

Background: Perforin is a critical component of immune homeostasis, responsible for clearing virally infected cells.
Results: The molecular details of calcium binding by the perforin C2 domain are revealed.
Conclusion: Calcium-mediated structural rearrangement activates perforin for membrane binding.
Significance: The C2 domain regulates membrane binding by calcium-dependent events, which have now been defined for a mammalian perforin C2.
Keywords: calcium-binding protein, crystal structure, immunology, NMR, x-ray crystallography, multisite ligand binding, protein-membrane interaction
Abstract
Natural killer cells and cytotoxic T-lymphocytes deploy perforin and granzymes to kill infected host cells. Perforin, secreted by immune cells, binds target membranes to form pores that deliver pro-apoptotic granzymes into the target cell. A crucial first step in this process is interaction of its C2 domain with target cell membranes, which is a calcium-dependent event. Some aspects of this process are understood, but many molecular details remain unclear. To address this, we investigated the mechanism of Ca2+ and lipid binding to the C2 domain by NMR spectroscopy and x-ray crystallography. Calcium titrations, together with dodecylphosphocholine micelle experiments, confirmed that multiple Ca2+ ions bind within the calcium-binding regions, activating perforin with respect to membrane binding. We have also determined the affinities of several of these binding sites and have shown that this interaction causes a significant structural rearrangement in CBR1. Thus, it is proposed that Ca2+ binding at the weakest affinity site triggers changes in the C2 domain that facilitate its interaction with lipid membranes.
Introduction
Cell-mediated immunity is an essential host cell defense system against invasion by viruses, bacteria, and other agents of disease. Cytotoxic lymphocytes, cytotoxic T-lymphocytes, and natural killer cells eliminate virally infected or oncogenic target cells. In concert with an immune response, the pore-forming protein perforin and the protease granzyme are released by cytotoxic T-lymphocytes into the synaptic cleft with target cells. Perforin binds target cell membranes and oligomerizes to form pores or lesions through which cytotoxic granzymes enter the cytosol of the target cell, with the subsequent initiation of apoptosis (1–5). Perforin-deficient cytotoxic lymphocytes are unable to kill target cells or to clear many viral infections in the host cells, resulting in immune dysregulation and the development of the serious disease familial hemophagocytic lymphohistiocytosis type II (2, 6).
Perforin consists of three domains as follows: an N-terminal membrane attack complex perforin-like (MACPF)4/cholesterol-dependent cytolysin pore-forming domain; a central epidermal growth factor (EGF) domain; and a C-terminal membrane- and Ca2+-binding C2 domain (Fig. 1A). The MACPF domain includes the central machinery of pore formation. Briefly, this domain mediates perforin oligomerization and deploys two membrane-spanning regions (transmembrane hairpin-1 (TMH)-1 and TMH-2) that enter the membrane as amphipathic β-hairpins. The central EGF domain forms a “shelf-like” assembly connecting the MACPF and C2 domains (Fig. 1A) (7). The role of this region remains to be fully understood. Finally, the C2 domain is critical for initial interaction with lipid membranes of the target cell.
FIGURE 1.

Crystal structure of murine perforin. A, crystal structure of murine perforin, PDB code 3NSJ (7), is shown with the C2 domain colored in magenta. The hydrophobic aromatic residues (Trp-427, Tyr-430, Tyr-486, and Trp-488) are shown in a stick representation where the side chain density of Tyr-486 is disordered. Two Ca2+ ions are indicated as magenta spheres. The MACPF and EGF domains are shown in gray. The shelf-like region is boxed with dashed line. Disulfide bonds between Cys-496 and Cys-509, and between Cys-524 and Cys-535 are represented by green and cyan sticks, respectively. The residues (410, 411, 461, 471, and 472), whose resonances were undetected in the current NMR study, are shown in yellow. B, sequence alignment of mouse perforin (Prf) C2 and the C2 quad mutant constructs used in the analyses carried out in this study. Amino acid sequences of the mouse perforin C2 domain (C2 WT) and the C2 quad mutants used for the current crystal (C2 quad(410–535)) and NMR studies (C2 quad(410–526)) together with SmC2P1 are aligned. The N-terminal signal sequence and the C-terminal additional sequences, including the hemagglutinin (HA) and/or His6 tag, are shown in gray. The four alanine residues substituted from the WT aromatic residues are illustrated in red. Five conserved Asp residues are highlighted in yellow. The positions of Asp-Asn mutations (Asp-429, Asp-435, Asp-483, Asp-490, and Asp-491) are colored in blue. Sequence numbers of the mouse perforin C2 domain and SmC2P1 are shown above and below the primary sequences, respectively. The positions of the CBRs are boxed. The signal sequence was cleaved during the export process at the position indicated by the arrow, resulting in an alanine overhang at the N terminus. Pairs of Cys residues that form disulfide bonds are highlighted in the same colors as in A. In the C2 quad(410–526), Cys-524 was mutated to serine (cyan) to remove a free thiol in the construct.
Most C2 domains are found in intracellular proteins and accordingly are functional with respect to membrane binding in the presence of low concentrations of Ca2+. The perforin C2 domain is unusual in that it requires high (>150 μm) concentrations of Ca2+. Perforin is thus activated to bind membranes only in the extracellular environment, where the Ca2+ concentration is estimated to be between 1 and 3 mm (8–10). It is suggested that the requirement for extracellular concentrations of Ca2+ provides a critical control point for perforin function and helps regulate unwanted perforin activity within the cell (11).
The perforin C2 domain comprises eight β-strands in a β-sandwich fold (7). Three Ca2+-binding regions (CBRs) contain conserved Asp residues and mediate the interaction with both Ca2+ and membranes. Different types of C2 domain bind different numbers of Ca2+ ions. For example, the synaptotagmin I C2B domain binds two Ca2+ ions (12, 13). In contrast, the synaptotagmin I C2A domain coordinates three Ca2+ ions (14, 15). However, some C2 domains, for example the PKC Apl II (16), can interact with membranes without first binding Ca2+.
In the original crystal structure of perforin (7), two Ca2+ ions were observed in the CBRs, which were scavenged from the environment and therefore presumably interact with the strongest binding sites. One Ca2+ ion (site II) was coordinated within CBR1 and CBR2 by conserved Asp residues (Asp-435 and Asp-483). The second Ca2+ ion was coordinated by a nonconserved Asp residue (Asp-490) and found outside the CBR3 in an unusual binding position that appears to be unique to the perforin C2 domain (Fig. 1). Currently, it is unknown how many Ca2+ ions are coordinated by perforin in the fully liganded state. However, the crystal structure of a related (39% identity) C2 domain-only protein (SmC2P1 from Scophthalmus maximus) (Fig. 1B) revealed three Ca2+ ions coordinated within the CBRs (17).
In the CBRs of the perforin C2 domain, two pairs of conserved and solvent-exposed aromatic residues in CBR1 (Trp-427 and Tyr-430) and CBR3 (Tyr-486 and Trp-488) have been shown to be critical for membrane binding, as substitution of all four residues with alanine resulted in complete loss of function in effector target cell-based and red blood cell lysis-based assays (17). It is likely that these four aromatic residues are thus essential for tight interaction of the perforin C2 domain with lipid membranes. By analogy with SmC2P1, it is suggested that Ca2+ binding in the CBRs induced a conformational change in CBR1 and subsequently that the relative positions of the four aromatic residues changed to orientations facilitating interaction with lipid membranes (17). However, there is no direct structural evidence of Ca2+-dependent rearrangements of the CBR1 in the perforin C2 domain, and the numbers of bound Ca2+ ions and their binding sites remain unclear.
To address these questions, we have investigated the interactions of perforin C2 domain with Ca2+ ions by both NMR spectroscopy and x-ray crystallography, using highly soluble and stable variants of mouse perforin C2 domain in which Trp-427, Tyr-430, Tyr-486, and Trp-488 were mutated to alanine. We propose a detailed Ca2+-binding mechanism of the perforin C2 domain and a role for bound Ca2+ in its interaction with lipid membranes.
