

Background: Hydrogen sulfide (H2S) modulates physiological processes in mammals.
Results: The reactivity of H2S toward disulfides (RSSR) and albumin sulfenic acid (RSOH) to form persulfides (RSSH) was assessed.
Conclusion: H2S is less reactive than thiols. Persulfides have enhanced nucleophilicity.
Significance: This kinetic study helps rationalize the contribution of the reactions with oxidized thiol derivatives to H2S biology.
Keywords: disulfide, hydrogen sulfide, kinetics, sulfhydryl, thiol, hydrodisulfide, persulfide, sulfenic acid
Abstract
Hydrogen sulfide (H2S) is increasingly recognized to modulate physiological processes in mammals through mechanisms that are currently under scrutiny. H2S is not able to react with reduced thiols (RSH). However, H2S, more precisely HS−, is able to react with oxidized thiol derivatives. We performed a systematic study of the reactivity of HS− toward symmetric low molecular weight disulfides (RSSR) and mixed albumin (HSA) disulfides. Correlations with thiol acidity and computational modeling showed that the reaction occurs through a concerted mechanism. Comparison with analogous reactions of thiolates indicated that the intrinsic reactivity of HS− is 1 order of magnitude lower than that of thiolates. In addition, H2S is able to react with sulfenic acids (RSOH). The rate constant of the reaction of H2S with the sulfenic acid formed in HSA was determined. Both reactions of H2S with disulfides and sulfenic acids yield persulfides (RSSH), recently identified post-translational modifications. The formation of this derivative in HSA was determined, and the rate constants of its reactions with a reporter disulfide and with peroxynitrite revealed that persulfides are better nucleophiles than thiols, which is consistent with the α effect. Experiments with cells in culture showed that treatment with hydrogen peroxide enhanced the formation of persulfides. Biological implications are discussed. Our results give light on the mechanisms of persulfide formation and provide quantitative evidence for the high nucleophilicity of these novel derivatives, setting the stage for understanding the contribution of the reactions of H2S with oxidized thiol derivatives to H2S effector processes.
Introduction
Beyond its classical conception as a highly toxic gas, hydrogen sulfide (H2S)4 is now considered a physiological modulator in mammals. It is produced through the enzymatic activity of cystathionine β-synthase and cystathionine γ-lyase and probably also of mercaptopyruvate S-transferase (1). At neutral pH, the species H2S, a weak acid (pKa = 7 (2)), is in equilibrium with its conjugate base, the anion hydrosulfide (HS−). Simultaneously with the description of its biological effects, efforts have been undertaken to elucidate the underlying molecular mechanisms, yielding slow progress. Hydrogen sulfide cannot react with thiols (RSH). Among a range of possible mechanisms that could operate in biological systems, the reaction of H2S, more precisely the hydrosulfide anion HS− in equilibrium with it, toward disulfides (RSSR) and sulfenic acids (RSOH) can be considered a plausible way of unleashing physiological consequences. Furthermore, these reactions produce persulfides (also known as hydrodisulfides or hydropersulfides),5 constituting possible mechanisms for the so-called “S-sulfhydration” (3–5).
Persulfides (RSSH) represent novel thiol modification products of increasing interest in signaling and catalysis. They are part of a class of chemical moieties known as sulfanes, in which sulfur is covalently linked to another sulfur and/or ionizable protons (6). The typically unstable persulfides possess electrophilic character. They also conserve the nucleophilic character of the original thiol, or actually increase it due to the presence of an adjacent sulfur containing unshared pairs of electrons, i.e. the α effect (3, 7, 8). Moreover, it has been proposed that these compounds are responsible for the biological effects initially assigned to H2S (9). Persulfides were detected years ago as naturally occurring products (6) and are proposed to have roles in sulfur transport (10) and enzymatic catalysis (11, 12). Indeed, animals have enzymes that produce and transfer persulfides, i.e. cystathionine γ-lyase, cystathionine β-synthase (13), sulfide:quinone oxidoreductase, and rhodanese (14), and others that decompose them, like persulfide dioxygenase or ETHE1 (15). Recently, persulfides have been produced in a diversity of systems and detected using analytical methods such as cold cyanolysis and dithiothreitol-dependent release of H2S, together with mass spectrometry and a variety of selective labeling methods (16–22).
