

ABSTRACT
Numerous experimental studies have demonstrated that CD8+ T cells contribute to immunity against influenza by limiting viral replication. It is therefore surprising that rigorous statistical tests have failed to find evidence of positive selection in the epitopes targeted by CD8+ T cells. Here we use a novel computational approach to test for selection in CD8+ T-cell epitopes. We define all epitopes in the nucleoprotein (NP) and matrix protein (M1) with experimentally identified human CD8+ T-cell responses and then compare the evolution of these epitopes in parallel lineages of human and swine influenza viruses that have been diverging since roughly 1918. We find a significant enrichment of substitutions that alter human CD8+ T-cell epitopes in NP of human versus swine influenza virus, consistent with the idea that these epitopes are under positive selection. Furthermore, we show that epitope-altering substitutions in human influenza virus NP are enriched on the trunk versus the branches of the phylogenetic tree, indicating that viruses that acquire these mutations have a selective advantage. However, even in human influenza virus NP, sites in T-cell epitopes evolve more slowly than do nonepitope sites, presumably because these epitopes are under stronger inherent functional constraint. Overall, our work demonstrates that there is clear selection from CD8+ T cells in human influenza virus NP and illustrates how comparative analyses of viral lineages from different hosts can identify positive selection that is otherwise obscured by strong functional constraint.
IMPORTANCE There is a strong interest in correlates of anti-influenza immunity that are protective against diverse virus strains. CD8+ T cells provide such broad immunity, since they target conserved viral proteins. An important question is whether T-cell immunity is sufficiently strong to drive influenza virus evolution. Although many studies have shown that T cells limit viral replication in animal models and are associated with decreased symptoms in humans, no studies have proven with statistical significance that influenza virus evolves under positive selection to escape T cells. Here we use comparisons of human and swine influenza viruses to rigorously demonstrate that human influenza virus evolves under pressure to fix mutations in the nucleoprotein that promote escape from T cells. We further show that viruses with these mutations have a selective advantage since they are preferentially located on the “trunk” of the phylogenetic tree. Overall, our results show that CD8+ T cells targeting nucleoprotein play an important role in shaping influenza virus evolution.
INTRODUCTION
Both arms of the adaptive immune system help control influenza virus replication: antibodies neutralize virus (1) and direct the clearance of infected cells (2), while CD8+ T cells kill infected cells that display viral peptides on their major histocompatibility complex (MHC) class I molecules (3, 4). While antibodies against the viral surface protein hemagglutinin (HA) provide the most potent protection when they are well matched to the virus strain (5–7), T cells offer broader protection against diverse strains since they tend to recognize epitopes in more conserved internal viral proteins such as nucleoprotein (NP) and matrix protein (M1) (3, 4, 8, 9).
Studies in both mice (10–14) and humans (9, 15, 16) have shown that preexisting influenza virus-specific CD8+ T cells reduce the severity of disease and enhance virus clearance. For instance, preexisting virus-specific CD8+ T cells were correlated with decreased symptoms in humans infected during the 2009 H1N1 pandemic (15). Similarly, T cells specific for NP were associated with a decreased incidence of symptomatic infection over a multiyear study of a large human cohort (9), and CD8 T-cell responses were correlated with recovery from severe H7N9 infection (16). Therefore, experimental and epidemiological work demonstrates that CD8+ T cells contribute to immunity against influenza.
Because humans are repeatedly infected with influenza over their lifetimes, one might expect viruses to be under evolutionary pressure to accumulate substitutions in epitopes targeted by immune memory. Indeed, there are numerous examples of the fixation of antibody escape mutations in HA (17, 18), consistent with the notion that this protein evolves under strong selection from antibodies. Several studies have also described influenza virus mutations that escape recognition by CD8+ T cells (19). In a mouse study, viral mutations arose that conferred T-cell escape in RAG-1-deficient mice expressing an influenza virus NP-specific T-cell receptor (TCR) (20). Rimmelzwaan and coworkers identified the fixation of mutations in NP of human H3N2 virus that mediated escape from CD8+ T cells by altering the epitope recognized by the T-cell receptor (21–23) or by abrogating binding of the epitope to MHC class I molecules (24). Valkenburg et al. described the emergence of CD8+ T-cell escape mutations in a persistently influenza-infected infant (25). These elegant studies demonstrate that influenza virus accumulates substitutions that escape CD8+ T cells as well as antibody-mediated immunity.
However, these studies do not prove that positive selection for CD8+ T-cell escape is an important driving force in the evolution of influenza virus, since many sites in the virus genome will fix substitutions given enough time (26–28). To rigorously establish the presence of positive selection, the field of molecular evolution has developed statistical tests to discern whether a subset of sites is evolving faster than expected. Most of these tests compute nonsynonymous and synonymous distances (referred to as dN and dS, respectively) and then test for sites with statistical evidence that the accumulation of nonsynonymous substitutions exceeds that of synonymous substitutions (dN/dS ratio of >1) (29, 30). These tests consistently find overwhelming evidence for positive selection in the antigenic sites of influenza virus hemagglutinin (31–33) but little evidence for positive selection in CD8+ T-cell epitopes (33). One study reported that CD8+ T-cell epitopes in NP have a higher dN/dS ratio than do other sites (34); however, that study made a pairwise comparison of two sequences only and included no tests for statistical significance. Below, we describe the use of several state-of-the-art tests to verify that CD8+ T-cell epitopes have neither an elevated frequency of sites with a dN/dS ratio of >1 nor an elevated rate of nonsynonymous substitutions. Therefore, by standard criteria, CD8+ T-cell epitopes are not under positive selection.
The results of these statistical tests for positive selection seem at odds with the extensive body of experimental work described above. We hypothesized that the discrepancy arises because known CD8+ T-cell epitopes are under strong functional constraint (34–37). If epitopes are highly constrained, then even strong positive selection might fail to elevate the rate of nonsynonymous substitutions in epitopes above that at less constrained nonepitope sites. To address this possibility, we developed new statistical tests that take advantage of the fact that some lineages of human influenza virus are paralleled by lineages of swine influenza virus that are not under selection from human CD8+ T cells. Using these tests, we show that CD8+ T-cell epitopes in NP evolve significantly faster in human influenza virus than in swine influenza virus. Furthermore, we show that substitutions in these epitopes are enriched on the trunk of the phylogenetic tree, indicating that viruses that acquire them have a selective advantage that promotes their evolutionary spread. Overall, our work provides clear statistical evidence that complements prior experimental studies showing that CD8+ T-cell epitopes are under selection in human influenza virus (22) and suggests that the failure of conventional tests to identify this selection is due to high levels of functional constraint in epitopes.
MATERIALS AND METHODS
Inference of phylogenetic trees and mutation counts.
M1 and NP protein-coding sequences were downloaded from the Influenza Virus Resource (http://www.ncbi.nlm.nih.gov/genomes/FLU/FLU.html) (38).
For human influenza virus, we assembled sequence sets by taking sequences for H1N1 (1918 to 1957), H2N2 (1957 to 1968), and H3N2 (1968 to 2013); if there were fewer than three sequences per year, we retained them all, and when there were more than three for a year, we randomly selected three to retain. For swine influenza virus, we similarly assembled sequence sets containing up to 3 sequences per subtype per year for H1N1 (1918 to 2013), H1N2 (1999 to 2013), and H3N2 (1998 to 2013). For swine influenza virus, the first available sequence is from 1933. We excluded sequences that were previously classified as being misannotated (39) or that were strong outliers based on molecular clock analysis using RAxML (40) and Path-O-Gen (http://tree.bio.ed.ac.uk/software/pathogen/). The sequence sets are in Tables S1 to S4 in the supplemental material.
