

ABSTRACT
Sendai virus (SeV) C protein inhibits the signal transduction pathways of interferon alpha/beta (IFN-α/β) and IFN-γ by binding to the N-terminal domain of STAT1 (STAT1ND), thereby allowing SeV to escape from host innate immunity. Here we determined the crystal structure of STAT1ND associated with the C-terminal half of the C protein (Y3 [amino acids 99 to 204]) at a resolution of 2.0 Å. This showed that two molecules of Y3 symmetrically bind to each niche created between two molecules of the STAT1ND dimer. Molecular modeling suggested that an antiparallel form of the full-length STAT1 dimer can bind only one Y3 molecule and that a parallel form can bind two Y3 molecules. Affinity analysis demonstrated anticooperative binding of two Y3 molecules with the STAT1 dimer, which is consistent with the hypothetical model that the second Y3 molecule can only target the STAT1 dimer in a parallel form. STAT1 with excess amounts of Y3 was prone to inhibit the dephosphorylation at Tyr701 by a phosphatase. In an electrophoretic mobility shift assay, tyrosine-phosphorylated STAT1 (pY-STAT1) with Y3 associated with the γ-activated sequence, probably as high-molecular-weight complexes (HMWCs), which may account for partial inhibition of a reporter assay from IFN-γ by Y3. Our study suggests that the full-length C protein interferes with the domain arrangement of the STAT1 dimer, leading to the accumulation of pY-STAT1 and the formation of HMWCs. In addition, we discuss the mechanism by which phosphorylation of STAT2 is inhibited in the presence of the C protein after stimulation by IFN-α/β.
IMPORTANCE Sendai virus, a paramyxovirus that causes respiratory diseases in rodents, possesses the C protein, which inhibits the signal transduction pathways of interferon alpha/beta (IFN-α/β) and IFN-γ by binding to the transcription factor STAT1. In virus-infected cells, phosphorylation of STAT1 at the Tyr701 residue is potently enhanced, although transcription by STAT1 is inert. Here, we determined the crystal structure of the N-terminal domain of STAT1 associated with the C-terminal half of the C protein. Molecular modeling and experiments suggested that the two C proteins bind to and stabilize the parallel form of the STAT1 dimer, which are likely to be phosphorylated at Tyr701, further inducing high-molecular-weight complex formation and inhibition of transcription by IFN-γ. We also discuss the possible mechanism of inhibition of the IFN-α/β pathways by the C protein. This is the first structural report of the C protein, suggesting a mechanism of evasion of the paramyxovirus from innate immunity.
INTRODUCTION
Sendai virus (SeV), a prototype of the family Paramyxoviridae, which includes important disease-causing viruses of humans and animals, such as measles virus, mumps virus, Nipah virus, canine distemper virus, Newcastle disease virus, and rinderpest virus. SeV has unique C proteins, which are translated from P and V mRNAs in a coding frame that is shifted from that of P and V proteins. C proteins comprise a nested set of four independently initiated and carboxyl-coterminal proteins—i.e., C (amino acids [aa] 1 to 204), Y1 (aa 24 to 204), Y2 (aa 30 to 204), and C′ (with an 11-aa addition to the N terminus of C)—where C is the major species type expressed in infected cells (reviewed in references 1 and 2).
C proteins are basically nonessential accessory proteins; thus, SeV can replicate minimally in cultured cells without C proteins, but C proteins are essential for efficient virus replication in cells and animals as well as for causing diseases. C proteins regulate viral RNA synthesis to reduce (IFN)-inducing RNA species (3–5) and to maintain protein kinase R in an inactive state (5), as well as controlling viral genome polarity (6, 7). In addition, C proteins are thought to regulate the viral particle formation (8, 9).
The important roles of C proteins include inhibition of signal transduction by associating with signal transducer and activator of transcription 1 (STAT1), particularly with the N-terminal domain of STAT1 (STAT1ND) (10, 11). IFN-α/β is unable to induce transcription from the IFN-stimulated response element (ISRE) sequence in the presence of C proteins. C proteins also inhibit signal transduction by IFN-γ (12–14). It was also demonstrated that STAT1 associated with C proteins forms high-molecular-weight complexes (HMWCs) (12). Furthermore, C proteins were reported to affect the phosphorylation level at the Tyr701 residue in STAT1 after IFN-α/β and IFN-γ stimulation; STAT1 phosphorylation is inhibited in the early phase of SeV infection, while phosphorylated STAT1 accumulates in the late phase of infection (15, 16).
STAT1 comprises an N-terminal domain (STAT1ND) and a core fragment, which are connected by a 15-aa linker peptide. The core fragment contains a coiled-coil domain, a DNA-binding domain, and an SH2 domain, followed by the phosphorylation site (Tyr701) and the transcriptional activation domain. Two STAT1NDs bind to form a dimer, and connected core fragments can adopt different arrangements. Thus, the STAT1 dimer forms two parallel forms in which the core fragments are arranged in parallel. In one parallel form, two SH2 domains of the STAT1 dimer are close to the N-terminal domains, while in the other parallel form they are distant from the N-terminal domains. The STAT1 dimer also forms an antiparallel form wherein the core fragments are positioned in the opposite direction (17). The transcriptional active form induced by IFN-γ is derived from a parallel form where a phosphorylated Tyr in one subunit binds to the SH2 domain in the other subunit, and two STAT1NDs dissociate to hold the target DNA between two core fragments in the nucleus (17, 18). It remains unknown how C proteins bind and affect the STAT1 molecule and inhibit transcriptional activation or why tyrosine-phosphorylated STAT1 (pY-STAT1) accumulates in cells when they are stimulated continuously by IFN-α/β and IFN-γ in the presence of C proteins.
In the present study, we determined the 2.0-Å-resolution structure of the complex of STAT1ND and the C-terminal region of the C protein (Y3 [aa 99 to 204]). Our results suggest that the flexibility between the N-terminal domain and the core fragment in STAT1 is greatly impaired by the binding of Y3. Binding of two C proteins to the STAT1ND dimer seems to force STAT1 to take one of the two parallel forms, which would be less likely to be dephosphorylated at Tyr701. We consider how C proteins inhibit transcription from pY-STAT1. Furthermore, we discuss how C proteins inhibit the phosphorylation of STAT2 under the stimulation of IFN-α/β.
MATERIALS AND METHODS
Cells and antibodies.
293T cells (human embryonic kidney cells expressing simian virus 40 T antigen) and A-431 cells (human epidermoid carcinoma-derived cells(were propagated in Dulbecco's modified minimum essential medium (DMEM) supplemented with 10% fetal bovine serum (Biological Industries, Kibbutz, Israel) and 100 U/ml penicillin–100 μg/ml streptomycin (Invitrogen). Mouse monoclonal antibodies against hemagglutinin (HA) tag (G036; Abcam), FLAG tag (M2; Sigma-Aldrich), STAT1 p91/84 (610185; BD Transduction Laboratories), and epidermal growth factor receptor (EGFR) (Ab-1; Thermo Scientific) and rabbit polyclonal antibodies against pY-STAT1 (sc-7988-R; Santa Cruz Biotech) and green fluorescent protein (GFP) (sc-8334; Santa Cruz Biotech) were used according to the manufacturers' instructions. Anti-SeV C rabbit antiserum was a gift from A. Kato (National Institute of Infectious Diseases, Japan).
Plasmid construction.
The cDNA fragments encoding N-terminally FLAG-tagged C (FL-C [aa 1 to 204]), Y1 (FL-Y1 [aa 24 to 204]), and Y3 (FL-Y3 [aa 99 to 204]) and N-terminally HA-tagged STAT1 N-terminal domain (HA-STAT1ND [aa 1 to 126]) were prepared by PCR and subcloned into the pCAGGS vector under the chicken β-actin promoter (19). Amino acids for the C protein and its derivatives are numbered with the N-terminal methionine of the C protein as 1. M150A, M150W, and R154A mutants of the C protein were generated by introducing mutations into the C cDNA using an AMAP site-directed mutagenesis kit (Amalgaam, Nagoya, Japan) or a KOD-Plus-Mutagenesis kit (Toyobo), and the mutants were inserted into the pCAGGS vector. The mutations were confirmed by DNA-sequencing analysis. pCAG-C, an expression plasmid for SeV C protein in the pCAGGS vector, was described previously (20). Reporter plasmids pISRE-EGFP and pGAS-EGFP, possessing ISRE and IFN-γ activation sequence (GAS) elements upstream of the enhanced GFP (EGFP) gene, respectively, were prepared from pISRE-luc and pGAS-luc, which were provided by B. Gotoh (Shiga University of Medical Science, Japan), by replacing their firefly luciferase gene with an EGFP gene (21, 22).