Experimental Procedures
Protein Expression and Purification
The C2 quad mutant (W427A/Y430A/Y486A/W488A) was cloned into the phagemid vector pComb3X for expression of the protein in the periplasmic space of Escherichia coli Top10F′ (Life Technologies, Inc.) with C-terminal HA and His6 tags (Fig. 1B). For crystallography, residues 410–535 of murine perforin (C2 quad(410–535)) were cloned into pComb3X by introduction of 5′ and 3′ asymmetric SfiI sites using standard molecular biology techniques. For NMR, residues 410–526 (C2 quad(410–526)) were cloned into pComb3X and, to avoid an unpaired cysteine in the construct, Cys-524 was mutated to serine by QuikChange mutagenesis according to the manufacturer's protocol (Stratagene). The clones for expression of the D429N, D435N, D483N, D490N, and D491N variants of C2 quad(410–526) were constructed using pComb3X as the template to introduce the mutation by overlap PCR with flanking 5′ EcoRI and 3′ NcoI sites. The amino acid sequences of the constructs used in this study are shown in Fig. 1B. For expression of all constructs, cells were grown overnight in 3 ml of SB (Super Broth) media contained 0.5% glucose (starting culture) at 37 °C and subsequently inoculated into 100 ml of SB media (subculture). The subculture was grown at 37 °C until an A600 of 0.5–0.6 and then transferred into 4 liters of SB media (main culture). The main culture was continuously grown at 37 °C to an A600 of 0.6. The temperature was reduced to 23 °C, and protein expression was induced by addition of 10 μm isopropyl β-d-1-thiogalactopyranoside (IPTG) followed by incubation for ∼20 h. Uniformly 15N- or 13C/15N-labeled proteins were produced by using a high cell density method (18) with modifications. Cells were grown using a similar procedure to that for expression of unlabeled protein, except that all SB media contained 0.5% glucose to prevent leaky protein expression, and the main culture was grown overnight at 37 °C without IPTG induction. After harvesting cells from 4 liters of main culture, cells were resuspended in 2 liters of minimal media containing 15NH4Cl as the sole nitrogen source or 15NH4Cl and [13C]glucose as the sole nitrogen and carbon sources for the expression of 15N- or 13C/15N-labeled proteins, respectively. Cells were incubated for 1 h at 23 °C to adapt to minimal media, and protein expression was induced by adding 10 μm IPTG. Cells were harvested ∼20 h after IPTG induction.
Purification of both the C2 quad(410–535) and C2 quad(410–526) mutants was performed by periplasmic protein extraction with osmotic shock (19). The cell pellets were resuspended in binding/resuspension buffer (50 mm Tris/Cl (pH 8.0), 300 mm NaCl, 20 mm imidazole, and 0.02% NaN3 (typical volume is 5 ml per 1 g of wet cells)). An osmotic shock buffer (50 mm Tris/Cl (pH 8.0), 150 mm NaCl, 1 m sucrose, and 2 mm EDTA) was added into the cell mixture with a 1:1 volume ratio and mixed well. After incubation at room temperature for 30 min, the osmotically shocked cells were collected by centrifugation at 10,000 rpm for 20 min. The pellets were resuspended in release buffer (5 mm MgSO4 and protease inhibitor (Roche Applied Science), typical volume 1 ml per 1 g of osmotic shocked cells) and incubated on ice for 30 min. The supernatant containing periplasmic proteins was collected by centrifugation at 15,000 rpm for 20 min and added to an equal volume of binding/resuspension buffer, and then ∼2% (v/v) Ni2+-charged chelating Sepharose (GE Healthcare) was added. After 1.5 h of incubation at 4 °C, the mixture was transferred into a column, and the supernatant was isolated from the resin by gravity flow. The resin was washed with 10 column volumes of binding/resuspension buffer followed by the same volume of wash buffer (50 mm Tris/Cl (pH 8.0), 300 mm NaCl, 40 mm imidazole, and 0.02% NaN3). The purified protein was eluted in 20 ml of elution buffer (50 mm Tris/Cl (pH 8.0), 300 mm NaCl, 300 mm imidazole, and 0.02% NaN3). The eluted fraction was applied onto a Superdex 75 10/300 GL or 16/60 size exclusion column (GE Healthcare) equilibrated in either crystallography buffer (25 mm HEPES (pH 7.4), 150 mm NaCl with 0.02% NaN3) or NMR buffer (20 mm Tris/Cl (pH 8.0), 300 mm NaCl, and 2 mm EDTA). The peak fractions were analyzed by SDS-PAGE and significant protein-containing fractions pooled.
Crystallographic Analysis
Crystallization was carried out by the hanging drop method with a 1:1 mixture of protein and mother liquor at 20 °C with the protein concentrated to 9.75 mg/ml. Apo-C2 quad(410–535) crystals were obtained in 0.1 m MES (pH 6.0), 0.2 m NaCl, and 20% (w/v) polyethylene glycol 2000 monomethyl ether. Crystals of holo-C2 quad(410–535) were obtained in 0.1 m HEPES sodium (pH 7.5), 0.2 m calcium chloride dihydrate, and 14% v/v polyethylene glycol 400. The crystals were flash-cooled in liquid nitrogen using 25% (v/v) glycerol as the cryoprotectant. Data sets were collected at the Australian Synchrotron MX2 beamline at 100K (20). The data were merged and processed using XDS (21), POINTLESS, and SCALA (22). Five percent of the data set was flagged as a validation set for calculation of the Rfree. Molecular replacement was carried out using the C2 domain of 3NSJ as a search probe (7). One molecule was found per asymmetric unit cell, and an initial model was generated using PHENIX. Model building was performed using COOT (23), and refinement was performed using PHENIX (24). Crystallographic and structural analysis was performed using the CCP4 suite (25), unless otherwise specified. The figures were generated using MacPyMOL (26), and the structural validation was performed using MolProbity (27). All atomic coordinates and structural factors were deposited in the PDB under codes 4Y1S (apo-C2 quad(410–535)) and 4Y1T (holo-C2 quad(410–535)).
NMR Spectroscopy
All NMR measurements were conducted in 20 mm HEPES buffer (pH 7.0) and 150 mm NaCl with 10% 2H2O at 25 °C on a Bruker Avance 600 MHz NMR spectrometer equipped with a cryoprobe. Backbone resonance assignments of C2 quad(410–526) were obtained from analysis of three-dimensional HNCA, HN(CO)CA, HNCACB, and CBCA(CO)NH spectra of a 0.5 mm uniformly 13C/15N-labeled sample. The side chain amide resonances of Asn and Gln were assigned by 15N NOESY-HSQC spectra (70 ms mixing time) of the same sample. Ca2+ titrations were monitored by 1H-15N SOFAST-HMQC spectra (28) of 0.15 mm uniformly 15N-labeled samples with 200-ms relaxation delay. Spectra were acquired at 13 different [Ca2+] up to 30 mm. The backbone resonance assignments in 30 mm [Ca2+] were verified by three-dimensional HNCA and CBCA(CO)NH spectra of a uniformly 13C/15N-labeled sample. The Ca2+ titration experiments induced nonlinear chemical shift perturbations (CSPs), so the Ca2+ concentration-dependent CSPs were calculated by Equations 1 and 2,
 |
where
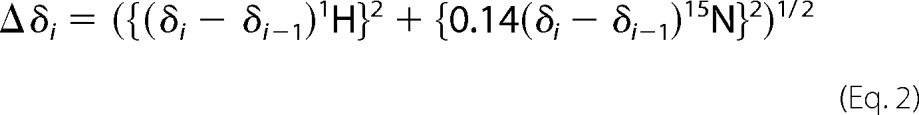 |
where n is the total number of titration points, and i is ith titration point at each [Ca2+] from 0 to 30 mm. δi is the chemical shift of either 1H or 15N at the ith [Ca2+]. Δδi is the CSP between the [Ca2+] of interest and the next lower [Ca2+]. If the chemical shifts at the next lower [Ca2+] were not detected because of exchange broadening, the observed chemical shifts at the nearest lower [Ca2+] were used for subtraction.
The dodecylphosphocholine (DPC) titration experiments were carried out by adding aliquots of a 1 m stock solution of 2H38-DPC (Sigma) in the same buffer. The chemical shift perturbations, Δδ, upon addition of DPC were calculated using {(Δδ1H)2 + (0.14Δδ15N)2}1/2 (29), where Δδ1H and Δδ15N are the chemical shift differences of the backbone amide proton and nitrogen resonances at 50 mm [DPC] compared with the absence of DPC. A mean dissociation constant, Kd, for the DPC micelles was obtained from the dependence of 1H chemical shifts of selected residues (Asp-429, Asp-485, Asp-489, and Asp-490) upon [DPC] for C2 quad(410–526), using Equation 3 (30)
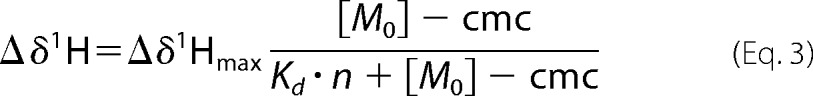 |
where Δδ1H is the difference between the backbone amide proton chemical shift at each [DPC] and the shift in the absence of DPC; Δδ1H max is the chemical shift difference at saturating [DPC]; [M0] is the monomeric [DPC]; cmc is the critical micelle concentration (1.5 mm (30)); and n is the aggregation number of DPC (54 (30)). The chemical shifts of backbone amide 15N and 1H and 13Cα and 13Cβ of the C2 quad(410–526) were deposited in the BMRB under accession number 25481.
