

ABSTRACT
The 4E10 antibody recognizes the membrane-proximal external region (MPER) of the HIV-1 Env glycoprotein gp41 transmembrane subunit, exhibiting one of the broadest neutralizing activities known to date. The neutralizing activity of 4E10 requires solvent-exposed hydrophobic residues at the apex of the complementarity-determining region (CDR) H3 loop, but the molecular basis for this requirement has not been clarified. Here, we report the cocrystal structures and the energetic parameters of binding of a peptide bearing the 4E10-epitope sequence (4E10ep) to nonneutralizing versions of the 4E10 Fab. Nonneutralizing Fabs were obtained by shortening and decreasing the hydrophobicity of the CDR-H3 loop (termed ΔLoop) or by substituting the two tryptophan residues of the CDR-H3 apex with Asp residues (termed WDWD), which also decreases hydrophobicity but preserves the length of the loop. The analysis was complemented by the first crystal structure of the 4E10 Fab in its ligand-free state. Collectively, the data ruled out major conformational changes of CDR-H3 at any stage during the binding process (equilibrium or transition state). Although these mutations did not impact the affinity of wild-type Fab for the 4E10ep in solution, the two nonneutralizing versions of 4E10 were deficient in binding to MPER inserted in the plasma membrane (mimicking the environment faced by the antibody in vivo). The conclusions of our structure-function analysis strengthen the idea that to exert effective neutralization, the hydrophobic apex of the solvent-exposed CDR-H3 loop must recognize an antigenic structure more complex than just the linear α-helical epitope and likely constrained by the viral membrane lipids.
IMPORTANCE The broadly neutralizing anti-HIV-1 4E10 antibody blocks infection caused by nearly all viral strains and isolates examined thus far. However, 4E10 (or 4E10-like) antibodies are rarely found in HIV-1-infected individuals or elicited through vaccination. Impediments to the design of successful 4E10 immunogens are partly attributed to an incomplete understanding of the structural and binding characteristics of this class of antibodies. Since the broadly neutralizing activity of 4E10 is abrogated by mutations of the tip of the CDR-H3, we investigated their impact on binding of the MPER-epitope at the atomic and energetic levels. We conclude that the difference between neutralizing and nonneutralizing antibodies of 4E10 is neither structural nor energetic but is related to the capacity to recognize the HIV-1 gp41 epitope inserted in biological membranes. Our findings strengthen the idea that to elicit similar neutralizing antibodies, the suitable MPER vaccine must be “delivered” in a membrane environment.
INTRODUCTION
The conserved membrane-proximal external region (MPER) domain has been proposed to function in gp41-mediated HIV-1 membrane fusion by perturbing the lipid packing of the viral envelope (1–3). This region is critical for viral entry and is highly conserved, as evidenced by the monoclonal antibody (MAb) 4E10, which binds to the MPER C-terminal helix and neutralizes up to 98% of global HIV-1 strains (4–6). Due to its exceptional neutralization breath, 4E10 has served as a model of broad protection against HIV infection, and consequently has been the subject of extensive structural and functional characterization, particularly for the purpose of vaccine design (reviewed in references 6 to 9).
The crystal structure of the 4E10 Fab in complex with a 13-residue-long peptide containing the gp41 core epitope 671NWFDIT676 was first determined by the Wilson group (10). The residues of this peptide in contact with the Fab (Trp-672, Phe-673, Ile-675, and Thr-676) all gathered at one face of the helix contacting the Fab. A second crystal structure of Fab 4E10 bound to an extended peptide epitope 671NWFDITNWLWYIK683-KKK was subsequently reported (11). This peptide (termed 4E10ep in the present study) displays an affinity 3 orders of magnitude higher than that of the shorter version (12). The enhanced affinity of the longer peptide is due to additional interactions between the extra helical turn of 4E10ep (Leu-679 and Trp-680) and the antibody. Based on the structural data available, the 4E10 epitope was lengthened to comprise the motif 672WFX(I/L)(T/S)XX(L/I)W680 with residues at Env positions 672, 673, 675, 676, 679, and 680, located on the hydrophobic face of the amphipathic helix comprising the “neutralizing face” of the epitope. These conclusions were reinforced when stabilization of the helical structure at the nonbinding face by covalently “stapling” residues 678 and 681 was shown to further enhance peptide affinity (13).
It has been proposed that the hydrophobic complementarity-determining region–H3 (CDR-H3) loop of 4E10 interacts with lipids of the viral envelope (10). This hypothesis was further elaborated after observing that binding of Fab and single-chain variable fragment of 4E10 to liposomes composed of viral lipids is governed by the hydrophobicity of its unusually long and highly flexible CDR-H3 loop (3, 14, 15). In particular, Scherer et al. (14) found a direct correlation between lipid binding and neutralizing activity for 4E10 IgGs bearing substituted Trp residues at the apex of the CDR-H3. For example, the double mutation WH100D/WH100BD (termed WDWD) exhibits poor affinity for lipids and undetectable activity in standardized viral neutralization assays. These authors concluded that hydrophobic residues of the apex region of CDR-H3 are required for MPER recognition through favorable lipid interactions in the context of the viral envelope.
In crystal structures of 4E10 Fab-peptide complexes, the long CDR-H3 loop required for neutralization extends beyond the bound helical peptide in an orientation that suggests its apex could contact the viral membrane and contribute to neutralization (10, 11). Using a recombinant Fv fragment (composed of VH + VL), it was shown that mutating a Trp residue of the apex does not change the binding mode of a scaffold protein containing the sequence of the core epitope peptide (16). Later, studies using the same Fv fragment suggested that the 4E10 paratope is flexible and undergoes a large restructuring process upon binding to peptide (17, 18). Specifically, this conformational change would involve a shift of ca. 12 Å of the CDR-H3 loop and especially of the apex segment (17). The possibility that the loop undergoes significant restructuring during peptide binding implies that mutations that abolish neutralization may alter the CDR-H3 conformational equilibrium and, through this mechanism, hamper peptide recognition.
Recently, a potential lipid-binding site has been described comprising residues of the CDR-H1 loop of the 4E10 Fab-peptide complex (13). This subsite is occupied by a phosphate ion under some crystallization conditions, suggestive of favorable noncovalent interactions between the antibody and the phospholipid head groups at the membrane interface. The neighboring CDR-H1 and CDR-H3 loops establish extensive interactions in the Fab structure, and therefore mutations blocking neutralization may also influence the lipid-binding pocket. Structural and mutagenesis studies addressing this potential binding site will be necessary to evaluate its functional role during viral neutralization by 4E10.
To determine the differences between neutralizing and nonneutralizing 4E10 Fabs at the molecular level, we have determined high-resolution crystal structures of two nonneutralizing Fabs bearing mutations in the CDR-H3 loops. The overall structure of the nonneutralizing antibodies and those of the wild-type (WT) Fab-peptide complexes previously reported (10, 11, 19) are nearly indistinguishable, except for some differences in the conformation of the CDR-H3 apex. Following strategies delineated in our previous studies, the structural data were complemented with a detailed kinetic and thermodynamic analysis of the 4E10ep binding mechanism, not only at equilibrium but also at the transition state (20, 21). We show that neutralizing and nonneutralizing Fabs recognize the helical 4E10ep by means of nearly identical mechanisms at both the energetic and the atomic levels. In particular, the novel crystal structure of the ligand-free Fab indicates that the CDR-H3 loop does not undergo significant conformational changes during recognition of the epitope. Comparison of the structures of Fab in the bound and free states revealed loss of flexibility in the complementarity-determining region (CDR) loops during peptide engagement, except for the apex region of the CDR-H3, which remained relatively flexible. In contrast to the common structural and energetic mechanism describing the binding of neutralizing and nonneutralizing Fab to 4E10ep in solution, the nonneutralizing forms of 4E10 Fab were defective in binding to an MPER that was anchored to the plasma membrane via the gp41 transmembrane region (22), which was otherwise efficiently recognized by the neutralizing WT Fab. Collectively, our data support the notion that the hydrophobic CDR-H3 apex governs recognition of the MPER epitope constrained in a membrane context, a process required for efficient neutralization of the virus.
MATERIALS AND METHODS
Materials.
The peptides used in the structural and affinity studies were synthesized in C-terminal carboxamide form by solid-phase methods using Fmoc (9-fluorenylmethoxy carbonyl) chemistry, purified by reversed-phase high-pressure liquid chromatography, and characterized by matrix-assisted time-of-flight mass spectrometry (purity > 95%). Peptides were routinely dissolved in dimethyl sulfoxide (spectroscopy grade), and their concentrations were determined by a bicinchoninic acid microassay (Pierce, Rockford, IL). The peptide, 4E10ep (NH3+-NWFDITNWLWYIK-KKK-COOH), bears residues 671 to 683 of HIV strain HXB2 Env. The following reagents were obtained through the National Institutes of Health (NIH) AIDS Reagent Program, Division of AIDS, DAIDS, NIAID, NIH: IgG1 MAb, and 4E10, from H. Katinger (Polymun Scientific GmbH, Vienna, Austria); pNL4-3.Luc.R-.E- from N. Landau (Aaron Diamond AIDS Research Center, The Rockefeller University); TZM-bl cells from J. C. Kappes, X. Wu, and Tranzyme Inc.; and plasmids encoding Env from the HIV-1 isolates: SS1196.1 clone 1 (SVPB9) from D. Montefiori and F. Gao (Duke University Medical Center), Bal.26 from J. Mascola (Vaccine Research Center, NIH), and JR-CSF from I. Chen (David Geffen School of Medicine, University of California Los Angeles) and Y. Koyanagi (Institute for Virus Research, Kyoto University). A plasmid encoding the engineered HIV-1 Env, SF162.LS, was kindly provided by N. Haigwood (Oregon National Primate Center, Beaverton, OR), and a plasmid encoding the VSV-G Env was provided by Patricia Villace (CSIC, Madrid, Spain). A hybridoma cell line producing the murine MAb 17/9, which recognizes the hemagglutinin (HA) tag sequence, DVPDYA, was kindly provided by R. Stanfield and I. A. Wilson (The Scripps Research Institute, La Jolla, CA); this IgG1 MAb was produced and purified from ascites fluid by Maine Biotechnology Services, Inc. (Portland, ME). Recombinant gp41 (r-gp41; HIV-1MN isolate) was purchased from ImmunoDiagnostics (Woburn, MA). A peptide bearing the MPER sequence residues 667 to 683 (MPER667-683; HXB2 numbering) from HIV strain JR-CSF gp41 [NH3+-ASLWNFDITKWLWYIK-SGK-(biotin)-CONH2] was purchased from United Biosystems (Herndon, VA) at >95% purity. Goat anti-human IgG-Fab antibody was purchased from Sigma (St. Louis, MO). Secondary antibodies conjugated to horseradish peroxidase (HRP), goat (anti-human kappa light-chain)-HRP, goat (anti-human IgG)-HRP, and goat (anti-murine IgG)-HRP, were purchased from Life Technologies (Carlsbad, CA). Experimental procedures described elsewhere (3, 23) were followed for the production and purification of Fabs.