The pCold ProS2 vector (TaKaRa) was modified by inserting a novel NdeI restriction enzyme site just after the His tag sequence using an AMAP site-directed mutagenesis kit. DNA fragments encoding Y3 (aa 99 to 204 in the C protein) and STAT1ND (aa 1 to 126 in STAT1) were cloned into the NdeI-XhoI sites of the modified pCold ProS2 vector. A DNA fragment encoding C-terminally truncated (aa 1 to 713) STAT1 [STAT1(1–713)] was cloned into the NdeI-BamHI sites of the modified pCold ProS2 vector. For the expression of T-cell protein tyrosine phosphatase (TC45), novel EcoRI and HindIII sites were inserted just downstream of the NdeI site of the modified pCold ProS2 using a KOD-Plus-Mutagenesis kit. A DNA fragment encoding TC45 was cloned into the EcoRI-PstI sites of this modified pCold ProS2 vector.
For the expression of STAT1ND and STAT1(1–713) fused with enhanced cyan fluorescent protein (ECFP) and Y3 fused with enhanced yellow fluorescent protein (EYFP), DNA fragments encoding ECFP or EYFP were amplified by PCR using a 5′ forward primer and a 3′ reverse primer with the thrombin recognition site. The fragments were cloned into the NdeI site of the expression vectors for STAT1ND, STAT1(1–713), and Y3.
Reporter assay.
For IFN-α/β signal transduction, subconfluent 293T cells in a 35-mm dish were transfected with pISRE-EGFP (1 μg) and one of the C expression plasmids (1 μg) using FuGENE HD reagent (Promega), and IFN-α (20 U/ml; Mochida Pharmaceutical, Tokyo, Japan) was included in the culture medium after 6 h. At 24 h posttransfection, cells were observed under a fluorescence microscope, and cell lysates were subsequently prepared and processed for Western blotting by using an anti-GFP antibody. For IFN-γ signal transduction, the reporter plasmid pGAS-EGFP and IFN-γ (250 U/ml; Roche Diagnostics) were used instead of pISRE-EGFP and IFN-α, respectively.
Immunoprecipitation and Western blotting.
293T cells in a 35-mm dish were transfected with the indicated plasmids. After 24 h, cells were solubilized in 0.5 ml of NP-40 lysis buffer (0.5% NP-40, 50 mM Tris-HCl [pH 7.6], 50 mM NaCl, 1 mM EDTA, Complete mini-protease inhibitor cocktail [Roche Diagnostics]). Cell lysates were immunoprecipitated with an anti-FLAG or HA antibody together with protein G Sepharose (GE Healthcare), and the immunoprecipitates were washed with cell lysis buffer three times and analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) followed by Western blotting using an anti-HA or anti-C antibody. Protein bands were detected by using horseradish peroxidase (HRP)-conjugated anti-mouse IgG antibody and Luminata Forte Western HRP substrate (Millipore), followed by analysis using a LumiCube imaging analyzer (Liponics, Tokyo, Japan). A part of the cell lysates was also processed for SDS-PAGE and Western blotting to confirm protein expression.
Protein preparations.
Expression of Y3 was carried out in Escherichia coli BL21(DE3)codonPlus RIL (Novagen) at 15°C for 24 h by induction with 0.2 mM IPTG (isopropyl-β-d-thiogalactopyranoside), and expression of STAT1ND, STAT1(1–713), and TC45 was carried out in E. coli BL21(DE3)/pLysS (Novagen) in a similar manner. All of the proteins possessing a His tag at the N terminus were purified by nickel affinity chromatography using His-Bind resin (Novagen) according to the supplier's instruction manual. Nickel affinity chromatography was also used for preparation of a Y3:STAT1ND complex after mixing the supernatant from Y3-expressed E. coli with that from STAT1ND-expressed E. coli. Then, the complex was separated from the STAT1ND dimer by gel filtration chromatography using Sephacryl S-200HR (GE Healthcare) equilibrated with 20 mM Tris-HCl buffer (pH 7.5) containing 100 mM imidazole, 1 mM EDTA, 1 mM dithiothreitol (DTT), and 5% glycerol. For the preparation of ECFP, ECFP-fused STAT1ND was cleaved by using the Novagen Thrombin cleavage capture kit (Novagen) according to the supplier's instruction manual, followed by nickel affinity chromatography and gel filtration chromatography.
Protein concentrations of Y3, STAT1ND, STAT1(1–713), TC45, and Y3:STAT1ND complex were determined by measuring the absorbance at 280 nm using the molar extinction coefficients 22,500, 23,900, 109,300, 42,000, and 46,400 M−1 cm−1, respectively. Protein concentrations of ECFP-fused STAT1ND and ECFP-fused STAT1(1–713) were determined by measuring the absorbance at 435 nm using the molar extinction coefficient 28,750 M−1 cm−1. Protein concentrations of EYFP-fused Y3 were determined by measuring the absorbance at 514 nm using the molar extinction coefficient 83,400 M−1 cm−1. The concentration of phosphorylated STAT1(1–713) was determined by a Bradford protein assay.
Crystallography.
Prior to crystallization, the Y3:STAT1ND complex solution was dialyzed against 20 mM Tris-HCl buffer (pH 7.6) containing 0.1 M imidazole, 5% glycerol, 1 mM dithiothreitol, and 1 mM EDTA and then concentrated to 10 mg/ml using Amicon Ultra (Millipore). The crystals of the Y3:STAT1ND complex were grown by the sitting-drop vapor-diffusion method with a 1:1 (vol/vol) ratio of protein solution to precipitant solution. Small and plate-like crystals were formed within 1 week by using 0.2 M Tris-HCl buffer (pH 8.0) containing 30% (wt/vol) polyethylene glycol 400 (Sigma-Aldrich) and 0.2 M CaCl2 as a precipitant solution. Crystals suitable for diffraction analysis were obtained by the microseeding technique. Diffraction intensities of the crystals were determined using synchrotron radiation from BL38B1 at SPring-8 (Harima, Japan). The X-ray diffraction was measured with a charge-coupled device (CCD) camera equipped at the station, and the intensities were integrated and scaled using the HKL2000 program (23). The tertiary structure of the Y3:STAT1ND complex was solved by the molecular replacement method using atomic coordinates of the N-terminal domain of human STAT1 (PDB code 1YVL) as a search model and the program Molrep in the CCP4 program suite (24). After the model had been refined by simulated annealing and conventional restrained refinement methods using the CNS program (25), an electron density map was prepared for model building by the program Coot (26). Based on the difference-of-electron-density map, a model of Y3 was built step by step. A subset of 5% of the reflections was used to monitor the free R factor (Rfree) (27). Each refinement cycle included refinement of the positional parameters and individual isotropic B-factors as well as revision of the model, which was visualized by the program Coot. Details of data collection and refinement statistics are shown in Table S1 in the supplemental material.
Affinity analysis.