Results
Expression and Purification of Perforin C2 Domain Mutants
Expression of the perforin C2 domain in isolation at levels required for crystallography and NMR spectroscopy has not been successful to date, despite extensive attempts over many years (17). We reasoned that the four aromatic residues in the CBRs (Trp-427, Tyr-430, Tyr-486, and Trp-488) (7) represent the major barrier to expression, possibly through driving association of the C2 domain with E. coli membranes during protein expression (Fig. 1A). By mutating these four hydrophobic aromatic residues to alanine (Fig. 1B), using the crystal structure of perforin to accurately define the domain boundaries, and exploiting periplasmic expression, two different C2 mutants W427A/Y430A/Y486A/W488A were expressed successfully (C2 quad(410–535) and C2 quad(410–526)). Two-dimensional 1H-15N SOFAST-HMQC spectra of the both constructs showed well resolved resonances, confirming that both were properly folded (data not shown). C2 quad(410–535) was used in crystallography experiments and C2 quad(410–526) for NMR spectroscopy, as it yielded better quality spectra than C2 quad(410–535).
Crystal Structure of C2 Quad(410–535)
The structure of apo-perforin remains unknown as the perforin structure contains the aforementioned two Ca2+ ions (7). Thus, we determined the crystal structure of C2 quad(410–535) in both the partially and fully calcium-liganded states (Fig. 2 and Table 1). In the 1.6 Å apo-C2 quad(410–535) structure (Fig. 2, A and B), a single Ca2+ ion was coordinated in the noncanonical position at residue Asp-490, which mirrors one of the two atoms bound in the full-length perforin structure. This Ca2+ ion was modeled as a 0.5 fractional occupancy, which indicates that, despite the presence of EDTA, some Ca2+ was scavenged during the purification process. The 2.6 Å holo-C2 quad(410–535) structure revealed five Ca2+ ions, four of which occupy the groove in the jaws of the C2 domain in addition to the noncanonical Ca2+ ion coordinated by Asp-490 (Fig. 2, A and C). In comparing the apo- and holo-C2 quad(410–535) structures, the most striking feature is the absence of the 427–431 portion of CBR1 loop (Fig. 2, A and B), consistent with predictions that this region in perforin is mobile in the absence of calcium (17).
FIGURE 2.

Crystal structure of apo- and holo-C2 quad(410–535) superimposed with the C2 domain from full-length perforin. A, C2 domain of 3NSJ (magenta) superimposed with both the apo-C2 (cyan) and holo-C2 (orange) quad(410–535). CBRs 1 and 3 are identified, and the corresponding colored spheres represent the Ca2+ ions for each structure. The view is rotated by 90° to view the C2 domain from the membrane perspective. CBR1 in the apo-C2 quad(410–535) structure is disordered, and the loop was not built into the density (cyan dashed line). B, superimposed structures of the apo-C2 (cyan) and holo-C2 (orange) quad(410–535) to illustrate the re-organization that occurs in the residues involved in Ca2+ coordination. The residues from Ala-427 to Ala-431 (CBR1, cyan dashed line) were not visible in the electron density map of the apo-structure and were not modeled into the final structure. Significant movement is observed in the CBR1, where residue Thr-432 moves through 6.8 Å, after which the CBR1 loop becomes well ordered and visible in the holo-C2 structure. Upon movement of the loop, Asp-429 is re-positioned to engage Ca2+ in positions I–III. C, superimposition of the C2 domain from murine perforin and the holo-C2 quad(410–535) further demonstrates the significant movement of residue Asp-429 over 11 Å. Throughout, key residues are represented in stick form labeled by residue number; arrows indicate movement, and disulfide bonds are represented as yellow sticks. Ca2+ is numbered as described previously (17), and the additional observed Ca2+ is numbered sequentially (position IV). D, key residues of the holo-C2 quad(410–535) structure making contact with Ca2+. E, schematic representation of Ca2+-binding interactions in the CBRs. The coordinates of the side chain carboxyl groups of conserved Asp residues and Asp-490 with Ca2+ ion at each site are indicated as dashed lines. The Ca2+ ions at each site are shown as orange spheres with corresponding site numbers. The weakest affinity site (III) is labeled in red.
TABLE 1.
Refinement statistics
Statistics for the highest resolution shell are shown in parentheses. R-free was calculated using 5% of randomly selected reflection, excluded from the refinement.
| Apo_C2 | Holo_C2 | |
|---|---|---|
| Data collection | ||
| Resolution range (Å) | 36.48–1.611 (1.669–1.611) | 42.38–2.666 (2.761–2.666) |
| Space group | P 21 21 21 | I 2 2 2 |
| Unit cell | ||
| a, b, c (Å) | 37.7, 43.211, 68.077 | 45.009, 59.828, 125.919 |
| α, β, γ (o) | 90, 90, 90 | 90, 90, 90 |
| Total reflections | 105,843 (9865) | 10,189 (930) |
| Unique reflections | 14,926 (1443) | 5115 (477) |
| Multiplicity | 7.1 (6.8) | 2.0 (1.9) |
| Completeness (%) | 99.87 (98.90) | 99.65 (96.95) |
| Mean I/σ(I) | 10.15 (3.94) | 21.25 (10.03) |
| Wilson B-factor | 11.78 | 26.68 |
| CC1/2 | 0.979 (0.86) | 0.997 (0.981) |
| CC* | 0.995 (0.962) | 0.999 (0.995) |
| Rmerge | 0.2159 (0.5473) | 0.071 (0.182) |
| Refinement | ||
| No. of molecules in asymmetric unit | 1 | 1 |
| R-work | 0.1661 (0.1929) | 0.1684 (0.2163) |
| R-free | 0.1852 (0.2491) | 0.2153 (0.2380) |
| No. of non-hydrogen atoms | 1120 | 1033 |
| Macromolecules | 985 | 965 |
| Ligands | 1 | 5 |
| Water | 134 | 63 |
| Protein residues | 122 | 126 |
| Root mean square (bonds) | 0.011 | 0.003 |
| Root mean square (angles) | 1.29 | 0.65 |
| Ramachandran favored (%) | 98 | 96 |
| Ramachandran allowed (%) | 2 | 4 |
| Ramachandran outliers (%) | 0 | 0 |
| Clashscore | 1.05 | 1.08 |
| Average B-factor | 17 | 24 |
| Macromolecules | 15.5 | 23.9 |
| Ligands | 17 | 22.4 |
| Solvent | 28.1 | 24.7 |
| MolProbity scorea | 0.09 (100th percentile) | 1.09 (100th percentile) |
a Data combine the clashscore, rotamer, and Ramachandran evaluations into a single score, normalized to be on the same scale as the x-ray resolution.
The most significant structural rearrangement driven by Ca2+ binding involves CBR1. This region contains the critical lipid-binding residues Trp-427 and Tyr-430 (both mutated to alanine in this protein) that make key contacts with Ca2+ ions in sites I–III. Here, Ca2+ binding is achieved through Thr-432 and Asp-429. Thr-432, which is the only Ca2+-binding residue visible in electron density in the apo-structure, moves 6.8 Å (Fig. 2B). Asp-429 moves 11.4 Å, from its position in the full-length perforin structure to engage Ca2+ (Fig. 2C), thus driving this significant structural rearrangement in CBR1. Elsewhere, conformational changes in the side chains of Asp-485 and Asp-491 in CBR3 position these residues so that they interact with sites I, II, and IV. Finally, a modest rearrangement of Asn-454 on CBR2 completes the coordination of site III (Fig. 2, D and E).
CBR3 undergoes only modest conformational changes upon calcium binding; indeed, the backbone atoms of the previously identified lipid-binding residues 486 and 488 (both alanine in this protein) remain essentially unchanged between the apo- and holo-forms. Thus, the key event driven by Ca2+ binding must be the conformational change in CBR1 and the repositioning of Trp-427 and Tyr-430 into an orientation suitable for interacting with membranes. These data are consistent with previous studies showing that the W427A and Y430A mutations caused the most substantial functional defects with respect to perforin membrane binding (17). In comparison with the SmC2P1 structure (PDB codes 3W56 and 3W57), similar positioning of Ca2+ ions at sites I–III was observed. This bonding pattern differs from that of MUNC13- C2B, which is the most structurally similar C2 domain characterized previously (7, 17, 31). Unlike full-length perforin and the structures of apo- and holo-C2 described here, SmC2P1 does not coordinate Ca2+ at the noncanonical position.
In the murine perforin structure, the CBR1 loop is stabilized via crystal contacts; our new data confirm the suggestions that CBR1 is largely mobile in the absence of Ca2+. Given that the site II Ca2+ is occupied in murine perforin (7), we reason this site is probably the strongest affinity Ca2+-binding site inside the jaws of the CBRs. However, Ca2+ binding to site II alone is clearly insufficient to drive conformational change in CBR1. Furthermore, because site IV makes no contact with CBR1, we reasoned that binding of Ca2+ to site I or III must represent the key event that brings Trp-427 and Tyr-430 into a position suitable for interacting with membranes. To further study these events, and to understand the order of Ca2+-binding events, we conducted NMR studies.