There are gaps in sequence availability in earlier years (most prominently, there are no sequences from between 1918 and the early 1930s). Therefore, we have a reduced power to identify substitutions in these early years. However, since our comparisons are between human and swine influenza viruses, and since both lineages have similarly sparse sequences in these early years, these gaps seem unlikely to systematically bias our study, although they may reduce its power.
We translated the sequences and inferred separate human and swine influenza virus phylogenies for each protein using Bayesian Evolutionary Analysis by Sampling Trees (BEAST) (41) with a strict molecular clock, a Jones-Taylor-Thornton (JTT) (42) model of substitution, and a constant population size demographic model. Figure 1 shows the maximum clade credibility trees rendered with FigTree (http://tree.bio.ed.ac.uk/software/figtree/). The trunk of each tree (dark lines in Fig. 1) was defined by tracing from the most recent sequence back to the oldest sequence. We used a stochastic mapping technique (43–45) implemented via the “MarkovJumps” feature in BEAST to estimate the posterior mean number of substitutions at each site for each phylogenetic tree and along the trunk of each tree. The times to the most recent common ancestor referred to in the Fig. 1 legend were estimated by a BEAST analysis of the joint swine and human influenza virus lineages. The dates of fixation of the CD8+ T-cell escape substitutions characterized by Rimmelzwaan and colleagues (21–24) refer to estimates obtained previously (see Fig. 2 supplement 2 in reference 37). Table S9 in the supplemental material lists all substitutions that are present along the trunk of at least 90% of the trees sampled from the posterior for each viral protein and lineage along the with posterior mean estimate of the date at which the substitution was fixed on the trunk.
FIG 1.

Phylogenetic trees for human and swine influenza virus M1 and NP. Shown are maximum clade credibility trees for NP and M1. The swine influenza virus lineage is in blue, and the human influenza virus lineage is in red. The dark blue and red lines represent the trunk. The dotted black lines indicate that human and swine influenza virus lineages share a recent common ancestor (estimated times to the most recent common ancestor are 13 and 6 years for M1 and NP, respectively).
FIG 2.
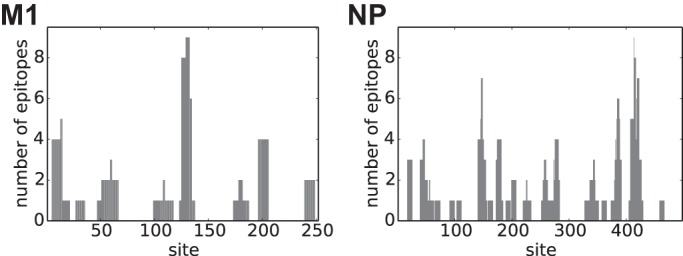
Distribution of CD8 T-cell epitopes in M1 and NP. The numbers of unique experimentally identified CD8+ T-cell epitopes to which each site contributes for M1 and NP are shown.
Identification of CD8+ T-cell epitope sites.
We downloaded all epitopes with an experimentally identified CD8+ T-cell response (source organism Influenza A virus and host Homo sapiens) from the Immune Epitope Database (http://www.iedb.org/) (46). We identified unique epitopes, as described in Results, using a previously described software package (https://github.com/jbloom/epitopefinder/) (47) and then determined the number of unique epitopes (Er) to which each site r contributes (Fig. 2). The epitopes and the counts of epitopes per site are in Tables S5 to S8 in the supplemental material.
Conventional dN/dS tests for positive selection.
We used DataMonkey (http://www.datamonkey.org/) (48) to perform two types of dN/dS analysis, the hierarchical Bayes method FUBAR (fast unconstrained Bayesian approximation) (30) and the maximum likelihood method FEL (fixed-effects likelihood) (29). The maximum clade credibility tree from BEAST was used as the input phylogeny for FUBAR and FEL, and a REV (general reversible model) codon substitution model was specified for FEL. For both methods, we calculated the percentage of sites for which the estimated dN/dS ratio was >1 and the percentage of sites for which there was strong statistical support for this ratio being >1 (posterior probability of >0.95 for FUBAR; P value of <0.05 for FEL).
Statistics for substitutions at each site in human and swine influenza virus lineages.
The posterior mean estimate of the number of nonsynonymous substitutions, Sr, at each site r was extracted from the BEAST trees. These estimates were used to compute the average substitution rates across all epitope sites (sites that fell into at least one epitope) and across all nonepitope sites, both for the entire tree and for the trunk alone. We also defined a statistic, F, which represents the average number of epitopes changed per substitution. This statistic is defined as
where Er is the number of unique epitopes to which site r contributes.
We performed statistical tests of whether we could reject the null hypothesis that there was no difference in the F statistics for human versus swine and for the trunk versus the tree. To do this, we calculated the ratios of these statistics for human versus swine or trunk versus tree and then created a null distribution by repeatedly recalculating the statistics after randomizing the epitope counts, Er, among sites. The P values represent the fraction of time that the randomized statistic is greater than the actual statistic in 104 randomizations.
Availability of data and computer codes.
Data and computer codes are available at https://github.com/hmmachko/TcellEpitopeComparisons.
RESULTS
Parallel human and swine influenza virus lineages reveal selection by CD8+ T cells.
Our goal is to determine whether epitopes targeted by human CD8+ T cells are under selection in influenza viruses that circulate in human hosts. The two most highly expressed influenza virus proteins are NP and M1 (49), and epitopes in these proteins are major targets of CD8+ T cells (3, 4, 8, 9). The NP and M1 proteins in contemporary human H3N2 influenza viruses have circulated in humans since at least 1918 (50, 51). For both genes, this unbroken lineage consists of H1N1 viruses from 1918 to 1957, H2N2 viruses from 1957 to 1968, and H3N2 viruses from 1968 to the present. The red lines in Fig. 1 show phylogenetic trees of NP and M1 from this human influenza virus lineage.
This human influenza virus lineage is closely paralleled by a swine influenza virus lineage descended from the common ancestor of the virus that caused concurrent pandemics in humans and swine in 1918 (50, 52). NP and M1 of this lineage have circulated exclusively in swine since 1918 (50, 52). The blue lines in Fig. 1 show phylogenetic trees of NP and M1 from this swine influenza virus lineage. The phylogenetic trees show that both human and swine influenza viruses undergo substantial genetic evolution in NP and M1; however, this fact alone does not reveal what forces drive this evolution. Influenza virus genetic evolution can be driven by positive selection, but it can also be driven by stochastic forces such as genetic hitchhiking or drift (26–28).