Despite increasing efforts dedicated to understand the reactivity of H2S, no systematic kinetic study of the reaction with oxidized thiol derivatives has been published, and few persulfide models suitable for biochemical characterization have been proposed (17–19, 22). The reaction toward disulfides could represent a significant consumption pathway for H2S, mainly in extracellular milieu and plasma, where the diversity and high amounts of disulfides present (23) make them a likely target. In plasma, human serum albumin (HSA)6 is the most abundant protein, and mixed disulfides of HSA constitute the predominant disulfides (23). HSA possesses 34 cysteines forming structural disulfides and only one reduced cysteine (Cys-34, HSA-SH) that is located in a cleft, hindering intra- and intermolecular reactions. These features allow a relatively stable sulfenic acid to be formed (HSA-SOH) (24). Sulfenic acids are transient intermediates generated in thiol oxidation pathways. They are typically unstable and decay mainly through reaction with a second thiol to form disulfides. Because HSA forms a long-lived and well characterized sulfenic acid (24, 25), it could open the way to kinetic measurements of the reactions with H2S. In addition, HSA could potentially also form a stable persulfide (HSA-SSH).
In this work, we studied the reactivity of H2S toward symmetrical low molecular weight (LMW) disulfides and mixed disulfides formed between HSA and LMW thiols, providing the first systematic study that allows comparison of the intrinsic reactivity of H2S with respect to thiols. In addition, we report for the first time the rate constant of the reaction of H2S with a sulfenic acid, the one formed in HSA. Finally, we obtained the HSA persulfide and determined the rate constant of two of its reactions, providing the first quantitative analysis of the nucleophilicity of persulfides and its comparison with that of thiols. Thus, our study contributes to the understanding of the biochemical reactivity of H2S and helps reveal the impact that its reactions with oxidized thiol derivatives to form persulfides could have in the biology of this recently recognized mediator.
Experimental Procedures
Preparation of Hydrogen Sulfide, LMW Thiol, and Disulfide Solutions
Stable Na2S solutions were prepared by two methods. (a) Stock solutions of Na2S·9H2O (Mallinckrodt Baker) were prepared daily in distilled water in sealed vials and used immediately. Concentrations were verified by iodometric titration and remained constant for several hours. (b) Anhydrous Na2S was purchased from Sigma, opened, and stored in a glove box (<1 ppm O2 and <1 ppm H2O). Stock solutions (100 mm) were prepared in the glove box using argon-bubbled nano-pure water pretreated with Chelex resin-100 and stored in dark glass vials with polytetrafluoroethylene septa at 4 °C. Gas-tight Hamilton syringes were used throughout the study. 5-Thio-2-nitrobenzoic acid (TNB) solutions were prepared as reported previously (26). LMW disulfides were purchased from Sigma or AppliChem. Glutathione disulfide (GSSG), cystine dimethyl ester (CysOMe-CysOMe), and hydroxyethyl disulfide (HED) stocks were prepared in distilled water; cystine (Cys-Cys) and homocystine (Hcy-Hcy) were diluted in 0.1 n NaOH; and 5,5′-dithiobis-(2-nitrobenzoic acid) (DTNB) stocks were prepared in 95% ethanol.