Crystallization of ligand-free Fab and Fab-peptide complexes.
Crystals of unbound WT 4E10 Fab, as well as crystals of WDWD or ΔLoop in complex with 4E10ep (NH3+-NWFDITNWLWYIK-KKK-COOH) bearing residues 671 to 683 of HIV strain HXB2 Env, were prepared by the hanging-drop method. The crystals of unbound WT Fab were obtained by mixing 0.5 μl of protein at 3 mg/ml with 0.5 μl of a solution containing 15% (wt/vol) PEG 20000 and 100 mM MES (morpholineethanesulfonic acid; pH 6.5). To obtain crystals of nonneutralizing Fabs in complex with peptide, antibody and epitope were first mixed at an approximate molar ratio of 1:1.3 (protein-peptide) and subsequently concentrated to 3 mg/ml. Suitable crystals of WDWD-4E10ep were obtained by mixing 2 μl of the solution containing protein and peptide with 2 μl of a solution composed of 200 mM ammonium acetate, 34% PEG 8000, and 100 mM Tris-HCl (pH 8.4). Similarly, cocrystals of ΔLoop Fab with epitope bound were obtained by mixing the complex with a solution composed of 200 mM ammonium acetate, 30% PEG 8000, and 100 mM Tris-HCl (pH 8.5). In both cases crystals grew to full size within a few days at 20°C. Suitable crystals were identified, harvested and briefly transferred to mother liquor supplemented with 25% (vol/vol) ethyleneglycol (unbound Fab) or 15% (vol/vol) glycerol (WDWD and ΔLoop Fabs in complex with 4E10ep), and then frozen and stored in a vessel containing liquid N2.
Data collection and structure refinement.
Diffraction data from a single crystal of the unbound Fab were collected using Beamline I02 of the Diamond Light Source (Oxfordshire, United Kingdom) under cryogenic conditions (100 K). Diffraction data of the WDWD and ΔLoop Fabs in complex with 4E10ep were collected in Beamline BL5A of the Photon Factory (Tsukuba, Japan) also under cryogenic conditions (100 K). Diffraction images were processed with the program MOSFLM and merged and scaled with the program SCALA of the CCP4 suite (24). The structures were determined by the method of molecular replacement using the coordinates of recombinant WT Fab previously determined by us (19) with the program PHASER (25). All models were refined with the program REFMAC5 (26) and manually built with COOT (27). The crystal of unbound WT Fab was partially twinned (ratio 70/30) as determined with the program REFMAC5 during the refinement stage. Validation was carried out with PROCHECK (28). Because of the weak electron density in the apex of the CDR-H3 (100WGWLG100D) in the crystal structure of unbound 4E10, additional validation steps were taken to rigorously evaluate the geometry of the residues modeled in this region. First, we verified no geometrical violations were found among the residues comprising the apex with the validation tools of CCP4 (29), PHENIX (30), and the PDB (31) with respect to bond lengths, bond angles, dihedral angles, chirality restrains, root mean square distances from planarity, psi-phi angles, rotamers, Cβ deviations, and geometrical clashes (including symmetry-related clashes). Second, we analyzed the top five automatic conformations of residues of the apex built by the program ARP-WARP (32). Although these conformations were built automatically (no human intervention), they showed great similarity to the conformation built by us (root mean square deviation [RMSD]-Cα = 0.8 ± 0.2). Third, the program RINGER was used to look into the conformational variability of the side chains of residues WH100, WH100B, and LH100D of the apex (GH100A and GH100D were not analyzed because they do not possess side chains). The individual propensity plots of each residue indicate that the side chains adopt a unique conformation above the standard electron density threshold value of 0.3 σ (33–35) (see Fig. S1 in the supplemental material). Collectively, the exhaustive validation procedure described above demonstrates that, although the region of the apex of CDR-H3 of the unbound 4E10 is highly dynamic (a general feature of the CDR-H3 apex of 4E10; see Fig. S1b in the supplemental material), the conformation modeled has adequate geometry and fits reasonably well the otherwise weak electron density present in that region. (Data collection and structure refinement statistics are given in Table 2.)
TABLE 2.
Data collection and refinement statisticsa
| Parameterb | ΔLoop + 4E10ep | WDWD + 4E10ep | WT unbound |
|---|---|---|---|
| Data collection | |||
| Space group | C2 | C2 | P21 |
| Unit cell | |||
| a, b, c (Å) | 157.5, 44.7, 85.3 | 157.3, 44.9, 86.2 | 44.7, 134.2, 81.9 |
| α, β, γ (°) | 90.0, 113.6, 90.0 | 90.0, 114.5, 90.0 | 90.0, 105.8, 90.0 |
| Resolution (Å) | 36.1–1.70 | 44.5–1.81 | 44.7–2.48 |
| Wavelength | 1.000 | 1.000 | 1.000 |
| No. of observations | 313,934 (31,932) | 265,738 (37,906) | 79,271 (11,742) |
| No. of unique reflections | 59,239 (7,703) | 50,189 (7,203) | 32,268 (4,692) |
| Rmerge (%) | 8.8 (39.1) | 9.9 (66.7) | 7.2 (21.5) |
| Rp.i.m. (%) | 4.2 (21.0) | 4.6 (31.1) | 5.6 (16.4) |
| CC1/2 | 1.00 (0.87) | 1.00 (0.78) | 0.99 (0.93) |
| I/σ | 12.5 (3.3) | 11.9 (2.8) | 9.7 (4.2) |
| Multiplicity | 5.3 (4.1) | 5.3 (5.3) | 2.5 (2.5) |
| Completeness (%) | 98.3 (88.5) | 99.7 (99.1) | 98.4 (98.7) |
| B-factor (Wilson plot) (Å2) | 11.8 | 16.4 | 17.1 |
| Refinement statistics | |||
| Resolution (Å) | 36.1–1.70 | 44.5–1.81 | 44.7–2.48 |
| Reflections (Rwork/Rfree) | 56,860/2,379 | 48,180/2,007 | 30,609/1,632 |
| Rwork/Rfree (%) | 14.7/17.8 | 16.3/18.8 | 18.3/23.1 |
| No. of atoms | |||
| Protein | 3,336 | 3,346 | 6,541 |
| Peptide | 159 | 129 | |
| Solvent | 590 | 412 | 135 |
| Other | 25 | 18 | |
| B-factor (Å2) | |||
| Protein | 24.7 | 27.7 | 25.1 |
| Peptide | 31.9 | 44.8 | |
| Solvent | 35.4 | 33.0 | 20.8 |
| Other | 34.9 | 36.7 | |
| Ramachandran plot (%) | |||
| Preferred | 89.9 | 91.2 | 88.7 |
| Allowed | 9.8 | 8.5 | 11.0 |
| Outliers | 0.3 | 0.3 | 0.3 |
| RMSD bond (Å) | 0.013 | 0.015 | 0.008 |
| RMSD angle (°) | 1.53 | 1.63 | 1.27 |
| Coordinate error (Å) | 0.08 | 0.10 | 0.13 |
| PDB entry code | 5CIN | 5CIL | 5CIP |
Statistical values given in parentheses refer to the highest-resolution bin.
Rmerge = Σhkl Σi |I(hkl)i − I(hkl)|/Σhkl Σi I(hkl); Rwork = Σhkl|F(hkl)o− [F(hkl)c]|/Σhkl F(hkl)o. Rfree was calculated as Rwork, where F(hkl)o values were taken from ca. 4 to 5% of data not included in the refinement. RMSD, root mean square deviation.
Differential scanning calorimetry (DSC).
The heat capacity of individual Fabs was measured using a VP-DSC system (MicroCal, Northampton, MA). All calorimetric scans were performed in a buffer composed of 10 mM NaH2PO4 (pH 7.5), 150 mM NaCl, and 10% glycerol. Protein samples at 10 μM were heated from 30 to 90°C at a scanning rate of 1°C min−1. The ORIGIN software package (MicroCal) was used for data collection and analysis. The buffer baseline was subtracted from the raw data, which were normalized by protein concentration to obtain thermodynamic parameters and then fitted to a to a non-two-state model.
SPR.
Temperature dependence surface plasmon resonance (SPR) experiments were performed on a Biacore T200 instrument (GE Healthcare). 4E10 Fabs, WT, WDWD, and ΔLoop were dialyzed against running buffer (10 mM HEPES-NaOH [pH 7.5], 150 mM NaCl, 3 mM EDTA, 0.1% [vol/vol] Tween 20). An antibody-capture method was used to obtain the binding response of the 4E10ep-Fab interaction. An anti-human IgG-Fab secondary antibody was immobilized on a Biacore CM5 chip (GE Healthcare) to surface densities of ∼15,000 response units (RU). For that purpose, the chip was first activated with N-hydroxysuccinimide and N-ethyl-N′-(3-dimethylaminopropyl) carbodiimide hydrochloride as previously described (20, 21, 36). After immobilizing the anti-human Fab antibody, the remaining active groups on the surface of the chip were blocked by flowing 100 µl of 1 M ethanolamine. The Fab variants were subsequently captured to surface densities of ∼1,400 RU. For this experiment, the lysine residues of the standard 4E10ep were substituted for Arg as previously described (NWFDITNWLWYIR683RRR) (4). This peptide was flowed over the immobilized Fabs at 30 μl/min in increasing concentrations from 7.8 to 500 nM. The association and dissociation phases were 120 and 200 s, respectively. Regeneration was achieved after completion of each sensorgram by injecting a solution of 10 mM glycine-HCl (pH 1.5) for 30 s at a flow rate of 30 μl/min. Association (kon) and dissociation (koff) rate constants were determined with the Biacore T200 evaluation software (GE Healthcare). The dissociation constant (KD) was calculated from the ratio of the rate constants (KD = koff/kon). Changes in enthalpy (ΔHvH) and entropy (ΔSvH) were calculated from the slope and intercept, respectively, of the temperature dependence of the dissociation constant using the van't Hoff approximation (36): lnKD = ΔHvH/RT + ΔSvH/R, where R is the gas constant and T is the absolute temperature. The activation energy parameters were obtained from the temperature dependence of the kinetic rate constant according to the Eyring equation (37): ln(kon/T) = −(ΔH‡/RT) ± (ΔS‡/R) + ln(kB/h), where kon is the kinetic rate constant, ΔH‡ is the activation enthalpy, R is the gas constant, T is the absolute temperature, ΔS‡ is the activation entropy, kB is the Boltzmann's constant, and h the Plank's constant.