The parallel lineages of human and swine influenza viruses enable us to perform an internally controlled analysis of whether CD8+ T cells represent an important selective force in driving influenza virus evolution, since human CD8+ T cells target epitopes in human but not swine influenza viruses. There are two reasons why we can be confident that swine influenza virus is not under selection from human CD8+ T cells. First, the MHC class I molecules that restrict CD8+ T-cell epitopes are highly variable among species; therefore, epitopes displayed to human CD8+ T cells will differ from those displayed to swine CD8+ T cells (53, 54) (note that our approach does not require the human and swine epitopes to be completely nonoverlapping; it simply assumes that the MHC alleles are sufficiently diverged so that not all epitopes targeted by humans are also targeted by swine). Second, swine influenza virus is under weaker selection from immune memory than is human influenza virus because pigs are infected only once or a few times during their short lives (55–59). Therefore, swine influenza virus is probably under less pressure from CD8+ T cells in general, and whatever pressure does exist will generally focus on different epitopes than those targeted by human T cells.
Experimentally identified human CD8+ T-cell epitopes.
We aimed to identify CD8+ T-cell epitopes targeted by individuals in the human population. There are two plausible ways to do this: by computationally predicting peptides that bind to MHC class I or by collating epitopes that have been experimentally identified as eliciting responses from CD8+ T cells isolated from humans. We chose to use experimentally identified epitopes since computational predictions are imperfect (60), and only a fraction of peptides that bind MHC class I molecules are targets of cytolytic CD8+ T cells (61, 62). We extracted all influenza virus epitopes from the Immune Epitope Database (46) that are between 8 and 12 amino acids in length with an experimentally identified human CD8+ T-cell response. We retained all epitopes that aligned to at least one strain from our human and swine influenza virus lineages with no more than one amino acid mismatch. We classified epitopes as redundant if they shared 8 or more amino acids and were in the same MHC class I group (63) (or supertype [64] if the group was not specified). We identified 133 unique epitopes in the seven proteins that did not reassort during the human influenza pandemic of 1957 or 1968 (NS1, NS2, PB2, PA, M1, M2, and NP). Of 133 epitopes, 62 were in NP (47%), and 29 were in M1 (22%), consistent with reports that these two proteins are major targets of CD8+ T cells (9). Figure 2 shows the number of epitopes to which each site in NP and M1 contributes; individual sites are involved in anywhere between zero and nine epitopes.
These experimentally identified epitopes probably do not represent an exhaustive list of all sites targeted by human T cells. In particular, some epitopes in historical strains may be overlooked since most studies use recent virus strains. However, since our analyses are internally controlled (we compare either human to swine influenza viruses or the trunk of the tree to side branches), missing some epitopes should not systematically bias our results.
Our approach identifies sites that contribute to epitopes in any of the influenza virus strains under consideration. Mutations of an epitope can mediate escape by abrogating peptide binding to MHC class I or by changing the sequence of the bound peptide such that it is no longer recognized by memory T cells. Both types of escape have been experimentally demonstrated for NP of human H3N2 virus. An example of escape by abrogation of MHC binding is the R384G mutation that fixed in 1993 (24). Three examples of escape of T-cell recognition but not MHC binding are the D421E/I425V mutations that fixed in 1979 (22, 23), the K103R mutation that fixed in 1980 (21), and the S259L mutation that fixed in 1990 (21). Additionally, mutations outside an epitope can affect its processing (65), although we are unaware of documented examples of extraepitopic escape mutations that have fixed in human influenza virus. Our analysis cannot distinguish among these types of escape, since most experiments identify epitopes without characterizing how prior or subsequent mutations affect their processing, MHC binding, and recognition by T cells.
We therefore classify sites according to the number of epitopes to which they contribute in any of our virus strains without attempting to determine whether the epitopes are present across all the virus strains. This approach is usually valid when mutations alter T-cell recognition without affecting processing or MHC binding, since epitopes that escape existing T cells via such mutations will often soon be targeted by new T cells (21). However, our approach is imperfect for mutations that abrogate binding to MHC molecules and so eliminate the epitope from all subsequent strains. However, since the NP and M1 homologs are closely related (the maximal protein sequence divergence among the sequences in Fig. 1 is just 12%), even if a site fixes just a single mutation that eliminates an epitope, this would represent a substantial elevation in its rate of evolution over the time frame of interest. Therefore, classification of sites by the number of epitopes as in Fig. 2 should identify positions in NP and M1 that will exhibit an increased rate of substitution if there is T-cell selection.
Conventional dN/dS tests fail to find positive selection in CD8+ T-cell epitopes.
One might hypothesize that immune selection from CD8+ T cells would lead to a greater proportion of epitope than nonepitope sites with a dN/dS ratio of >1. We therefore used two state-of-the-art methods (a hierarchical Bayes approach [29] and a maximum likelihood approach [30]) to identify sites in human and swine influenza viruses with dN/dS ratios of >1 and partitioned the sites based on whether or not they were involved in at least one experimentally identified epitope. As shown in Fig. 3A and B, the proportion of sites with a dN/dS ratio of >1 was actually lower for epitope than for nonepitope sites in human influenza virus. Additionally, we calculated the proportion of sites with strong statistical evidence of a dN/dS ratio of >1 (Fig. 3C and D). In no instance is there a greater proportion of epitope than nonepitope sites with a dN/dS ratio of >1. Our findings are consistent with data from previous work that failed to find evidence for positively selected sites in NP or M1 (33). Thus, overall, state-of-the-art dN/dS tests fail to identify enhanced positive selection in T-cell epitopes in NP.
FIG 3.

Conventional dN/dS tests do not detect positive selection in T-cell epitopes. State-of-the-art methods for detecting positive selection fail to find any enrichment in sites with a dN/dS ratio of >1 at epitopes. (A and B) Percentages of sites estimated to have a dN/dS ratio of >1 by using the hierarchical Bayesian approach implemented in FUBAR (A) and the maximum likelihood approach implemented in FEL (B). (C) Percentages of sites for which FUBAR reports a posterior probability of >0.95 that the dN/dS ratio is >1. (D) Percentages of sites for which FEL reports a dN/dS ratio of >1 with a P value of <0.05.
Epitope sites do not evolve faster than nonepitope sites, although the rate is higher on the trunk of the human influenza virus NP lineage.
We next estimated the number of nonsynonymous substitutions at each site in NP and M1 for both the full swine and influenza virus trees in Fig. 1 and for the “trunks” of these trees (dark lines in Fig. 1). The rationale for examining the trunk separately is that we expect beneficial substitutions to be enriched on the trunk since they will confer a selective advantage that favors the propagation of sequences that contain them. Consistent with this idea, studies of human H3N2 influenza virus HA have found that presumably beneficial substitutions that alter antigenicity are enriched on the trunk versus the entire tree (66, 67).
Figure 4 shows the ratios of the substitution rates at epitope versus nonepitope sites. In none of the cases (NP or M1, swine or human influenza virus, or trunk or tree) is the ratio substantially greater than 1 when taken over the whole tree, so in no case are the epitope sites evolving faster than the nonepitope ones. This result helps explain why the dN/dS tests fail to find evidence for positive selection in the epitopes: for NP and M1 (unlike for HA), epitope sites simply do not evolve faster than their nonepitope counterparts.
FIG 4.
