

Background: The effects of silent mutations on pre-mRNA splicing are poorly understood.
Results: Silent mutations can significantly influence exon inclusion and are under purifying selection.
Conclusion: Splicing and coding pressures have co-evolved to maintain sufficient exon inclusion levels.
Significance: Modified species alignment approaches can be used to identify silent mutations that may alter exon inclusion.
Keywords: alternative splicing, evolution, genomics, RNA processing, RNA splicing, spliceosome
Abstract
Alternative splicing diversifies mRNA transcripts in human cells. This sequence-driven process can be influenced greatly by mutations, even those that do not change the protein coding potential of the transcript. Synonymous mutations have been shown to alter gene expression through modulation of splicing, mRNA stability, and translation. Using a synonymous position mutation library in SMN1 exon 7, we show that 23% of synonymous mutations across the exon decrease exon inclusion, suggesting that nucleotide identity across the entire exon has been evolutionarily optimized to support a particular exon inclusion level. Although phylogenetic conservation scores are insufficient to identify synonymous positions important for exon inclusion, an alignment of organisms filtered based on similar exon/intron architecture is highly successful. Although many of the splicing neutral mutations are observed to occur, none of the exon inclusion reducing mutants was found in the filtered alignment. Using the modified phylogenetic comparison as an approach to evaluate the impact on pre-mRNA splicing suggests that up to 45% of synonymous SNPs are likely to alter pre-mRNA splicing. These results demonstrate that coding and pre-mRNA splicing pressures co-evolve and that a modified phylogenetic comparison based on the exon/intron architecture is a useful tool in identifying splice altering SNPs.
Introduction
Pre-mRNA splicing is an essential process necessary for both proper gene expression and the generation of transcript diversity throughout metazoans (1). Intron removal, directed by sequence signals within the pre-mRNA, is catalyzed by the spliceosome, a large ribonuclear protein complex (2). The interaction of splicing regulatory elements and their trans-acting binding partners (hnRNPs, SR proteins, and small nuclear ribonucleoproteins)2 determines where and how splicing takes place (3). Mutations within binding sites of these trans-acting factors will likely alter the identity of the resulting mRNA (4).
The presence of sequence elements that influence splicing throughout the transcript suggests that organisms that rely on high fidelity splicing will be under evolutionary pressure to maintain optimal splice signals within the pre-mRNA molecule (5). Indeed, it has previously been shown that splicing regulatory elements exhibit positive selection, that intron/exon boundaries have a decreased frequency of single nucleotide polymorphisms (SNPs), and that certain codons are generally preferred around intron/exon junctions (6–8). These observations suggest that the spliceosome has to recognize exons by using sequences that are co-evolving with the amino acid sequence code to generate an RNA molecule that both translates and properly splices. However, it is difficult to uncouple sequence requirements imposed by evolutionary coding pressures from sequence requirements necessary to generate the appropriate mRNA through splicing.
Splicing requires sequence elements from both the intron and the exon for proper intron excision. Within the exon, those sequence elements are restricted in sequence by the need for correct protein coding. Given the diversity of the genetic code in the third or wobble position, it has been possible in a few cases to separate splicing constraints from coding constraints (9, 10). Here, we characterize the contribution of splicing evolutionary pressures through the identification of synonymous mutations that specifically alter the efficiency of intron removal. We created a library of synonymous mutations across exon 7 of the SMN1 gene to identify positions that influence exon inclusion using deep-sequencing approaches. Phylogenetic comparisons with organisms that have a similar SMN1 exon 7 architecture demonstrated a selection against exon inclusion-reducing mutants, clearly demonstrating that evolutionary pressures exist at wobble positions to uphold efficient splicing of this crucial exon. A survey of human synonymous SNPs showed that 45% contain potentially splice altering nucleotide substitutions. These observations suggest that a large proportion of synonymous SNPs can cause defects in splicing. Thus, filtering multiple species alignments by exon architectural similarity represents a novel strategy to identify exonic splicing regulatory elements and synonymous mutations that disrupt proper pre-mRNA splicing.
Experimental Procedures
Cell Culture and Transfection of HeLa Cells
HeLa cells used in this work were maintained at 37 °C in a monolayer in Dulbecco's high glucose modified Eagle's medium (Invitrogen) supplemented with 10% fetal bovine serum, 4 mm l-glutamate, and 1 mm Na-pyruvate. Their cell confluence was kept to ∼80% or less before splitting cells. Cells were transfected according to the manufacturer's specifications for Lipofectamine 2000 (Invitrogen) for plate sizes of 10-cm 6-well plates with 3-cm wells, or 15-cm plates.
Creation/Transfection of SMN1 Exon 7 Library Mutations
We used previously described SMN mini-genes containing exons 6, 7, and 8 with shortened introns between each exon (11–13). To generate a library of synonymous mutations across exon 7, we created a series of mutagenesis primers to make a site-directed saturation mutagenesis library of exon 7. This library is comprised of synonymous site-directed mutations spanning the 54-nucleotide exon 7 region of the SMN1 gene. All possible combinations of synonymous mutations were generated within a sliding 2-codon window such that the gene amino acid sequence remained the same and all possible synonymous mutations were created.
Sets of oligonucleotide primers were used to generate the mutations to each synonymous position in exon 7. To generate the library with fewer per-base errors, oligonucleotide sets used were kept as short as possible and the mutagenesis reactions were split into three separate pooled reactions (sequences of oligonucleotides used are available upon request). The first reaction contained a pool of 8 forward and 8 reverse oligonucleotides (oligos 1–8), the second reaction contained a pool of 10 forward and 10 reverse oligonucleotides (oligos 9–18), and the third reaction contained a pool of 12 forward and 12 reverse oligonucleotides (oligos 19–30), all of which contain a double randomized NNN NNN codon at its center such that the two amino acids are simultaneously mutated (but only to synonymous mutations).
PCR amplifications for each mutagenesis pool of SMN1 exon 7 were performed in 50-μl reactions containing: 50 ng of the plasmid based pCi SMN1 mini-gene template, 5 units of Pfu Ultra-High Fidelity DNA polymerase (Stratagene); 400 μm dNTPs; 1× Pfu Ultra-High Fidelity reaction buffer; and 0.03 μm of each complementary mutant primer pair. Primer extension and PCR amplification reactions were carried out by: 10 min denaturation at 95 °C, followed by 16 cycles of 15 s at 95 °C, 40 s at 55 °C, 3 min at 72 °C, and a final step of 10 min at 72 °C. The 50-μl PCR products were digested with 30 units of DpnI for 3 h at 37 °C to remove the methylated template plasmid. 5 μl of the digestion reaction was used for transformation of DH5α cells and plated onto LB-agar plates (100 μg/ml of ampicillin). At this point, a plasmid library containing saturated site-directed mutated target regions is generated. Colonies were selected and suspended in 5 ml of LB medium (100 μg/ml of ampicillin) and incubated at 37 °C overnight. 15% glycerol stocks were created, and larger 200-ml flasks of LB medium (100 μg/ml of ampicillin) were inoculated and incubated at 37 °C overnight. Plasmid libraries were purified from the large cultures using the Nucleobond Midi-prep Purification system. Plasmid libraries were then combined in equal molar amounts to make one master library.