The binding affinity of EYFP-fused Y3 to ECFP-fused STAT1ND or ECFP-fused STAT1(1–713) was measured using fluorescence resonance energy transfer (FRET). All of the fluorescence spectra, recorded from 455 nm to 600 nm, were acquired using an Enspire 2300 plate reader (PerkinElmer). The initial solution was prepared in a well of a 96-well plate (flat bottom; Corning) with a volume of 0.25 ml of a broad-range buffer (100 mM 3,3-dimethylglutaric acid, 100 mM Tris, and 100 mM 2-amino-2-methyl-1,3-propandiol adjusted to pH 8.0) containing 100 mM imidazole, 100 mM NaCl, 1.0 mM EDTA, 1 mM dithiothreitol, 5% glycerol, 0.8 mg/ml bovine serum albumin (BSA), and 0.2 μM ECFP-fused STAT1ND or ECFP-fused STAT1(1–713) at 30°C. Five-microliter aliquots containing the given concentrations of EYFP-fused Y3 (0.5 to 17.5 μM) were successively added to the solution, followed by pipetting, extraction of a 5-μl aliquot of the reaction mixture, and recording of the fluorescence intensity data.
In this study, we defined the dissociation constants for the first and second EYFP-fused Y3 to each of the Y3-binding sites in the ECFP-fused STAT1ND dimer as Kd1 and Kd2, respectively:
| (1) |
| (2) |
In these equations, X represents ECFP-fused STAT1ND or ECFP-fused STAT1(1–713), while Y represents EYFP-fused Y3. Assuming that the quenching ratio after the binding of Y to X2 homodimer is the same as that after the binding of Y to X2·Y heterotrimer, quenched fluorescence intensity (ΔI) is represented by equation 3:
| (3) |
In this equation, [X2]t represents the sum of concentrations of X2 homodimer complexed and uncomplexed with Y, ΔImax means the maximum quenched intensity per mole of X2 homodimer, and [Y] represents the concentration of uncomplexed Y, which can be determined as a solution satisfying equations 4 and 5:
| (4) |
| (5) |
The Kd1 and Kd2 values were obtained by the nonlinear least-squares method together with ΔImax.
In vitro dephosphorylation assay.
In vitro phosphorylation of STAT1(1–713) was done according to a published procedure with minor modification (28). Briefly, A-431 cells were scraped and Dounce homogenized for five strokes in ice-cold lysis buffer (10 mM HEPES-HCl [pH 7.5], 150 mM NaCl, 0.5% Triton X-100, 10% glycerol, 1 mM Na3VO4, 10 mM EDTA, Complete Mini proteinase inhibitor cocktail). The lysates were then cleared by centrifugation and diluted 5 times with lysis buffer. EGFR precipitates were obtained from the diluted lysates using anti-EGFR antibody and protein G Sepharose. Immediately before an in vitro kinase reaction, the protein G Sepharose-bound EGFR was washed once with kinase buffer (20 mM Tris-HCl [pH 8.0], 50 mM KCl, 0.3 mM Na3VO4, 2 mM DTT) and then suspended in 0.4 ml of this buffer. Phosphorylation reactions were carried out in a final volume of 1 ml in a kinase buffer containing 2 mM DTT, 5 mM ATP, 5 mM MnCl2, 4 mg STAT1(1–713) and the protein G Sepharose-bound EGFR at 4°C for more than 20 h. The reaction mixture was then loaded onto an Affi-Gel heparin gel (Bio-Rad) column (7 by 1.0 cm). The column was washed with 50 mM HA buffer (20 mM Tris-HCl [pH 8.0], 1 mM EDTA, 2 mM DTT), and then pY-STAT1 was eluted with a linear gradient of 0 to 400 mM KCl.
Assays were performed as described previously (29). Briefly, STAT1(1–713) prepared from E. coli was phosphorylated at Tyr701 by using EGFR prepared from A-431 cell extracts. pY-STAT1(1–713) (0.6 μM) and the given concentrations of Y3 were mixed in 100 mM imidazole buffer (pH 7.6) containing 100 mM NaCl, 1 mM EDTA, and 1 mM DTT and preincubated for more than 60 min at 37°C. Dephosphorylation was initiated by the addition of purified TC45 (10 nM). Samples were taken at various time points and analyzed by Western blotting using a rabbit antibody against pY-STAT1 as a primary antibody and an HRP-conjugated anti-rabbit IgG goat polyclonal antibody as a secondary antibody. Protein bands were visualized as described above. Band signals were quantified with MultiGauge (Fujifilm Medical). Standard deviations were calculated on the basis of data obtained at least three times.
EMSA.
For the electrophoretic mobility shift assay (EMSA), the oligonucleotide 5′-CGACATTTCCCGTAAATCTG-3′ (GAS is underlined) and its complement chain were synthesized. The 3′ end of each strand was labeled by using a biotin 3′-end DNA labeling kit (Thermo Scientific), and the labeled DNA was purified according to the manufacturer's protocol. To generate a double-stranded DNA probe, a mixture of two strands was heated at 95°C for 5 min and gradually cooled at 2°C/min.
A given concentration of pY-STAT1(1–713) was incubated with 0.5 nM biotin-labeled DNA probe in 20 mM Tris-HCl buffer (pH 7.5) containing 150 mM NaCl, 1 mM DTT, 5 mM MgCl2, 50 ng/ml poly(dI-dC), and 1 mg/ml BSA for 20 min at room temperature. In the case of addition of Y3, pY-STAT1 was preincubated with Y3 for 10 min before mixing with the DNA probe. Protein-DNA complexes were resolved on a 6% nondenaturing polyacrylamide gel and transferred onto a nylon membrane (Biodyne B; Pall). After fixation by heating at 90°C for 2 h, the biotin-labeled probe was detected by using a LightShift chemiluminescent EMSA kit (Thermo Scientific) following the manufacturer's instructions.
Protein structure accession number.
The atomic coordinates and structure factors of the Y3:STAT1ND complex have been deposited in the Protein Data Bank under accession no. 3WWT.
RESULTS AND DISCUSSION
Association of the Y3 protein with STAT1ND.
Previously, it was shown that the constitutive expression of Y3 (aa 99 to 204), an N-terminally truncated mutant of the C protein, inhibited signal transduction of the Jak/STAT pathway starting from IFN-α/β in HeLa cells (30). We also confirmed the inhibition of signal transduction in cells transiently expressing Y3. 293T cells were transfected with the reporter plasmid pISRE-EGFP together with a plasmid to express the C protein and its deletion mutants, Y1 and Y3, and IFN-α was added to the culture medium (Fig. 1A). The EGFP expression from pISRE-EGFP in the presence of IFN-α was restricted by the coexpression of FLAG-tagged Y3 (FL-Y3) (Fig. 1A). However, the extent of the inhibition was lower than those of FL-C and FL-Y1. This may be due partly to a lower level of accumulation of FL-Y3.
FIG 1.

Suppression of signal transduction from IFN-α/β and IFN-γ by Y3 and complex formation of Y3 with STAT1ND. (A) For IFN-α/β signal transduction analysis, subconfluent 293T cells were transfected with pISRE-EGFP and an expression plasmid for FL-C, FL-Y1, FL-Y3, or no protein (−), and IFN-α (20 U/ml) was included in the culture medium after 6 h. At 24 h posttransfection, cells were processed for Western blotting using an anti-GFP antibody. A gel image and quantified graph from three independent experiments are shown. The error bar indicates the standard deviation. (B) For IFN-γ signal transduction analysis, reporter plasmids pGAS-EGFP and IFN-γ (250 U/ml) were used instead of pISRE-EGFP and IFN-α, respectively. Cells were analyzed as described in panel A. (C and D) 293T cells were transfected with the indicated plasmids and solubilized in cell lysis buffer after 24 h. Cell lysates were immunoprecipitated (IP) with an anti-FLAG antibody (αFLAG) together with protein G Sepharose, and the immunoprecipitates were analyzed by SDS-PAGE followed by Western blotting (WB) using an anti-HA antibody (αHA). A part of the cell lysates was processed for SDS-PAGE and Western blotting to confirm protein expression using the anti-HA antibody and anti-FLAG antibody. (E) Analytical size exclusion chromatograms of Y3 (dotted line) and STAT1ND in the presence (black line) or absence (gray line) of Y3. (F) Fractions eluted from the size exclusion chromatogram for STAT1ND with Y3 in panel E were analyzed using SDS-PAGE and silver staining.