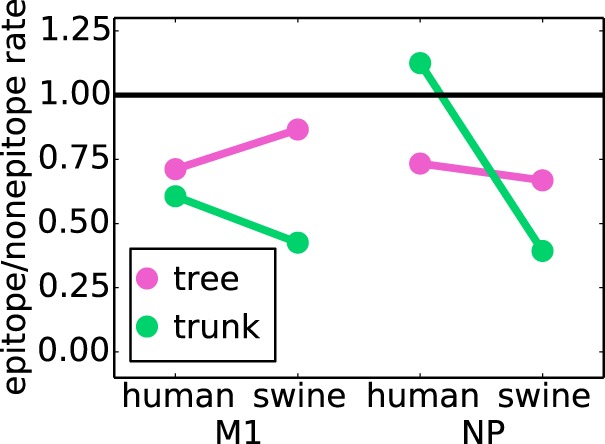
Epitope sites do not evolve faster than nonepitope sites. The ratios of the average nonsynonymous substitution rate of epitope sites to that of nonepitope sites are shown. When the substitution rate is computed across the entire tree, the epitope sites always have a lower substitution rate than the nonepitope sites. However, along the trunk of the tree for NP from human influenza virus, the substitution rate in epitopes is slightly higher than the substitution rate at nonepitopes.
However, further examination of the data in Fig. 4 reveals an interesting trend. Although there are no instances where epitope sites are evolving substantially faster than nonepitope ones, the ratio of epitope to nonepitope substitution rates is higher for NP of human influenza virus than for NP of swine influenza virus. This trend is particularly pronounced along the trunk of the tree, which is exactly where we would expect to see the largest increase in rates if epitope substitutions confer a selective advantage. To test if this trend was indicative of statistically significant CD8+ T-cell selection in NP, we undertook a more nuanced analysis, as described below.
Substitutions alter more NP epitopes in human than in swine influenza virus, especially on the trunk.
The above-described analyses simply subdivided sites as epitope or nonepitope sites based on whether they fall into at least one experimentally identified epitope. However, as shown in Fig. 2, some sites are involved in far more unique epitopes than others. To account for this, we defined a new statistic (which we denote F) that gives the average number of unique epitopes that are altered per substitution. Figure 5A shows this F statistic for the trunk and entire tree for NP and M1 of both human and swine influenza viruses. There appears to be little difference in this F statistic for M1 between the trunk and the entire tree and between human and swine influenza viruses. However, for NP, F is greater for human influenza virus than for swine influenza virus, with the increase being much larger for the trunk than for the entire tree. This result indicates that the average substitution alters more NP epitopes in human influenza virus than in swine influenza virus and that this trend is especially pronounced on the trunk of the tree, exactly as we would expect under selection for epitope-altering substitutions.
FIG 5.
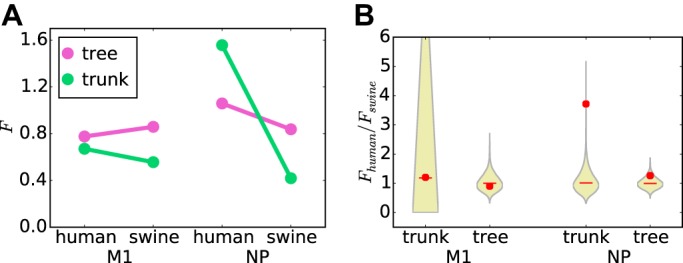
The average substitution changes more NP epitopes in human influenza than swine influenza virus. The F statistic is the average number of epitopes altered per substitution. (A) The average substitution alters a similar number of epitopes in M1 proteins of both human and swine influenza viruses and for both the entire tree and the trunk alone. However, for NP, the average substitution alters substantially more epitopes in human than in swine influenza virus, particularly along the trunk of the tree. (B) The increased rate of epitope-altering substitutions in NP along the trunk of human versus swine influenza virus is statistically significant. The violin plots show the null distribution of the ratio of F for human influenza virus to that for swine influenza virus, with the median shown by the red lines and the actual value shown by red circles. The P values (fraction of the null distribution that is greater than or equal to the actual value) are 0.49 for the M1 trunk, 0.65 for the M1 tree, 0.0002 for the NP trunk, and 0.098 for the NP tree.
To test if the trend is statistically significant, we computed the ratio of F for human influenza virus to that for swine influenza virus and then calculated a null distribution by repeatedly randomizing the epitopes among sites. For M1, the actual ratio falls near the center of the null distribution (Fig. 5B), confirming that there is no enhancement of the rate of fixation of epitope-altering substitutions in M1 of human influenza virus. However, for NP, the actual ratio falls near the top the null distribution for both the trunk and the tree (Fig. 5B). In particular, for the trunk, there is strong statistical support (P = 0.0002) for rejecting the null hypothesis that epitope-altering substitutions are equally likely to fix in human and swine influenza viruses. This result indicates that there is selection for substitutions that alter CD8+ T-cell epitopes in NP of human influenza virus.
To corroborate this statistical finding of an enhanced rate of epitope-altering substitutions along the trunk of human versus swine influenza virus, we undertook a further subanalysis of the small subset of epitopes for which specific T-cell escape mutations have been identified. The vast majority of epitopes that comprise the analysis shown in Fig. 5 were identified by relatively high-throughput studies that characterized peptides eliciting T-cell responses without identifying escape mutations. However, a series of meticulous studies by Rimmelzwaan and coworkers (21, 22, 24) identified sites of specific escape mutations for a small number of epitopes. Table 1 shows that substitutions are fixed at all of these sites along the trunk of the human influenza virus tree but that swine influenza virus has no fixed substitutions at any of these sites. This finding lends further support to the idea that human T cells exert positive selection on human but not swine influenza virus.
TABLE 1.
Trunk substitutions for the small subset of NP epitopes where specific escape substitutions have been experimentally validateda
| Position in NP | Experimental evidence (reference) | No. of trunk substitutions for human influenza virus (substitution[s] and yr of isolation) |
|---|---|---|
| 103 | K103R escapes recognition by a HLA-B*1503-restricted T cell (21) | 2 (K103R in 1981, R103K in 1997) |
| 259 | S259L escapes recognition by a HLA-B*4002-restricted T cell (21) | 2 (L259S in 1972, S259L in 1990) |
| 384 | R384G abrogates MHC binding in HLA-B*2705 (24) | 1 (R384G in 1990) |
| 421 | D421E escapes recognition by an HLA-B*3501-restricted T cell (22) | 1 (D421E in 1977) |
| 425 | I425V escapes recognition by an HLA-B*3501-restricted T cell (22) | 2 (I425V in 1975, V425I in 1999) |
There were no trunk substitutions for swine influenza virus at any of these five sites.
We next tested whether selection for epitope-altering substitutions in human influenza virus NP was stronger on the trunk than on the rest of the tree, as would be expected if such substitutions confer a selective advantage. We computed the ratio of the F statistic for the trunk to that for the entire tree and generated null distributions for this statistic by randomizing the epitopes among sites (Fig. 6). For M1, the actual ratios are near the center of the null distributions. However, for NP, the actual ratio is near the top of the null distribution (P = 0.003) for human influenza virus and near the bottom for swine influenza virus (P = 0.01). This result demonstrates that epitope-altering substitutions in NP are significantly enriched on the trunk of the human influenza virus lineage, suggesting that viruses that fix these mutations are more fit than other strains. The depletion of epitope-altering substitutions in the swine lineage can also be given a clear explanation: if the known CD8+ T-cell epitopes in NP are in functionally constrained regions of the protein (as has been suggested by a variety of experimental studies [34–37]), then substitutions in these epitopes will often be deleterious in swine influenza virus lineages that experience no human CD8+ T-cell selection and so will be relatively depleted on the trunk. Thus, overall, the results described above provide strong statistical evidence of positive selection in the CD8+ T-cell epitopes of human influenza virus NP.