Cell lysate production.
According to our previously described protocol (22), 293T cells were transiently transfected with 1 μg of plasmid DNA encoding recombinant MPER proteins, MPER-TM1 and MPER-PGDFR (described below and in reference 22), using the XtremeGENE 9 transfection reagent (Roche, Basel, Switzerland), according to the manufacturer's instructions, at a ratio of 1:6 (i.e., μg of DNA to μl of transfection reagent). Cells were cultured in six-well plates (Sarstedt, Numbrecht, Germany) in Dulbecco's modified Eagle medium (DMEM; Life Technologies) supplemented with 10% (vol/vol) fetal calf serum (FCS; Life Technologies) and 1 mM l-glutamine (Life Technologies) at 37°C and 5% CO2. After 48 h, the cells were washed four times in phosphate-buffered saline (PBS; Life Technologies) and recovered from the plate with 1 mM Na2EDTA-NaOH (pH 8.0) (Bioshop, Burlington, Ontario, Canada). Cells were pelleted by centrifugation for 5 min at 350 × g, resuspended in 200 μl of lysis buffer comprising a protease inhibitor cocktail (cOmplete ULTRA tablets, mini, EDTA-Free; Roche) diluted 1:12 in 250 mM sucrose and 0.5 mM Na2EDTA-NaOH (pH 8.0). Cell lysates were produced by 30 passages through a 22-gauge needle, followed by two 15-s pulses with a Virsonic sonicator (VirTis, Gardiner, NY). The total protein content was quantified using Bradford reagent (Bio-Rad protein assay; Bio-Rad, Mississauga, Canada) according to the manufacturer's instructions; lysates were stored at −80°C.
Enzyme-linked immunosorbent assays (ELISAs).
Wells of high-binding microtiter plates (Corning Inc., Corning, NY) were coated overnight at 4°C with 35 μl/well of one of the following diluted in Tris-buffered saline (TBS; 50 mM Tris-HCl [pH 7.5], 150 mM NaCl): (i) cell lysate (the equivalent of 10 μg of protein), (ii) r-gp41 (50 ng), (iii) biotinylated MPER667–683 peptide (400 ng), or (iv) TBS containing 2% (wt/vol) bovine serum albumin (TBS-BSA; Sigma-Aldrich). The wells were blocked for 1 h at room temperature with TBS-BSA and then washed twice with TBS containing 0.1% (vol/vol) Tween 20 (TBS-T), washed once with TBS, and incubated for 2 h at room temperature with the indicated dilutions of 4E10 IgG1, 17/9 IgG1, or one of the 4E10 Fabs (WT, WDWD, or ΔLoop) diluted in TBS-T containing 5% (wt/vol) nonfat dried milk (5% NFDM; Bio-Rad). After five washes with TBS-T followed by one wash with TBS, bound 4E10 IgG or 4E10 Fabs were detected with goat (anti-human kappa light-chain IgG)-HRP, and bound 17/9 IgG1 was detected with goat (anti-murine IgG)-HRP, diluted 1:500 and 1:1,000, respectively, in 5% NFDM. After 1 h of incubation at room temperature, the plates were washed five times in TBS-T and once in TBS. Bound HRP was detected using ABTS (2′2′-azino-bis-3-ethylbenzthiazoline-6-sulfonic acid; Sigma-Aldrich, St. Louis, MO) at 400 ng/ml in developer solution comprising a 1:1.5 ratio of citrate (0.1 M, pH 2.4) and phosphate (0.2 M, pH 9.2) buffers, along with 0.03% (vol/vol) H2O2. The absorbance was measured at 405 to 490 nm using a Tecan Infinite M200 Pro microplate reader (Tecan, Mannedorf, Switzerland).
HIV-1 envelope neutralization assays.
Neutralization assays were performed using a single-round infection assay with envelope (Env)-pseudotyped virus (PsV) as described previously (38). Briefly, PsVs were produced by cotransfection of 293T cells with (i) an Env-negative HIV genomic vector that carries a firefly luciferase reporter gene (pNL4.3.Luc.R.E-) and (ii) a plasmid carrying an HIV envelope gene expression cassette. 293T cells (2 × 106 cells) were cotransfected with plasmid DNA at ratios of 1:3 to 1:20 μg of pNL4.3.Luc.R.E- plasmid DNA to μg of HIV Env+ plasmid DNA and 54 μg of polyethyleneimine (Polysciences, Warrington, PA). Plasmids encoding Env from HIV-1 isolates included SF162.LS, SS1196.1 clone 1 (SVPB9), or JR-CSF. A plasmid expressing the Env from vesicular stomatitis virus was employed to produce a negative-control PsV. PsVs were harvested from cell-free supernatants at 72 h posttransfection, passed through a 0.45-μm-pore-size filter, diluted 1:10 in 10× PBS (Life Technologies), divided into aliquots, and stored at −80°C.
PsV infection is measured using a luciferase-based assay in TZM-bl cells (39), with neutralization being measured as a reduction in luciferase activity following single-round infection. TZM-bl cells were seeded overnight (10,000/well) in flat-bottom 96-well plates (Corning) in a total volume of 200 μl of DMEM supplemented with 10% heat-inactivated FCS and 1 mM l-glutamine (DMEM-ΔFCS). Threefold serial dilutions of the appropriate MAb or Fab concentration were prepared in DMEM-ΔFCS in a total volume of 150 μl and then added in triplicate to the wells of a separate round-bottom 96-well plate (Corning). PsVs were added at 200 50% tissue culture infective doses/well in a total volume of 50 μl of DMEM-ΔFCS supplemented with DEAE-dextran hydrochloride (DEAE-dextran; Sigma-Aldrich) at 25 μg/ml, and the plates were incubated for 1 h at 37°C and 5% CO2. After incubation, the medium was removed from the wells of the TZM-bl-seeded 96-well plates, and PsV-MAb/Fab samples were transferred to the plates. Control wells comprising TZM-bl cells only (cell control) and virus-only (virus control) controls were also included. After a 72-h incubation, the medium was removed from wells, 50 μl of Glo lysis buffer (Promega, Madison, WI) was added to each well, and lysis was allowed to proceed at room temperature with gentle rocking for 5 min. One-Glo luciferase substrate (Promega) (50 μl/well) was added, followed by incubation for 5 min at room temperature. Samples were transferred (50 μl/sample) to 96-well, white solid plates (Thermo Fisher Scientific, Waltham, MA), and the luciferase activity was measured using a Tecan Infinite M200 Pro multimode plate reader (Tecan). All values are reported as relative luminescence units (RLU), and neutralization was calculated with respect to cell and virus control wells using the following formula: 1 − [(RLUsample wells – RLUcell-only wells)/(RLUvirus-only wells – RLUcell-only wells)] × 100. The 50% inhibitory concentration (IC50) was calculated as the antibody/Fab concentration producing a 50% reduction in RLU compared to the level in the virus control wells after subtraction of the cell control RLU.
PDB accession numbers.
Atomic coordinates and structure factors for the crystal structures have been deposited in the Protein Data Bank (PDB) under accession codes 5CIN (ΔLoop Fab), 5CIL (WDWD Fab), and 5CIP (unbound WT Fab).
RESULTS
Crystal structures of nonneutralizing 4E10 Fabs.
Modification of the flexible, hydrophobic CDR-H3 loop may result in impaired neutralizing 4E10 activity (3, 14–16). To investigate this phenomenon at the molecular level, we determined the crystal structure of two different nonneutralizing 4E10 Fabs mutated at the CDR-H3 loop, which were generated as follows: (i) truncating the hydrophobic apex by replacing the WH100-GH100A-WH100B-LH100C sequence with a Ser-Gly dipeptide (termed ΔLoop) and (ii) substituting the aromatic WH100 and WH100B residues for polar-charged Asp residues, hence reducing the hydrophobicity while preserving the length of the loop (termed WDWD and previously described by Scherer et al. [14]). Recombinant Fabs were expressed in Escherichia coli without loss of performance or change of structure with respect to the Fab obtained by papain cleavage of IgG (19). The three recombinant Fabs (WT, WDWD, and ΔLoop) displayed the typical β-rich structure of the immunoglobulin fold in solution, as demonstrated by the position of the circular dichroism minima at 217 nm (see Fig. S2 in the supplemental material). However, compared to WT antibodies (IgG and its Fab fragment) the ΔLoop and WDWD Fabs did not exhibited neutralizing activity in a standard assay (Table 1). The 4E10 IgG exhibits higher neutralization potency than that of the Fab fragment (6.5-fold), an observation in good agreement with results reported in a previous study (4.4-fold) (40). The molecular basis of this gap in neutralization potency is still unclear, although it might reflect the effect of avidity at the two available 4E10 sites in the Env trimer (41).
TABLE 1.