Transfection and Sequencing Library Preparation
The approach for the generation of sequencing libraries is outlined in Fig. 1. To generate a library of spliced mRNAs, HeLa cells at ∼80% confluence in 10-cm plates were transfected with 22 μg of the master plasmid library using Lipofectamine 2000 for 6 h in serum-free medium at 37 °C. After 6 h, the Lipofectamine medium was removed and medium-containing serum was added. The cells were incubated another 18 h at 37 °C. Total RNA was purified from the cells using TRIzol reagent (Ambion) according to the manufacturer's recommendations and treated with DNase (Invitrogen). Reverse transcription using SuperScript II reverse polymerase (Invitrogen) was carried out using library plasmid-specific primers (pCI backbone forward primer: GCTAACGCAGTCAGTGCTTC; pCI backbone reverse primer: GTATCTTATCATGTCTGCTCG) and 4 μg of RNA. The cDNA reaction was then cleaned up using a Qiagen PCR purification kit according to the manufacturer's recommendations. Approximately 0.5 μg of purified cDNA was then amplified using primers specific to exon 6 and exon 8 (output cDNA forward primer: CCCTACACGACGCTCTTCCGATCTCATGAGTGGCTATCATACTGGC; output cDNA reverse primer: CCTGCTGAACCGCTCTTCCGATCTGTCATTTAGTGCTGCTCTATGC; Seq. tail forward primer: AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCT; Seq. tail reverse primer: CAAGCAGAAGACGGCATACGAGATCGGTCTCGGCATTCCTGCTGAACCGCTCTTCCGATCT) that also had tails containing the initial segment of Illumina-specific sequencing primers in a 50-μl reaction containing 400 nm dNTPs using proofreading DNA polymerase. PCR amplification reactions were carried out by: 5 min denaturation at 95 °C, 3 cycles of: 15 s denaturing at 95 °C, 30 s primer annealing at 55 °C, 20 s of extension at 72 °C, and a final extension of 5 min at 72 °C. This initial PCR library amplification was then run out on a 1.5% agarose gel to remove exon exclusion events and gel purified using a Qiagen Gel extraction kit. The purified PCR product was then amplified again for 12 cycles using the same PCR cycle conditions as described above, except using a full-length Illumina sequencing primer. The products of this reaction were separated by 1.5% agarose gel electrophoresis and the PCR product band was purified using the Qiagen gel extraction kit according to the manufacturer's recommendations. This final library was analyzed for quality on an Agilent Bioanalyzer 2100, quantified using a Nanodrop (ND-1000 Thermo), and then diluted to 10 nm and submitted for sequencing on the Illumina Hi-seq platform. The spliced mRNA pool is referred to as the “output pool.”
FIGURE 1.

Mutation scheme for the SMN1 exon 7 library preparation. A, all possible silent mutations were generated per in-frame hexamer across the exon. For example, the first two codons depicted are GGT TTC. All three mutations were made in GGT resulting in GGN and combined with all silent mutations in TTC (TTT), resulting in eight combinations including the wild-type sequence. B, the library of mutations were sequenced and transfected into HeLa cells. The RNA resulting from the transfection was purified and sequenced. The relative abundance of each mutation was compared between the two pools to generate fitness index values.
To generate an equivalent sequencing library of the expression plasmid pool, the same procedure (minus transfection and RNA purification) was carried on the plasmid pool using primers specific to introns 6 and 7 (input DNA forward primer: CCCTACACGACGCTCTTCCGATCTGCTATTTTTTTTAACTTCCTTTATTTTC; input DNA reverse primer: CCTGCTGAACCGCTCTTCCGATCTGTAAGATTCACTTTCATAATGCTG). The expression plasmid pool is referred to as the “input pool.” PCR analysis of SMN exon 7 mutants for validation and comparison of our sequencing experiment was carried out using primers as previously described (11, 17).
Bioinformatic Analysis of SMN1 Exon 7 Library Mutations
We received 54,780,073 single-end reads of 100 nt from our sequencing run. Using Bowtie these reads were aligned to a custom index made from the genomic segment of SMN1 spanning exons 6 to 8 (14), from the spliced mRNA sequence spanning exons 6, 7, and 8, and from all library mutants made. Thus, the alignment index contained an entry for each library mutation to control for possible mutations or sequencing errors. Only reads that contained the wild-type sequence or library mutations were aligned. Approximately three fourths of the reads were aligned to the custom index. Another 8% of the reads corresponded to exon exclusion reads, and the remaining reads did not align.
The total read count for each library mutation was used to determine the relative representation of each mutation within in the sequenced input and output pools. To evaluate changes in exon 7 inclusion, we calculated WT-normalized differences in mutant output and input ratios according to “fitness index” = (output/WT output)/(input/WT input). These ratios are defined as the fitness index. The average fitness index for a position is the average of the fitness indices for all mutations at that position.
For statistical analysis we compared the output and input reads for each mutation to the non-mutated reads using a Fisher's Exact test and determined the significance using a Bonferroni correction for comparison between the 138 mutations. In addition to the Fisher's Exact test, we applied a biological cutoff for significance of the fitness index value. Twenty percent exon 7 inclusion is consistent with mRNA expression reports for SMA type I. An observable difference in the exon inclusion phenotypes between SMA types I and III has been reported (12, 15–17). In SMA type III there is ∼70% inclusion of exon 7 compared with WT levels, versus the 20% observed in type I. Therefore, we set our biological significance cutoff at this 70% value.
Our analysis relied on the circumstantially validated assumptions that all plasmids transfect and transcribe with the same efficiency and that the resulting RNAs are similarly stable in HeLa cells. A decrease in transcription or transcriptional pausing causing a loss of reads is unlikely, as significant numbers of reads were generated and analyzed corresponding to each mutation (average cDNA reads 209,891/mutation, median: 39,288). This suggests that each mutation was represented within the input plasmid pool, successfully transfected, and successfully expressed. The lowest read count observed for a single mutant was 30 output reads for the mutations in positions 24 and 27. This position is known for being a splicing regulator (Tra2-β1/SRSF10) binding site, suggesting that the loss in reads was due to the loss of an enhancer binding sequence (18). Consistently, low output reads correlated with a loss of a known positive splicing regulator, the gain of a negative splicing regulator, or increasing known local RNA secondary structures. It is not known whether mRNAs generated from the reporter plasmids are translated, and therefore are potential NMD targets. However, because all exon 7 mutations are located within 50 nt of the last exon-exon junction, it is unlikely that the resulting mRNA would be targeted by NMD (19). It is also unknown whether the stability of exon 7 included mRNAs generated from the mini-gene constructs is different from mRNAs lacking exon 7.
Comparative Alignment Analysis of SMN1 Exon 7 Mutations and Synonymous SNPs
The position of SMN1 exon 7 was identified through the UCSC Genome Browser. The UCSC Table Browser was then used to download the PhyloP Score by position across the exon (position chr5: 70,247,768 to 70,247,821) using the Comparative Genomics Group and the Conservation Track in the Vertebrate 46 way alignment table SMN1. PhyloP conservation scores allow a measure of conservation by position. Multiple alignment data were generated by taking this same genomic position into the Esembl! database (GRCh37) and selecting the comparative genomics (text option) tool for all 36 eutherian mammals in the Ensembl! Genome Sequence Viewer. This was also used to validate intron/exon architecture. Intron/exon architecture similarity was determined through sequence analysis using the comparative genomics tool. Organisms with similar exon length (±6 nt) and splice site sequences were compared. Species that exhibit similar exon/intron architecture were kept in the alignment. Species exhibiting differing exon/intron architecture were excluded from the alignment. In the case of the SMN alignment, all primates were included in the intron/exon architecture filter, however, this excluded multiple species from rodents to the alpaca. The same approach was used for filtering SNP-based species alignments.