FIG 6.
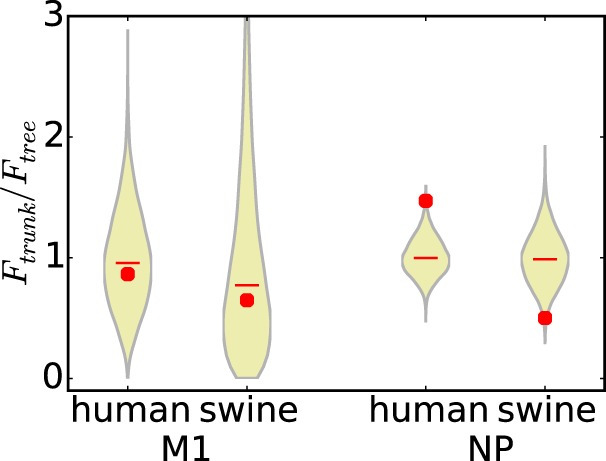
Epitope-altering substitutions in human influenza virus NP are enriched on the trunk of the tree. Ratios of the average number of epitopes altered per substitution (F) for the trunk to that for the entire tree are shown. The violin plots show the actual values versus the null distributions, as described in the legend of Fig. 5B. There is no difference between the trunk and the entire tree for M1 (P values are 0.59 for M1 of human influenza virus and 0.57 for M1 of swine influenza virus). However, for NP, epitope-altering substitutions are significantly enriched on the trunk of the human influenza virus tree relative to the rest of the tree (P = 0.003) and significantly depleted on the trunk of the swine influenza virus lineage (P = 0.01 for depletion) relative to the rest of the tree.
DISCUSSION
We describe the first rigorous statistical evidence that CD8+ T-cell epitopes are under positive selection in human influenza virus. Our work adds to a growing body of evidence suggesting an important role for T-cell immunity in shaping influenza virus evolution. Previous studies showed that T cells help protect against human influenza virus (9, 15) and detailed specific instances of T-cell escape (21–25). Our work shows that T-cell selection increases the rate at which mutations are fixed in epitopes of NP and indicates that viruses with these substitutions have a selective advantage that makes them more likely to fall along the trunk of the phylogenetic tree.
Our results also explain why conventional dN/dS tests fail to detect positive selection in CD8+ T-cell epitopes. Known human CD8+ T-cell epitopes tend to be under strong functional constraint (34–37). It is unclear whether this is because T cells inherently target conserved epitopes, because repeated infections preferentially boost T cells that target conserved epitopes, or because there is a bias toward experimentally identifying conserved epitopes. However, in any case, the fact that known epitopes are under strong constraint means that even fairly strong positive selection may not enhance the nonsynonymous substitution rate to a level detectable by conventional dN/dS tests. This contrasts with antibody epitopes in HA, where the ability of dN/dS tests to detect antibody-mediated positive selection is probably augmented by the fact that antigenic sites are disproportionately tolerant of point mutations (68).
The novel approach that we developed ameliorates this problem by comparing the evolution of epitopes of human and swine influenza viruses or the entire phylogenetic tree and only its trunk. These comparisons should better control for site-to-site variation in functional constraint, since comparisons are always made between homologous sites that should be subject to similar functional constraints. Admittedly, there may also be other differences in functional constraints between human and swine influenza viruses beyond T cells, but unless these differential constraints are systematically biased toward occurring at T-cell epitopes, they should not alter the fundamental validity of our approach. By making comparisons in this way, we demonstrated clear positive selection in CD8+ T-cell epitopes in NP, both in human versus swine influenza viruses and in the trunk versus the entire phylogenetic tree.
One interesting aspect of our study is that we found positive selection in NP but not M1. This finding is consistent with a recent large-scale study that found that NP was the only protein for which the presence of preexisting memory T cells correlated with decreased rates of symptomatic infections (9). However, our study does not preclude an important role of T cells targeting M1, which contains an immunodominant HLA-A2 epitope spanning residues 58 to 66 (69, 70). One study argued that T cells targeting this epitope are ineffectual (70), although this interpretation is disputed (71, 72). However, experiments have also shown that this epitope is under strong constraint (34). If an epitope is completely intolerant of mutations, it will of course be unable to accumulate substitutions regardless of the strength of selection. It remains unclear if our failure to detect positive selection in M1 reflects a lack of effective immunity targeting this protein or strong constraints that simply prevent the fixation of escape mutations.
It is well established that antibodies are strong drivers of repeated selective sweeps in the evolution of human influenza virus (66, 73). The fact that we can detect positive selection by CD8+ T cells even in the presence of these antibody-driven selective sweeps demonstrates the importance of T-cell immunity in driving viral evolution. The existence of such selection is consistent with modeling studies showing that T-cell immunity that reduces the infectious period can strongly favor viral escape (74).
There is considerable interest in developing vaccines that elicit stronger T-cell immunity to better protect against diverse influenza virus strains (3). Our demonstration of the evolutionary importance of T-cell selection suggests that this interest is well founded. In addition, our results suggest that attempts to forecast the seasonal evolution of influenza (75, 76) could benefit from examining changes in T-cell as well as antibody epitopes.
Supplementary Material
ACKNOWLEDGMENTS
The research reported here was supported by the NIGMS of the National Institutes of Health under grants R01GM102198 (to J.D.B.) and MIDAS U01GM110721 (to T.B. and J.D.B). H.M.M. was supported in part by training grant T32AI083203 from the NIAID of the National Institutes of Health.
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.01571-15.