Analysis of Ca2+-binding Mode of the C2 Quad Mutant
We undertook Ca2+ titration experiments in solution, monitored by NMR. The 1H-15N SOFAST-HMQC spectrum of the C2 quad(410–535) used for crystallography showed a number of random coil chemical shifts in the central region of the spectrum, mostly arising from additional non-native residues at the C terminus introduced during cloning (Fig. 1B). Therefore, we created a new construct in which the C-terminal residues were removed after residue 526 (C2 quad(410–526)) and only a hexa-His tag was attached at the C terminus (Fig. 1B). The 1H-15N SOFAST-HMQC spectrum of this new construct was less cluttered in the central region (supplemental Fig. S1A).
Complete backbone resonance assignments of C2 quad(410–526) (supplemental Fig. S1B) were obtained by conventional triple-resonance experiments, except for proline residues, two residues at the N terminus (410 and 411) and residues 461, 471, and 472. Residues 461, 471, and 472 are located in loop regions in the crystal structure (Fig. 1A) and are subject to exchange broadening among more than two conformers and/or exchange with water molecules, resulting in undetectably broad resonances under our solution conditions.
Prior to Ca2+ titrations, protein samples were treated with EDTA to ensure that residual Ca2+ ions were completely removed, and then the EDTA was removed. The 13 spectra with different Ca2+ concentrations are superimposed in Fig. 3. The chemical shifts of many resonances in the CBRs changed smoothly with increasing Ca2+, indicating that the exchange between free and bound Ca2+ is fast on the chemical shift time scale (Fig. 3A). However, several of these chemical shift changes were nonlinear, as exemplified by the resonances of Gly-428 or Asp-483. This reflects the fact that different Ca2+-bound states have different backbone chemical shifts, and several states contribute simultaneously to the observed chemical shifts at any given [Ca2+]. Complex titration profiles were also observed for the resonances of Ala-484 and Asp-490 (Fig. 3, B and C).
FIGURE 3.

Titrations of C2 quad(410–526) with Ca2+. A, overlay of 1H-15N SOFAST-HMQC spectra of 150 μm 15N-labeled C2 quad(410–526) recorded with increasing [Ca2+]. Spectra were processed identically and plotted with the same contour levels. Resonance colors correspond to different [Ca2+] as shown at the top left corner. Resonances of interest in the main text are labeled. The chemical shift change patterns are shown as black arrows, and intensity changes are roughly represented by a gradient. The resonances of Thr-431 decreased its intensity with increasing [Ca2+] and disappeared by 0.6 mm [Ca2+]. The resonances of Trp-453 and Asp-485 were significantly broadened at several [Ca2+] in the middle of titrations. B and C, expanded spectral regions showing resonances of Ala-484 and Asp-490, respectively.
The calcium titrations were conducted at a protein concentration of 0.15 mm and a [Ca2+] range of 0–30 mm. The initial chemical shift perturbations (Fig. 3, B and C) were largely complete by 0.3 mm Ca2+, implying Kd values <0.05 mm. The next transition is evident at around 3 mm, reflecting Kd values <0.4 mm for those binding events, whereas the chemical shift perturbations at the highest [Ca2+] were still incomplete at 30 mm, implying a Kd value of 5–10 mm for the final Ca2+-binding event. At the typical physiological Ca2+ concentration of the extracellular space, ∼1–3 mm (8–10), the weakest Ca2+-binding site would therefore not be fully occupied.
As expected, the number of Ca2+-binding sites is five based on the crystal structure of holo-C2 quad(410–535), and the Kd values at several of those sites are expected to be similar to one another. Therefore, the changes in backbone amide chemical shifts are separately summarized up to 3 mm and from 3 to 30 mm [Ca2+] in Fig. 4A. The residues perturbed by ≥0.2 ppm at 3 and 30 mm [Ca2+] are mapped on the crystal structure of holo-C2 quad(410–535) in red and orange, respectively, in Fig. 4B. The significantly perturbed residues up to 3 mm [Ca2+] are found in the vicinity of the five Ca2+ ions, indicating that the Ca2+-binding mode of the C2 quad mutant in solution (C2 quad(410–526)) is the same as in the crystal (C2 quad(410–535)). In addition, significant additional CSPs were subsequently observed in CBR1 and CBR3 up to 30 mm [Ca2+].
FIGURE 4.

Concentration-dependent CSPs of C2 quad(410–526) upon binding to Ca2+. A, CSP values upon Ca2+ binding were calculated up to 30 mm [Ca2+] using Equations 1 and 2 (see “Experimental Procedures”) and plotted for each residue on the primary sequence of the C2 quad(410–526). CSPs of >0.2 ppm are colored and labeled. Bars with CSPs up to 3 mm and with CSPs from 3 to 30 mm [Ca2+] are represented in red and orange, respectively. The resonance of Thr-434 was undetected at 3 mm [Ca2+] because of exchange broadening. Asterisks mark resonances not detected because of exchange broadening beyond 0.3 mm [Ca2+]. Dashed lines indicate CSP values of ≥0.2 ppm. The positions of CBRs are shown below the residue number. B, residues with significant CSPs upon Ca2+ binding are mapped on the crystal structure of holo-C2 quad(410–535) using the same color representations as in A. The positions of CBRs are labeled. Residues with undetected resonances beyond 0.3 mm [Ca2+] are shown in yellow and labeled. Right and left figures are related by a 180° rotation around vertical axis. Blue spheres indicate bound Ca2+ ions.
As noted above, most of the perturbed resonances were detected at all [Ca2+], but some disappeared with increasing [Ca2+] (e.g. Thr-431 in Fig. 3A). This is a consequence of intermediate exchange among two or more Ca2+-free and multiple Ca2+-bound states for these resonances. Several of those resonances reappeared at high [Ca2+] (e.g. Asn-455 and Asp-485 in Fig. 3A), indicating that they are in a single major conformation at the highest concentrations examined. By contrast, the resonances of Ala-430, Thr-431, Thr-432, and Ala-486, shown in yellow in Fig. 4B, disappeared with increasing [Ca2+]. These residues presumably sample more than one conformation even at the highest [Ca2+], which is close to saturation of all Ca2+ binding, and these conformers are exchanging with one another on an intermediate time scale that gives rise to peak broadening. These residues are involved in the quad mutation sites, which contain the hydrophobic aromatic residues Tyr-430 and Trp-486 in the wild-type perforin C2 domain.
Detailed Ca2+-binding Mechanism of the C2 Quad Mutant
To analyze details of the Ca2+-binding mechanism of C2 quad(410–526), we mutated the conserved Ca2+-binding Asp residues and nonconserved Asp-490 to Asn (D429N, D435N, D483N, D491N, and D490N) and repeated the Ca2+ titration experiments. Two distinct areas of Ca2+ titration spectra for each mutant are shown in Fig. 5 (extended spectra are shown in supplemental Figs. S2–S5). The Ca2+ titration spectra of the mutants showed significant differences from those of C2 quad(410–526), confirming that these Asp residues bind Ca2+ ions, in agreement with the crystal structure of the holo-C2 quad(410–535).
FIGURE 5.
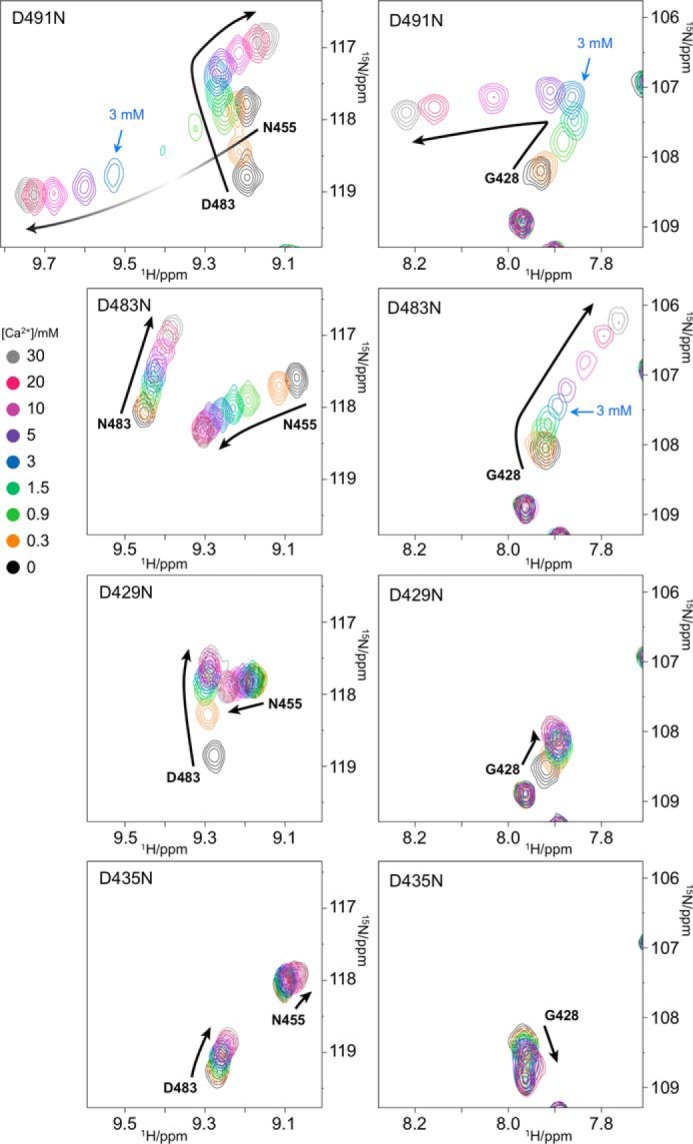
Contribution of four conserved Asp residues Asp-491, Asp-483, Asp-429, and Asp-435 to Ca2+ binding. Overlay of a portion of 1H-15N SOFAST-HMQC spectra of 150 μm 15N-labeled C2 D491N, D483N, D429N, and D435N mutants (from the top to bottom) recorded with increasing [Ca2+]. Spectra were processed identically and plotted with the same contour levels. Resonance colors correspond to a different [Ca2+] shown at left. Resonances of interest in the main text are labeled. The chemical shift change patterns are shown as black arrows, and intensity changes are roughly represented by gradient. Different spectral regions are shown in the left and right panels.