Synonymous SNPs associated with human disease were taken from the literature and from the Entrez dbSNP web browser and added to synonymous human SNPs downloaded using the Ensembl! PerlAPI (20). The 40 disease associated SNPs and 40 Ensembl! SNPs were randomly selected and analyzed using the Ensembl! genome browser comparative genomics alignment tools as was done for the SMN exon analysis. Eighty more human synonymous SNPs from non-coding exons, plus an additional 82 synonymous SNPs with a single exon were also acquired using the Ensembl! PerlAPI and analyzed in the same manner (supplemental Table S1). Filtered organisms were compared by sequence conservation in an 11-nucleotide window around the SNP position. The numbers of the classified putative splice altering SNPs and neutral SNPs were compared using a χ2 test of independence.
Results
Analysis of Library Mutations
To determine the influence of synonymous mutations on exon inclusion, we used the well studied SMN1 mini-gene, spanning exons 6–8 (13, 17, 21) in which exon 7 can be included or excluded depending on the splicing signals within the pre-mRNA. Each set of two neighboring codons in exon 7 was mutated to every possible combination of silent mutations within the context of a hexamer, a minimal binding platform for splicing regulatory proteins (Fig. 1A) (22). The resulting library of plasmids was transfected into HeLa cells and plasmid-specific mRNAs were analyzed by deep sequencing (Fig. 1B). Relative exon inclusion indexes were determined by calculating the abundance of mutations present in the exon 7-included mRNA population, normalized to the abundance in the input plasmid control. Presumably, differences in inclusion indexes reflect differences in exon 7 inclusion levels (Table 1). To test this assumption a small number of mutants (Table 2 and Fig. 2) were tested individually for exon inclusion levels using standard RT-PCR approaches. The near linear relationship between the different experimental approaches demonstrate that changes in the fitness indexes are largely driven by altered pre-mRNA splicing efficiencies. If a mutation was observed more frequently in the spliced mRNA pool than the input plasmid pool, that mutation was considered to be beneficial to exon inclusion. If a mutation was observed less frequently in the spliced mRNA pool compared with the input plasmid pool, that mutation was considered to be inhibitory to exon inclusion.
TABLE 1.
All mutations analyzed
Summary of results obtained for all library mutations analyzed. The mutation positions are from the first position of SMN1 exon 7. The wild-type and mutant nucleotides are separated by “>”. Read counts and exon inclusion ratios were found and calculated as described under “Experimental Procedures”. Mutants highlighted in grey were found to cause a significant reduction in the fitness index value.
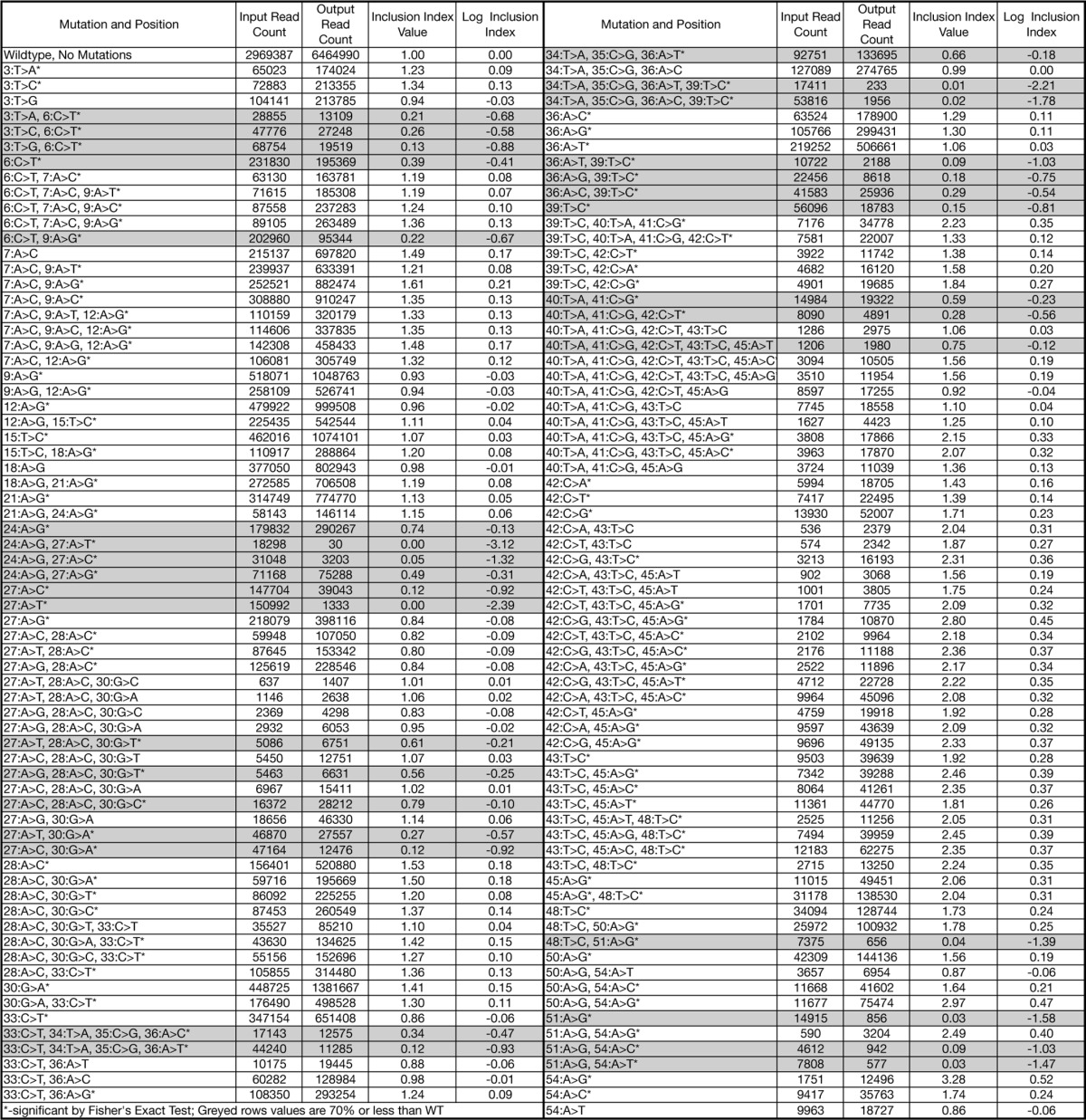
TABLE 2.
Validated mutations
The mutations in this table have been previously reported in the literature or were validated from individual clones isolated from our library pools. Comparison to our data set shows that previous results are replicated faithfully (compare the similarity in changes between fitness index value or log transform with the previously published inclusion values).
| Mutation and position | Inclusion index value | Log inclusion index value | Previously analyzed % inclusion for exon 7 | Citations |
|---|---|---|---|---|
| Wild-type, no mutations | 1 | 0 | 75–100% | 11, 12, 20, 23, 31, 32, see Fig. 2 |
| 3:T>G | 0.94 | −0.03 | 96% | 31 |
| 3:T>G, 6:C>T | 0.13 | −0.88 | 6% | 31 |
| 6:C>T | 0.39 | −0.41 | 26% | 23 |
| 6:C>T, 7:A>C | 1.19 | 0.08 | 100% | 26, 32 |
| 7:A>C | 1.49 | 0.17 | 100% | 26, 32 |
| 27:A>C | 0.12 | −0.92 | 46% | See Fig. 2 |
| 27:A>T | 0.00 | −2.40 | 11% | See Fig. 2 |
| 27:A>G | 0.84 | −0.08 | 89% | See Fig. 2 |
| 27:A>T, 30:G>A | 0.27 | −0.57 | 76% | See Fig. 2 |
| 27:A>G, 30:G>A | 1.14 | 0.06 | 93% | See Fig. 2 |
| 39:T>C | 0.15 | −0.81 | 33% | See Fig. 2 |
| 39:T>C | 0.15 | −0.81 | 24% | 20 |
| 39:>C, 42:C>A | 1.58 | 0.20 | 91% | See Fig. 2 |
| 39:T>C, 42:C>G | 1.85 | 0.27 | 85% | See Fig. 2 |
| 42:C>A | 1.43 | 0.16 | 84% | See Fig. 2 |
| 42:C>G | 1.71 | 0.23 | 87% | See Fig. 2 |
| 45:A>G | 2.06 | 0.31 | 93% | 31 |
| 54:A>G | 3.28 | 0.52 | 100% | 20 |
FIGURE 2.