REFERENCES
- 1.Knossow M, Gaudier M, Douglas A, Barrère B, Bizebard T, Barbey C, Gigant B, Skehel JJ. 2002. Mechanism of neutralization of influenza virus infectivity by antibodies. Virology 302:294–298. doi: 10.1006/viro.2002.1625. [DOI] [PubMed] [Google Scholar]
- 2.Jegaskanda S, Job ER, Kramski M, Laurie L, Isitman G, de Rose R, Winnall WR, Stratov I, Brooks AG, Reading PC, Kent SJ. 2013. Cross-reactive influenza-specific antibody-dependent cellular cytotoxicity antibodies in the absence of neutralizing antibodies. J Immunol 190:1837–1848. doi: 10.4049/jimmunol.1201574. [DOI] [PubMed] [Google Scholar]
- 3.Valkenburg SA, Rutigliano JA, Ellebedy AH, Doherty PC, Thomas PG, Kedzierska K. 2011. Immunity to seasonal and pandemic influenza A viruses. Microbes Infect 13:489–501. doi: 10.1016/j.micinf.2011.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rimmelzwaan GF, Kreijtz JHCM, Bodewes R, Fouchier RAM, Osterhaus ADME. 2009. Influenza virus CTL epitopes, remarkably conserved and remarkably variable. Vaccine 27:6363–6365. doi: 10.1016/j.vaccine.2009.01.016. [DOI] [PubMed] [Google Scholar]
- 5.Virelizier JL. 1975. Host defenses against influenza virus: the role of anti-hemagglutinin antibody. J Immunol 115:434–439. [PubMed] [Google Scholar]
- 6.Clements ML, Betts RF, Tierney EL, Murphy BR. 1986. Serum and nasal wash antibodies associated with resistance to experimental challenge with influenza A wild-type virus. J Clin Microbiol 24:157–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Couch RB, Kasel JA. 1983. Immunity to influenza in man. Annu Rev Microbiol 37:529–549. doi: 10.1146/annurev.mi.37.100183.002525. [DOI] [PubMed] [Google Scholar]
- 8.Yewdell JW, Bennink JR, Smith GL, Moss B. 1985. Influenza A virus nucleoprotein is a major target antigen for cross-reactive anti-influenza A virus cytotoxic T lymphocytes. Proc Natl Acad Sci U S A 82:1785–1789. doi: 10.1073/pnas.82.6.1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hayward AC, Wang L, Goonetilleke N, Fragaszy EB, Bermingham A, Andrew C, Oliver D, Millett ER, Nazareth I, Nguyen-Van-Tam JS, Watson JM, Zambon M, Flu Watch Group, Johnson AM, McMichael AJ. 2015. Natural T cell-mediated protection against seasonal and pandemic influenza. Am J Respir Crit Care Med 191:1422–1431. doi: 10.1164/rccm.201411-1988OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Guo H, Santiago F, Lambert K, Takimoto T, Topham DJ. 2011. T cell-mediated protection against lethal 2009 pandemic H1N1 influenza virus infection in a mouse model. J Virol 85:448–455. doi: 10.1128/JVI.01812-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Laidlaw BJ, Decman V, Ali MAA, Abt MC, Wolf AI, Monticelli LA, Mozdzanowska K, Angelosanto JM, Artis D, Erikson J, Wherry EJ. 2013. Cooperativity between CD8+ T cells, non-neutralizing antibodies, and alveolar macrophages is important for heterosubtypic influenza virus immunity. PLoS Pathog 9:e1003207. doi: 10.1371/journal.ppat.1003207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kreijtz JHCM, Bodewes R, van Amerongen G, Kuiken T, Fouchier RAM, Osterhaus ADME, Rimmelzwaan GF. 2007. Primary influenza A virus infection induces cross-protective immunity against a lethal infection with a heterosubtypic virus strain in mice. Vaccine 25:612–620. doi: 10.1016/j.vaccine.2006.08.036. [DOI] [PubMed] [Google Scholar]
- 13.Taylor PM, Askonas BA. 1986. Influenza nucleoprotein-specific cytotoxic T-cell clones are protective in vivo. Immunology 58:417–420. [PMC free article] [PubMed] [Google Scholar]
- 14.Epstein SL, Lo CY, Misplon JA, Bennink JR. 1998. Mechanism of protective immunity against influenza virus infection in mice without antibodies. J Immunol 160:322–327. [PubMed] [Google Scholar]
- 15.Sridhar S, Begom S, Bermingham A, Hoschler K, Adamson W, Carman W, Bean T, Barclay W, Deeks JJ, Lalvani A. 2013. Cellular immune correlates of protection against symptomatic pandemic influenza. Nat Med 19:1305–1312. doi: 10.1038/nm.3350. [DOI] [PubMed] [Google Scholar]
- 16.Wang Z, Wan W, Qiu C, Quiñones-Parra S, Zhu Z, Loh L, Tian D, Ren Y, Hu Y, Zhang X, Thomas PG, Inouye M, Doherty PC, Kedzierska K, Xu J. 2015. Recovery from severe H7N9 disease is associated with diverse response mechanisms dominated by CD8+ T cells. Nat Commun 6:6833. doi: 10.1038/ncomms7833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Smith DJ, Lapedes AS, de Jong JC, Bestebroer TM, Rimmelzwaan GF, Osterhaus ADME, Fouchier RAM. 2004. Mapping the antigenic and genetic evolution of influenza virus. Science 305:371–376. doi: 10.1126/science.1097211. [DOI] [PubMed] [Google Scholar]
- 18.Both GW, Sleigh MJ, Cox NJ, Kendal AP. 1983. Antigenic drift in influenza virus H3 hemagglutinin from 1968 to 1980: multiple evolutionary pathways and sequential amino acid changes at key antigenic sites. J Virol 48:52–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rimmelzwaan GF, Boon AC, Voeten JT, Berkhoff EG, Fouchier RA, Osterhaus AD. 2004. Sequence variation in the influenza A virus nucleoprotein associated with escape from cytotoxic T lymphocytes. Virus Res 103:97–100. doi: 10.1016/j.virusres.2004.02.020. [DOI] [PubMed] [Google Scholar]
- 20.Price GE, Ou R, Jiang H, Huang L, Moskophidis D. 2000. Viral escape by selection of cytotoxic T cell-resistant virus variants in vivo. J Exp Med 191:1853–1867. doi: 10.1084/jem.191.11.1853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Berkhoff EGM, Geelhoed-Mieras MM, Fouchier RAM, Osterhaus ADME, Rimmelzwaan GF. 2007. Assessment of the extent of variation in influenza A virus cytotoxic T-lymphocyte epitopes by using virus-specific CD8+ T-cell clones. J Gen Virol 88:530–535. doi: 10.1099/vir.0.82120-0. [DOI] [PubMed] [Google Scholar]
- 22.Boon ACM, de Mutsert G, Graus YMF, Fouchier RAM, Sintnicolaas K, Osterhaus ADME, Rimmelzwaan GF. 2002. Sequence variation in a newly identified HLA-B35-restricted epitope in the influenza A virus nucleoprotein associated with escape from cytotoxic T lymphocytes. J Virol 76:2567–2572. doi: 10.1128/jvi.76.5.2567-2572.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Boon AC, de Mutsert G, van Baarle D, Smith DJ, Lapedes AS, Fouchier RA, Sintnicolaas K, Osterhaus AD, Rimmelzwaan GF. 2004. Recognition of homo- and heterosubtypic variants of influenza A viruses by human CD8+ T lymphocytes. J Immunol 172:2453–2460. doi: 10.4049/jimmunol.172.4.2453. [DOI] [PubMed] [Google Scholar]
- 24.Voeten JT, Bestebroer TM, Nieuwkoop NJ, Fouchier RA, Osterhaus AD, Rimmelzwaan GF. 2000. Antigenic drift in the influenza A virus (H3N2) nucleoprotein and escape from recognition by cytotoxic T lymphocytes. J Virol 74:6800–6807. doi: 10.1128/JVI.74.15.6800-6807.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Valkenburg SA, Quiñones-Parra S, Gras S, Komadina N, McVernon J, Wang Z, Halim H, Iannello P, Cole C, Laurie K, Kelso A, Rossjohn J, Doherty PC, Turner SJ, Kedzierska K. 2013. Acute emergence and reversion of influenza A virus quasispecies within CD8+ T cell antigenic peptides. Nat Commun 4:2663. doi: 10.1038/ncomms3663. [DOI] [PubMed] [Google Scholar]