Preparation of HSA Derivatives
HSA (Sigma) was delipidated; its thiol was reduced with β-mercaptoethanol, and the concentrations of protein and thiol were quantified as described previously (26). Mixed disulfides were prepared by incubating reduced HSA (∼1 mm) overnight with the disulfide of interest (up to 20 mm) in acetate buffer (100 mm, pH 4.5, 0.1 mm diethylenetriaminepentaacetic acid (DTPA), 4 °C) or tris buffer (100 mm, pH 7.4, 0.1 mm DTPA, 4 °C). The pH of the buffer was chosen so as to favor maximum displacement of equilibria toward mixed disulfide formation as follows: acidic pH when the pKa of the leaving thiol was higher than that of HSA-SH (8.1 (23, 25)) as in the case of Hcy-Hcy, GSSG, and HED, and basic pH when the pKa was lower (DTNB, CysOMe-CysOMe) (27). The mixed disulfides were subjected to gel filtration in a PD-10 column (GE Healthcare) equilibrated with phosphate buffer (100 mm, pH 7.4, 0.1 mm DTPA), and the yield of disulfide was quantified by subtraction of HSA-SH concentration before and after treatment with LMW disulfides. HSA preparations enriched in sulfenic acid (HSA-SOH) were prepared by incubation of reduced HSA (0.6 mm) with H2O2 (4 mm, 4 min, 37 °C), kept on ice, and used daily. The concentration of HSA-SOH was quantified by reaction with TNB (24). Sulfanes in HSA were produced following different procedures, depending on the experiment. For most determinations, mixed HSA disulfides were incubated with equimolar Na2S in phosphate buffer (100 mm, pH 7.4, 0.1 mm DTPA, 25 °C) for a specified period, followed by gel filtration. Persulfides for kinetic studies toward 4,4′-dithiodipyridine (DTDPy) were prepared by incubating 6 μm HSA-SOH with equimolar Na2S in phosphate buffer (100 mm, pH 7.4, 0.1 mm DTPA, 25 °C) during variable periods of time. Persulfides for kinetic studies toward peroxynitrite were prepared by reacting HSA-TNB with Na2S. HSA-SSH was quantified based on the amount of TNB released and purified by gel filtration on Sephadex G-10 equilibrated with phosphate buffer (30 mm, pH 7.4). Solutions were kept under argon and on ice to minimize decay.
Kinetics of Hydrogen Sulfide Reactions with Disulfides by Continuous UV-Visible Measurements
Reactions were initiated by the addition of low amounts (30–500 μm) of Na2S to disulfides in pseudo-first order excess in borate (100 mm, pH 9.5, 0.1 mm DTPA) or phosphate (100 mm, pH 7.4, 0.1 mm DTPA) buffer using sealed quartz cuvettes with minimal head space. UV-visible spectra were registered in a Varian Cary 50 spectrophotometer at 25 °C, and increases in absorbance at 350 nm due to the appearance of sulfane sulfur (28) were fitted to single exponential functions to obtain pseudo-first order rate constants (kobs) for Cys-Cys, CysOMe-CysOMe, GSSG, HED, and Hcy-Hcy. The reaction with DTNB was followed from TNB formation at 412 nm and fitted to double exponential functions. The second order rate constants were determined from the slopes of plots of kobs versus disulfide concentration. The pH-independent rate constants were calculated by correction for HS− taking into account the pH and the pKa of H2S.
Kinetics of Hydrogen Sulfide Reactions with Disulfides by Discrete Hydrogen Sulfide Measurements
Reaction mixtures containing excess disulfides in phosphate buffer (100 mm, pH 7.4, 0.1 mm DTPA) were incubated in closed vials with minimal head space at 25 °C. Aliquots were removed using a gas-tight syringe and mixed with 4 mm zinc acetate to quench the reaction. The H2S concentration of the aliquots was determined using the methylene blue method (29). In the case of LMW disulfides, concentration decreases were fitted to single exponential functions to obtain the corresponding kobs. In the case of mixed HSA disulfides, mixtures for methylene blue formation were centrifuged to remove the precipitated protein, and the rate constants were calculated using the method of initial rates.
