacterial consortium of Rhodococcus sp. T3-1, Delftia sp. T3-6 and Sphingobium sp. MEA3-1. Lett Appl Microbiol 59:35–42. [DOI] [PubMed] [Google Scholar]
- 3.Chen Q, Wang CH, Deng SK, Wu YD, Li Y, Yao L, Jiang JD, Yan X, He J, Li SP. 2014. Novel three-component Rieske non-heme iron oxygenase system catalyzing the N-dealkylation of chloroacetanilide herbicides in sphingomonads DC-6 and DC-2. Appl Environ Microbiol 80:5078–5085. doi: 10.1128/AEM.00659-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Xiao N, Jing B, Ge F, Liu X. 2006. The fate of herbicide acetochlor and its toxicity to Eisenia fetida under laboratory conditions. Chemosphere 62:1366–1373. doi: 10.1016/j.chemosphere.2005.07.043. [DOI] [PubMed] [Google Scholar]
- 5.Dearfield KL, McCarroll NE, Protzel A, Stack HF, Jackson MA, Waters MD. 1999. A survey of EPA/OPP and open literature on selected pesticide chemicals: II. Mutagenicity and carcinogenicity of selected chloroacetanilides and related compounds. Mutat Res 443:183–221. [DOI] [PubMed] [Google Scholar]
- 6.Coleman S, Linderman R, Hodgson E, Rose RL. 2000. Comparative metabolism of chloroacetamide herbicides and selected metabolites in human and rat liver microsomes. Environ Health Perspect 108:1151–1157. doi: 10.2307/3434827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhu JS, Qiao XW, Wang J, Qin S. 2004. Degradation and the influencing factors of acetochlor in soils. J Agro-Environ Sci 23:1025–1029. [Google Scholar]
- 8.Souissi Y, Bourcier S, Ait-Aissa S, Maillot-Maréchal E, Bouchonnet S, Genty C, Sablier M. 2013. Using mass spectrometry to highlight structures of degradation compounds obtained by photolysis of chloroacetamides: case of acetochlor. J Chromatogr A 1310:98–112. doi: 10.1016/j.chroma.2013.07.091. [DOI] [PubMed] [Google Scholar]
- 9.Zhang J, Zheng JW, Liang B, Wang CH, Cai S, Ni YY, He J, Li SP. 2011. Biodegradation of chloroacetanilide herbicides by Paracoccus sp. FLY-8 in vitro. J Agric Food Chem 59:4614–4621. doi: 10.1021/jf104695g. [DOI] [PubMed] [Google Scholar]
- 10.Li Y, Chen Q, Wang CH, Cai S, He J, Huang X, Li SP. 2013. Degradation of acetochlor by consortium of two bacterial strains and cloning of a novel amidase gene involved in acetochlor-degrading pathway. Bioresour Technol 148:628–631. doi: 10.1016/j.biortech.2013.09.038. [DOI] [PubMed] [Google Scholar]
- 11.Wang F, Zhou J, Li Z, Dong W, Hou Y, Huang Y, Cui Z. 2015. Involvement of the cytochrome P450 system EthBAD in the N-deethoxymethylation of acetochlor by Rhodococcus sp. strain T3-1. Appl Environ Microbiol 81:2182–2188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang F, Hou Y, Zhou J, Li Z, Huang Y, Cui Z. 2014. Purification of an amide hydrolase DamH from Delftia sp. T3-6 and its gene cloning, expression, and biochemical characterization. Appl Microbiol Biotechnol 98:7491–7499. [DOI] [PubMed] [Google Scholar]
- 13.Boonrattanakij N, Lu MC, Anotai J. 2009. Kinetics and mechanism of 2,6-dimethyl-aniline degradation by hydroxyl radicals. J Hazard Mater 172:952–957. doi: 10.1016/j.jhazmat.2009.07.079. [DOI] [PubMed] [Google Scholar]
- 14.Chao MW, Kim MY, Ye W, Ge J, Trudel LJ, Belanger C, Wogan GN. 2012. Genotoxicity of 2,6- and 3,5-dimethylaniline in cultured mammalian cells: the role of reactive oxygen species. Toxicol Sci 130:48–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sambrook J, Russell DW. 2001. Molecular cloning: a laboratory manual, 3rd ed Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 16.Endo R, Kamakura M, Miyauchi K, Fukuda M, Ohtsubo Y, Tsuda M, Nagata Y. 2005. Identification and characterization of genes involved in the downstream degradation pathway of γ-hexachlorocyclohexane in Sphingomonas paucimobilis UT26. J Bacteriol 187:847–853. doi: 10.1128/JB.187.3.847-853.