- 26.Pybus OG, Rambaut A, Belshaw R, Freckleton RP, Drummond AJ, Holmes EC. 2007. Phylogenetic evidence for deleterious mutation load in RNA viruses and its contribution to viral evolution. Mol Biol Evol 24:845–852. [DOI] [PubMed] [Google Scholar]
- 27.Simonsen L, Viboud C, Grenfell BT, Dushoff J, Jennings L, Smit M, Macken C, Hata M, Gog J, Miller MA, Holmes EC. 2007. The genesis and spread of reassortment human influenza A/H3N2 viruses conferring adamantane resistance. Mol Biol Evol 24:1811–1820. doi: 10.1093/molbev/msm103. [DOI] [PubMed] [Google Scholar]
- 28.Nelson MI, Simonsen L, Viboud C, Miller MA, Taylor J, St George K, Griesemer SB, Ghedi E, Sengamalay NA, Spiro DJ, Volkov I, Grenfell BT, Lipman DJ, Taubenberger JK, Holmes EC. 2006. Stochastic processes are key determinants of short-term evolution in influenza A virus. PLoS Pathog 2:e125. doi: 10.1371/journal.ppat.0020125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kosakovsky Pond SL, Frost SDW. 2005. Not so different after all: a comparison of methods for detecting amino acid sites under selection. Mol Biol Evol 22:1208–1222. doi: 10.1093/molbev/msi105. [DOI] [PubMed] [Google Scholar]
- 30.Murrell B, Moola S, Mabona A, Weighill T, Sheward D, Kosakovsky Pond SL, Scheffler K. 2013. FUBAR: a fast, unconstrained Bayesian approximation for inferring selection. Mol Biol Evol 30:1196–1205. doi: 10.1093/molbev/mst030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bush RM, Fitch WM, Bender CA, Cox NJ. 1999. Positive selection on the H3 hemagglutinin gene of human influenza virus A. Mol Biol Evol 16:1457–1465. doi: 10.1093/oxfordjournals.molbev.a026057. [DOI] [PubMed] [Google Scholar]
- 32.Shih AC-C, Hsiao T-C, Ho M-S, Li W-H. 2007. Simultaneous amino acid substitutions at antigenic sites drive influenza A hemagglutinin evolution. Proc Natl Acad Sci U S A 104:6283–6288. doi: 10.1073/pnas.0701396104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kosakovsky Pond SL, Poon AFY, Leigh Brown AJ, Frost SDW. 2008. A maximum likelihood method for detecting directional evolution in protein sequences and its application to influenza A virus. Mol Biol Evol 25:1809–1824. doi: 10.1093/molbev/msn123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Berkhoff EGM, de Wit E, Geelhoed-Mieras MM, Boon ACM, Symons J, Fouchier RAM, Osterhaus ADME, Rimmelzwaan GF. 2005. Functional constraints of influenza A virus epitopes limit escape from cytotoxic T lymphocytes. J Virol 79:11239–11246. doi: 10.1128/JVI.79.17.11239-11246.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Assarsson E, Bui H-H, Sidney J, Zhang Q, Glenn J, Oseroff C, Mbawuike IN, Alexander J, Newman MJ, Grey H, Sette A. 2008. Immunomic analysis of the repertoire of T-cell specificities for influenza A virus in humans. J Virol 82:12241–12251. doi: 10.1128/JVI.01563-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rimmelzwaan GF, Berkhoff EGM, Nieuwkoop NJ, Fouchier RAM, Osterhaus ADME. 2004. Functional compensation of a detrimental amino acid substitution in a cytotoxic-T-lymphocyte epitope of influenza A viruses by comutations. J Virol 78:8946–8949. doi: 10.1128/JVI.78.16.8946-8949.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gong LI, Suchard MA, Bloom JD. 2013. Stability-mediated epistasis constrains the evolution of an influenza protein. eLife 2:e00631. doi: 10.7554/eLife.00631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bao Y, Bolotov P, Dernovoy F, Kiryutin B, Zaslavsky L, Tatusova T, Ostell J, Lipman D. 2008. The Influenza Virus Resource at the National Center for Biotechnology Information. J Virol 82:596–601. doi: 10.1128/JVI.02005-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Krasnitz M, Levine AJ, Rabadan R. 2008. Anomalies in the influenza virus genome database: new biology or laboratory errors? J Virol 82:8947–8950. doi: 10.1128/JVI.00101-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stamatakis A. 2006. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22:2688–2690. doi: 10.1093/bioinformatics/btl446. [DOI] [PubMed] [Google Scholar]
- 41.Drummond AJ, Suchard MA, Xie D, Rambaut A. 2012. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol Biol Evol 29:1969–1973. doi: 10.1093/molbev/mss075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jones DT, Taylor WR, Thornton JM. 1992. The rapid generation of mutation data matrices from protein sequences. Comput Appl Biosci 8:275–282. [DOI] [PubMed] [Google Scholar]
- 43.Minin VN, Suchard MA. 2008. Counting labeled transitions in continuous-time Markov models of evolution. J Math Biol 56:391–412. [DOI] [PubMed] [Google Scholar]
- 44.O'Brien JD, Minin VN, Suchard MA. 2009. Learning to count: robust estimates for labeled distances between molecular sequences. Mol Biol Evol 26:801–814. doi: 10.1093/molbev/msp003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lemey P, Minin VN, Bielejec F, Pond SLK, Suchard MA. 2012. A counting renaissance: combining stochastic mapping and empirical Bayes to quickly detect amino acid sites under positive selection. Bioinformatics 28:3248–3256. doi: 10.1093/bioinformatics/bts580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vita R, Overton JA, Greenbaum JA, Ponomarenko J, Clark JD, Cantrell JR, Wheeler DK, Gabbard JL, Hix D, Sette A, Peters B. 2015. The immune epitope database (IEDB) 3.0. Nucleic Acids Res 43:D405–D412. doi: 10.1093/nar/gku938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gong LI, Bloom JD. 2014. Epistatically interacting substitutions are enriched during adaptive protein evolution. PLoS Genet 10:e1004328. doi: 10.1371/journal.pgen.1004328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Delport W, Poon AFY, Frost SDW, Kosakovsky Pond SL. 2010. Datamonkey 2010: a suite of phylogenetic analysis tools for evolutionary biology. Bioinformatics 26:2455–2457. doi: 10.1093/bioinformatics/btq429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kummer S, Flottmann M, Schwanhausser B, Sieben C, Veit M, Selbach M, Klipp E, Herrmann A. 2014. Alteration of protein levels during influenza virus H1N1 infection in host cells: a proteomic survey of host and virus reveals differential dynamics. PLoS One 9:e94257. doi: 10.1371/journal.pone.0094257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dos Reis M, Hay AJ, Goldstein RA. 2009. Using non-homogeneous models of nucleotide substitution to identify host shift events: application to the origin of the 1918 ‘Spanish’ influenza pandemic virus. J Mol Evol 69:333–345. doi: 10.1007/s00239-009-9282-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cox NJ, Subbarao K. 2000. Global epidemiology of influenza: past and present. Annu Rev Med 51:407–421. doi: 10.1146/annurev.med.51.1.407. [DOI] [PubMed] [Google Scholar]