Neutralization of primary isolate viruses by 4E10 IgG, WT, and mutant Fabsa
| Antibody | IC50 (nM) |
|||
|---|---|---|---|---|
| Tier 1A (SF162.LS) | Tier 1B (SS1196.1) | Tier 2 (JRCSF) | Control (VSV-G) | |
| 4E10 IgG | 21 | 21 | 20 | >200 |
| 4E10 WT Fab | 146 | 155 | 106 | >200 |
| 4E10 WDWD Fab | >600 | >600 | >600 | >200 |
| 4E10 ΔLoop Fab | >600 | >600 | >600 | >200 |
The data shown are from one of two experiments, both of which gave similar results.
High-resolution crystal structures of WDWD or ΔLoop in complex with 4E10ep were determined at 1.81 and 1.70 Å, respectively (Table 2). The two complexes crystallized in the same space group as that reported for the WT Fab obtained either from papain-treated IgG or by heterologous expression in E. coli (11, 19). The superposition of the three crystal structures showed that they are nearly indistinguishable from each other (Fig. 1). The root mean square deviation (RMSD) values between the coordinates of WT and WDWD and between WT and ΔLoop were 0.20 and 0.29 Å, respectively. A significant difference was found in the conformation of the CDR-H3 apex of WT compared to that of ΔLoop, possibly because the latter construct is two residues shorter in this region. This conformational change brings the apex of the CDR-H3 of ΔLoop Fab closer to the peptide, generating an H-bond between residue Trp680 of the peptide and the backbone oxygen of the residue GlyH100A of ΔLoop (distance = 2.7 Å) (Fig. 2; see also Table S1 in the supplemental material). A similar H-bond was observed in one copy of the crystal structure of WT Fab in complex with a peptide containing α-aminoisobutyric acid at position Trp678 (11).
FIG 1.

Crystal structures of neutralizing and nonneutralizing 4E10 Fabs. (A) Superposition of the backbone atoms of WT (gray), WDWD (white), and ΔLoop (black). The RMSD of the backbone coordinates of the heavy chain of WT with those of WDWD was 0.20 Å, and that determined with those of ΔLoop was 0.29 Å. The arrows indicate differences in the conformation of the apex region of the CDR-H3 loop. The 4E10ep bound to WT, WDWD, and ΔLoop Fab is shown in orange, magenta, and blue, respectively. (B to D) Close-up view of the conformation of the CDR-H3 loop (residues 95-100J) of WT (B), WDWD (C), and ΔLoop (D) with respect to the peptide. Residues with side chains within the region 100-100D are depicted.
FIG 2.

Interaction network of 4E10ep peptide with neutralizing and nonneutralizing 4E10 Fabs. (A to C) Diagram for WT (A), WDWD (B), and ΔLoop (C). Residues corresponding to the Fab (upper half) and the peptide (bottom half) are labeled in black and magenta, respectively. Water molecules are labeled in cyan. H-bonds and their distances are depicted by green lines. Residues engaging in direct or water-mediated H-bonds are shown in a ball-and-stick representation. Residues displaying nonpolar and van der Waals interactions are represented by spiked semicircles. The red arrow in panel C indicates the unique Fab-protein interaction observed in the complex with ΔLoop. The figure was generated with LIGPLOT+ (57).
Other polar and nonpolar interactions between the peptide and WT Fab were very well conserved in the WDWD and ΔLoop Fabs (Fig. 2). Key H-bonds between peptide residues Asn671, Trp672, Asp674, and Thr676 and the Fabs (see Table S1 in the supplemental material), as well as the nonpolar interaction surface involving the residues Asn671, Trp672, Phe673, Ile675, Thr676, Leu679, and Trp680 of the peptide, are well preserved in the three crystal structures (see Tables S2 and S3 in the supplemental material). Moreover, the values of buried surface area calculated with PISA (42) corresponding to the interaction surface between the peptide and the heavy or the light chain of the Fab remain essentially unchanged in the three crystal structures (Tables S2 and S3 in the supplemental material). The total buried surface areas for WT, WDWD, and ΔLoop were within 2% of each other: 1487, 1482, and 1511 Å2, respectively.
The shape complementarity coefficient (Sc), a parameter that quantifies the complementarity between protein/protein interfaces (39), was also very similar among the three Fabs. The Sc values calculated for the interface formed between the heavy chain and 4E10ep in the three crystal structures were in the narrow range of 0.77 to 0.78, whereas those between the light chain and the peptide were in the range 0.80 to 0.82. The potential lipid-binding site located in the CDR-H1 loop also remained unchanged in the three crystal structures. We note that a water molecule takes the position occupied by the phosphate ion reported in the PDB structure with entry code 4NHC (13; data not shown).
Mechanism of binding and stability of neutralizing and nonneutralizing 4E10 Fabs.
The structural data gathered thus far has not satisfactorily explained the poor neutralization performance of the two CDR-H3 mutants, given the minimal structural differences among the Fabs. To further characterize the molecular basis for these large differences in neutralization potency, we performed a comparative functional and thermodynamic analysis using WT, ΔLoop, and WDWD Fabs.
DSC measurements demonstrated that the CDR-H3 mutations did not affect the stability of the ligand-free forms of the Fabs (Fig. 3). The melting temperatures (Tm) determined from the midpoint unfolding transition of WT Fab, WDWD, and ΔLoop were 70, 72, and 72°C, respectively. Therefore, decreasing the hydrophobicity of CDR-H3 at the apex translates into a modest increase in the thermostability of the Fabs (ΔTm = 2°C).
FIG 3.

Thermal stability by DSC. DSC thermograms for WT, WDWD, and ΔLoop are shown as indicated in the panels. Unfolding of the three Fabs shows a single thermal transition between 30 and 90°C. The midpoint of the thermal unfolding (Tm) and the associated enthalpy (ΔH) are also given. The values of Tm and ΔH were obtained by curve fitting with the program ORIGIN 7.0.
Kinetic and thermodynamic parameters governing 4E10ep binding were next examined by SPR as previously described (20, 21). Figure 4 and Fig. S3 in the supplemental material show the sensorgrams corresponding to the binding of the 4E10ep to immobilized WT, WDWD, and ΔLoop Fabs, illustrating the effect of mutations in the apex of the CDR-H3 loop on the association (kon) and dissociation (koff) rate constants. The values of kon and koff for WT Fab at room temperature were 5.2 × 104 M−1 s−1 and 1.1 × 10−3 s−1, respectively, corresponding to a dissociation constant (KD) of 21 nM (Fig. 4A and Table 3). The small value of koff indicates slow dissociation of the peptide from the Fab, a finding consistent with tight binding of the epitope-peptide to the antibody under these experimental conditions. The values of kon, koff, and KD were also determined for the WDWD and ΔLoop Fabs (Fig. 4A and Table 3). Ablation of the hydrophobic CDR-H3 tip did not appreciably affect peptide affinity (KD = 18 nM). Compared to the WT Fab, the slower kon in ΔLoop was compensated by a slower koff, suggesting that the intact loop establishes faster interactions with the peptide, but at the same time its release from the paratope is also accelerated. The slower dissociation rate observed for the ΔLoop Fab could be explained by the stronger H-bond between the GlyH100A(O) of the Fab and Trp680(Nξ) of the peptide (Fig. 2; see also Table S1 in the supplemental material). The double mutation introduced in WDWD Fab decreased the affinity slightly (KD = 41 nM) because of a faster koff with respect to the pWT Fab, which was not compensated with a faster kon (Table 3). Although these differences are arguably small, the data suggest that the more hydrophobic loop of WT Fab facilitated binding of the 4E10ep more effectively than the polar WDWD.
FIG 4.

Kinetic and thermodynamic characterization of Fab-peptide binding by SPR. (A) Sensorgrams corresponding to the binding of 4E10ep (analyte) to a surface decorated with WT, WDWD, or ΔLoop Fabs (immobilization levels were ∼1,400 RU). The concentration of peptide injected in each run is indicated. Black and red curves correspond to the experimental data and best fit (using the Biacore T200 evaluation software), respectively. (B) Evolution of the thermodynamic parameters along the reaction coordinate. Thermodynamic parameters corresponding to the transition state and at equilibrium were obtained from the temperature dependence of kon and KD using the Eyring and van't Hoff equations, respectively. The change in Gibbs energy (ΔG), change in enthalpy (ΔH), and change in entropy (−TΔS) are shown in blue, red, and black, respectively. Values are given in Table 4.
TABLE 3.
Kinetic parameters of binding of Fab 4E10 to a 4E10ep varianta
| Fab | kon (M−1 s−1) | koff (s−1) | KD (nM) |
|---|---|---|---|
| WT | 5.2 × 104 | 1.1 × 10−3 | 21 |
| ΔLoop | 2.9 × 104 | 5.3 × 10−4 | 18 |
| WDWD | 4.7 × 104 | 1.9 × 10−3 | 41 |
NWFDITNWLWYIR-RRR. Lys residues in 4E10ep are replaced with Arg residues.
The van't Hoff thermodynamic parameters for the WT Fab and the mutated CDR-H3 Fabs were obtained from the temperature dependence of KD (Fig. 4B and Table 4). The change of enthalpy associated with the binding of WT Fab was negative (ΔHvH < 0, exothermic reaction), whereas the contribution from the entropy energetic term (−TΔSvH, calculated at 25°C) was small and positive. These values suggested that binding was essentially driven by favorable noncovalent interactions between Fab and peptide. The thermodynamic parameters obtained at equilibrium for the nonneutralizing WDWD and ΔLoop Fabs exhibited the same thermodynamic signature (Table 4), although the entropic cost in the case of the WDWD mutant was slightly higher (−TΔΔSvH = 1.5 kcal mol−1), which could account for the decrease in affinity observed for this Fab.
TABLE 4.
Thermodynamic parameters of binding of Fab 4E10 to a 4E10ep varianta
| Fab | ΔGvH (kcal mol−1) | ΔHvH (kcal mol−1) | −TΔSvH (kcal mol−1) | ΔG‡ (kcal mol−1) | ΔH‡ (kcal mol−1) | −TΔS‡ (kcal mol−1) |
|---|---|---|---|---|---|---|
| WT | –10.5 | –11.5 | 1.0 | 11.1 | 29.4 | –18.3 |
| ΔLoop | –10.6 | –12.4 | 1.7 | 11.4 | 25.8 | –14.5 |
| WDWD | –10.1 | –12.6 | 2.5 | 11.2 | 18.6 | –7.4 |
NWFDITNWLWYIR-RRR. “‡” refers to the transition state (see the text).