Analysis of library mutants with a single mutation. A, correlation between fitness index values derived from the high-throughput analysis and exon inclusion levels of individual mutants as determined by PCR analysis. B, average fitness index values for library mutants with mutations at a single hexamer position. The gray line represents the significance cutoff. Mutations that caused a significant decrease in the fitness index value are highlighted in red. All other single mutant positions are in blue. The inset is an example of the nucleotide-specific mutational effects observed. Specific mutations at a given position can display large differences in the fitness index as illustrated by position 27. C, conservation scores at synonymous mutation positions across SMN1 exon 7 based on placental mammal PhyloP scores. Bars in red denote high conservation at the same position where mutations cause significant decreases in the fitness index. Bars in orange show the positions where conservation is low and where mutations cause significant decreases in the fitness index.
The statistical significance of an observed change in relative mutant abundance was compared with the wild-type (WT) representation using a Fisher's Exact test with Bonferroni correction. In addition a biological filter was imposed based on previous observations that mRNA levels of SMA type III patients display exon 7 inclusion levels at ∼70% of that observed for healthy individuals (12, 15). All mutants that caused a decrease in the fitness index to 70% or less of the unchanged WT value were considered to have a negative effect on exon inclusion. Of 138 mutations tested, 32 met the criteria for significant changes in the fitness index (Table 1).
The fact that only 32 of the 138 mutations evaluated caused significant fitness index reductions demonstrates that the majority of the mutations tested did not appear to cause striking changes in exon inclusion. Most mutations that caused significant changes negatively affected the fitness index. Interestingly, the fitness index-altering mutations were observed across the entire SMN exon 7. This observation suggests that optimization of exon inclusion levels for SMN was achieved at multiple exonic locations, providing a sufficient degree of flexibility to respond to localized coding pressures.
Single Library Mutations
When single nucleotide mutations were analyzed we observed both increased and decreased splicing efficiencies (Fig. 2B). Of the 32 mutations that caused significant decreases in the fitness index value, 6 of them were single nucleotide mutations (Table 1). Several of the single point mutations that caused the largest decreases in the fitness index have previously been reported as prominent splice altering mutations. Of 8 mutations previously evaluated in the literature, all are faithfully reproduced using our pooled deep sequencing approach (Table 2). Similarly, some of the mutations in our data that increased the fitness index value were previously reported in the literature to either destabilize an inhibitory RNA secondary structures or to increase the strength of the splice sites, i.e. alter the sequence to be closer to the splicing consensus sequence as measured by maximum entropy score (Fig. 3) (23). This agreement with previously published work further demonstrates that our deep sequencing approach reliably evaluates changes in splicing efficiencies. For example, when the highly conserved residue at position 6 was mutated to a synonymous codon (cytidine to thymidine), thus emulating the known difference in sequence between SMN1 and SMN2, a 2.6-fold decrease in the fitness index was observed, in agreement with exon inclusion differences observed for SMN1 and SMN2 (4, 12, 24–27) (Table 1). Increases in fitness index values due to mutations may also be due to pre-mRNA or mature mRNA stability issues.
FIGURE 3.
Analysis of library mutants at the 5′ splice site of SMN1 exon 7/intron 7. A, fitness index values for mutants at the last positions of exon 7. Mutations that caused a significant decrease in the fitness index value are highlighted in red. All other mutations are in blue. B, maximum entropy scores for the wild-type (WT) 5′ splice site and the mutations at position 54, which is the last exonic nucleotide. Greater maximum entropy score correlate with more efficient splice site usage. The slash in the sequences denotes the exon-intron junction.
Furthermore, single nucleotide mutant influences on the fitness index values are nucleotide specific. For example, changing nucleotide position 27 from an adenosine to thymidine or cytidine caused a ∼200- or ∼8-fold decrease in exon inclusion, respectively (Fig. 2B, inset). However, when mutated to guanosine no significant change in the fitness index was observed.
To test the hypothesis that impermissible nucleotide changes at synonymous positions correlate with evolutionary conservation, we compared the mutated positions with their conservation scores as measured by PhyloP (28, 29). PhyloP uses a 46-vertebrate species alignment to assign positive values to positions that are conserved and gives lower or negative values to swiftly evolving or less conserved positions. Presumably, if a synonymous position is important for splicing, it is expected that its nucleotide identity would be conserved. This expectation is met when evaluating nucleotides at positions 6 and 51 (Fig. 2C). Based on the PhyloP score both positions are highly conserved, which is in agreement with the hypothesis that the nucleotide identity may be evolutionarily fixed due to selective pressures for correct splicing. However, as data from mutations at positions 27 and 39 indicate, this splicing correlated conservation is not typical in our dataset (Fig. 2C). Several of the synonymous mutation positions that had significant fitness index changes exhibit very low PhyloP scores. We conclude that the PhyloP score at a given wobble position does not directly predict the influence of the nucleotide on pre-mRNA splicing.
One potential explanation for the lack of a direct correlation between the PhyloP score and the fitness index value at synonymous positions is the fact that the exon/intron architecture may vary between species (Fig. 4A). As such, the evolutionary pressures to maintain a sequence for a particular splicing pattern can be different between species, eliminating positional conservation restrictions imposed by splicing. Our analysis clearly shows that different nucleotide changes at the same position can range from neutral to high impact, demonstrating that some nucleotide changes are permissible whereas others are not (Fig. 2B, inset, and Table 1). As PhyloP only measures the frequency of a nucleotide substitution irrespective of patterns or trends of mutations at a specific position, it is expected that in some cases the conservation score cannot adequately reflect splicing pressures. To circumvent these PhyloP limitations we filtered species alignments based on the requirement of similar intron/exon architecture around SMN1 exon 7. Using the Ensembl! genomic alignment tool, we hand filtered species alignments based on SMN exon 7 architectural conservation. This filtering approach reduced the number of aligned species to 17, demonstrating that even among the more closely related placental vertebrates large differences within the exon/intron architecture exist. The filtered alignments were then evaluated for the presence or absence of synonymous mutations that were introduced by our high-throughput approach (Fig. 4B). Interestingly, none of the synonymous mutations that caused significant decreases in the fitness index were found in this alignment data, whereas synonymous mutants that do not decrease the fitness index value were admissible (Fig. 4B). This perfect correlation between admissible/inadmissible synonymous mutations and their fitness index values suggests that the exon/intron architecture filtered conservation evaluation at synonymous positions can be utilized to determine exon positions important for efficient splicing, at least in the context of SMN exon 7.