A similar experiment was performed using pGAS-EGFP and IFN-γ, and the Y3 peptide was also found to inhibit signal transduction from IFN-γ, although the inhibition was incomplete (ca. 50% inhibition) (Fig. 1B).
In a coimmunoprecipitation experiment, the HA-tagged STAT1 N terminus (aa 1 to 126 [HA-STAT1ND]) was pulled down by FL-Y3 as well as by FL-C and FL-Y1, thereby indicating that Y3 also binds to STAT1ND (Fig. 1C). The expression of FL-Y3 alone was barely detected (Fig. 1D), probably because of its degradation or translocation into the nucleus, as previously shown (21), but FL-Y3 expression appeared to increase with the coexpression of HA-STAT1ND (Fig. 1D). These observations suggest that HA-STAT1ND and FL-Y3 form a complex in the cells, thereby improving the stability of the proteins in the cytosol.
The faint upper band of HA-STAT1ND was observed in Fig. 1C and D. This is probably due to a protein modification, the precise nature of which was not determined.
Crystal structure of the Y3:STAT1ND complex.
The Y3 and STAT1ND peptides purified from Escherichia coli were mixed, and fractions containing both of them were separated by gel filtration column chromatography (Fig. 1E and F). We obtained crystals from the purified complex (see Table S1 in the supplemental material) and determined the crystal structure of Y3-bound STAT1ND at a resolution of 2.0 Å. An asymmetric unit in the crystal included one molecule each of Y3 and STAT1ND. A heterotetrameric structure comprising two Y3 and two STAT1ND molecules could be prepared with a crystallographic 2-fold symmetry axis. Two molecules of Y3 symmetrically bound to each niche that was created between two subunits in the STAT1ND dimer (Fig. 2A). The molecular mass of the heterotetrameric Y3:STAT1ND complex observed in the crystal (63 kDa) was consistent with that estimated by gel filtration (∼71 kDa) (Fig. 1E). One Y3:STAT1ND tetramer directly or indirectly interacted with adjacent tetramers via several water molecules or Ca2+ ions derived from the precipitant solution, which suggests that these interactions are induced by the crystal-packing effect.
FIG 2.

Crystal structure of the Y3:STAT1ND complex and comparison between Y3-bound and Y3-free STAT1ND dimers. (A) The tetrameric structure of two STAT1ND and two Y3 molecules is shown in a ribbon representation. Each structure of Y3 is colored in blue and green, respectively, and each subunit of the STAT1ND dimer is colored in yellow and magenta, respectively. All of the structural diagrams presented in this article were made by using PyMOL (38). (B) Structure of a Y3 monomer derived from panel A with a distinct view angle to show each α-helix separately. (C) Superimposed structures of Y3-bound and Y3-free STAT1ND dimers. Each subunit of the Y3-bound STAT1ND dimer is colored in yellow and in magenta, respectively, and two subunits of the Y3-free STAT1ND dimer are shown in white. The Cα atoms of one subunit (shown in yellow) of the Y3-bound STAT1ND dimer were maximally fitted to those of one subunit of the Y3-free STAT1ND dimer. Carbon atoms of E29, N82, K85, and E96 from Y3-bound STAT1ND shown in the stick diagram are colored in green, and those from Y3-free STAT1ND shown in the stick diagram are colored in white. The direction of the view is the same as that in panel A. (D) An enlarged view of panel C centered on the dimer interface is shown. The dotted lines represent hydrogen bonds.
Y3 comprises six α-helices (α1 to α6), which can be separated into two layers. Each of the layers is formed by α1-, α3-, and α4-helices and α2-, α5-, and α6-helices, respectively (Fig. 2A and B), and the bilayer structure is maintained mostly via hydrophobic interactions. No electron densities were observed in the loop between the α4- and α5-helices or in the side chains of the two N-terminal residues of the α5-helix (Lys168 and Asp169), thereby suggesting that the α4- and α5-helices of Y3 are connected through a flexible loop. In addition, the electron densities of the residues from Ser196 to Glu203 were relatively weak, and the electron density of Glu204 was not observed. Among the consecutive His residues fused to the N-terminal amino acid residue of Y3 (Met99), two His residues adjacent to Met99, which are located in the N-terminal part of the α1-helix, exhibited clear electron densities. These two His residues correspond to the Leu97 and His98 residues in the intact C protein, respectively.
The monomer of Y3-bound STAT1ND comprises seven α-helices. The monomeric structure of Y3-bound STAT1ND did not differ from that of Y3-free STAT1ND (17) (Fig. 2C), except for the loop connecting the α3- and α4-helices (root mean square deviation [RMSD] of 0.552 Å for 111 Cα atoms). Y3-bound STAT1ND formed a dimeric structure via the same binding interface as that observed in the Y3-free STAT1ND dimer. However, one subunit in the STAT1ND dimer was closer to the other subunit because of the binding of Y3. For example, the distance between the Cα atoms of two Asn82 residues and that between the Cα atoms of two Glu96 residues in the Y3-bound STAT1ND dimer decreased by 1.02 and 4.33 Å, respectively, compared with the distances in the Y3-free STAT1ND dimer. The closer arrangement of the two STAT1ND subunits allows the negatively charged carboxyl group of Glu29 in one subunit to interact with the positively charged amino group of Lys85 in the other subunit (Fig. 2D). In addition, the side chain of Asn82 reinforces the interaction between the two STAT1ND subunits via intramolecular interaction with the side chain of Lys85. These findings suggest that the STAT1ND dimer is stabilized and is difficult to dissociate into monomers in the presence of the Y3 protein.
Interactions between Y3 and STAT1ND.
A number of charged or polar amino acids from Y3 and STAT1ND form the extended hydrogen-bond network at the binding interfaces between Y3 and STAT1ND, together with some water molecules (Fig. 3A). The residues that participate in the network come from the α1-, α4-, α5-, and α6-helices of Y3, the α5- and α6-helices of one monomer in the STAT1ND dimer, and the α2-helix of the other monomer in the STAT1ND dimer. Hydrophilic interactions mainly occur at the interface between Y3 and STAT1ND, which indicates that the binding affinity of the C protein to STAT1 is increased in a hypotonic condition, as previously reported (12).
FIG 3.

Binding interface between STAT1ND and Y3. (A) Each subunit of the STAT1ND dimer shown in the ribbon representation is colored as in Fig. 2. Carbon atoms shown in the stick diagram from STAT1ND and Y3 are colored in green and white, respectively. Water molecules are shown as red spheres. (B) The contact area between one subunit of the STAT1ND dimer and Y3 and that between the other subunit and Y3 on the molecular surface are shown in yellow and magenta, respectively. Y3 is colored in blue. The direction of the view is the same as that in panel A. (C) The molecular surface of the STAT1ND dimer is shown with its hydrophobic property colored in green. The L64, D65, Y68, I83, R84, and K87 residues of STAT1ND (green) and the M150 residue of Y3 (white) are shown in the stick diagram. The direction of the view is almost similar to that of the view in panel A. (D) Structure around the pocket created on the STAT1ND molecular surface. Carbon atoms from the Y3-bound STAT1ND dimer are colored in green, and those from the Y3-free STAT1ND dimer are colored in white. Carbon atoms from Y3 are colored in cyan. The direction of the view is the same as that in panel A.
The contact area between one molecule of Y3 and one subunit in the STAT1ND dimer significantly differs from that between one molecule of Y3 and the other subunit in the STAT1ND dimer (930 and 220 Å2, respectively) (Fig. 3B), which indicates the importance of the former interaction for the association between Y3 and STAT1ND.