Kinetics of Hydrogen Sulfide Reactions with Disulfides by Amperometric Hydrogen Sulfide Measurements
Na2S (10 μm) was injected into phosphate buffer (50 mm, pH 7.4) and its concentration was followed using a selective H2S electrode connected to a free radical analyzer (World Precision Instruments). The electrode response was continuously recorded. When it reached a maximum, different concentrations of HSA disulfides were injected. Decay of H2S at 25 °C followed first order kinetics, and the kobs were plotted versus concentration of the protein derivative.
Kinetics of Hydrogen Sulfide Reaction with the Sulfenic Acid in HSA Using a Competition Approach
The reaction was evaluated through competition with TNB, using a procedure similar to a previously reported one (24). Sulfenic acid-enriched HSA (∼0.5 μm HSA-SOH) was mixed with excess TNB (66 μm) in phosphate buffer (100 mm, pH 7.4, 0.1 mm DTPA) in the absence or presence of Na2S (up to 100 μm). The decay of TNB was recorded at 412 nm and fitted to exponential plus straight line functions (24). The second order rate constant for the reaction of H2S with HSA-SOH was calculated from the slopes of plots of kobs versus Na2S concentration.
Sulfane Quantification
The protein fraction was separated by gel filtration, and sulfane sulfur was quantified by cold cyanolysis. Samples were incubated with cyanide, and the thiocyanate formed was determined by the absorbance of the complex formed with ferric ions at 460 nm (28).
Tag-switch Assay
Labeling of persulfides in HSA preparations (dot blots) and fixed cells was performed following an established protocol (19, 30). 2-(Methylsulfonyl)1,3-benzothiazole was purchased from Santa Cruz Biotechnology. CN-biotin was synthesized as described previously (19, 30).
Detection of HSA Persulfide with Tris(2,4-dimethyl-5-sulfophenyl)phosphine (TDMSP)
HSA-SSH (20 μm) was prepared from HSA-TNB by incubation with Na2S at a 1:1.5 molar ratio, purified on Sephadex G-10, and incubated with a 20-fold excess of TDMSP at room temperature for 30 min. The reaction mixture was sprayed into ultra-high resolution ESI-TOF Bruker Daltonik (Bremen, Germany) maXis5G, an ESI-TOF MS capable of resolution of at least 60,000 full width at half-maximum. Detection was performed in negative-ion mode, and the source voltage was 4.0 kV. The flow rates were 180 μl/h. The instrument was calibrated by direct infusion of the Agilent ESI-TOF low concentration tuning mixture, which provided an m/z range of singly charged peaks up to 2700 Da in both ion modes.
Detection of HSA Persulfide by Ultra-high Resolution ESI-TOF MS
HSA-TNB was mixed with equimolar Na2S for 15 min at room temperature, desalted on Sephadex G-10, mixed with acetonitrile (1:1, v/v) containing 0.5% formic acid, and continuously sprayed into maXis5G MS. Detection was performed in positive ion mode, and the source voltage was 4.5 kV. The flow rates were 180 μl/h. Spectra were post-processed using Data Analysis software (Bruker Daltonics).
Identification of the Persulfidated Cys Residue
HSA-SSH prepared from HSA-TNB was collected into vials containing 100 mm biotin-maleimide to ensure the immediate blocking of both persulfides and eventually thiols. After a 2-h incubation at 37 °C, the protein was desalted again on Sephadex G-10 and mixed with proteomics grade porcine trypsin (Sigma, 1:100 w/w) overnight at 37 °C. The reaction was stopped by adjusting the pH to 3, and 100 μl of the peptide mixture was mixed with 100 μl of PierceTM streptavidin-Agarose beads (Thermo Fisher Scientific). The unbound peptides were discarded, and the beads were washed three times with phosphate buffer (50 mm, pH 7.4). The bound peptides were those modified by biotin-maleimide, but in the case of peptides initially containing persulfide, the reaction with biotin-maleimide creates a disulfide bond. These peptides were eluted by addition of 100 μl of 10 mm DTT. After centrifugation, the eluted peptides were analyzed by LC-MS. Reverse phase HPLC (Dionex, Thermo Fisher Scientific) was performed using a C18 column and a combination of water (solvent A) and acetonitrile (solvent B), both containing 0.5% formic acid. The following elution protocol was used: 5 min of 10% B and then 45 min of continuous rise to 90% B. In addition, the peptides were directly injected into maXis5G using in-source collision-induced dissociation tandem MS approach (source voltage was 4.5 kV; source temperature was 250 °C, and collision energy was 90 eV).