Differences in the binding mechanism arising from fluctuating loop conformations could be more evident at the transition state, i.e., before reaching the final conformation of the complex. Hence, the activation free energy (ΔG‡ [the double dagger symbol refers to the transition state]) of each Fab-peptide complex was determined from the temperature dependence of kon (Fig. 4B and Table 3). The transition state in the three Fabs was governed by the unfavorable enthalpy term (ΔH‡ > 0), reflecting the destruction of noncovalent interactions during the rate-determining step. The unfavorable enthalpy term was partially counterbalanced by favorable entropic contributions (−TΔS‡ < 0). Due to the differences in mass and charge in the CDR-H3 loop of the mutated Fabs with respect to WT, we expected to see dissimilar activation barriers for binding reactions requiring conformational changes of the loop. However, the resulting ΔG‡ values were strikingly similar for the three Fabs (Table 4). Therefore, the shared thermodynamic signature at the transition state in the three constructs examined suggests that rearrangement of water molecules at the contact interface, rather than major alterations of CDR-H3 loop conformation, plays a key role on the activation of the binding process.
Crystal structure of 4E10 Fab in the unbound state.
The thermodynamic data described above argue against major conformational changes occurring in the 4E10 paratope during 4E10ep binding. This conclusion differs from that reported with the ligand-free Fv fragment (17). In that study it was shown that a large segment of the CDR-H3 loop (including the apex region) adopts a significantly different conformation in the unbound state, resulting in occlusion of the paratope. To solve this paradox and to allow direct structural comparison between the Fab-peptide complex and those previously reported in the literature (10, 11, 13), we determined the crystal structure of unbound 4E10 Fab (Fig. 5).
FIG 5.

Comparison of the ligand-bound and ligand-free forms of 4E10 Fab. Sausage (tubular) representation of B-factors of ligand-free (A) and ligand-bound (B) 4E10 Fab. Higher B-factors are depicted by red and thick tubes. For the unbound form of 4E10, the blue-white-red gradient corresponds to B-factor values of 12.6, 36.6, and 100.9 Å2, respectively. For the peptide-bound form, the blue-white-red gradient corresponds to B-factor values of 21.8, 40.0, and 90.2 Å2, respectively. These values correspond to the minimum, a midpoint, and the maximum B-factor values of each crystal structure. Both molecules are oriented to display the paratope CDR loops at the front plane. In panel B, the bound peptide is represented with a green ribbon. (C) Per-residue real-space correlation coefficient (RSCC; a measure of the fit between the model and the experimental electron density) of the six hypervariable loops of 4E10 Fab in the unbound (red) and bound (black) state. Lower correlation values indicate less agreement between the model and the experimental electron density, suggesting the regions with lower RSCC are more dynamically disordered. The RSCC values were calculated using PHENIX (30).
Single crystals of unbound WT 4E10 Fab were obtained in a solution containing 15% PEG 20,000 and 100 mM MES at pH 6.5. The best diffracting crystal achieved a resolution of 2.48 Å in space group P21, which was different from the space group (C2), where the crystal of the Fab in complex with peptide was obtained (Table 2). Consequently, the packing forces experienced by protein chains of the unbound and the bound Fabs were different. Despite this difference, the structures of unbound and bound Fab were essentially identical (RMSD = 0.32 Å) (Fig. 5).
The electron density corresponding to residues of the apex of CDR-H3 (WH100-GH100D) was weak, indicative of dynamic disorder (Fig. 5C and Fig. 6A). For that reason, we took additional steps to validate the geometry and the conformation of the residues of the apex modeled in this structure (see Materials and Methods for a complete description). Importantly, the conformation of the CDR-H3 loop, in its entire length, is nearly identical to that of the bound form of WT but clearly different from that of the unbound form of the FV fragment (Fig. 6B). We also note that the putative lipid-binding site located in CDR-H1 also remains virtually unchanged with respect to the Fab with peptide bound (data not shown).
FIG 6.

Conformation of CDR-H3 in the unbound form. (A) Sigma-A weighted electron density map of the CDR-H3 of unbound WT Fab (σ = 1.0). The electron density for the residues of the apex (100WGWLG100D) is only visible at low contouring levels (σ ∼ 0.5 [data not shown]) but is sufficient to allow modeling of the residues. (B) Comparison of conformations of the CDR-H3 in the unbound form of Fab (orange), in the reported Fab-peptide complex (PDB code 2FX7, black), and in the reported structure of the unliganded Fv fragment (PDB code 4LLV, blue). The sequence numbers are given.
Crystallographic models contain dynamic information in the form of B-factors and dynamic contact networks (43), and thus the comparison of models of the same protein in the bound and unbound state may reveal regions of altered dynamics. The antibody uses the CDRs H1 (25SGGSFSTYAL34), H2 (50GVIPLLTITNYA60), H3 (95EGTTGWGWLGKPIGAFAH102), and L3 (89QQYGQSLST97) to engage 4E10ep (11). The CDR-L2 (50GASSRPS56) does not engage with the peptide, whereas CDR-L1 (24RASQSVGNNKLAW35) only makes minor contacts (11). Comparison of bound and unbound 4E10 Fab models revealed a reduction in the backbone dynamics (B-factors) of several CDR loops in the bound state according to their involvement in peptide binding (Fig. 5A and B; see also Fig. S4 in the supplemental material).
To strengthen the conclusions about the dynamic behavior of the CDR loops of Fab upon binding, we calculated the real-space correlation coefficient (RSCC) (44) with PHENIX (30) (Fig. 5C). The RSCC values for residues of the CDRs H1, H2, H3, and L3 are greater in the bound form than in the unbound form. This observation is explained by a better fitting of the electron density to the model, which can be qualitatively interpreted as a reduction in dynamic disorder. These differences cannot be attributed to the resolution of the crystal structures, since the average value of RSCC for the entire structure of bound (RSCC = 0.97) and unbound Fab (RSCC = 0.95) was comparable to each other. Although RSCC values for all residues of the CDR-H3 increased upon binding of the peptide, it is also certain that residues of the apex (99GWGWL100C) displayed a noticeable residual disorder, as judged from the values of RSCC (<0.90). In contrast to the CDR loops described above, CDRs L1 and L2 exhibited very little reduction of mobility, a finding consistent with their minor role in binding. Similar results are observed in the average plots of six other crystal structures of 4E10 with various peptides bound (see Fig. S1b in the supplemental material).
These results have proved that the binding process resulted in minor conformational adjustments mostly involving a change of mobility of the CDR loops after the antibody comes into contact with the epitope. Importantly, our results indicate that the hydrophobic cavity accommodating the peptide remains intact even in the absence of peptide.
PsV neutralization and binding to the MPER in the context of the plasma membrane.
The results presented above describing the structure and energetics of peptide binding to the 4E10 Fabs indicated that mutation of the CDR-H3 apex do not alter the ability of the antibody to interact with the peptide. However, a PsV-based neutralization assay (Table 1) showed that the Fab mutants had lost all neutralization activity compared to that of WT IgG and WT Fab. The results displayed on Fig. 7 confirmed the inability of the nonneutralizing Fab to bind to PsV particles under the experimental conditions that otherwise sustain dose-dependent binding of the WT-4E10 Fab and the anti-gp120 PG9 antibody, the latter being used as a positive control of Env expression. Thus, the dramatic differences in biological activity were mirrored by differences in binding of the Fabs to the immobilized PsVs, further supporting that preservation of a solvent-exposed, flexible and hydrophobic CDR-H3 loop apex is required for binding and neutralization in the biological context.
FIG 7.
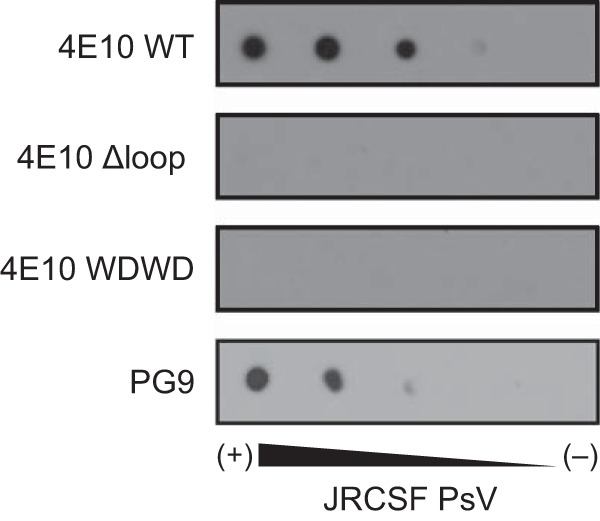
JRCSF PsV recognition as determined by dot blot analysis. Decreasing amounts of PsVs (from left to right) were spotted onto Hybond-C nitrocellulose. The nitrocellulose was then blocked with 5% fat-free milk in PBS (blocking buffer) for 1 h and incubated for 1 additional hour with antibodies (0.2 μg/ml) in blocking buffer at room temperature. The membranes were washed three times for 10 min each time with PBS. Spots were revealed using an HRP-conjugated antibody.
The results in Fig. 8 strengthen the conclusion that availability of the WT CDR-H3 apex contributes to additional interactions with membrane components as previously suggested (10, 14, 22, 45–48). The DNA vaccine candidates, MPER-TM1 and MPER-PDGFR (22), express fusion proteins comprising: (i) an N-terminal HA tag, (ii) the MPER, (iii) either the transmembrane domain of gp41 followed by 27-AA of the cytoplasmic domain (TM1) or that of the platelet-derived growth-factor receptor (PDGFR). These fusions are expressed in the context of the plasma membrane; however, the PDGFR transmembrane domain has been shown to reduce exposure of the 4E10 epitope (22) and, to a lesser extent, the 10E8 epitope (N. Gulzar, unpublished data). The titrations shown in Fig. 8A indicate that IgG and Fab WT engaged effectively with the 4E10 epitope presented in the context of the plasma membrane (EC50s of ca. 0.1 and 1 nM, respectively). In comparison, in the range of measured concentrations, binding to MPER-TM1 was negligible for the WDWD and ΔLoop mutants (Fig. 8A, bottom panels). None of the antibodies bound directly to the plasma membrane of cells devoid of an MPER construct (squares and dotted lines), demonstrating that the binding scored for the WT IgG and Fab under our experimental conditions was strictly dependent on the presence of the MPER epitope.