In the D491N mutant (Fig. 5), significant differences in the perturbation curves were observed for Gly-428, Asn-455, and Ala-484 in comparison with the Ca2+ titration spectra of C2 quad(410–526) (Fig. 3). Because the crystal structure of holo-C2 quad(410–535) shows that the carboxyl group of Asp-491 coordinates two Ca2+ ions at sites I and IV (Fig. 2), mutation of Asp-491 to Asn substantially reduced the Ca2+ binding affinity at these sites. A turning point in the titration curves was still observed at 3 mm [Ca2+], as seen for Gly-428 and Asp-483, indicating that the weakest affinity site still exists in the D491N mutant. Therefore, the weakest affinity site is either site II or III. Because site II was occupied by Ca2+ scavenged from the buffer in the crystal structure of full-length perforin (7), we argue that site III must be the weakest affinity site.
Site III was confirmed as the weakest affinity site by examination of the D483N mutant. Ca2+ titration spectra of the D483N mutant showed that the resonances of Gly-428 and Asn-483 continued to shift beyond 3 mm [Ca2+], suggesting that the weakest affinity site is maintained in this mutant (Fig. 5). The crystal structure of the holo-C2 quad(410–535) showed that the carboxyl group of Asp-483 interacts with two Ca2+ ions at sites I and II but not site III (Fig. 2), so the observation of chemical shift perturbations at [Ca2+] of >3 mm in this mutant supports the conclusion that site III is the weakest affinity site. This was also supported by Ca2+ titration experiments of the D429N and D435N mutants. The carboxyl groups of both Asp-429 and Asp-435 coordinate a Ca2+ ion at site III (Fig. 2). In the Ca2+ titration spectra of the D429N mutant, similar chemical shift perturbation profiles to those for the D491N mutant were observed for resonances of Gly-428, Asn-455, and Asp-483 at low [Ca2+], but beyond 3 mm [Ca2+], in contrast, the chemical shift perturbations were very small, indicating loss of the weakest affinity site (Fig. 5). Similarly, the chemical shift changes of the D435N mutant at high [Ca2+] (>3 mm) were as small as those of the D429N mutant (Fig. 5), confirming the absence of the weakest affinity site.
Asp-429 coordinates three Ca2+ ions at sites I–III (Fig. 2). However, the D429N mutant may still be able to coordinate a single Ca2+ ion at site II, as its chemical shift perturbation patterns were similar to those of the D491N mutant at low [Ca2+]. As mentioned previously, the D491N mutation does not affect Ca2+ binding at site II. In the D435N mutant, significantly different chemical shift perturbation patterns were observed for residues Gly-428, Ala-484, Trp-453, Asn-455, and Asp-483, showing that the Asp-435 mutation affects a different Ca2+-binding site from those of Asp-491 and Asp-429 (Fig. 5). This is in good agreement with the crystal structure of holo-C2 quad(410–535), in which the side chain carboxyl of Asp-435 coordinates Ca2+ at sites II and III but not sites I and IV (Fig. 2). The D435N mutant is still capable of coordinating Ca2+ ions at sites I and IV (and the noncanonical position), and the observed titration curves reflect Ca2+ binding events only at these sites. In the crystal structure of holo-C2 quad(410–535) (Fig. 2), all of the backbone amides in CBR2 are >10 Å away from sites I and IV, implying that the resonances of backbone amides in the CBR2 should be less affected by the Ca2+-binding events at these sites. Indeed, the resonances of Trp-453, Asn-454, and Asn-455 in the CBR2 were almost unchanged over the entire Ca2+ titration in the D435N mutant.
Asp-490 coordinates a single Ca2+ ion at a noncanonical position (site V) outside CBR3 (Fig. 2). Ca2+ titrations of the D490N mutant showed similar chemical shift perturbation patterns to those of the C2 quad(410–526) (supplemental Fig. S6), except that several resonances appeared to be affected by local conformational differences associated with the point mutation. At 30 mm [Ca2+], the spectrum of the D490N mutant was similar to that of C2 quad(410–526) (supplemental Fig. S7), suggesting that Ca2+ bound at the noncanonical position had no significant effect on the saturated Ca2+-binding state of the C2 quad mutant.
Interaction of C2 Quad Mutant with Membranes and Role of Ca2+ Ions
Because of the replacement of four hydrophobic aromatic amino acid residues with alanine, the C2 quad mutant is considered to have lost its membrane binding capacity (17). However, NMR is capable of detecting weak interactions that cannot be readily monitored by other methods. To analyze the interaction of the C2 quad mutant with membranes, dodecylphosphocholine (DPC) titration experiments were performed. DPC titration spectra of C2 quad(410–526) at three different [Ca2+] (0, 2, and 30 mm) are shown in Fig. 6 (extended spectra are shown in supplemental Figs. S8–S10). Considering the comparatively high protein concentrations and the expected Kd value of Ca2+ at the weakest affinity site (>5 mm), complete Ca2+ binding was observed at ∼30 mm [Ca2+], even though Ca2+ binding at the weakest affinity site commenced at ∼3 mm [Ca2+]. In the presence of 30 mm [Ca2+] (Fig. 6A), several backbone amide resonances were linearly perturbed with increasing [DPC]. Not only did chemical shifts change, but the intensities of resonances were also attenuated as a consequence of interaction with the DPC micelles, the critical micelle concentration of which is ∼1.5 mm. The chemical shift perturbations upon addition of 50 mm [DPC] in the presence of 30 mm [Ca2+] are plotted on the primary sequence of the C2 quad(410–526) in Fig. 6D. Residues with chemical shift perturbations of >0.05 ppm are mapped on the surface of the crystal structure of holo-C2 quad(410–535) (Fig. 6E). The perturbed residues were localized to a surface containing the CBRs. In addition, the side chain amide resonance of Asn-454 (Asn-454Δ) was also stronglyaffected by DPC titration, disappearing beyond 3 mm [DPC] (Fig. 6A). The side chain of this residue is exposed and oriented downward in the right view in Fig. 6E. These results strongly suggest a specific interaction with DPC micelles. Significantly changed 1H chemical shifts (Δδ1H) of Asp-429, Asp-485, Asp-489, and Asp-490 (Fig. 6D) were fitted to Equation 3 (see under “Experimental Procedures”) (Fig. 7A), and the dissociation constant (Kd) of C2 quad(410–526) against DPC micelles was determined as ∼1 mm.
FIGURE 6.

Interaction of C2 quad(410–526) with DPC micelles. A, overlay of a portion of 1H-15N SOFAST-HMQC spectra of 150 μm 15N-labeled C2 quad(410–526) recorded with increasing [DPC] in the presence of 30 mm [Ca2+]. Spectra were processed identically and plotted with the same contour levels. Resonance colors correspond to different [DPC] as shown at bottom right of the spectra. Significantly perturbed resonances upon addition of DPC are labeled and traced by dotted arrows. B and C, same as A except that spectra were recorded in the absence of Ca2+ (B) and with 2 mm [Ca2+] (C), respectively. Only the boxed area in A is represented. D, CSPs values upon addition of the DPC in the presence of 30 mm [Ca2+] were calculated using {(Δδ1H)2 + (0.14Δδ15N)2}1/2 and plotted for each residue on the primary sequence of C2 quad(410–526). The bars with chemical shift perturbations of >0.05 ppm (dashed line) are shown in magenta and labeled. Asterisks mark undetected resonances because of exchange broadening beyond 0.3 mm [Ca2+]. The positions of CBRs are shown below the residue number. E, residues with CSPs of >0.05 ppm are mapped on the surface of the crystal structure of holo-C2 quad(410–535) using the same color representations as in D. The positions of CBRs are labeled. The side chain of residue Asn-454 is shown as a stick model. Residues with undetected resonances beyond 0.3 mm [Ca2+] are shown in yellow. Right and left figures are related by a 90° rotation around the horizontal axis. Blue spheres mark bound Ca2+ ions.