Reactivity of HSA Persulfide toward Peroxynitrite
Peroxynitrite was synthesized as described previously (31). Kinetic data were obtained by recording time-resolved UV-visible spectra using a modified μSFM-20 Bio-Logic stopped-flow module equipped with a J&M TIDAS high speed diode array spectrometer with combined deuterium and tungsten lamps (200–1015 nm wavelength range). The syringes were controlled by separate drives, allowing the variation of the ratio of mixing volumes used in the kinetic runs. Data were analyzed in the Specfit/32TM program. At least five kinetic runs were recorded for all conditions. Final concentration of peroxynitrite was 9 μm, and the concentrations of HSA-SSH and HSA-SH were in the range of 2–60 μm. The yield of nitrotyrosine was determined by measuring absorbance at 430 nm (ϵ = 4400 m−1 cm−1) after alkalinization to pH 10, 5 min after mixing 183 μm protein with 500 μm peroxynitrite in phosphate buffer (50 mm, pH 7.4).
Reactivity of HSA Persulfide toward DTDPy
An Applied Photophysics RX2000 stopped-flow accessory coupled to a Varian 50 spectrophotometer was used. Preparations enriched in HSA-SOH were incubated with equimolar Na2S in one syringe and mixed at increasing time points with excess DTDPy contained in the other syringe, in phosphate buffer (100 mm, pH 7.4, 0.1 mm DTPA) at 25 °C. The time course of thiopyridone appearance was followed at 324 nm (ϵ = 21,400 m−1 cm−1). The traces were fitted to double exponential functions to obtain the corresponding kobs.
Computational Modeling of the Reaction Mechanism of HS− with a Representative Symmetrical LMW Disulfide (Zwitterionic Cys-Cys) in Aqueous Solution
The structures of the reactant and product complexes and the corresponding transition state, including two explicit water molecules, were fully optimized and verified by inspection of the eigenvalues of the Hessian matrix using the density functional theory (DFT) methodology at the IEFPCM/ωB97X-D/6-31+G(d) (32–34) level of theory, as implemented in Gaussian09 Revision D.01 (35). Water was also described as a continuum solvent (ϵ = 78.5), placing solutes in molecularly shaped cavities constructed using Bondi's radii (36). The reaction path was followed by the intrinsic reaction coordinate (37) calculated at the same level of theory with the Hessian-based Predictor-Corrector integrator algorithm (38), 50 points each side of the transition state with a step size of 0.05 Bohr. Thermochemistry at 298 K and 1 atm was assessed at the same level of theory, applying usual approximations of statistical thermodynamics and unscaled zero point vibrational energies, as implemented in the Gaussian09 suite (35).
Computational Comparison of the Global Reactivity and Nucleophilicity of HS− and Cys Persulfide with Respect to LMW Thiolates
The structures of five LMW thiolates of known basicity (cysteine methyl ester; cysteinylglycine; β-mercaptoethanol; Cys, and Hcy) as well as cysteine persulfide anion (CysSS−) were determined at the same level of theory in aqueous solution as described above. The energy of the highest occupied Kohn-Sham (KS) orbital, EHOMO, was taken as an indicator of the nucleophilicity of each species. The corresponding global reactivity was quantified by the chemical hardness (η), calculated under the finite differences approximation with Equation 1 (39),
where ELUMO represents the energy of the lowest unoccupied KS orbital.