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.de Bruijn FJ. 1992. Use of repetitive (repetitive extragenic palindromic and enterobacterial repetitive intergeneric consensus) sequences and the polymerase chain reaction to fingerprint the genomes of Rhizobium meliloti isolates and other soil bacteria. Appl Environ Microbiol 58:2180–2187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.O'Sullivan DJ, Klaenhammer TR. 1993. Rapid mini-prep isolation of high-quality plasmid DNA from Lactococcus and Lactobacillus spp. Appl Environ Microbiol 59:2730–2733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kaufmann ME. 1998. Pulsed-field gel electrophoresis. Mol Bacteriol 15:33–50. doi: 10.1385/0-89603-498-4:33. [DOI] [PubMed] [Google Scholar]
- 20.Castoe TA, Poole AW, Gu W, Jason de Koning AP, Daza JM, Smith EN, Pollock DD. 2010. Rapid identification of thousands of copperhead snake (Agkistrodon contortrix) microsatellite loci from modest amounts of 454 shotgun genome sequence. Mol Ecol Resour 10:341–347. doi: 10.1111/j.1755-0998.2009.02750.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ansorge WJ. 2009. Next-generation DNA sequencing techniques. New Biotechnol 25:195–203. doi: 10.1016/j.nbt.2008.12.009. [DOI] [PubMed] [Google Scholar]
- 22.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. 1990. Basic Local Alignment Search Tool. J Mol Biol 215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 23.Tang B, Wang Q, Yang M, Xie F, Zhu Y, Zhuo Y, Wang S, Gao H, Ding X, Zhang L, Zhao G, Zheng H. 2013. ContigScape: a Cytoscape plugin facilitating microbial genome gap closing. BMC Genomics 14:289. doi: 10.1186/1471-2164-14-289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang S, He J, Cui Z, Li S. 2007. Self-formed adaptor PCR: a simple and efficient method for chromosome walking. Appl Environ Microbiol 73:5048–5051. doi: 10.1128/AEM.02973-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ramagli LS. 1999. Quantifying protein in 2-D PAGE solubilization buffers. Methods Mol Biol 112:99–103. [DOI] [PubMed] [Google Scholar]
- 26.Song Y, Cui J, Zhang H, Wang G, Zhao FJ, Shen Z. 2013. Proteomic analysis of copper stress responses in the roots of two rice (Oryza sativa L.) varieties differing in Cu tolerance. Plant Soil 366:647–658. doi: 10.1007/s11104-012-1458-2. [DOI] [Google Scholar]
- 27.Yan JX, Wait R, Berkelman T, Harry RA, Westbrook JA, Wheeler CH, Dunn MJ. 2000. A modified silver staining protocol for visualization of proteins compatible with matrix-assisted laser desorption/ionization and electrospray ionization-mass spectrometry. Electrophoresis 21:3666–3672. doi:. [DOI] [PubMed] [Google Scholar]
- 28.Larkin M, Blackshields G, Brown N, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG. 2007. Clustal W and Clustal X version 2.0. Bioinformatics 23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- 29.Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. 2011. MEGA5: Molecular Evolutionary Genetics Analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28:2731–2739. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kovach ME, Elzer PH, Hill DS, Robertson GT, Farris MA, Roop RM Jr, Peterson KM. 1995. Four new derivatives of the broad-host-range cloning vector pBBR1MCS, carrying different antibiotic-resistance cassettes. Gene 166:175–176. doi: 10.1016/0378-1119(95)00584-1. [DOI] [PubMed] [Google Scholar]
- 31.Martínez-García E, Jatsenko T, Kivisaar M, de Lorenzo V. 2014. Freeing Pseudomonas putida KT2440 of its proviral load strengthens endurance to environmental stresses. Environ Microbiol 17:76–90. [DOI] [PubMed] [Google Scholar]