FIGURE 7.

Effects of the bound Ca2+ to the C2 quad(410–526) upon interaction with DPC micelles. A, changes in chemical shifts, Δδ1H, of indicated amide proton resonances of the C2 quad(410–526) are plotted against [DPC]. Solid lines show the fits of the data to Equation 3 (see “Experimental Procedures”). Filled triangles, diamonds, circles, and squares showed Δδ1H values of residues Asp-429, Asp-489, Asp-490 and Asp-485, respectively. The average dissociation constant (Kd) was determined as 1.1 ± 0.3 mm. B–F, overlay of a portion of 1H-15N SOFAST-HMQC spectra of 150 μm 15N-labeled C2 D491N (B), D429N (C), D435N (D), D483N (E), and D490N (F) mutants in the presence of 30 mm [Ca2+] recorded with increasing [DPC]. Spectra were processed identically and plotted with the same contour levels. Resonance colors correspond to different [DPC] shown in the same color on the bottom right side of spectra. In the D491N mutant, no significant CSPs upon adding DPC were observed in the resonances of backbone amides, but a side chain amide resonance of Asn-454Δ was clearly perturbed. In the D429N, D435N, and D483N mutants, resonances indicative of degradation or aggregation gradually emerged with increasing [DPC]. In the D490N mutant, the same resonances of the C2 quad(410–526) were perturbed with increasing [DPC].
To investigate the effect of Ca2+ ions on the interaction with membranes, DPC titration experiments were performed at different [Ca2+]. In the absence of Ca2+ (Fig. 6B), resonances indicative of unfolded or disordered protein appeared in the central region of the spectrum at 3 mm [DPC], and these resonances intensified at 10 mm [DPC]. These data imply that the interaction with DPC micelles partially unfolds the structure of the C2 quad(410–526) in the absence of Ca2+.
To assess how many Ca2+ ions are required for interaction with membranes, we undertook a DPC titration in the presence of 2 mm [Ca2+], in which the weakest affinity site should be largely unoccupied (Fig. 6C). No CSPs were observed except for the C-terminal residues. In addition, the C2 quad(410–52) was partially unfolded at 10 mm [DPC], and adding increasing [DPC] resulted in loss of the original well resolved resonances.
Importance of the Weakest Ca2+-binding Site for Interaction with Lipid Membranes
The titration of C2 quad(410–526) with DPC demonstrated that occupancy of Ca2+ at the weakest affinity site is critical for interactions with membranes (Fig. 6). The importance of the weakest affinity site was confirmed by DPC titrations with the D491N, D429N, D435N, D483N, and D490N mutants in the presence of 30 mm [Ca2+], as shown in Fig. 7, B–F (extended spectra are shown in supplemental Figs. S11–S15). DPC titration of the D491N mutant in the presence of 30 mm [Ca2+] showed no significant perturbations of the backbone amide resonances. However, the side chain amide resonance of Asn-454Δ was clearly perturbed (Fig. 7B). The CSP of Asn-454Δ was also observed in C2 quad(410–526) at 30 mm [Ca2+]. In addition, no unfolded resonances were observed up to 50 mm [DPC]. Therefore, the D491N mutant can still bind to DPC micelles in the proper orientation. The much smaller chemical shift change indicates that the affinity of DPC micelles became much weaker because of the loss of Ca2+-binding sites I and IV.
By contrast, resonances indicative of unfolding caused by nonspecific interaction with DPC micelles were observed in the D429N and D435N mutants in the presence of 30 mm [Ca2+] with increasing [DPC] (Fig. 7, C and D). Both mutants lose the weakest affinity site, resulting in partial Ca2+-bound states even at 30 mm [Ca2+]. Indeed, in the presence of 50 mm [DPC], the spectra of both mutants are very similar to the C2 quad(410–526) at 2 mm [Ca2+], in which the weakest affinity site should be unoccupied (Fig. 6C). These data support the critical role of Ca2+ binding at the weakest affinity site in enabling the C2 quad mutant to interact with lipid membranes. Similar destabilization was observed as the D483N mutant interacted with DPC micelles, which maintains the weakest affinity site (Fig. 7E). However, this mutant lost Ca2+-binding sites II and possibly I, indicating that sequential Ca2+ occupancy at Ca2+-binding sites II and III is important for proper interaction of the C2 quad mutant with DPC micelles. The DPC titration of the D490N mutant showed similar chemical shift perturbation patterns to that of the C2 quad(410–526) (Fig. 7F), indicating the Ca2+ binding to Asp-490 is not important for interaction with membranes.
Discussion
Our studies demonstrate that the C2 quad mutant binds multiple Ca2+ ions in the CBRs, the positions of which were identified in the crystal structure. Substitution of the four aromatic residues Trp-427, Tyr-430, Tyr-486, and Trp-488 with alanine resulted in a loss of perforin activity (17), although these four residues are not involved in Ca2+ binding among known C2 structures (14–16, 32–37). Indeed, the mutations of all four residues to alanine did not influence Ca2+ binding as confirmed by thermodynamic stability (17). The C2 quad mutant is therefore a suitable model for analyzing Ca2+ binding to the perforin C2 domain.
The crystal structures of the apo- and holo-C2 quad(410–535) (Fig. 2, C and D) demonstrate good agreement with previous results identified through mutational analyses and inferred from the C2 domain-only protein, SmC2P1 from S. maximus (17, 38). These structures have identified the exact positions of the coordinated Ca2+ ions and demonstrated that the C2 domain is capable of binding five Ca2+ ions. These structures also provided further evidence for the noncanonical Ca2+ ion coordinated at Asp-490, which was observed previously in the full-length perforin crystal structure but is known to be nonessential for perforin function (38). CBR1 is highly mobile in the absence of Ca2+, placing it in a conformation that is suboptimal for binding membranes. Upon Ca2+ binding, CBR1 undergoes a very large movement, and the “jaws” of the C2 domain coordinate four Ca2+ ions within the hydrophobic groove, predominantly mediated by Asp residues. Furthermore, Ca2+ binding is a critical regulatory step in the hydrophobically dependent membrane binding of perforin, bringing the CBR1 and CBR3 into close proximity forming a hydrophobic cleft/groove. Although the structures of the C2 domains do not reveal any obvious rearrangements that could possibly trigger conformational change within other domains of perforin, in particular the MACPF domain, our holo-C2 quad(410–535) structure is the first evidence of the locations of Ca2+ in the C2 domain of perforin and the first indication of an additional canonical Ca2+ coordination position, site IV.
As demonstrated by our NMR titrations with Ca2+, the affinity of one Ca2+-binding site is significantly weaker compared with other sites. From mutation analyses of the four conserved Asp residues Asp-429, Asp-435, Asp-483, and Asp-491 in the CBRs, we defined the weakest affinity site to be site III. The crystal structure of the holo-C2 quad(410–535) showed that the side chain carboxyl group of Asp-429 in CBR1 coordinates three Ca2+ ions at sites I–III, whereas this residue, together with the flanking two residues, is disordered in the apo-form (Fig. 2). The crystal structure of the perforin C2 domain revealed that the CBR1 domain swings out from the Ca2+-binding pockets, and the carboxyl side chain of Asp-429 is directed toward solvent (7). Comparing the holo-C2 quad(410–535) and the perforin C2 domain, significant structural differences are seen only in the relative orientation of CBR1 (Fig. 2). A previous study (17) suggested that bound Ca2+ ions induced conformational rearrangement of CBR1, which then facilitated re-positioning of the four key hydrophobic residues to interact with lipid membranes.
The crystal structure of the holo-C2 quad(410–535) showed that Asp-435 binds two Ca2+ ions at sites II and III, and Asp-491 coordinates Ca2+ ions at site I (and site IV), which is consistent with the crystal structure of SmC2P1 (17). In contrast, Asp-429 interacts with three Ca2+ ions at sites I–III, which differs from SmC2P1, where this residue coordinates Ca2+ ions only at sites I and II. Based on Ca2+ titration results by NMR, Asp-429 will be able to coordinate two Ca2+ ions at sites I and II at low [Ca2+]. At higher than extracellular [Ca2+], in contrast, Asp-429 is capable of coordinating one more Ca2+ ion at site III; subsequently, a conformational rearrangement of CBR1 is induced. The coordination of Ca2+ ion at site III by the carboxyl group of Asp-429 is also seen in other C2 domains, for example synaptotagmin I (12, 14) and the PKC-β (35), both of which bind three Ca2+ ions in the CBRs.