An Nω atom in the Arg154 residue of Y3 interacts with a Nδ atom and backbone carbonyl oxygen in the His58 residue of STAT1ND (Fig. 3A). This indicates that the affinity between Y3 and STAT1ND is weakened at an acidic pH because electrostatic repulsion would be generated through protonation of the His58 residue in STAT1ND. However, despite extensive efforts, we could not clarify the binding affinity between STAT1ND and Y3 in acidic conditions. In addition, the side chain in the Met150 residue of Y3 is inserted into a pocket created on the molecular surface of STAT1ND, the wall of which is mainly formed by charged residues, such as Asp65, Tyr68, Arg84, and Lys87, and the bottom is formed by Leu64 and Ile83 (Fig. 3C). The methyl group in the Met150 residue of Y3 is located in close proximity to the side chain of Ile83 at the bottom of the pocket. The intrusion of the side chain of the Met150 residue of Y3 into the pocket causes the movement of the side chain of the Tyr68 residue in STAT1ND toward that of the Gln80 residue (Fig. 3D). According to the movement of the side chain of Tyr68, the side chain of Arg84 in one subunit of the STAT1ND dimer, which interacts with Tyr68, and that of Glu16 in the other subunit, which makes a salt bridge with Arg84, move toward the side chain of Tyr68.
To validate the roles of the Met150 and Arg154 residues in the binding of Y3 with STAT1ND, coimmunoprecipitation experiments were conducted using C protein mutants in which Met150 was replaced by Ala (M150A) or Trp (M150W) and Arg154 was replaced by Ala (R154A), respectively. When FL-Y3 proteins with or without these mutations were expressed in cells together with HA-STAT1ND (Fig. 4A), wild-type FL-Y3 was pulled down by HA-STAT1ND but not any of the mutants. When HA-tagged full-length STAT1 and the C protein were used instead of their truncated forms, similar results were obtained by immunoprecipitation: i.e., the C protein was found at high levels after immunoprecipitation, whereas its mutants were found only at low levels (Fig. 4B). These results indicate that the Met150 and Arg154 residues of the C protein significantly affect the capacity to bind with STAT1.
FIG 4.

Effects of the M150 and R154 residues of the C protein on binding with STAT1. (A and B) 293T cells were transfected with the indicated plasmids and solubilized in cell lysis buffer after 24 h. Cell lysates were immunoprecipitated with an anti-FLAG (A) or anti-HA (B) antibody together with protein G Sepharose, and the immunoprecipitates were analyzed by SDS-PAGE followed by Western blotting using an anti-HA (A) or anti-C (B) antibody. A part of the cell lysates was processed for SDS-PAGE and Western blotting to confirm protein expression using anti-HA, anti-FLAG, and anti-C antibodies. (C) Accumulated EGFP expressed from pISRE-EGFP or pGAS-EGFP reporter plasmid after addition of IFN-α or IFN-γ was shown by Western blotting in the presence of FL-Y3 protein mutants, as indicated in the figure. Expression of FL-Y3 protein mutants in one experiment was also shown. (D and E) The experiments were repeated at least three times, and band signals of accumulated EGFP were quantified with the MultiGauge software. The average values were plotted on the graph, and the error bar indicates the standard deviation. The P value was calculated on the basis of the Mann-Whitney U test.
This conclusion is supported by the significantly lower efficiency of inhibition of the IFN-α and IFN-γ signal transduction pathways by the FL-Y3 proteins with these mutations (Fig. 4C, D, and E). On the other hand, when we employed the full-length C mutants instead of the N-terminally truncated Y3 mutants for the reporter assay for IFN-α and IFN-γ, the C mutants suppressed the reporter expression almost completely, similar to the wild-type C protein (data not shown). The low extent of binding of the mutated C proteins may be sufficient to block the signal transduction pathways.
An amino acid sequence comparison among the C proteins with the residues that corresponded to Met150 and Arg154 in the SeV C protein showed that they were completely conserved in viruses belonging to the genus Respirovirus, which suggests the importance of these residues. The FL-Y3-R154A mutant protein suppressed IFN-γ signaling but not IFN-α signaling (Fig. 4D and E). This discrepancy may be acceptable because the mechanisms of inhibition by the C protein may differ between the two signaling pathways (31).
Molecular modeling of the Y3:STAT1 complex.
Despite intensive efforts, full-length STAT1 or STAT1(1–713), which contains both an N-terminal domain and core fragment, has not been cocrystallized with Y3, probably because of the low solubility of the complex. To investigate the effect of Y3 on the positional relationship in the domains of STAT1, we conducted gel filtration chromatography and showed that Y3-bound STAT1 was eluted at almost the same volume as that of Y3-free STAT1, indicating that Y3-bound STAT1 as well as Y3-free STAT1 forms a dimer in solution (data not shown). In addition, stoichiometric oligomer analysis of the Y3:STAT1(1–713) complex was performed using SDS-PAGE and diluted protein standards, and the results strongly suggested that an equivalent mole of Y3 and STAT1(1–713) was included in the complex, which indicates that two molecules of Y3 bind to one STAT1 dimer (data not shown).
Unphosphorylated STAT1 dimerizes via the interaction between the two N-terminal domains of the subunits (17). Moreover, it is considered that the relative arrangement between the N-terminal domain and core fragment of STAT1 is remarkably variable because of the high flexibility of the linker peptide (Fig. 5A), and the unphosphorylated STAT1 dimer has been proposed to exist in equilibrium between parallel and antiparallel forms (17).
FIG 5.

Structure of STAT1 and predicted models of Y3-bound STAT1. (A) Linear representation of the domains of human STAT1. ND, N-terminal domain; CCD, coiled-coil domain; DBD, DNA-binding domain; LD, linker domain; SH2D, SH2 domain; TAD, transcription activation domain. (B and C) A STAT1(1–683) dimer taking an antiparallel form associated with one Y3 molecule (B) and one taking a parallel form associated with two Y3 molecules are modeled. Each domain in the STAT1 structure is colored corresponding to that in panel A, and Y3 is colored in blue. These crystal structures were generated as shaded ribbons using coordinates from the Protein Data Bank. (The PDB accession number for the unphosphorylated STAT1 dimer is 1YVL).
We generated model structures based on the current Y3:STAT1ND structure and the structures of STAT1(1–683) (17). One conformation of the STAT1 dimer, which has an antiparallel form, can be attached via only one molecule of Y3 to one binding site in the STAT1ND dimer (Fig. 5B). The other binding site in the STAT1ND dimer is covered by the SH2 domain of STAT1, thereby preventing Y3 from binding to the site. Another conformation of the STAT1 dimer, which has one of two possible parallel forms, can be attached via two molecules of Y3 using two binding sites in the STAT1ND dimer (Fig. 5C). In contrast, the other conformation of the STAT1 dimer, which has another parallel form, cannot be attached via Y3 because both binding sites are covered by SH2 domains.
Affinity analysis.
To estimate the dissociation constants between STAT1 and Y3, we performed an affinity analysis by using FRET. For this purpose, ECFP was fused to the N terminus of STAT1ND or STAT1(1–713), while EYFP was fused to the N terminus of Y3. If ECFP is close to EYFP (within about 100 Å), emission from ECFP at 475 nm, which is generated after excitation of the ECFP chromophore at 435 nm, is decreased, while emission from EYFP at 527 nm is increased (32). Gel filtration analysis showed that ECFP-fused STAT1ND could bind to EYFP-fused Y3 (Fig. 6A). FRET was observed in a titration experiment, in which EYFP-fused Y3 was successively added to the solution of ECFP-fused STAT1ND at the known concentration, whereas it was not observed between unfused ECFP and EYFP-fused Y3 (Fig. 6B and C). Two dissociation constants were determined from the decreased rates of the emission intensity at 475 nm. One dissociation constant (Kd1) between the first Y3 molecule and the STAT1ND dimer was determined to be 0.68 ± 0.17 μM, while the other constant (Kd2) between the second Y3 molecule and the STAT1ND dimer was 0.49 ± 0.08 μM (Fig. 6D). Assuming that the binding affinity of the STAT1ND dimer for the first Y3 is the same as that for the second Y3, then Kd2 must be 4-fold higher than Kd1. Current data indicate that two Y3 molecules bind to the STAT1ND dimer cooperatively. On the other hand, as for the dissociation constants between the nearly full-length STAT1(1–713) dimer and Y3, Kd1 and Kd2 were determined to be 0.022 ± 0.016 and 0.51 ± 0.35 μM, respectively (Fig. 6E). The affinity of STAT1(1–713) for the first Y3 is higher than that of STAT1ND, suggesting that other domains of STAT1 help the association with Y3. Furthermore, Kd2 is significantly higher than Kd1 in the case of STAT1(1–713). This suggests that the second Y3 can target only the STAT1 dimer taking a parallel form, while the first Y3 can target either the STAT1 dimer taking a parallel form or that taking an antiparallel form. In addition, binding of the first Y3 may stabilize the antiparallel form.