Computational Prediction of the pKa of Cysteine Persulfide
Using the same level of theory described above, a prediction of the pKa shift (ΔpKa) corresponding to CysSSH with respect to CysSH was obtained by applying the proton-exchange method proposed by Yu et al. (40) with the calculated free-energy, ΔGexch, corresponding to Reaction 1,
from which ΔpKa is obtained as shown in Equation 2,
Cell Experiments
Human umbilical vein endothelial cells (HUVEC) (PromoCell) were grown in endothelial cell growth medium 2 (ready-to-use, PromoCell) on ibidi dishes (ibidi, Martinsried, Germany) at 5% CO2 and 20% oxygen until they reached >50% confluency. Human neuroblastoma cells (SH-SY5Y, ECACC, Sigma) were grown in Ham's F-12/Eagle's minimum essential medium (EBSS) (1:1) medium supplemented with 2 mm glutamine, 1% non-essential amino acids + 15% fetal bovine serum FBS/FCS, on ibidi dishes. Inhibition of endogenous production of H2S was performed by using aminooxyacetic acid (2 mm) in SH-SY5Y cultures or propargylglycine (2 mm) in HUVEC cultures. Fluorescence microscopy was carried out using Carl Zeiss Axiovert 40 CLF inverted microscope, equipped with green fluorescent filters and AxioCam ICm1. Images were post-processed in ImageJ software.
Data Processing
Data were processed using OriginPro 8 (Microcal Software). Numerical fitting of data to reaction sequences was performed with Gepasi (41).
Results and Discussion
Kinetics of Reaction of Hydrogen Sulfide with Symmetric LMW Disulfides
To evaluate the reaction of H2S toward LMW disulfides, kinetic measurements were performed. Parallel approaches were used in most cases. Under pseudo-first order conditions with disulfides in excess, exponential decays in H2S concentration were observed (Fig. 1A). The reactions are likely to reach equilibrium situations, but under our conditions the consumption of H2S was close to total, probably because of the excess disulfide reagent and because of further reactions of the products. The observed rate constants showed a linear dependence on disulfide concentration (Fig. 1B). The second order rate constants for six disulfides are summarized in Table 1. The values were in the order of 10−2 to 100 m−1 s−1 for alkyl disulfides. Because HS− is the species responsible for reactivity, the rate constants determined at pH 7.4 were corrected to obtain the pH-independent values, which represent the rate constants that would be measured if all the H2S was ionized to HS−. Results obtained by initial rate processing or by UV-visible absorbance measurements were consistent with H2S consumption measurements.
FIGURE 1.
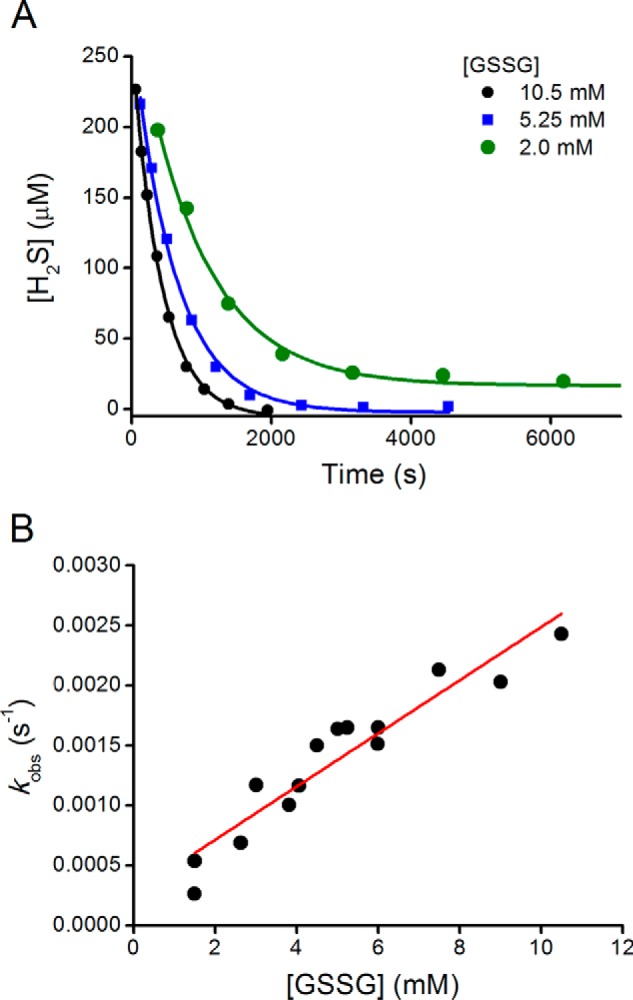