The importance of the conformational rearrangement of CBR1 for the C2 domain interaction with membranes was demonstrated by DPC titration experiments. It is clear that bound Ca2+ stabilizes the structure of the C2 quad mutant in its interaction with DPC micelles. These data are in good agreement with previous evidence indicating that perforin is stabilized in the presence of Ca2+, as determined by a thermal stability assay (17). Importantly, stabilization of the C2 quad mutant by Ca2+ is highly dependent on the [Ca2+], and complete stabilization requires full occupancy of Ca2+ ion at the weakest affinity site (site III). If the weakest affinity site is unoccupied, DPC micelles interact with the C2 quad mutant nonspecifically, leading to partial unfolding. The Ca2+ titration spectra showed that Ca2+ binding at the weakest affinity site starts around 3 mm [Ca2+], which is well matched with physiological extracellular [Ca2+]. However, the Ca2+-free state at the weakest affinity site is still dominant around these [Ca2+]. Because the exchange between the free and the Ca2+-bound forms is fast in the NMR time scale, the NMR spectra are observed as a population average of the free and the Ca2+-bound forms. Therefore, observation of the fully Ca2+-bound state requires excess [Ca2+] (∼30 mm), at which the Ca2+-bound state is dominant at the weakest affinity site III. A previous study (38) demonstrated that the Ca2+ concentration required for full activation of perforin in a sheep red blood cell lysis assay is ∼250 μm, which is less than the physiological extracellular [Ca2+]. In the case of native perforin, the interaction with membrane should be tighter than the C2 quad mutant. Once Ca2+ binds to the weakest affinity site, the native C2 domain can interact with the membrane, which may occur when a small proportion of the protein is Ca2+-bound, rather than requiring the dominant population to be in a fully Ca2+-bound state. This allows for oligomerization of perforin and activity at less than physiological extracellular [Ca2+].
The importance of occupancy of Ca2+ ion at site III was confirmed by the results of DPC titration of the D491N mutant, in which the C2 quad(410–526) was still stable at 30 mm [Ca2+] even when the Ca2+-binding site I was not occupied. By contrast, the site III-deficient mutants, D429N and D435N, were unstable in the presence of DPC micelles even at 30 mm [Ca2+]. In addition, the NMR results for the D483N mutant clearly indicated that the proper conformational rearrangement of CBR1 is required for Ca2+ binding at site III as well as site II. Our results are consistent with previous studies that Asp-429, Asp-435, and Asp-483, but not Asp-491, are critical residues for plasma membrane binding and cell lysis (38).
The conformational rearrangement of CBR1 enables the C2 quad(410–526) to interact with DPC micelles thorough the CBRs. Based on our observations, we propose the following mechanism of perforin C2 domain interaction with lipid membranes. At less than extracellular [Ca2+], although the C2 domain coordinates two Ca2+ ions in the CBRs, the CBR1 does not change conformation and is in the “inactive form.” At higher than extracellular [Ca2+], three Ca2+ ions are bound within the CBRs. The Ca2+ binding at site III, which is the weakest affinity site, induces a conformational rearrangement of CBR1 that leads to the “active form,” facilitating interaction with membranes (Fig. 8). The affinity of the C2 quad(410–526) for DPC micelles is very low (∼1 mm), which is insufficient for functional activities of perforin. Indeed, no perforin activity was observed in the full-length perforin quad mutant (17) because of the lack of four hydrophobic aromatic residues that are crucial for proper interaction with membranes. On the basis of the Ca2+ titration results, the regions, including the quad mutations, will not be fixed as a single conformer even at close to saturating concentrations of Ca2+, which may reduce the ability of the C2 quad(410–526) to interact with the membrane. As indicated above, however, the mutation of four key hydrophobic aromatic residues to alanine does not affect Ca2+ binding to the perforin C2 domain. Therefore, the Ca2+-dependent conformational switch from the inactive form to the active form should also occur in the native perforin C2 domain. The conformational plasticity in the regions including the quad mutations may be necessary for the initial interaction with membranes. Subsequently, in the case of native perforin C2 domain, the four key hydrophobic aromatic residues would be rearranged into the orientation required for tight binding to lipid membranes. Importantly, higher extracellular [Ca2+] promotes membrane binding of the C2 domain on granule exocytosis, whereas low [Ca2+] prevents premature activation of perforin (3, 38–40). Our crystal and NMR data are in excellent agreement with these biological results.
FIGURE 8.

Model of Ca2+-induced activation of perforin C2 domain interaction with membranes. Left, in the absence of Ca2+ ions, the relative orientation of CBR1 is not properly arranged to interact with membranes, resulting in aggregation or degradation with interacting membranes (inactive form). Right, at higher [Ca2+], the carboxyl group of Asp-429 interacts with Ca2+ at site III, and CBR1 changes the relative orientation to readily interact with membranes properly (active form). This rearrangement of CBR1 allows the re-positioning of key aromatic residues to enable membrane binding. The CBR1 is drawn in magenta, and Asp-429 and Asp-435 are shown as stick models. The crystal structures of the apo- and holo-C2 quad(410–535) are used as the models of inactive and active forms, respectively. The orientation of the C2 domain and membranes is based on the results of DPC titration to the C2 quad(410–526), such that the surface of the C2 quad(410–526) where CSPs were observed upon DPC micelles binding faces to headgroups of the lipid membrane.
In conclusion, we have clarified the mechanism of the perforin C2 domain interaction with membranes and the role of Ca2+ in that process. Our results represent the first observation of structural details regarding the interaction of the perforin C2 domain with lipid membranes and will facilitate further understanding of perforin function.
Author Contributions
H. Y., P. J. C., J. C. W., and R. S. N. designed the experiments. H. Y., P. J. C., E. W. W. L., and R. H. P. L. performed the experiments. J. A. T., I. V., and all authors contributed to interpretation of the data. H. Y., P. J. C., J. C. W., and R. S. N. wrote the manuscript. All authors reviewed the results and approved the final version of the manuscript.
Supplementary Material
Acknowledgment
This research was undertaken on the MX2 beamline at the Australian Synchrotron, Victoria, Australia.
This work was supported in part by Australian National Health and Medical Research Council Grant 1042858. The authors declare that they have no conflicts of interest with the contents of this article.

This article contains supplemental Figs. S1–S15.
The atomic coordinates and structure factors (codes 4Y1S and 4Y1T) have been deposited in the Protein Data Bank (http://wwpdb.org/).
Submitted to the BMRB Database under code 25481.
- MACPF
- membrane attack complex perforin-like
- CBR
- Ca2+-binding region
- DPC
- dodecylphosphocholine
- IPTG
- isopropyl β-d-1-thiogalactopyranoside
- PDB
- Protein Data Bank
- CSP
- chemical shift perturbation.