FIG 6.

Affinity analysis. (A) Analytical-exclusion chromatograms of ECFP-fused STAT1ND (gray line) and EYFP-fused Y3 (black line). The broken line shows a 1:1 mixture of ECFP-fused STAT1ND and EYFP-fused Y3. A 15 μM concentration of ECFP-fused STAT1ND was preincubated with or without an equimolar concentration of EYFP-fused Y3 for 10 min, followed by gel filtration chromatography using a Superdex 75 10/300 GL high-performance liquid chromatography (HPLC) column. (B and C) Fluorescence emission spectra by direct titration to 0.2 μM ECFP-fused STAT1ND (B) or 0.05 μM unfused ECFP (C) with EYFP-fused Y3 at 0.16, 0.32, 0.48, 0.64, 0.80, 0.96, and 1.12 μM (gray line). Two arrows represent decreased fluorescence intensity at 475 nm and increased fluorescence intensity at 527 nm. rfu, relative fluorescence units. (D and E) The quenched fluorescence intensities (ΔI) of ECFP-fused STAT1ND (D) or STAT1(1–713) (E) at 475 nm caused by the addition of EYFP-fused Y3 are divided by the concentration of each STAT1 dimer. The values are then plotted against the concentration of free EYFP-fused Y3. The gray curves drawn represent the best fits to the data.
In FRET analysis, we also used the purified Y3-M150A and Y3-R154A proteins instead of the wild-type Y3 protein. The dissociation constant was high (Kd of >15 μM), and we could not perform detailed analysis (data not shown).
Effects of C proteins on STAT1 dephosphorylation.
It is suggested that the domain arrangement in the STAT1 dimer is important for the phosphorylation and dephosphorylation of Tyr701 (33, 34). The STAT1 dimer in the antiparallel form would be readily dephosphorylated by cellular phosphatase. However, it would be difficult for the dimer in parallel forms to be dephosphorylated by phosphatase because the phosphorylated Tyr701 residue in one monomer interacts with the SH2 domain in the other. The parallel forms are considered to be the conformations that can be phosphorylated by membrane kinases (17). According to the molecular modeling of Y3 and STAT1 described above, it is predicted that the STAT1 dimer with one Y3 molecule, which has an antiparallel form, tends to be dephosphorylated, whereas the STAT1 dimer with two Y3 molecules, which has a parallel form, is resistant to dephosphorylation. We investigated these possibilities.
The pY-STAT1 dimer translocated into the nucleus is dephosphorylated by TC45, which is one of the nuclear protein tyrosine phosphatases (29). To clarify how Y3 affects the dephosphorylation of pY-STAT1 by TC45, we performed an in vitro dephosphorylation assay using Y3, STAT1(1–713), and TC45 purified from E. coli (Fig. 7). The dephosphorylation assay was initiated by adding TC45 to pY-STAT1(1–713), which was phosphorylated in vitro beforehand. When the assay was performed in the presence of Y3 with half the concentration of pY-STAT1(1–713), 80% of the substrate was dephosphorylated after 30 min. In contrast, when the assay was performed in the presence of Y3 with a 4-fold concentration of pY-STAT1(1–713), only 40% of the substrate was dephosphorylated after 30 min. The differences at 30 min were significant (Fig. 7). These results suggest that the presence of a low concentration of C protein promotes the dephosphorylation of pY-STAT1 by TC45, whereas a high concentration of C protein inhibits dephosphorylation. These results are consistent with the observation that STAT1 phosphorylation at Tyr701 is inhibited in SeV-infected cells, but leaky phosphorylation continues and STAT1 finally becomes phosphorylated at a high level in the late phase of infection (35).
FIG 7.

In vitro dephosphorylation assay of pY-STAT1 with a given concentration of Y3. Dephosphorylation rates of pY-STAT1(1–713) catalyzed by TC45 were measured under the condition of a high concentration of Y3 (a 4-fold-higher concentration than that of pY-STAT1 [▲]), a low concentration of Y3 (half of that of pY-STAT1 [■]), and the absence of Y3 (◆). The residual ratio is the ratio of band signals for pY-STAT1(1–713) at various time points to that at 0 min determined by Western blotting. Statistical significance was evaluated with the Student t test based on the values at 30 min. *, P < 0.05; **, P < 0.01; ***, P < 0.005.
Association of the pY-STAT1 dimer with GAS in the presence of Y3.
We performed an EMSA by using pY-STAT1(1–713) and a biotin-labeled GAS probe. As a result, a shifted band was observed in the presence of pY-STAT1(1–713), and the bands were further shifted upward by the addition of Y3 (Fig. 8A).
FIG 8.

Electrophoretic mobility shift assay. (A) pY-STAT1(1–713) and a given concentration of Y3 were incubated, followed by the addition of a 0.5 nM concentration of a biotin-labeled DNA probe containing a single GAS element. Protein-DNA complexes were resolved on a 6% nondenaturing polyacrylamide gel and transferred onto a nylon membrane. The biotin-labeled probe was detected by using a LightShift chemiluminescent EMSA kit. The positions of the electrophoretic origin and unbound probe are shown. (B and C) Two tandem GAS elements (2×GAS probe) and a given concentration of pY-STAT1(1–713) and Y3 (molar ratio of 1:10) (B) or pY-STAT1(1–713) alone (C) was incubated and analyzed as in panel A to compare the affinities of pY-STAT1 with the GAS element in the presence of different concentrations of Y3.
Gel filtration analysis suggested that pY-STAT1(1–713) associated with Y3 formed multimers or HMWCs (Fig. 9). pY-STAT1 is known to be equilibrated among various multimers, in which one pY-STAT1 dimer interacts with another dimer through free N-terminal domains in each dimer (36). The HMWCs of pY-STAT1 appears to bind to the probe (Fig. 8A). The binding affinity of pY-STAT1:Y3 complex with the probe was almost equivalent to that of Y3-free pY-STAT1 (Fig. 8B and C), indicating that Y3 does not inhibit the binding of pY-STAT1 to the probe.
FIG 9.
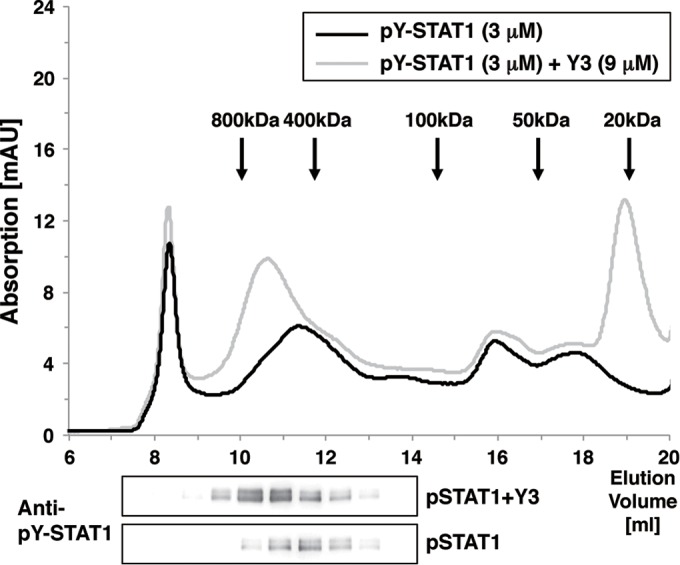
Oligomeric analysis of pY-STAT1 with or without Y3. pY-STAT1(1–713) (3 μM) was preincubated with or without 9 μM Y3 for 10 min. A portion of the solution was injected into a Superdex 200 10/300 GL HPLC column equilibrated with 0.1 M Tris-HCl buffer (pH 8.0) containing 100 mM NaCl at room temperature at a flow rate of 0.7 ml/min. The pY-STAT1 contained in each fraction was resolved by SDS-PAGE and detected by using an anti-pY-STAT1 antibody.