FIGURE 4.

Sequence alignment of SMN1 exon 7 with 17 animals with similar intron/exon architecture. A, a graphical representation of the method used to filter species alignments based on conservation of intron/exon architecture. Species E and F would not be included in an architecture-filtered alignment. B, the alignment of similar mammals that pass the intron/exon architecture filter to human SMN1 exon 7. The mutations that caused significant decreases in the fitness index value are listed above their position in the alignment with the specific hexamer mutations in red. The alignment below the hexamers shows the silent mutation positions that diverge from the human sequence. The mutations that did not cause changes in the fitness index value are highlighted in green. The color-coding used to differentiate the effects of the observed mutants is shown in the key on the figure bottom. The category “nonsynonymous alignment mutations” refers to positions in the multispecies alignment where observed nucleotide substitutions would be considered nonsynonymous mutations in the human exon.
Hexamer Library Mutations
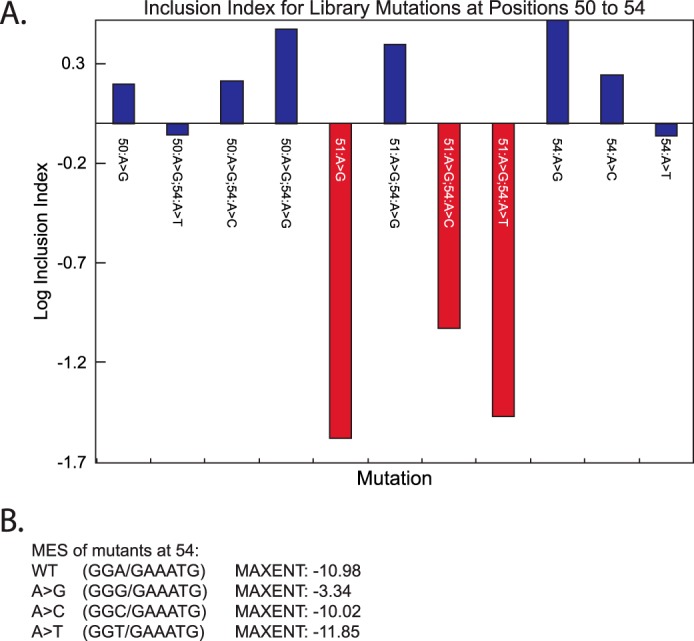
Mutations of more than one nucleotide position within a hexamer appear to either rescue or to exacerbate the fitness index changes seen in the single mutant variations (Fig. 5 and Table 1). By themselves, mutations 24:A>G, and 27:A>C/T cause significant decreases in the fitness index value, but surrounding positions (21, 28, and 30) do not (Fig. 5A). When mutated in combination, synergistic decreases in the fitness index are observed, even in the case of the mutation 24:A>G with 27:A>G (not significant on its own, but significant with position 24). Conversely, when paired with mutations at positions 28, 29, or 30, the affects of position 27 are decreased, although not always to the point of being not significant (except in the case of 27:A>C, 30:G>A where there was no appreciable change). These results suggest that neighboring mutations within the context of a hexamer can influence exon inclusion in multiple ways. These data and others (Figs. 3 and 5) suggest that specific internal sequences in the exon are vital for optimal exon inclusion in a hexamer specific context.
FIGURE 5.
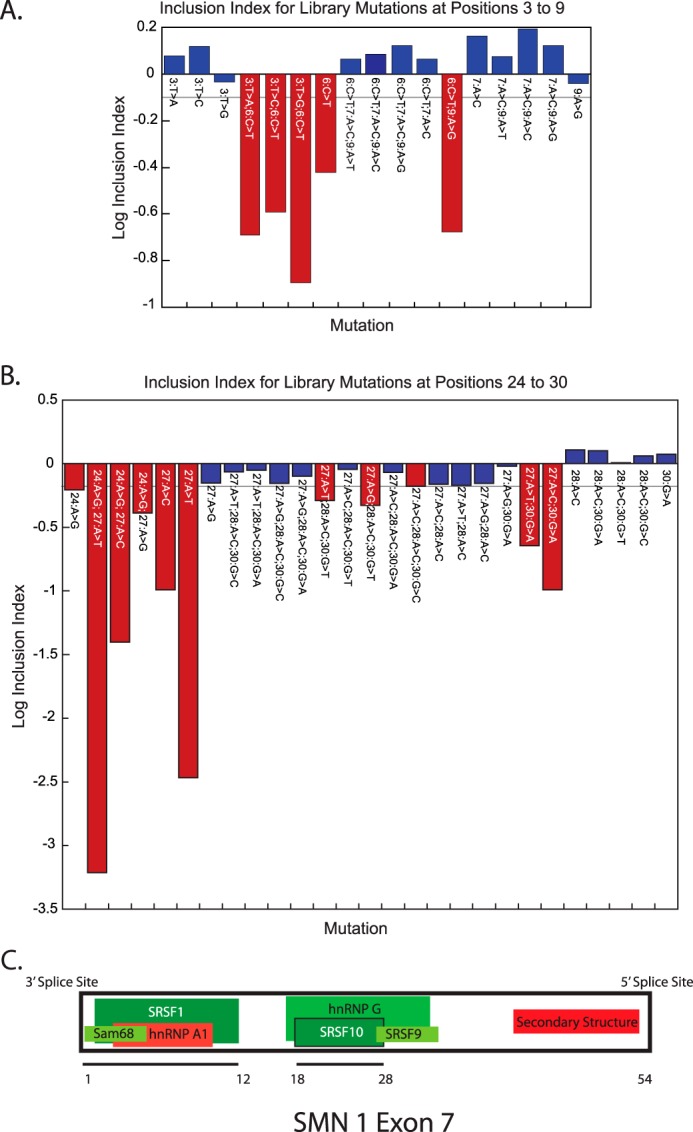
Compensatory, additive, and synergistic mutational effects within hexamers. A, the bars represent the fitness index values at positions 3 through 9. Mutations with significant fitness index changes are highlighted in red, whereas all others are in blue. The gray line represents the significance cutoff. B, the bars represent the fitness index values for mutations made between position 24 and 30. Color-coding follows the same convention as in A. C, model of known protein interactions across SMN exon 7. Boxes here depict a schematic of SMN exon 7 and proteins that have been shown to interact with the exon and with each other to modulate exon 7 inclusion. Elements that are assumed to increase exon inclusion have been shaded in green, whereas those that decrease inclusion are shown in red (22, 30).
The extensive amount of study devoted to the SMN1 gene allowed multiple built-in controls for our analysis by correlating changes in regions that are known to be important binding sites for splicing regulatory elements with changes observed in the fitness index (Fig. 5C) (4, 13, 24–27, 30–32). At position 3, none of the 3 possible single silent mutations alter the fitness index appreciably (Fig. 5B). However, when combined with the C to T mutation at position 6 exon skipping is exasperated, consistent with the notion that the binding site for splicing regulators are disrupted or that more stable RNA secondary structures are formed (4, 13, 24, 25, 27, 30–32). The same synergy is also observed when mutations in positions 6 and 9 are combined. However, if position 7 is mutated along with positions 6 or 9, the reduction in exon inclusion is no longer significant. This result reproduces the finding that the A to C mutation at position 7 rescues the C to T mutation at position 6 either through inhibiting the binding of Sam68, or by preventing the binding of another inhibitory protein (33, 34). Similar examples of synergistic or compensatory mutations are observed to occur at the Tra2-β1 (SRSF10) binding site (positions 18–28), at the 3′ inhibitory secondary structure (positions 33–42), and at the 5′ splice site (21, 23, 32, 35) (Figs. 2, 3, and 5, Table 1). These mutations all involve sequence-specific compensatory mutations that can relieve the decrease in the fitness index caused by other mutations.