FIG 8.

Alterations in the CDR H3 of 4E10 Fab impacts the binding of Fab-s to its cognate epitope in the context of the plasma membrane. (A) Cell vesicles displaying the MPER-TM1 protein product (solid line), and “empty” control vesicles (dashed lines) were probed with 4E10 IgG and Fabs (WT, WDWD, and ΔLoop) at the indicated concentrations. (B) Cell vesicles bearing recombinant MPER proteins (MPER-TM1, black columns; MPER-PDGFR, gray columns) were probed with 20 nM 4E10 IgG and Fabs and 20 nM 17/9 IgG (which binds to an HA tag located at the N terminus of the MPER constructs and is used to assess cell surface expression). Absorbance values were normalized to the cell surface expression of the constructs (i.e., the absorbance of 4E10IgG or Fabs/the absorbance of 17/9IgG). (C) Binding of 20 nM 4E10 IgG and Fabs to recombinant gp41, the MPER667–683 peptide, and assay controls (“empty” 293T cell lysates, BSA, and ovalbumin). For all experiments, the absorbance at 405 to 490 nm was recorded. Error bars represent standard error of the mean. The data shown are from one of two (see Fig. 8B and C) or three (see Fig. 8A) experiments, all of which yielded similar results.
Our previous work (22) showed that, compared to the MPER-TM1 construct, binding of 4E10 IgG to the MPER-PDGFR construct was significantly reduced; this was also observed to a lesser extent for the bNAb, 10E8, whose epitope overlaps with that of 4E10 (N. Gulzar and J. K. Scott, unpublished data), but not for bNAb 2F5 (49), whose epitope is on the N-terminal helix of the MPER (22). These results reflect the ability of the transmembrane domain to affect the positioning of bNAb epitopes in the C-terminal helix of the MPER but not in its N-terminal helix. The binding of the WT and mutant Fabs, particularly the ΔLoop Fab, to the MPER-TM1 and MPER-PDGFR constructs was assessed in relation to 4E10 IgG. Figure 8B shows that, as expected, binding to the MPER-PDGFR construct by the 4E10 IgG and WT Fab was significantly reduced, whereas the mutant WDWD and ΔLoop Fabs bound negligibly to both constructs. The influence of membrane was finally inferred from control ELISAs performed using recombinant gp41 ectodomain or MPER667–683 peptide adsorbed to plates (Fig. 8C). Experiments in the absence of membranes showed for the three Fabs comparable levels of binding to these antigens, which does not correlate with their neutralizing capacities (Table 1).
DISCUSSION
The HIV-1 bNAbs isolated to date provide templates for rational vaccine design, facilitate the understanding of immune pathways and molecular processes that confer broad protection, and constitute the basis for immunotherapies with the potential to effectively prevent and treat HIV-1 infection (6, 50, 51). The 4E10 bNAb targets the conserved MPER C-terminal helix and displays near pan-neutralizing activity in standard panels of HIV strains and isolates (4, 5, 52–56). Thus, 4E10 constitutes a bNAb model particularly useful to study the molecular basis underlying HIV neutralization breath mediated through MPER recognition.
The neutralizing activity of 4E10 is strictly dependent on aromatic residues of the CDR-H3 loop (14, 15). Here, we have studied the mechanistic basis of this requirement by characterizing structurally and functionally WDWD and ΔLoop, two Fab variants mutated at the critical apex region of the CDR-H3 loop. Thus, we have performed crystallographic, kinetic, and thermodynamic studies on neutralization-active and neutralization-inactive Fabs and compared their 4E10 epitope binding mechanisms at the molecular level. The recombinant Fabs were obtained by heterologous expression in the cytoplasm of Escherichia coli, avoiding aberrant forms that may occur upon refolding of inclusion bodies.
The crystal structures of 4E10 Fab WT, WDWD, and ΔLoop determined in complex with the epitope-peptide revealed combining sites and bound helical peptides essentially indistinguishable from each other (Fig. 1). From these results, we have concluded that mutations in the CDR-H3 apex abolishing neutralization neither affect the collective set of interactions between Fab and the peptide (Fig. 2; see also Tables S1 to S3 in the supplemental material) nor change the configuration of the proposed lipid-binding subsite in significant ways (data not shown). The only visible differences were restricted to the CDR-H3 loop apex, which remains exposed to solvent, i.e., does not contact the bound peptide (Fig. 1). The mutated loop region in WDWD displays essentially the same conformation as that in WT Fab and very similar interactions (buried surface area and H-bond pattern) despite the large differences in the loop hydrophobicity (Fig. 2; see also Tables S1 to S3 in the supplemental material). In contrast, the backbone of the shorter loop of ΔLoop approaches the helical peptide as a result of the deletion of two residues of the CDR-H3 apex (Fig. 1), thereby strengthening an H-bond between the Fab and Trp680 of the peptide (Fig. 2; see also Table S1 in the supplemental material). Thus, the structural data demonstrate that an antibody bearing a short and nonhydrophobic CDR-H3 loop is capable of engaging with the helical MPER epitope as much or even more effectively than the WT Fab. These observations may have important implications for the design of anti-MPER vaccines.
It has been proposed that the long CDR-H3 loop enhances paratope flexibility to effectively capture the membrane-inserted MPER (22) by means of an induced-fit mechanism. Consistent with this idea, crystallographic studies on the unliganded 4E10 variable domain Fv revealed large conformational changes of CDR-H1 and, more prominently, of CDR-H3 (Fig. 6B). The position of CDR-H3 in the unliganded Fv shields the epitope-binding pocket, creating an electropositive and unspecific lipid-binding subsite (16–18) that is different from the one later proposed by Bird et al. (13). Based on the structural data obtained with the Fv fragment, it was proposed that a large portion of the CDR-H3 undergoes a large conformational change from the unbound to the bound state (see Fig. 6B). If this was the case, mutations blocking the neutralizing activity could also impact the conformational equilibrium of the CDR-H3 in the unbound form and thus alter the overall epitope-binding function of the paratope.
In contrast to that prediction, the binding affinities of 4E10ep at equilibrium for the three Fabs were strikingly similar (Fig. 4A and Table 3). Similarly, the free energy changes at the rate-determining step (transition state) of the three versions of the Fab were nearly identical to each other (Fig. 4B and Table 3). Given the differences in size and polarity of CDR-H3 in WT and mutated Fabs, this result does not support a major restructuring of the loop during the activation stage of peptide binding.
To tackle the disagreement between the reported unliganded structure of the Fv fragment (17), and our thermodynamic data obtained with a Fab construct, we determined the crystal structure of WT Fab in the absence of peptide. Consistent with the thermodynamic analysis, the crystal structure of the ligand-free 4E10 Fab displayed a paratope structurally similar to that found in the solved Fab-peptide complexes (Fig. 5 and 6). Notwithstanding the overall similarity, higher degree of dynamic disorder was detected at the CDR loops implicated in peptide binding in the ligand-free Fab (Fig. 5; see also Fig. S4 in the supplemental material). Specifically, peptide engagement reduced the mobility of CDRs H1, H2, H3, and L3 to a noticeable extent. The flexibility of the CDR-H3 loop, critical for neutralization, decreased markedly, although we note that significant residual disorder remained in its apex region even in the presence of bound peptide. In contrast, the flexibility of CDRs L1 and L2 did not change significantly between the bound and unbound forms of the Fab. These findings support the crystal structure of the peptide-free Fab described in our work as a relevant reference to describe the conformation of 4E10 Fab in the unliganded state.
Thus, the kinetic, thermodynamic, and structural data gathered herein indicated limited conformational changes of the Fab upon epitope binding (essentially the immobilization of certain CDR loops), suggesting a flexible lock-and-key binding mechanism. The underlying reason explaining the conformational differences between the CDR-H3 (and CDR-H1) in the unbound forms of the short Fv and the longer Fab constructs (movement of backbone atoms of up to 14 Å in WH100) (17, 18) (Fig. 6B) was not clarified at this time. We suggest that the difference could be explained by the low pH required to crystallize the Fv construct (pH 4.6) and/or to different structural stabilities of these two specimens. In the unbound structure of Fv, the residues of the CDR-H3 move toward the empty pocket of the peptide-binding site, establishing numerous hydrophobic contacts with other CDR-H3 loops of other molecules of the asymmetric unit, and thus achieving maximum stability and minimizing their exposure to the low pH environment of the aqueous solution (17). In contrast, although the crystal structures of unbound and bound forms of Fab were obtained each in a different space group (thus experiencing different structural constraints and packing forces), the CDR-H3 loop remains in a similar conformation. In addition, the thermal stability (as judged from the Tm values) for the smaller 4E10 Fv fragments (Tm = 52°C) (18) is significantly lower than those of the Fab fragments (Tm = ∼70°C; Fig. 3). Therefore, the combination of lower stability, extreme crystallization conditions, and crystal packing forces may all have induced a nonnative conformation of the CDR-H3 loop in the crystal structure of the Fv fragment.
It has been speculated that length, hydrophobicity, and conformational flexibility of the 4E10 CDR-H3 loop apex may govern the interactions with the viral membrane (10, 11). The structural and thermodynamic results obtained with WDWD formally demonstrate that length and flexibility are not sufficient to elicit viral neutralization. Thus, maintaining an accessible hydrophobic patch at the apex of the flexible CDR-H3 emerges as a stringent requirement for the neutralizing activity of 4E10. Assuming that a common mechanism (i.e., conformational stabilization) applies to all CDR loops involved in binding, the high mobility of CDR-H3 indicates the existence of yet additional interactions with antigen that are not satisfied by contacts with 4E10ep in solution.