On the other hand, the EGFP expression from pGAS-EGFP in the presence of IFN-γ was partially restricted by almost 50% in the coexpression of FL-Y3, while it was mostly restricted in that of FL-C and FL-Y1 (Fig. 1B). pY-STAT1 in the HMWCs may not be fully active for transcription.
In the presence of full-length C protein, pY-STAT1 formed aggregates, and no binding with GAS was observed in EMSA (12). Actually, the C protein readily forms insoluble aggregates in purification from E. coli. The ability of C or Y1 to form HMWCs of pY-STAT1 is likely stronger than that of Y3. The N-terminal half of the C protein may accelerate the HMWC formation of pY-STAT1 and augment STAT1 inhibition.
Possible mechanism responsible for inhibition of STAT activities by the C protein.
Based on these observations, we considered the underlying mechanism that the C protein inhibits the STAT1 activity after SeV infection (Fig. 10). In the early phase of SeV infection, the concentration of the C protein would be low; thus, one molecule of the C protein would bind to the STAT1 dimer, thereby inducing domain rearrangement to an antiparallel form. This association would extend the period when STAT1 is in an antiparallel form, facilitating dephosphorylation by phosphatase (33, 34). In contrast, in the late phase of SeV infection, the concentration of the C protein would be higher and two molecules of the C protein would bind to the STAT1 dimer, thereby leading to domain rearrangement to a parallel form. The Kd2 value suggests that the second C protein associates with one C-bound STAT1 dimer in an inefficient way, coupling with domain arrangement to a parallel form (Fig. 9, dotted arrow). This association would extend the period when STAT1 is present in the parallel form, thereby inhibiting dephosphorylation (33, 34) and causing the accumulation of pY-STAT1 (15). Concomitantly, pY-STAT1 with C would form HMWCs, probably through the interaction of a phosphorylated Tyr in one dimer with an SH2 domain in another dimer or due to the aggregation through the N-terminal half of the C protein, leading to the inhibition of the IFN-γ pathway.
FIG 10.

Transition of the domain arrangement of the STAT1 dimer in the presence of C protein. Structural models of the STAT1 dimer are shown, and the components of STAT1 are indicated in the inset. The transition of the STAT1 dimer by the C protein is shown by a red arrow, and inefficient transition from a one-C-bound form to a two-C-bound form is shown by a red dotted arrow.
The C protein also inhibited the IFN-α/β pathways. Although the C protein inhibits the phosphorylation of STAT2, there have been no reports showing the direct interaction between STAT2 and C protein (12, 16, 37). In addition, we failed to detect any interactions between the N-terminal domain of STAT2 (STAT2ND) and Y3 or C protein by coimmunoprecipitation and gel filtration, despite the high sequence identity between STAT1ND and STAT2ND (identity of 46.9%). A comparison of the amino acid sequences of STAT1ND and STAT2ND showed that His58 in STAT1ND, which interacts with Arg154 in the C protein, is replaced by Phe, thereby resulting in the loss of one or two hydrogen bonds with the C protein. In addition, Asp59 of STAT1ND, the carboxyl group of which interacts with Nε of Lys183 in the C protein, is replaced by His in STAT2ND, which suggests that the interaction between STAT2 and the C protein is weakened by electrostatic repulsion.
Although the C protein is known to inhibit the phosphorylation of STAT2, inhibition does not occur when STAT1 is absent or when the C protein has lost its STAT1-binding ability (16). This indicates that the C protein acts on STAT2 by binding to STAT1. The structure of the STAT1:STAT2 heterodimer remains unknown. Assuming that the behavior of the STAT1:STAT2 heterodimer is similar to that of the STAT1 homodimer, the C protein appears to bind to the molecular surface produced by dimerization between the N-terminal domains of STAT1 and STAT2. It is important to note that the C protein would bind to only one side of the molecular surface formed by STAT1ND and STAT2ND so that it can make extensive contact with STAT1ND but not with STAT2ND. Therefore, the STAT1:STAT2 heterodimer bound to the C protein would be induced to assume an antiparallel form. This may facilitate the dephosphorylation of the STAT1:STAT2 heterodimer. In this study, we found that the R154A mutant of FL-Y3 inhibited the signal transduction by IFN-γ but not that by IFN-α/β. This may imply that the binding affinity of FL-Y3 with the STAT1:STAT2 heterodimer is lost due to replacement of Arg154 of Y3 by Ala, whereas the binding affinity with the STAT1 homodimer is not completely lost. To clarify this hypothesis, an investigation of the behavior of the STAT1:STAT2 heterodimer in the presence of the C protein is now in progress.
Supplementary Material
ACKNOWLEDGMENTS
We are grateful to the beamline staff at SPring-8, Japan for their kind help with X-ray data collection. We also thank Mamoru Satoh and Takashi Oda, Yokohama City University, for the small-angle X-ray scattering experiment and valuable advice. We thank Yuko Eda for excellent technical assistance and the staff of the Analysis Center of Life Science, Hiroshima University for the use of their facilities. This work was partly carried out at the Joint Usage/Research Center (RIRBM), Hiroshima University. Synchrotron radiation experiments were performed at SPring-8 with the approval of the Japan Synchrotron Radiation Research Institute (JASRI) under proposal no. 2013B1065.
This study was funded by the Inamori Foundation (to K.O.), GlaxoSmithCline Japan (to K.O.), the Takeda Science Foundation (to K.O.), and the Japan Society for the Promotion of Science (to T.S. and K.O.).
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.01887-15.
REFERENCES
- 1.Lamb RA, Parks GD. 2013. Paramyxoviridae: the viruses and their replication, p 957–995. In Knipe DM, Howley PM (ed), Fields virology, 6th ed, vol 1 Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 2.Nagai Y, Takakura A, Irie T, Yonemitsu Y, Gotoh B. 2011. Sendai virus: evolution from mouse pathogen to a state-of-the-art tool in virus research and biotechnology, p 115–173. In Samal SK. (ed), The biology of paramyxoviruses. Caister Academic Press, Norfolk, United Kingdom. [Google Scholar]
- 3.Irie T, Nagata N, Igarashi T, Okamoto I, Sakaguchi T. 2010. Conserved charged amino acids within Sendai virus C protein play multiple roles in the evasion of innate immune responses. PLoS One 5:e10719. doi: 10.1371/journal.pone.0010719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Komatsu T, Takeuchi K, Yokoo J, Gotoh B. 2004. C and V proteins of Sendai virus target signaling pathways leading to IRF-3 activation for the negative regulation of interferon-beta production. Virology 325:137–148. doi: 10.1016/j.virol.2004.04.025. [DOI] [PubMed] [Google Scholar]
- 5.Takeuchi K, Komatsu T, Kitagawa Y, Sada K, Gotoh B. 2008. Sendai virus C protein plays a role in restricting PKR activation by limiting the generation of intracellular double-stranded RNA. J Virol 82:10102–10110. doi: 10.1128/JVI.00599-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Irie T, Nagata N, Yoshida T, Sakaguchi T. 2008. Paramyxovirus Sendai virus C proteins are essential for maintenance of negative-sense RNA genome in virus particles. Virology 374:495–505. doi: 10.1016/j.virol.2008.01.004. [DOI] [PubMed] [Google Scholar]