When multiple mutations within a hexamer are analyzed using the intron/exon architecture filtered alignment method described above, none of the mutations that cause negative changes in the fitness index were represented in organisms with similar intron/exon architecture. The alignment showed 11 possible synonymous mutations, all of which appeared in our analysis as non-significant changes to the fitness index (Fig. 4B). This observation supports the conclusion that splicing-detrimental hexamer mutants are selected against due to splicing-related sequence constraints.
Analysis of Synonymous SNPs
The filtered phylogenetic comparison approach may allow the identification of genetic mutations that have a high probability to alter pre-mRNA splicing. To test this hypothesis we applied the modified alignment analysis (Fig. 4A) to evaluate whether disease-associated synonymous SNPs are likely to be associated with inducing changes in pre-mRNA splicing. Forty human disease-associated synonymous SNPs and 40 randomly selected synonymous SNPs from the Ensembl! Perl API were combined as a representation of genomic SNPs in coding regions (20, 36). The genomic sequence within a ±5 nt window around the SNP was compared with the homologous sequence of aligned organisms containing similar exon/intron architecture. This sequence window was analyzed for the occurrence of the human SNP nucleotide and surrounding nucleotide changes (supplemental Table S1).
The SNPs were divided into two categories based on the appearance of the evaluated SNP: neutral SNPs and putative splice altering SNPs. There were also non-conserved SNPs that had fewer than 5 organisms in the species alignment after the exon/intron architecture filtering. These cases were not considered further due to limited alignment information. Neutral SNPs were defined as SNPs that were found in the alignment one or more times without additional nucleotide changes in the surrounding sequence. The presence of sequences identical to such human SNPs in other species suggests that they are not selected against and, thus, are likely to be splicing neutral. Putative splice altering SNPs were defined as SNP sequences that did not occur in the alignment or only occurred in conjunction with other mutations in the surrounding hexamer sequence. As was argued by our experimental analysis, such a representation of human SNP sequence across species suggests that nucleotide changes at this position may alter exon inclusion.
In 15 of 80 SNPs there were fewer than 5 organisms that maintained intron/exon architecture conservation (Table 3). Another 36 alignments had multiple occurrences of the human SNP sequence without mutations in the surrounding hexamers, suggesting that these synonymous position mutations were not selected against (Table 3). In 10 of 80 cases the synonymous SNP was not observed in any aligned organism, providing circumstantial evidence that the SNP may alter splicing efficiencies, i.e. the SNP is selected against for that particular intron/exon architecture. In 19 cases the SNP mutation was observed in the aligned species, however, only in conjunction with additional nucleotide changes within the 5-nucleotide flanking window (Table 3). Thus, 45% of the SNPs surveyed are potentially splice altering SNPs. These observations suggest that in many cases of sequence evolution, nucleotide changes within the hexamer context are driven by the need to uphold splicing efficiency.
TABLE 3.
Summary of SNPs analyzed by filtering phylogenetic alignments for similar intron/exon architecture
“Splice altering SNPs” represent SNPs that did not occur in the alignment of organisms with similar intron/exon architecture or, if they occurred, only occurred alongside other mutations within a 6-mer (possible compensatory mutations). “Neutral SNPs” were SNPs that had SNP occurrences without mutations in the surrounding hexamer. The classes of coding exons and exons derived from non-coding genes or single exon genes' exons were found to be independent by a χ2 test (p < 0.05). The SNP identities for all groups are listed in supplemental Table S1.
| Data set | Splice altering SNPs | Neutral SNPs |
|---|---|---|
| Disease associated and random synonymous SNPs | 45% (29) | 55% (36) |
| Randomly selected noncoding exonic or single exon SNPs | 30% (34) | 70% (80) |
| p < 0.05 between the two SNP datasets | ||
To determine whether synonymous SNPs in coding exons exhibit unique selection pressures, we prepared a dataset of 162 SNPs from noncoding exons or from single exon genes (supplemental Table S1). These exons should either only be under evolutionary selection for correct splicing or, in the case of single exon genes, should only be under selection for optimal protein coding. Because of their lack of protein coding constraints, noncoding exons would be expected to have more freedom to mutate nucleotides throughout the exon, not just in silent positions. For these exons it is expected that the selection on synonymous substitution positions would be reduced. Single exon genes, on the other hand, should have a different selective pressure on their synonymous mutation positions that are strictly based on protein coding within the reading frame. We analyzed these 162 control SNPs in the same manner as described above and found that single exon and non-coding genes had a significantly lower number of putative splice altering SNPs at synonymous positions, as expected from the relaxation of evolutionary pressures (Table 3). We conclude that applying the exon/intron architecture filtered approach of phylogeny aids in the identification of exonic sequence that may be necessary for exon inclusion.
Discussion
The work described above outlines a unique approach to identify exonic positions that influence alternative pre-mRNA splicing. Through the creation of a library of synonymous mutations impacting splicing of a single exon coupled with deep sequencing, we found that 23% of those synonymous mutations affected exon inclusion significantly in SMN exon 7. Several of the SMN1 exon 7 mutations evaluated behaved as expected from previous work, highlighting the reliability of our experimental approach. We expected that synonymous positions that significantly alter exon inclusion when mutated would be conserved. However, such positions were not well conserved by measure of PhyloP scores across 46 vertebrates. This prompted us to filter the species used for the evolutionary comparison based on similar intron/exon architecture. With this filter in place, none of the exon inclusion reducing mutations was found in the species alignment. Additionally, mutations that decreased exon inclusion were often rescued back to wild-type inclusion levels by other mutations within a 5-nt radius, suggesting an ability to modulate exon inclusion through the use of compensatory mutations within a hexameric sequence space. The selection of the hexamer as a sequence space for testing combinatorial mutations was based on the size of known RNA-binding protein footprints (22). Although this framework may have been limiting, its use here clearly demonstrates the importance of incorporating analyses of local RNA sequence contexts when evaluating exon inclusion efficiencies and evolutionary flexibility.
The modified alignment approach was used to demonstrate that up to 45% of verified synonymous SNPs might be splice altering. We propose that the exon/intron architecture-based method of species alignment is much more likely to attribute functional significance to sequence elements involved in splicing.
Evolutionary Selection Against Splicing Mutants
Our systematic approach to determine the effect of synonymous mutations on exon inclusion allowed us to comparatively interrogate alignment data in a splicing centric context. The finding that mutations that cause significant reduction of the fitness index were not represented in multiple comparative alignments points to an influence of nucleotide selection by splicing. The presence of splicing regulatory elements at many of these conserved synonymous positions further suggests that there has been positive selection throughout evolution for specific splicing events. Positive selection due to splicing within splicing regulatory elements and around intron/exon junctions has been previously suggested and documented, but only recently experimentally analyzed by testing for missense and nonsense SNPs associated with disease (4, 7, 37–39). However, positive selection is also at work in the genetic code, masking changes that may have occurred due to splicing. Starting with a library of mutants at synonymous positions and filtering phylogenetic data based on the exon/intron architecture allowed us to determine the interesting compensatory nature of evolution to favor a specific functional transcript and to find a way to add information through small nucleotide changes.