- 52.Brockwell-Staats C, Webster RG, Webby RJ. 2009. Diversity of influenza viruses in swine and the emergence of a novel human pandemic influenza A (H1N1). Influenza Other Respir Viruses 3:207–213. doi: 10.1111/j.1750-2659.2009.00096.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Renard C, Hart E, Sehra H, Beasley H, Coggill P, Howe K, Harrow J, Gilbert J, Sims S, Rogers J, Ando A, Shigenari A, Shiina T, Inoko H, Chardon P, Beck S. 2006. The genomic sequence and analysis of the swine major histocompatibility complex. Genomics 88:96–110. doi: 10.1016/j.ygeno.2006.01.004. [DOI] [PubMed] [Google Scholar]
- 54.Lienert K, Parham P. 1996. Evolution of MHC class I genes in higher primates. Immunol Cell Biol 74:349–356. doi: 10.1038/icb.1996.62. [DOI] [PubMed] [Google Scholar]
- 55.Sheerar MG, Easterday BC, Hinshaw VS. 1989. Antigenic conservation of H1N1 swine influenza viruses. J Gen Virol 70:3297–3303. doi: 10.1099/0022-1317-70-12-3297. [DOI] [PubMed] [Google Scholar]
- 56.Noble S, McGregor MS, Wentworth DE, Hinshaw VS. 1993. Antigenic and genetic conservation of the haemagglutinin in H1N1 swine influenza viruses. J Gen Virol 74:1197–1200. doi: 10.1099/0022-1317-74-6-1197. [DOI] [PubMed] [Google Scholar]
- 57.Garten RJ, Davis CT, Russell CA, Shu B, Lindstrom S, Balish A, Sessions WM, Xu X, Skepner E, Deyde V, Okomo-Adhiambo M, Gubareva L, Barnes J, Smith CB, Emery SL, Hillman MJ, Rivailler P, Smagala J, de Graaf M, Burke DF, Fouchier RAM, Pappas C, Alpuche-Aranda CM, López-Gatell H, Olivera H, López I, Myers CA, Faix D, Blair PJ, Yu C, Keene KM, Dotson PD, Boxrud D, Sambol AR, Abid SH, St George K, Bannerman T, Moore AL, Stringer DJ, Blevins P, Demmler-Harrison GJ, Ginsberg M, Kriner P, Waterman S, Smole S, Guevara HF, Belongia EA, Clark PA, Beatrice ST, Donis R, Katz J, et al. 2009. Antigenic and genetic characteristics of swine-origin 2009 A(H1N1) influenza viruses circulating in humans. Science 325:197–201. doi: 10.1126/science.1176225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wei CJ, Boyington JC, Dai K, Houser KV, Pearce MB, Kong W-P, Yang Z-Y, Tumpey TM, Nabel GJ. 2010. Cross-neutralization of 1918 and 2009 influenza viruses: role of glycans in viral evolution and vaccine design. Sci Transl Med 2:24ra21. doi: 10.1126/scitranslmed.3000799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Vincent AL, Lager KM, Ma W, Lekcharoensuk P, Gramer MR, Loiacono C, Richt JA. 2006. Evaluation of hemagglutinin subtype 1 swine influenza viruses from the United States. Vet Microbiol 118:212–222. doi: 10.1016/j.vetmic.2006.07.017. [DOI] [PubMed] [Google Scholar]
- 60.Roomp K, Antes I, Lengauer T. 2010. Predicting MHC class I epitopes in large datasets. BMC Bioinformatics 11:90. doi: 10.1186/1471-2105-11-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Deng Y, Yewdell JW, Eisenlohr LC, Bennink JR. 1997. MHC affinity, peptide liberation, T cell repertoire, and immunodominance all contribute to the paucity of MHC class I-restricted peptides recognized by antiviral CTL. J Immunol 158:1507–1515. [PubMed] [Google Scholar]
- 62.Yewdell JW. 2006. Confronting complexity: real-world immunodominance in antiviral CD8+ T cell responses. Immunity 25:533–543. doi: 10.1016/j.immuni.2006.09.005. [DOI] [PubMed] [Google Scholar]
- 63.Anthony Nolan Research Institute. 2015. HLA nomenclature. Anthony Nolan Research Institute, London, United Kingdom: http://hla.alleles.org/nomenclature/naming.html. [Google Scholar]
- 64.Sidney J, Peters B, Frahm N, Brander C, Sette A. 2008. HLA class I supertypes: a revised and updated classification. BMC Immunol 9:1. doi: 10.1186/1471-2172-9-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Allen TM, Altfeld M, Yu XG, O'Sullivan KM, Lichterfeld M, Le Gall S, John M, Mothe BR, Lee PK, Kalife ET, Cohen DE, Freedberg KA, Strick DA, Johnston MN, Sette A, Rosenberg ES, Mallal SA, Goulder PJR, Brander C, Walker BD. 2004. Selection, transmission, and reversion of an antigen-processing cytotoxic T-lymphocyte escape mutation in human immunodeficiency virus type 1 infection. J Virol 78:7069–7078. doi: 10.1128/JVI.78.13.7069-7078.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bedford T, Suchard MA, Lemey P, Dudas G, Gregory V, Hay AJ, McCauley JW, Russell CA, Smith DJ, Rambaut A. 2014. Integrating influenza antigenic dynamics with molecular evolution. eLife 3:e01914. doi: 10.7554/eLife.01914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bedford T, Rambaut A, Pascual M. 2012. Canalization of the evolutionary trajectory of the human influenza virus. BMC Biol 10:38. doi: 10.1186/1741-7007-10-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Thyagarajan B, Bloom JD. 2014. The inherent mutational tolerance and antigenic evolvability of influenza hemagglutinin. eLife 3:e03300. doi: 10.7554/eLife.03300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Boon ACM, de Mutsert G, Graus YMF, Fouchier RAM, Sintnicolaas K, Osterhaus ADME, Rimmelzwaan GF. 2002. The magnitude and specificity of influenza A virus-specific cytotoxic T-lymphocyte responses in humans is related to HLA-A and -B phenotype. J Virol 76:582–590. doi: 10.1128/JVI.76.2.582-590.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Keskin DB, Reinhold BB, Zhang GL, Ivanov AR, Karger BL, Reinherz EL. 2015. Physical detection of influenza A epitopes identifies a stealth subset on human lung epithelium evading natural CD8 immunity. Proc Natl Acad Sci U S A 112:2151–2156. doi: 10.1073/pnas.1423482112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.van de Sandt CE, Rimmelzwaan GF. 2015. Immunodominant responses to the influenza virus M1 58-66 epitope: stealth or protection? Proc Natl Acad Sci U S A 112:E2417. doi: 10.1073/pnas.1503245112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Keskin DB, Reinhold BR, Zhang GL, Ivanov AR, Karger BL, Reinherz EL. 2015. Reply to van de Sandt and Rimmelzwaan: matching epitope display with functional avidity. Proc Natl Acad Sci U S A 112:E2418. doi: 10.1073/pnas.1503931112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Rambaut A, Pybus OG, Nelson MI, Viboud C, Taubenberger JK, Holmes EC. 2008. The genomic and epidemiological dynamics of human influenza A virus. Nature 453:615–619. doi: 10.1038/nature06945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gog JR, Rimmelzwaan GF, Osterhaus ADME, Grenfell BT. 2003. Population dynamics of rapid fixation in cytotoxic T lymphocyte escape mutants of influenza A. Proc Natl Acad Sci U S A 100:11143–11147. doi: 10.1073/pnas.1830296100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Luksza M, Lassig M. 2014. A predictive fitness model for influenza. Nature 507:57–61. doi: 10.1038/nature13087. [DOI] [PubMed] [Google Scholar]
- 76.Neher RA, Russell CA, Shraiman BI. 2014. Predicting evolution from the shape of genealogical trees. eLife 3:e03568. doi: 10.7554/eLife.03568. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.