- 7.Irie T, Okamoto I, Yoshida A, Nagai Y, Sakaguchi T. 2014. Sendai virus C proteins regulate viral genome and antigenome synthesis to dictate the negative genome polarity. J Virol 88:690–698. doi: 10.1128/JVI.02798-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hasan MK, Kato A, Muranaka M, Yamaguchi R, Sakai Y, Hatano I, Tashiro M, Nagai Y. 2000. Versatility of the accessory C proteins of Sendai virus: contribution to virus assembly as an additional role. J Virol 74:5619–5628. doi: 10.1128/JVI.74.12.5619-5628.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kurotani A, Kiyotani K, Kato A, Shioda T, Sakai Y, Mizumoto K, Yoshida T, Nagai Y. 1998. Sendai virus C proteins are categorically nonessential gene products but silencing their expression severely impairs viral replication and pathogenesis. Genes Cells 3:111–124. doi: 10.1046/j.1365-2443.1998.00170.x. [DOI] [PubMed] [Google Scholar]
- 10.Garcin D, Latorre P, Kolakofsky D. 1999. Sendai virus C proteins counteract the interferon-mediated induction of an antiviral state. J Virol 73:6559–6565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gotoh B, Takeuchi K, Komatsu T, Yokoo J, Kimura Y, Kurotani A, Kato A, Nagai Y. 1999. Knockout of the Sendai virus C gene eliminates the viral ability to prevent the interferon-alpha/beta-mediated responses. FEBS Lett 459:205–210. doi: 10.1016/S0014-5793(99)01241-7. [DOI] [PubMed] [Google Scholar]
- 12.Takeuchi K, Komatsu T, Yokoo J, Kato A, Shioda T, Nagai Y, Gotoh B. 2001. Sendai virus C protein physically associates with Stat1. Genes Cells 6:545–557. doi: 10.1046/j.1365-2443.2001.00442.x. [DOI] [PubMed] [Google Scholar]
- 13.Kato A, Ohnishi Y, Kohase M, Saito S, Tashiro M, Nagai Y. 2001. Y2, the smallest of the Sendai virus C proteins, is fully capable of both counteracting the antiviral action of interferons and inhibiting viral RNA synthesis. J Virol 75:3802–3810. doi: 10.1128/JVI.75.8.3802-3810.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Young DF, Didcock L, Goodbourn S, Randall RE. 2000. Paramyxoviridae use distinct virus-specific mechanisms to circumvent the interferon response. Virology 269:383–390. doi: 10.1006/viro.2000.0240. [DOI] [PubMed] [Google Scholar]
- 15.Gotoh B, Komatsu T, Takeuchi K, Yokoo J. 2003. The C-terminal half-fragment of the Sendai virus C protein prevents the gamma-activated factor from binding to a gamma-activated sequence site. Virology 316:29–40. doi: 10.1016/S0042-6822(03)00590-7. [DOI] [PubMed] [Google Scholar]
- 16.Gotoh B, Takeuchi K, Komatsu T, Yokoo J. 2003. The STAT2 activation process is a crucial target of Sendai virus C protein for the blockade of alpha interferon signaling. J Virol 77:3360–3370. doi: 10.1128/JVI.77.6.3360-3370.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mao X, Ren Z, Parker GN, Sondermann H, Pastorello MA, Wang W, McMurray JS, Demeler B, Darnell JE Jr, Chen X. 2005. Structural bases of unphosphorylated STAT1 association and receptor binding. Mol Cell 17:761–771. doi: 10.1016/j.molcel.2005.02.021. [DOI] [PubMed] [Google Scholar]
- 18.Reich NC. 2013. STATs get their move on. JAKSTAT 2:e27080. doi: 10.4161/jkst.27080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Niwa H, Yamamura K, Miyazaki J. 1991. Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene 108:193–199. doi: 10.1016/0378-1119(91)90434-D. [DOI] [PubMed] [Google Scholar]
- 20.Sugahara F, Uchiyama T, Watanabe H, Shimazu Y, Kuwayama M, Fujii Y, Kiyotani K, Adachi A, Kohno N, Yoshida T, Sakaguchi T. 2004. Paramyxovirus Sendai virus-like particle formation by expression of multiple viral proteins and acceleration of its release by C protein. Virology 325:1–10. doi: 10.1016/j.virol.2004.04.019. [DOI] [PubMed] [Google Scholar]
- 21.Irie T, Yoshida A, Sakaguchi T. 2013. Clustered basic amino acids of the small Sendai virus C protein Y1 are critical to its Ran GTPase-mediated nuclear localization. PLoS One 8:e73740. doi: 10.1371/journal.pone.0073740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sakaguchi T, Irie T, Kawabata R, Yoshida A, Maruyama H, Kawakami H. 2011. Optineurin with amyotrophic lateral sclerosis-related mutations abrogates inhibition of interferon regulatory factor-3 activation. Neurosci Lett 505:279–281. doi: 10.1016/j.neulet.2011.10.040. [DOI] [PubMed] [Google Scholar]
- 23.Otwinowski Z, Minor W. 1997. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol 276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 24.Collaborative Computational Project, Number 4. 1994. The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr 50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 25.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. 1998. Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr 54:905–921. [DOI] [PubMed] [Google Scholar]
- 26.Emsley P, Cowtan K. 2004. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 27.Brunger AT. 1992. Free R value: a novel statistical quantity for assessing the accuracy of crystal structures. Nature 355:472–475. doi: 10.1038/355472a0. [DOI] [PubMed] [Google Scholar]
- 28.Bromberg J, Chen X. 2001. STAT proteins: signal tranducers and activators of transcription. Methods Enzymol 333:138–151. doi: 10.1016/S0076-6879(01)33052-5. [DOI] [PubMed] [Google Scholar]
- 29.ten Hoeve J, de Jesus Ibarra-Sanchez M, Fu Y, Zhu W, Tremblay M, David M, Shuai K. 2002. Identification of a nuclear Stat1 protein tyrosine phosphatase. Mol Cell Biol 22:5662–5668. doi: 10.1128/MCB.22.16.5662-5668.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kato A, Ohnishi Y, Hishiyama M, Kohase M, Saito S, Tashiro M, Nagai Y. 2002. The amino-terminal half of Sendai virus C protein is not responsible for either counteracting the antiviral action of interferons or down-regulating viral RNA synthesis. J Virol 76:7114–7124. doi: 10.1128/JVI.76.14.7114-7124.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gotoh B, Takeuchi K, Komatsu T. 2004. Inhibition of the gamma interferon response by a Sendai virus C protein mutant with no STAT1-binding ability. FEBS Lett 567:291–296. doi: 10.1016/j.febslet.2004.05.001. [DOI] [PubMed] [Google Scholar]
- 32.Schaap M, Hancock R, Wilderspin A, Wells G. 2013. Development of a steady-state FRET-based assay to identify inhibitors of the Keap1-Nrf2 protein-protein interaction. Protein Sci 22:1812–1819. doi: 10.1002/pro.2384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhong M, Henriksen MA, Takeuchi K, Schaefer O, Liu B, ten Hoeve J, Ren Z, Mao X, Chen X, Shuai K, Darnell JE Jr. 2005. Implications of an antiparallel dimeric structure of nonphosphorylated STAT1 for the activation-inactivation cycle. Proc Natl Acad Sci U S A 102:3966–3971. doi: 10.1073/pnas.0501063102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mertens C, Zhong M, Krishnaraj R, Zou W, Chen X, Darnell JE Jr. 2006. Dephosphorylation of phosphotyrosine on STAT1 dimers requires extensive spatial reorientation of the monomers facilitated by the N-terminal domain. Genes Dev 20:3372–3381. doi: 10.1101/gad.1485406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Komatsu T, Takeuchi K, Yokoo J, Gotoh B. 2002. Sendai virus C protein impairs both phosphorylation and dephosphorylation processes of Stat1. FEBS Lett 511:139–144. doi: 10.1016/S0014-5793(01)03301-4. [DOI] [PubMed] [Google Scholar]
- 36.Wenta N, Strauss H, Meyer S, Vinkemeier U. 2008. Tyrosine phosphorylation regulates the partitioning of STAT1 between different dimer conformations. Proc Natl Acad Sci U S A 105:9238–9243. doi: 10.1073/pnas.0802130105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Garcin D, Marq JB, Strahle L, le Mercier P, Kolakofsky D. 2002. All four Sendai virus C proteins bind Stat1, but only the larger forms also induce its mono-ubiquitination and degradation. Virology 295:256–265. doi: 10.1006/viro.2001.1342. [DOI] [PubMed] [Google Scholar]
- 38.DeLano WL. 2002. The PyMOL molecular graphics system. DeLano Scientific, San Carlos, CA. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.