References
- 1. Tschopp J., Masson D., Stanley K. K. (1986) Structural/functional similarity between proteins involved in complement- and cytotoxic T-lymphocyte-mediated cytolysis. Nature 322, 831–834 [DOI] [PubMed] [Google Scholar]
- 2. Trapani J. A., Smyth M. J. (2002) Functional significance of the perforin/granzyme cell death pathway. Nat. Rev. Immunol. 2, 735–747 [DOI] [PubMed] [Google Scholar]
- 3. Voskoboinik I., Smyth M. J., Trapani J. A. (2006) Perforin-mediated target-cell death and immune homeostasis. Nat. Rev. Immunol. 6, 940–952 [DOI] [PubMed] [Google Scholar]
- 4. Froelich C. J., Orth K., Turbov J., Seth P., Gottlieb R., Babior B., Shah G. M., Bleackley R. C., Dixit V. M., Hanna W. (1996) New paradigm for lymphocyte granule-mediated cytotoxicity–target cells bind and internalize granzyme B, but an endosomolytic agent is necessary for cytosolic delivery and subsequent apoptosis. J. Biol. Chem. 271, 29073–29079 [DOI] [PubMed] [Google Scholar]
- 5. Lopez J. A., Susanto O., Jenkins M. R., Lukoyanova N., Sutton V. R., Law R. H., Johnston A., Bird C. H., Bird P. I., Whisstock J. C., Trapani J. A., Saibil H. R., Voskoboinik I. (2013) Perforin forms transient pores on the target cell plasma membrane to facilitate rapid access of granzymes during killer cell attack. Blood 121, 2659–2668 [DOI] [PubMed] [Google Scholar]
- 6. Voskoboinik I., Whisstock J. C., Trapani J. A. (2015) Perforin and granzymes: function, dysfunction and human pathology. Nat. Rev. Immunol. 15, 388–400 [DOI] [PubMed] [Google Scholar]
- 7. Law R. H., Lukoyanova N., Voskoboinik I., Caradoc-Davies T. T., Baran K., Dunstone M. A., D'Angelo M. E., Orlova E. V., Coulibaly F., Verschoor S., Browne K. A., Ciccone A., Kuiper M. J., Bird P. I., Trapani J. A., et al. (2010) The structural basis for membrane binding and pore formation by lymphocyte perforin. Nature 468, 447–451 [DOI] [PubMed] [Google Scholar]
- 8. Fogh-Andersen N., Christiansen T. F., Komarmy L., Siggaard-Andersen O. (1978) Measurement of free calcium ion in capillary blood and serum. Clin. Chem. 24, 1545–1552 [PubMed] [Google Scholar]
- 9. Dvorak M. M., Siddiqua A., Ward D. T., Carter D. H., Dallas S. L., Nemeth E. F., Riccardi D. (2004) Physiological changes in extracellular calcium concentration directly control osteoblast function in the absence of calciotropic hormones. Proc. Natl. Acad. Sci. U.S.A. 101, 5140–5145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Aivar P., Valero M., Bellistri E., Menendez de la Prida L. (2014) Extracellular calcium controls the expression of two different forms of ripple-like hippocampal oscillations. J. Neurosci. 34, 2989–3004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Brennan A. J., Chia J., Browne K. A., Ciccone A., Ellis S., Lopez J. A., Susanto O., Verschoor S., Yagita H., Whisstock J. C., Trapani J. A., Voskoboinik I. (2011) Protection from endogenous perforin: glycans and the C terminus regulate exocytic trafficking in cytotoxic lymphocytes. Immunity 34, 879–892 [DOI] [PubMed] [Google Scholar]
- 12. Ubach J., Zhang X., Shao X., Südhof T. C., Rizo J. (1998) Ca2+ binding to synaptotagmin: how many Ca2+ ions bind to the tip of a C2-domain? EMBO J. 17, 3921–3930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Fernandez I., Araç D., Ubach J., Gerber S. H., Shin O., Gao Y., Anderson R. G., Südhof T. C., Rizo J. (2001) Three-dimensional structure of the synaptotagmin 1 C2B-domain synaptotagmin 1 as a phospholipid binding machine. Neuron 32, 1057–1069 [DOI] [PubMed] [Google Scholar]
- 14. Shao X., Fernandez I., Südhof T. C., Rizo J. (1998) Solution structures of the Ca2+-free and Ca2+-bound C2A domain of synaptotagmin I: does Ca2+ induce a conformational change? Biochemistry 37, 16106–16115 [DOI] [PubMed] [Google Scholar]
- 15. Sutton R. B., Davletov B. A., Berghuis A. M., Südhof T. C., Sprang S. R. (1995) Structure of the first C2 domain of synaptotagmin I: a novel Ca2+/phospholipid-binding fold. Cell 80, 929–938 [DOI] [PubMed] [Google Scholar]
- 16. Pepio A. M., Fan X., Sossin W. S. (1998) The role of C2 domains in Ca2+-activated and Ca2+-independent protein kinase Cs in Aplysia. J. Biol. Chem. 273, 19040–19048 [DOI] [PubMed] [Google Scholar]
- 17. Traore D. A., Brennan A. J., Law R. H., Dogovski C., Perugini M. A., Lukoyanova N., Leung E. W., Norton R. S., Lopez J. A., Browne K. A., Yagita H., Lloyd G. J., Ciccone A., Verschoor S., Trapani J. A., Whisstock J. C., Voskoboinik I. (2013) Defining the interaction of perforin with calcium and the phospholipid membrane. Biochem. J. 456, 323–335 [DOI] [PubMed] [Google Scholar]
- 18. Marley J., Lu M., Bracken C. (2001) A method for efficient isotopic labeling of recombinant proteins. J. Biomol. NMR 20, 71–75 [DOI] [PubMed] [Google Scholar]
- 19. Conroy P. J., Law R. H., Gilgunn S., Hearty S., Caradoc-Davies T. T., Lloyd G., O'Kennedy R. J., Whisstock J. C. (2014) Reconciling the structural attributes of avian antibodies. J. Biol. Chem. 289, 15384–15392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. McPhillips T. M., McPhillips S. E., Chiu H. J., Cohen A. E., Deacon A. M., Ellis P. J., Garman E., Gonzalez A., Sauter N. K., Phizackerley R. P., Soltis S. M., Kuhn P. (2002) Blu-Ice and the distributed control system: software for data acquisition and instrument control at macromolecular crystallography beamlines. J. Synchrotron Radiat. 9, 401–406 [DOI] [PubMed] [Google Scholar]
- 21. Kabsch W. (2010) XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Evans P. (2006) Scaling and assessment of data quality. Acta Crystallogr. D Biol. Crystallogr. 62, 72–82 [DOI] [PubMed] [Google Scholar]
- 23. Emsley P., Lohkamp B., Scott W. G., Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., et al. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Winn M. D., Ballard C. C., Cowtan K. D., Dodson E. J., Emsley P., Evans P. R., Keegan R. M., Krissinel E. B., Leslie A. G., McCoy A., McNicholas S. J., Murshudov G. N., Pannu N. S., Potterton E. A., Powell H. R., et al. (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 67, 235–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. DeLano W. L. (2010) The PyMOL Molecular Graphics System, Version 1.3, Schrödinger, LLC, New York [Google Scholar]
- 27. Chen V. B., Arendall W. B. 3rd, Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., Richardson D. C. (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Schanda P., Kupce E., Brutscher B. (2005) SOFAST-HMQC experiments for recording two-dimensional heteronuclear correlation spectra of proteins within a few seconds. J. Biomol. NMR. 33, 199–211 [DOI] [PubMed] [Google Scholar]
- 29. Williamson M. P. (2013) Using chemical shift perturbation to characterise ligand binding. Prog. Nucl. Magn. Reson. Spectrosc. 73, 1–16 [DOI] [PubMed] [Google Scholar]
- 30. Hornemann S., von Schroetter C., Damberger F. F., Wüthrich K. (2009) Prion protein-detergent micelle interactions studied by NMR in solution. J. Biol. Chem. 284, 22713–22721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Shin O. H., Lu J., Rhee J. S., Tomchick D. R., Pang Z. P., Wojcik S. M., Camacho-Perez M., Brose N., Machius M., Rizo J., Rosenmund C., Südhof T. C. (2010) Munc13 C2B domain is an activity-dependent Ca2+ regulator of synaptic exocytosis. Nat. Struct. Mol. Biol. 17, 280–288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Perisic O., Fong S., Lynch D. E., Bycroft M., Williams R. L. (1998) Crystal structure of a calcium-phospholipid binding domain from cytosolic phospholipase A2. J. Biol. Chem. 273, 1596–1604 [DOI] [PubMed] [Google Scholar]
- 33. Ubach J., García J., Nittler M. P., Südhof T. C., Rizo J. (1999) Structure of the Janus-faced C2B domain of rabphilin. Nat. Cell Biol. 1, 106–112 [DOI] [PubMed] [Google Scholar]
- 34. Verdaguer N., Corbalan-Garcia S., Ochoa W. F., Fita I., Gómez-Fernández J. C. (1999) Ca2+ bridges the C2 membrane-binding domain of protein kinase Cα directly to phosphatidylserine. EMBO J. 18, 6329–6338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sutton R. B., Sprang S. R. (1998) Structure of the protein kinase Cβ phospholipid-binding C2 domain complexed with Ca2+. Structure 6, 1395–1405 [DOI] [PubMed] [Google Scholar]
- 36. Essen L. O., Perisic O., Lynch D. E., Katan M., Williams R. L. (1997) A ternary metal binding site in the C2 domain of phosphoinositide-specific phospholipase C-δ1. Biochemistry 36, 2753–2762 [DOI] [PubMed] [Google Scholar]
- 37. Arcaro A., Volinia S., Zvelebil M. J., Stein R., Watton S. J., Layton M. J., Gout I., Ahmadi K., Downward J., Waterfield M. D. (1998) Human phosphoinositide 3-kinase C2β, the role of calcium and the C2 domain in enzyme activity. J. Biol. Chem. 273, 33082–33090 [DOI] [PubMed] [Google Scholar]
- 38. Voskoboinik I., Thia M. C., Fletcher J., Ciccone A., Browne K., Smyth M. J., Trapani J. A. (2005) Calcium-dependent plasma membrane binding and cell lysis by perforin are mediated through its C2 domain: a critical role for aspartate residues 429, 435, 483, and 485 but not 491. J. Biol. Chem. 280, 8426–8434 [DOI] [PubMed] [Google Scholar]
- 39. Podack E. R., Young J. D., Cohn Z. A. (1985) Isolation and biochemical and functional characterization of perforin 1 from cytolytic T-cell granules. Proc. Natl. Acad. Sci. U.S.A. 82, 8629–8633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Young J. D., Nathan C. F., Podack E. R., Palladino M. A., Cohn Z. A. (1986) Functional channel formation associated with cytotoxic T-cell granules. Proc. Natl. Acad. Sci. U.S.A. 83, 150–154 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.