- 32.Li J, Huang Y, Hou Y, Li X, Cao H, Cui Z. 2013. Novel gene clusters and metabolic pathway involved in 3,5,6-trichloro-2-pyridinol degradation by Ralstonia sp. strain T6. Appl Environ Microbiol 79:7445–7453. doi: 10.1128/AEM.01817-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Glazebrook J, Walker GC. 1991. Genetic techniques in Rhizobium meliloti. Methods Enzymol 204:398–418. doi: 10.1016/0076-6879(91)04021-F. [DOI] [PubMed] [Google Scholar]
- 34.Liu H, Wang SJ, Zhang JJ, Dai H, Tang H, Zhou NY. 2011. Patchwork assembly of nag-like nitroarene dioxygenase genes and the 3-chlorocatechol degradation cluster for evolution of the 2-chloronitrobenzene catabolism pathway in Pseudomonas stutzeri ZWLR2-1. Appl Environ Microbiol 77:4547–4552. doi: 10.1128/AEM.02543-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sasaki M, Maki JI, Oshiman KI, Matsumura Y, Tsuchido T. 2005. Biodegradation of bisphenol A by cells and cell lysate from Sphingomonas sp. strain AO1. Biodegradation 16:449–459. doi: 10.1007/s10532-004-5023-4. [DOI] [PubMed] [Google Scholar]
- 36.Lin YM, Li LY, Hu JW, Huang XF, Zhou C, Jia M, Li ZB. 2014. Photometric determination of ammonia nitrogen in slaughterhouse wastewater with Nessler's reagent: effects of different pretreatment methods. Adv Mater 955:1241–1244. [Google Scholar]
- 37.Gillings M, Holley M. 1997. Repetitive element PCR fingerprinting (rep-PCR) using enterobacterial repetitive intergenic consensus (ERIC) primers is not necessarily directed at ERIC elements. Lett Appl Microbiol 25:17–21. doi: 10.1046/j.1472-765X.1997.00162.x. [DOI] [PubMed] [Google Scholar]
- 38.Zhang T, Zhang J, Liu S, Liu Z. 2008. A novel and complete gene cluster involved in the degradation of aniline by Delftia sp. AN3. J Environ Sci 20:717–724. doi: 10.1016/S1001-0742(08)62118-X. [DOI] [PubMed] [Google Scholar]
- 39.Gai Z, Wang X, Liu X, Tai C, Tang H, He X, Xu P. 2010. The genes coding for the conversion of carbazole to catechol are flanked by IS6100 elements in Sphingomonas sp. strain XLDN2-5. PLoS One 5:e10018. doi: 10.1371/journal.pone.0010018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Thotsaporn K, Sucharitakul J, Wongratana J, Suadee C, Chaiyen P. 2004. Cloning and expression of p-hydroxyphenylacetate 3-hydroxylase from Acinetobacter baumannii: evidence of the divergence of enzymes in the class of two-protein component aromatic hydroxylases. Biochim Biophys Acta 1680:60–66. doi: 10.1016/j.bbaexp.2004.08.003. [DOI] [PubMed] [Google Scholar]
- 41.Van der Geize R, Yam K, Heuser T, Wilbrink MH, Hara H, Anderton MC, Sim E, Dijkhuizen L, Davies JE, Mohn WW, Eltis LD. 2007. A gene cluster encoding cholesterol catabolism in a soil actinomycete provides insight into Mycobacterium tuberculosis survival in macrophages. Proc Natl Acad Sci U S A 104:1947–1952. doi: 10.1073/pnas.0605728104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Brünker P, Sterner O, Bailey JE, Minas W. 2001. Heterologous expression of the naphthocyclinone hydroxylase gene from Streptomyces arenae for production of novel hybrid polyketides. Antonie Van Leeuwenhoek 79:235–245. doi: 10.1023/A:1012037329949. [DOI] [PubMed] [Google Scholar]
- 43.Galán B, Díaz E, Prieto MA, García JL. 2000. Functional analysis of the small component of the 4-hydroxyphenylacetate 3-hydroxylase of Escherichia coli W: a prototype of a new flavin: NAD(P)H reductase subfamily. J Bacteriol 182:627–636. doi: 10.1128/JB.182.3.627-636.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Thompson JD, Higgins DG, Gibson TJ. 1994. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gu T, Zhou C, Sørensen SR, Zhang J, He J, Yu P, Yan X, Li S. 2013. The novel bacterial N-demethylase PdmAB is responsible for the initial step of N,N-dimethyl-substituted phenylurea herbicide degradation. Appl Environ Microbiol 79:7846–7856. doi: 10.1128/AEM.02478-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Levenberg B, Hayaishi O. 1959. A bacterial pterin deaminase. J Biol Chem 234:955–961. [PubMed] [Google Scholar]