The Fab binding activity in dot blot and cell lysate ELISA analyses supports the idea that additional CDR-H3 interactions can be fulfilled in the context of the membrane (Fig. 7 and 8). In the cell lysate ELISA, the MPER-TM1 construct was used to constrain the MPER and position it on the membrane surface. In contrast to what was observed for 4E10ep binding in solution, the capacity of the Fabs to bind MPER-TM1 was correlated with their neutralization activity. Because the range of concentrations allowing for effective Fab-MPER-TM1 binding was sufficiently low, direct association with membranes was negligible, as reflected by the lack of binding to “empty” cell lysates (Fig. 8). Importantly, binding by WT 4E10 IgG and Fab to the MPER-PDGFR construct was weak relative to MPER-TM1 (Fig. 8). Since the PDGFR transmembrane region has been shown to improperly constrain the MPER (22), this result supports the necessity of proper tethering by the transmembrane domain to optimally expose the MPER in the context of the plasma (or viral) membrane. Thus, overall, the results from this and previous cell lysate ELISAs (22) strongly suggest that the hydrophobic CDR-H3 loop enables direct binding to an MPER epitope that achieves its neutralization-competent antigenic structure through membrane insertion and tethering via an appropriate transmembrane domain (9, 22, 45, 46, 48).
Together with the realization that antibodies bearing shorter and less-hydrophobic CDR-H3 loops may display equal or even higher affinity toward helical 4E10ep, these findings should have important implications for anti-MPER vaccine development. Moreover, our results also suggest that a vaccine devoid of lipid might elicit high-affinity antibodies against the MPER C-terminal helix. However, since antibodies with short-polar CDR-H3 loops are arguably easier to produce, the generated responses are likely to comprise nonneutralizing antibodies for the most part. On the other hand, accessibility to the MPER epitope inserted in membranes appears to correlate with the neutralization competence for 4E10-related antibodies. In contrast, however, the bNAb, 10E8 (4), binds to an MPER epitope that overlaps with the 4E10 epitope. Although it bears a long (22-residue) CDR-H3, it does not appear to bind significantly to lipids; yet it, too binds tightly to the MPER-TM1 in the context of the plasma membrane (Gulzar and Scott, unpublished) and less strongly to the MPER-PDGFR construct. It would be interesting to produce MPER667–683 peptide-binding, nonneutralizing mutants of this antibody as well, to determine whether it, too, will bind poorly to the MPER-TM1, as has also been shown for nonneutralizing, CDR-H3 mutants of 2F5, another MPER-specific bNAb (22). Taken together, the data presented here further support the vaccine design strategy of replicating the neutralization-competent structure of the MPER on the viral membrane surface to elicit HIV-1 neutralizing antibodies.
Supplementary Material
ACKNOWLEDGMENTS
We acknowledge financial support by the National Institutes of Health (1R01AI097051-01 to J.K.S. and J.L.N.). This study was also supported by a Grant-in-Aid for Scientific Research A (grant 25249115 to K.T.) and a Grant-in-Aid for Scientific Research C (grant 15K06962 to J.M.M.C.) from the Japan Society for the Promotion of Science. E.R. is a recipient of a predoctoral fellowship from the Basque Government. Access to Beamline BL-5A was granted by the Photon Factory Advisory Committee (proposals 2014G190 and 2013G738).
We thank D. Albesa-Jove and M. E. Guerin (Biophysics Unit, CSIC, UPV-EHU) for expert advice and assistance with data collection at the Diamond Light Source in the United Kingdom. We thank M. Kiyoshi and S. Tashiro for excellent advice and support with SPR data collection and analysis, M. Garcia for technical assistance, and J. P. Julien for critical reading of the manuscript.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.01793-15.
REFERENCES
- 1.Salzwedel K, West JT, Hunter E. 1999. A conserved tryptophan-rich motif in the membrane-proximal region of the human immunodeficiency virus type 1 gp41 ectodomain is important for Env-mediated fusion and virus infectivity. J Virol 73:2469–2480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Apellaniz B, Ivankin A, Nir S, Gidalevitz D, Nieva JL. 2011. Membrane-proximal external HIV-1 gp41 motif adapted for destabilizing the highly rigid viral envelope. Biophys J 101:2426–2435. doi: 10.1016/j.bpj.2011.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Apellaniz B, Rujas E, Carravilla P, Requejo-Isidro J, Huarte N, Domene C, Nieva JL. 2014. Cholesterol-dependent membrane fusion induced by the gp41 membrane-proximal external region-transmembrane domain connection suggests a mechanism for broad HIV-1 neutralization. J Virol 88:13367–13377. doi: 10.1128/JVI.02151-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Huang J, Ofek G, Laub L, Louder MK, Doria-Rose NA, Longo NS, Imamichi H, Bailer RT, Chakrabarti B, Sharma SK, Alam SM, Wang T, Yang Y, Zhang B, Migueles SA, Wyatt R, Haynes BF, Kwong PD, Mascola JR, Connors M. 2012. Broad and potent neutralization of HIV-1 by a gp41-specific human antibody. Nature 491:406–412. doi: 10.1038/nature11544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Binley JM, Wrin T, Korber B, Zwick MB, Wang M, Chappey C, Stiegler G, Kunert R, Zolla-Pazner S, Katinger H, Petropoulos CJ, Burton DR. 2004. Comprehensive cross-clade neutralization analysis of a panel of anti-human immunodeficiency virus type 1 monoclonal antibodies. J Virol 78:13232–13252. doi: 10.1128/JVI.78.23.13232-13252.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kwong PD, Mascola JR. 2012. Human antibodies that neutralize HIV-1: identification, structures, and B cell ontogenies. Immunity 37:412–425. doi: 10.1016/j.immuni.2012.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Burton DR, Desrosiers RC, Doms RW, Koff WC, Kwong PD, Moore JP, Nabel GJ, Sodroski J, Wilson IA, Wyatt RT. 2004. HIV vaccine design and the neutralizing antibody problem. Nat Immunol 5:233–236. doi: 10.1038/ni0304-233. [DOI] [PubMed] [Google Scholar]
- 8.Zwick MB. 2005. The membrane-proximal external region of HIV-1 gp41: a vaccine target worth exploring. AIDS 19:1725–1737. doi: 10.1097/01.aids.0000189850.83322.41. [DOI] [PubMed] [Google Scholar]
- 9.Montero M, van Houten NE, Wang X, Scott JK. 2008. The membrane-proximal external region of the human immunodeficiency virus type 1 envelope: dominant site of antibody neutralization and target for vaccine design. Microbiol Mol Biol Rev 72:54–84. doi: 10.1128/MMBR.00020-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cardoso RM, Zwick MB, Stanfield RL, Kunert R, Binley JM, Katinger H, Burton DR, Wilson IA. 2005. Broadly neutralizing anti-HIV antibody 4E10 recognizes a helical conformation of a highly conserved fusion-associated motif in gp41. Immunity 22:163–173. doi: 10.1016/j.immuni.2004.12.011. [DOI] [PubMed] [Google Scholar]
- 11.Cardoso RM, Brunel FM, Ferguson S, Zwick M, Burton DR, Dawson PE, Wilson IA. 2007. Structural basis of enhanced binding of extended and helically constrained peptide epitopes of the broadly neutralizing HIV-1 antibody 4E10. J Mol Biol 365:1533–1544. doi: 10.1016/j.jmb.2006.10.088. [DOI] [PubMed] [Google Scholar]
- 12.Brunel FM, Zwick MB, Cardoso RMF, Nelson JD, Wilson IA, Burton DR, Dawson PE. 2006. Structure-function analysis of the epitope for 4E10, a broadly neutralizing human immunodeficiency virus type 1 antibody. J Virol 80:1680–1687. doi: 10.1128/JVI.80.4.1680-1687.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bird GH, Irimia A, Ofek G, Kwong PD, Wilson IA, Walensky LD. 2014. Stapled HIV-1 peptides recapitulate antigenic structures and engage broadly neutralizing antibodies. Nat Struct Mol Biol 21:1058–1067. doi: 10.1038/nsmb.2922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Scherer EM, Leaman DP, Zwick MB, McMichael AJ, Burton DR. 2010. Aromatic residues at the edge of the antibody combining site facilitate viral glycoprotein recognition through membrane interactions. Proc Natl Acad Sci U S A 107:1529–1534. doi: 10.1073/pnas.0909680107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Alam SM, Morelli M, Dennison SM, Liao HX, Zhang R, Xia SM, Rits-Volloch S, Sun L, Harrison SC, Haynes BF, Chen B. 2009. Role of HIV membrane in neutralization by two broadly neutralizing antibodies. Proc Natl Acad Sci U S A 106:20234–20239. doi: 10.1073/pnas.0908713106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xu HY, Song LK, Kim M, Holmes MA, Kraft Z, Sellhorn G, Reinherz EL, Stamatatos L, Strong RK. 2010. Interactions between lipids and human anti-HIV antibody 4E10 can be reduced without ablating neutralizing activity. J Virol 84:1076–1088. doi: 10.1128/JVI.02113-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Finton KA, Larimore K, Larman HB, Friend D, Correnti C, Rupert PB, Elledge SJ, Greenberg PD, Strong RK. 2013. Autoreactivity and exceptional CDR plasticity (but not unusual polyspecificity) hinder elicitation of the anti-HIV antibody 4E10. PLoS Pathog 9:e1003639. doi: 10.1371/journal.ppat.1003639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Finton KA, Friend D, Jaffe J, Gewe M, Holmes MA, Larman HB, Stuart A, Larimore K, Greenberg PD, Elledge SJ, Stamatatos L, Strong RK. 2014. Ontogeny of recognition specificity and functionality for the broadly neutralizing anti-HIV antibody 4E10. PLoS Pathog 10:e1004403. doi: 10.1371/journal.ppat.1004403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Apellaniz B, Rujas E, Serrano S, Morante K, Tsumoto K, Caaveiro JMM, Jimenez MA, Nieva JL. 2015. The atomic structure of the HIV-1 gp41 transmembrane domain and its connection to the immunogenic membrane-proximal external region. J Biol Chem 290:12999–13015. doi: 10.1074/jbc.M115.644351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kiyoshi M, Caaveiro