Loss of full-length SMN transcript has been shown to be problematic for multiple organisms and its splicing regulation seems to be relatively consistent across species (31). Many examples of mutations affecting splicing and causing disease have been described (4, 20, 37, 40, 41). Despite this, SNPs are poorly characterized when it comes to splicing. They are frequently misattributed as missense or frameshift mutations due to the SNP not being found in close proximity to an intron/exon junction (4, 40, 42). Additionally, synonymous SNPs have only recently been studied in earnest as causes of disease after being found to follow non-neutral evolution (8–10, 37, 41). Our work demonstrated that 23% of synonymous mutation positions in the evaluated coding exon play a role in their inclusion, comparable with previous work (38, 43). The ability to predict how a synonymous SNP may alter pre-mRNA maturation could prove invaluable in the determination of molecular causes of genetic disease. Our method using splicing centric filtered alignments helps to assign the importance of a particular SNP in changing pre-mRNA splicing.
Functional Filtering of Alignments to Enrich for Splicing Related Information
Previous discovery and analysis of splicing regulatory elements and splice altering SNPs relied on conservation data. Our analysis suggests that the limitations of this approach will cause important splicing regulatory positions to be missed. An initial comparison between our mutation outcomes and PhyloP conservation scores did not support the notion that fixation of synonymous exon positions are driven by splicing constraints. In part, this observation is explained by the fact that PhyloP scoring is not able to predict the nucleotide-specific alterations in exon inclusion that occurred in our dataset. Our analysis shows that the use of sequence identity analysis instead of divergence from a specific nucleotide is important when evaluating splicing. Limiting our comparative alignment to similar intron/exon architectures presumably enriched our search space for organisms that regulate exons in a similar fashion, a notion supported by our SNP analysis, which demonstrated that up to 45% of exonic synonymous SNPs are likely to be splice-altering SNPs.
Here, we evaluated the influence of nucleotide identities at synonymous positions on the overall splicing efficiency within the context of the 54-nt long SMN exon 7. We chose this exon mainly because its disease association sparked extensive studies that identified many exonic positions as regulator binding sites that have been shown to be important for splicing, thus providing a knowledge base essential to explain any observed fitness index differences (4, 13, 24–27, 30–32). At 54 nucleotides in length, SMN exon 7 is significantly smaller than the average length of a human exon (∼120 nt). Thus, it is possible that the density of splicing regulatory sites embedded within SMN exon 7 is greater than for human exons of average size. Given these considerations, it is possible that average sized human exons may be exposed to different purifying selection pressures.
Synonymous mutations can alter the ability of an mRNA to code for protein through creation of regulatory defects. They have been shown to alter protein-folding abilities, to change RNA stability, and to induce exon loss (8, 41, 44). These observations suggest that selective pressures to maintain efficient splicing work in tandem with selective pressures to uphold all aspects of the genetic code. In our attempt to uncouple these influences we evaluated the influence of synonymous mutations on pre-mRNA splicing. It is also appreciated that a codon usage bias exists. Lower organisms experience a correlation between codon usage and tRNA copy number, suggesting an optimization for particular codons for efficient translation (45). Interestingly, codon usage in mammals does not correlate with tRNA abundance or tRNA gene copy number, an observation distinctly different from other taxa (8, 45). This deviation, along with this work, suggest that whereas codon bias may influence the final sequence of an exon, it is part of a larger evolutionary picture that includes influences from other processes, such as pre-mRNA splicing, to properly produce functional proteins.
Author Contributions
Experimental design was conceived by K. J. H. and W. F. M. Cloning and construction of plasmids was carried out by L. L. and W. H. Library prep and data analysis was done by W. F. M and A. G. Manuscript preparation was completed by W. F. M. and K. J. H.
Supplementary Material
Acknowledgments
We thank W. Augustine Dunn III and Anke Busch for their discussions and advice, and Tom Caldwell for technical assistance.
This work was supported, in whole or in part, by National Institutes of Health Grant R01 GM62287 (to K. J. H.) and F31 Ca171791 (to A. G.). We declare no competing interests or conflicts of interest in the writing or publishing of this paper.

This article contains supplemental Table S1.
- hnRNP
- heterogeneous nuclear ribonucleoprotein
- SMN
- survival of motor neuron
- SMA
- spinal muscular atrophy
- nt
- nucleotide
- PhyloP
- part of the PHAST package (PHylogenetic Analysis with Space/Time models) used through UCSC Genome Browser to evaluate phylogenetic conservation.
References
- 1.Nilsen T. W., and Graveley B. R. (2010) Expansion of the eukaryotic proteome by alternative splicing. Nature 463, 457–463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Black D. L. (2003) Mechanisms of alternative pre-messenger RNA splicing. Annu. Rev. Biochem. 72, 291–336 [DOI] [PubMed] [Google Scholar]
- 3.Hertel K. J. (2008) Combinatorial control of exon recognition. J. Biol. Chem. 283, 1211–1215 [DOI] [PubMed] [Google Scholar]
- 4.Cartegni L., Chew S. L., and Krainer A. R. (2002) Listening to silence and understanding nonsense: exonic mutations that affect splicing. Nat. Rev. Genet. 3, 285–298 [DOI] [PubMed] [Google Scholar]
- 5.Fox-Walsh K. L., and Hertel K. J. (2009) Splice-site pairing is an intrinsically high fidelity process. Proc. Natl. Acad. Sci. U.S.A. 106, 1766–1771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Willie E., and Majewski J. (2004) Evidence for codon bias selection at the pre-mRNA level in eukaryotes. Trends Genet. 20, 534–538 [DOI] [PubMed] [Google Scholar]
- 7.Parmley J. L., Chamary J. V., and Hurst L. D. (2006) Evidence for purifying selection against synonymous mutations in mammalian exonic splicing enhancers. Mol. Biol. Evol. 23, 301–309 [DOI] [PubMed] [Google Scholar]
- 8.Chamary J. V., Parmley J. L., and Hurst L. D. (2006) Hearing silence: non-neutral evolution at synonymous sites in mammals. Nat. Rev. Genet. 7, 98–108 [DOI] [PubMed] [Google Scholar]
- 9.Goren A., Ram O., Amit M., Keren H., Lev-Maor G., Vig I., Pupko T., and Ast G. (2006) Comparative analysis identifies exonic splicing regulatory sequences: the complex definition of enhancers and silencers. Mol. Cell 22, 769–781 [DOI] [PubMed] [Google Scholar]
- 10.Pagani F., Raponi M., and Baralle F. E. (2005) Synonymous mutations in CFTR exon 12 affect splicing and are not neutral in evolution. Proc. Natl. Acad. Sci. U.S.A. 102, 6368–6372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Geib T., and Hertel K. J. (2009) Restoration of full-length SMN promoted by adenoviral vectors expressing RNA antisense oligonucleotides embedded in U7 snRNAs. PLoS ONE 4, e8204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Madocsai C., Lim S. R., Geib T., Lam B. J., and Hertel K. J. (2005) Correction of SMN2 Pre-mRNA splicing by antisense U7 small nuclear RNAs. Mol. Ther. 12, 1013–1022 [DOI] [PubMed] [Google Scholar]