- 47.Goszczynski S, Paszczynski A, Pasti-Grigsby MB, Crawford RL, Crawford DL. 1994. New pathway for degradation of sulfonated azo dyes by microbial peroxidases of Phanerochaete chrysosporium and Streptomyces chromofuscus. J Bacteriol 176:1339–1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ewers J, Rubio MA, Knackmuss HJ, Freier-Schröder D. 1989. Bacterial metabolism of 2,6-xylenol. Appl Environ Microbiol 55:2904–2908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hofrichter M, Bublitz F, Fritsche W. 1995. Cometabolic degradation of o-cresol and 2,6-dimethylphenol by Penicillium frequentans Bi7/2. J Basic Microbiol 35:303–313. doi: 10.1002/jobm.3620350505. [DOI] [PubMed] [Google Scholar]
- 50.McIntire WS, Bohmont C. 1987. The chemical and stereochemical course of oxidation of 4-ethylphenol and other 4-alkylphenols by p-cresol methylhydroxylase, p 677–686. In Edmondson DE, McCormick DB (ed), Flavins and flavoproteins IX. Walter de Gruyter, Berlin, Germany. [Google Scholar]
- 51.Reeve CD, Carver MA, Hopper DJ. 1990. Stereochemical aspects of the oxidation of 4-ethylphenol by the bacterial enzyme 4-ethylphenol methylenehydroxylase. Biochem J 269:815–819. doi: 10.1042/bj2690815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Alfieri A, Fersini F, Ruangchan N, Prongjit M, Chaiyen P, Mattevi A. 2007. Structure of the monooxygenase component of a two-component flavoprotein monooxygenase. Proc Natl Acad Sci U S A 104:1177–1182. doi: 10.1073/pnas.0608381104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Miyazaki R, Sato Y, Ito M, Ohtsubo Y, Nagata Y, Tsuda M. 2006. Complete nucleotide sequence of an exogenously isolated plasmid, pLB1, involved in γ-hexachlorocyclohexane degradation. Appl Environ Microbiol 72:6923–6933. doi: 10.1128/AEM.01531-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Feng X, Ou LT, Ogram A. 1997. Plasmid-mediated mineralization of carbofuran by Sphingomonas sp. strain CF06. Appl Environ Microbiol 63:1332–1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shintani M, Urata M, Inoue K, Eto K, Habe H, Omori T, Yamane H, Nojiri H. 2007. The Sphingomonas plasmid pCAR3 is involved in complete mineralization of carbazole. J Bacteriol 189:2007–2020. doi: 10.1128/JB.01486-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Stolz A. 2014. Degradative plasmids from sphingomonads. FEMS Microbiol Lett 350:9–19. doi: 10.1111/1574-6968.12283. [DOI] [PubMed] [Google Scholar]
- 57.Liang B, Wang G, Zhao Y, Chen K, Li S, Jiang J. 2011. Facilitation of bacterial adaptation to chlorothalonil-contaminated sites by horizontal transfer of the chlorothalonil hydrolytic dehalogenase gene. Appl Environ Microbiol 77:4268–4272. doi: 10.1128/AEM.02457-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wang C, Chen Q, Wang R, Shi C, Yan X, He J, Hong Q, Li S. 2014. A novel angular dioxygenase gene cluster encoding 3-phenoxybenzoate 1′,2′-dioxygenase in Sphingobium wenxiniae JZ-1. Appl Environ Microbiol 80:3811–3818. doi: 10.1128/AEM.00208-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Takeo M, Maeda Y, Maeda J, Nishiyama N, Kitamura C, Kato DI, Negoro S. 2012. Two identical nonylphenol monooxygenase genes linked to IS6100 and some putative insertion sequence elements in Sphingomonas sp. NP5. Microbiology 158:1796–1807. doi: 10.1099/mic.0.055335-0. [DOI] [PubMed] [Google Scholar]
- 60.Dogra C, Raina V, Pal R, Suar M, Lal S, Gartemann KH, Holliger C, van der Meer JR, Lal R. 2004. Organization of lin genes and IS6100 among different strains of hexachlorocyclohexane-degrading Sphingomonas paucimobilis: evidence for horizontal gene transfer. J Bacteriol 186:2225–2235. doi: 10.1128/JB.186.8.2225-2235.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.