JMM, Miura E, Nagatoishi S, Nakakido M, Soga S, Shirai H, Kawabata S, Tsumoto K. 2014. Affinity improvement of a therapeutic antibody by structure-based computational design: generation of electrostatic interactions in the transition state stabilizes the antibody-antigen complex. PLoS One 9:e87099. doi: 10.1371/journal.pone.0087099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kiyoshi M, Caaveiro JMM, Kawai T, Tashiro S, Ide T, Asaoka Y, Hatayama K, Tsumoto K. 2015. Structural basis for binding of human IgG1 to its high-affinity human receptor Fc[gamma]RI. Nat Commun 6:6866. doi: 10.1038/ncomms7866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Montero M, Gulzar N, Klaric KA, Donald JE, Lepik C, Wu S, Tsai S, Julien JP, Hessell AJ, Wang S, Lu S, Burton DR, Pai EF, Degrado WF, Scott JK. 2012. Neutralizing epitopes in the membrane-proximal external region of HIV-1 gp41 are influenced by the transmembrane domain and the plasma membrane. J Virol 86:2930–2941. doi: 10.1128/JVI.06349-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Julien JP, Huarte N, Maeso R, Taneva SG, Cunningham A, Nieva JL, Pai EF. 2010. Ablation of the complementarity-determining region H3 apex of the anti-HIV-1 broadly neutralizing antibody 2F5 abrogates neutralizing capacity without affecting core epitope binding. J Virol 84:4136–4147. doi: 10.1128/JVI.02357-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Evans P. 2006. Scaling and assessment of data quality. Acta Crystallogr D 62:72–82. doi: 10.1107/S0907444905036693. [DOI] [PubMed] [Google Scholar]
- 25.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. 2007. Phaser crystallographic software. J Appl Crystallogr 40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Murshudov GN, Vagin AA, Dodson EJ. 1997. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D 53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 27.Emsley P, Lohkamp B, Scott WG, Cowtan K. 2010. Features and development of Coot. Acta Crystallogr D 66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. 1993. PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Crystallogr 26:283–291. doi: 10.1107/S0021889892009944. [DOI] [Google Scholar]
- 29.Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AGW, McCoy A, McNicholas SJ, Murshudov GN, Pannu NS, Potterton EA, Powell HR, Read RJ, Vagin A, Wilson KS. 2011. Overview of the CCP4 suite and current developments. Acta Crystallogr D 67:235–242. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. 2010. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D 66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gore S, Velankar S, Kleywegt GJ. 2012. Implementing an X-ray validation pipeline for the Protein Data Bank. Acta Crystallogr D 68:478–483. doi: 10.1107/S0907444911050359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Langer G, Cohen SX, Lamzin VS, Perrakis A. 2008. Automated macromolecular model building for X-ray crystallography using ARP/wARP version 7. Nat Protoc 3:1171–1179. doi: 10.1038/nprot.2008.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fraser JS, Clarkson MW, Degnan SC, Erion R, Kern D, Alber T. 2009. Hidden alternative structures of proline isomerase essential for catalysis. Nature 462:669–673. doi: 10.1038/nature08615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lang PT, Ng HL, Fraser JS, Corn JE, Echols N, Sales M, Holton JM, Alber T. 2010. Automated electron-density sampling reveals widespread conformational polymorphism in proteins. Protein Sci 19:1420–1431. doi: 10.1002/pro.423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Miyafusa T, Caaveiro JMM, Tanaka Y, Tsumoto K. 2013. Dynamic elements govern the catalytic activity of CapE, a capsular polysaccharide-synthesizing enzyme from Staphylococcus aureus. FEBS Lett 587:3824–3830. doi: 10.1016/j.febslet.2013.10.009. [DOI] [PubMed] [Google Scholar]
- 36.Sakamoto S, Caaveiro JMM, Sano E, Tanaka Y, Kudou M, Tsumoto K. 2009. Contributions of interfacial residues of human interleukin15 to the specificity and affinity for its private α-receptor. J Mol Biol 389:880–894. doi: 10.1016/j.jmb.2009.04.050. [DOI] [PubMed] [Google Scholar]
- 37.Cleland WW, Northrop DB. 1999. Energetics of substrate binding, catalysis, and product release. Methods Enzymol 308:3–27. doi: 10.1016/S0076-6879(99)08003-9. [DOI] [PubMed] [Google Scholar]
- 38.Seaman MS, Janes H, Hawkins N, Grandpre LE, Devoy C, Giri A, Coffey RT, Harris L, Wood B, Daniels MG, Bhattacharya T, Lapedes A, Polonis VR, McCutchan FE, Gilbert PB, Self SG, Korber BT, Montefiori DC, Mascola JR. 2010. Tiered categorization of a diverse panel of HIV-1 Env pseudoviruses for assessment of neutralizing antibodies. J Virol 84:1439–1452. doi: 10.1128/JVI.02108-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lawrence MC, Colman PM. 1993. Shape complementarity at protein-protein interfaces. J Mol Biol 234:946–950. doi: 10.1006/jmbi.1993.1648. [DOI] [PubMed] [Google Scholar]
- 40.Klein JS, Gnanapragasam PNP, Galimidi RP, Foglesong CP, West AP, Bjorkman PJ. 2009. Examination of the contributions of size and avidity to the neutralization mechanisms of the anti-HIV antibodies b12 and 4E10. Proc Natl Acad Sci U S A 106:7385–7390. doi: 10.1073/pnas.0811427106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Leaman DP, Lee JH, Ward AB, Zwick MB. 2015. Immunogenic display of purified chemically cross-linked HIV-1 spikes. J Virol 89:6725–6745. doi: 10.1128/JVI.03738-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Krissinel E, Henrick K. 2007. Inference of macromolecular assemblies from crystalline state. J Mol Biol 372:774–797. doi: 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- 43.van den Bedem H, Bhabha G, Yang K, Wright PE, Fraser JS. 2013. Automated identification of functional dynamic contact networks from X-ray crystallography. Nat Methods 10:896–U110. doi: 10.1038/nmeth.2592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jones TA, Zou JY, Cowan SW, Kjeldgaard M. 1991. Improved methods for building protein models in electron-density maps and the location of errors in these models. Acta Crystallogr A 47:110–119. doi: 10.1107/S0108767390010224. [DOI] [PubMed] [Google Scholar]
- 45.Huarte N, Lorizate M, Maeso R, Kunert R, Arranz R, Valpuesta JM, Nieva JL. 2008. The broadly neutralizing anti-human immunodeficiency virus type 1 4E10 monoclonal antibody is better adapted to membrane-bound epitope recognition and blocking than 2F5. J Virol 82:8986–8996. doi: 10.1128/JVI.00846-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lorizate M, Cruz A, Huarte N, Kunert R, Perez-Gil J, Nieva JL. 2006. Recognition and blocking of HIV-1 gp41 pre-transmembrane sequence by monoclonal 4E10 antibody in a raft-like membrane environment. J Biol Chem 281:39598–39606. doi: 10.1074/jbc.M605998200. [DOI] [PubMed] [Google Scholar]
- 47.Sanchez-Martinez S, Lorizate M, Katinger H, Kunert R, Nieva JL. 2006. Membrane association and epitope recognition by HIV-1 neutralizing anti-gp41 2F5 and 4E10 antibodies. AIDS Res Hum Retroviruses 22:998–1006. doi: 10.1089/aid.2006.22.998. [DOI] [PubMed] [Google Scholar]
- 48.Sun ZY, Oh KJ, Kim M, Yu J, Brusic V, Song L, Qiao Z, Wang JH, Wagner G, Reinherz EL. 2008. HIV-1 broadly neutralizing antibody extracts its epitope from a kinked gp41 ectodomain region on the viral membrane. Immunity 28:52–63. doi: 10.1016/j.immuni.2007.11.018. [DOI] [PubMed] [Google Scholar]
- 49.Ofek G, McKee K, Yang YP, Yang ZY, Skinner J, Guenaga FJ, Wyatt R, Zwick MB, Nabel GJ, Mascola JR, Kwong PD. 2010. Relationship between antibody 2F5 neutralization of HIV-1 and hydrophobicity of its heavy chain third complementarity-determining region. J Virol 84:2955–2962. doi: 10.1128/JVI.02257-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Burton DR, Mascola JR. 2015. Antibody responses to envelope glycoproteins in HIV-1 infection. Nat Immunol 16:571–576. doi: 10.1038/ni.3158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Klein F, Mouquet H, Dosenovic P, Scheid JF, Scharf L, Nussenzweig MC. 2013. Antibodies in HIV-1 vaccine development and therapy. Science 341:1199–1204. doi: 10.1126/science.1241144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Muster T, Steindl F, Purtscher M, Trkola A, Klima A, Himmler G, Ruker F, Katinger H. 1993. A conserved neutralizing epitope on gp41 of human immunodeficiency virus type 1. J Virol 67:6642–6647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Muster T, Guinea R, Trkola A, Purtscher M, Klima A, Steindl F, Palese P, Katinger H. 1994. Cross-neutralizing activity against divergent human immunodeficiency virus type 1 isolates induced by the gp41 sequence ELDKWAS. J Virol 68:4031–4034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Purtscher M, Trkola A, Gruber G, Buchacher A, Predl R, Steindl F, Tauer C, Berger R, Barrett N, Jungbauer A, et al. 1994. A broadly neutralizing human monoclonal antibody against gp41 of human immunodeficiency virus type 1. AIDS Res Hum Retroviruses 10:1651–1658. doi: 10.1089/aid.1994.10.1651. [DOI] [PubMed] [Google Scholar]
- 55.Stiegler G, Kunert R, Purtscher M, Wolbank S, Voglauer R, Steindl F, Katinger H. 2001. A potent cross-clade neutralizing human monoclonal antibody against a novel epitope on gp41 of human immunodeficiency virus type 1. AIDS Res Hum Retroviruses 17:1757–1765. doi: 10.1089/08892220152741450. [DOI] [PubMed] [Google Scholar]
- 56.Kunert R, Ruker F, Katinger H. 1998. Molecular characterization of five neutralizing anti-HIV type 1 antibodies: identification of nonconventional D segments in the human monoclonal antibodies 2G12 and 2F5. AIDS Res Hum Retroviruses 14:1115–1128. doi: 10.1089/aid.1998.14.1115. [DOI] [PubMed] [Google Scholar]
- 57.Wallace AC, Laskowski RA, Thornton JM. 1995. Ligplot: a program to generate schematic diagrams of protein ligand interactions. Prot Eng 8:127–134. doi: 10.1093/protein/8.2.127. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.