- 13.Lim S. R., and Hertel K. J. (2001) Modulation of survival motor neuron pre-mRNA splicing by inhibition of alternative 3′ splice site pairing. J. Biol. Chem. 276, 45476–45483 [DOI] [PubMed] [Google Scholar]
- 14.Langmead B., Trapnell C., Pop M., and Salzberg S. L. (2009) Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mailman M. D., Heinz J. W., Papp A. C., Snyder P. J., Sedra M. S., Wirth B., Burghes A. H., and Prior T. W. (2002) Molecular analysis of spinal muscular atrophy and modification of the phenotype by SMN2. Genet. Med. 4, 20–26 [DOI] [PubMed] [Google Scholar]
- 16.Monani U. R., Lorson C. L., Parsons D. W., Prior T. W., Androphy E. J., Burghes A. H., and McPherson J. D. (1999) A single nucleotide difference that alters splicing patterns distinguishes the SMA gene SMN1 from the copy gene SMN2. Hum. Mol. Genet. 8, 1177–1183 [DOI] [PubMed] [Google Scholar]
- 17.Lorson C. L., Hahnen E., Androphy E. J., and Wirth B. (1999) A single nucleotide in the SMN gene regulates splicing and is responsible for spinal muscular atrophy. Proc. Natl. Acad. Sci. U.S.A. 96, 6307–6311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hofmann Y., Lorson C. L., Stamm S., Androphy E. J., and Wirth B. (2000) Htra2-β 1 stimulates an exonic splicing enhancer and can restore full-length SMN expression to survival motor neuron 2 (SMN2). Proc. Natl. Acad. Sci. U.S.A. 97, 9618–9623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Maquat L. E. (2004) Nonsense-mediated mRNA decay: splicing, translation and mRNP dynamics. Nat. Rev. Mol. Cell Biol. 5, 89–99 [DOI] [PubMed] [Google Scholar]
- 20.Sauna Z. E., and Kimchi-Sarfaty C. (2011) Understanding the contribution of synonymous mutations to human disease. Nat. Rev. Genet. 12, 683–691 [DOI] [PubMed] [Google Scholar]
- 21.Singh N. N., Singh R. N., and Androphy E. J. (2007) Modulating role of RNA structure in alternative splicing of a critical exon in the spinal muscular atrophy genes. Nucleic Acids Res. 35, 371–389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Graveley B. R., and Hertel K. J. (2005) SR Proteins. in Encyclopedia of Life Sciences, John Wiley and Sons, Ltd., Chichester [Google Scholar]
- 23.Singh R. N. (2007) Evolving concepts on human SMN Pre-mRNA splicing. RNA Biol. 4, 7–10 [DOI] [PubMed] [Google Scholar]
- 24.Lorson C. L., and Androphy E. J. (2000) An exonic enhancer is required for inclusion of an essential exon in the SMA-determining gene SMN. Hum. Mol. Genet. 9, 259–265 [DOI] [PubMed] [Google Scholar]
- 25.Miyaso H., Okumura M., Kondo S., Higashide S., Miyajima H., and Imaizumi K. (2003) An intronic splicing enhancer element in survival motor neuron (SMN) pre-mRNA. J. Biol. Chem. 278, 15825–15831 [DOI] [PubMed] [Google Scholar]
- 26.Lefebvre S., Bürglen L., Reboullet S., Clermont O., Burlet P., Viollet L., Benichou B., Cruaud C., Millasseau P., and Zeviani M. (1995) Identification and characterization of a spinal muscular atrophy-determining gene (see comments). Cell 80, 155–165 [DOI] [PubMed] [Google Scholar]
- 27.Kashima T., and Manley J. L. (2003) A negative element in SMN2 exon 7 inhibits splicing in spinal muscular atrophy. Nat. Genet. 34, 460–463 [DOI] [PubMed] [Google Scholar]
- 28.Pollard K. S., Hubisz M. J., Rosenbloom K. R., and Siepel A. (2010) Detection of nonneutral substitution rates on mammalian phylogenies. Genome Res, 20, 110–121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pollard K. S., Salama S. R., King B., Kern A. D., Dreszer T., Katzman S., Siepel A., Pedersen J. S., Bejerano G., Baertsch R., Rosenbloom K. R., Kent J., and Haussler D. (2006) Forces shaping the fastest evolving regions in the human genome. PLoS Genet. 2, e168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lorson C. L., Rindt H., and Shababi M. (2010) Spinal muscular atrophy: mechanisms and therapeutic strategies. Hum. Mol. Genet. 19, R111–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Singh N. N., Androphy E. J., and Singh R. N. (2004) The regulation and regulatory activities of alternative splicing of the SMN gene. Crit. Rev. Eukaryot. Gene Expr. 14, 271–285 [DOI] [PubMed] [Google Scholar]
- 32.Singh N. N., Androphy E. J., and Singh R. N. (2004) In vivo selection reveals combinatorial controls that define a critical exon in the spinal muscular atrophy genes. RNA 10, 1291–1305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cartegni L., Hastings M. L., Calarco J. A., de Stanchina E., and Krainer A. R. (2006) Determinants of exon 7 splicing in the spinal muscular atrophy genes, SMN1 and SMN2. Am. J. Hum. Genet. 78, 63–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pedrotti S., Bielli P., Paronetto M. P., Ciccosanti F., Fimia G. M., Stamm S., Manley J. L., and Sette C. (2010) The splicing regulator Sam68 binds to a novel exonic splicing silencer and functions in SMN2 alternative splicing in spinal muscular atrophy. EMBO J. 29, 1235–1247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hoffman B. E., and Grabowski P. J. (1992) U1 snRNP targets an essential splicing factor, U2AF65, to the 3′ splice site by a network of interactions spanning the exon. Genes Dev. 6, 2554–2568 [DOI] [PubMed] [Google Scholar]
- 36.Sherry S. T., Ward M. H., Kholodov M., Baker J., Phan L., Smigielski E. M., and Sirotkin K. (2001) dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 29, 308–311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Teng M., Wang Y., Wang G., Jung J., Edenberg H. J., Sanford J. R., and Liu Y. (2011) Prioritizing single-nucleotide variations that potentially regulate alternative splicing. BMC Proc. 5, S40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sterne-Weiler T., Howard J., Mort M., Cooper D. N., and Sanford J. R. (2011) Loss of exon identity is a common mechanism of human inherited disease. Genome Res. 21, 1563–1571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ke S., Zhang X. H., and Chasin L. A. (2008) Positive selection acting on splicing motifs reflects compensatory evolution. Genome Res. 18, 533–543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Krawczak M., Reiss J., and Cooper D. N. (1992) The mutational spectrum of single base-pair substitutions in mRNA splice junctions of human genes: causes and consequences. Hum. Genet. 90, 41–54 [DOI] [PubMed] [Google Scholar]
- 41.Sauna Z. E., Kimchi-Sarfaty C., Ambudkar S. V., and Gottesman M. M. (2007) The sounds of silence: synonymous mutations affect function. Pharmacogenomics 8, 527–532 [DOI] [PubMed] [Google Scholar]
- 42.Faustino N. A., and Cooper T. A. (2003) Pre-mRNA splicing and human disease. Genes Dev. 17, 419–437 [DOI] [PubMed] [Google Scholar]
- 43.Chen R., Davydov E. V., Sirota M., and Butte A. J. (2010) Non-synonymous and synonymous coding SNPs show similar likelihood and effect size of human disease association. PLoS ONE 5, e13574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kimchi-Sarfaty C., Oh J. M., Kim I. W., Sauna Z. E., Calcagno A. M., Ambudkar S. V., and Gottesman M. M. (2007) A “silent” polymorphism in the MDR1 gene changes substrate specificity. Science 315, 525–528 [DOI] [PubMed] [Google Scholar]
- 45.Plotkin J. B., and Kudla G. (2011) Synonymous but not the same: the causes and consequences of codon bias. Nat. Rev. Genet. 12, 32–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.