Kinetics of hydrogen sulfide consumption in the presence of glutathione disulfide. A, Na2S (250 μm) was mixed with 2.0, 5.25, and 10.5 mm GSSG in phosphate buffer (100 mm, pH 7.4, 0.1 mm DTPA, 25 °C) in closed vials. Aliquots were analyzed for H2S content with the methylene blue method. B, observed rate constants for the reaction of HS− with GSSG obtained from the fitting of time courses to single exponential equations.
TABLE 1.
Second order rate constants for the reactions of hydrogen sulfide with symmetric LMW disulfides in 100 mm phosphate buffer at 25 °C
Reaction is as follows: HS− + RSSR → RSS− + RSH.
| Disulfide | pKa of the thiol | k pH 7.4 | k pH-independent |
|---|---|---|---|
| m−1 s−1 | m−1 s−1 | ||
| DTNB | 4.4a | (8.61 ± 0.04) × 102 | (1.20 ± 0.01) × 103 |
| CysOMe-CysOMe | 6.45b | 3.3 ± 0.7 | 5 ± 1 |
| Cys-Cys | 8.29c | (6 ± 1) × 10−1 | (9 ± 2) × 10−1 |
| GSSG | 8.94c | (1.6 ± 0.1) × 10−1 | (2.2 ± 0.1) × 10−1 |
| Hcy-Hcy | 9.1c | (3.2 ± 0.4) × 10−1 | (4.5 ± 0.6) × 10−1 |
| HED | 9.6c | (3.92 ± 0.03) × 10−2 | (5.48 ± 0.04) × 10−2 |
As precedent, rate constants of (6.2 ± 0.7) × 10−2 and 1.3 × 10−2 m−1 s−1 have been reported for the reaction of cystine with excess H2S in 50 mm borate buffer at pH 10.0 or NaOH 0.1 m, respectively (42). These values are lower, by factors of 15–70, than the pH-independent rate constant determined in this work, (9 ± 2) × 10−1 m−1 s−1. The difference can be explained because at alkaline pH values the predominant species is cystine2− and not the zwitterion, as in our conditions. In addition, HS− may ionize to S2−. A change in the charge in the reacting species could affect the reaction rate constant by 2 orders of magnitude (43). Recently, Vasas et al. (44) kinetically characterized the reaction of H2S with DTNB. The second order rate constant reported at pH 7.4 is 881 m−1 s−1, consistent with our determination (861 m−1 s−1, see Table 1). They also report that the kinetics of reaction with cysteine and glutathione are significantly slower.
Kinetics of Reaction of Hydrogen Sulfide toward Asymmetric Disulfides of HSA Formed with LMW Thiols
Second order rate constants were obtained for the reactions of H2S with several mixed disulfides (Table 2) by measuring initial rates of H2S consumption using the methylene blue method (Fig. 2A). The values were in the order of 10−1–101 m−1 s−1 for the disulfides formed between HSA and alkyl thiols. Controls were performed using HSA-SH to rule out the reduction of structural disulfides as well as other decay pathways of H2S such as autoxidation. Rate constants were also determined by following the disappearance of H2S amperometrically, with similar results (Fig. 2B).
TABLE 2.
Second order rate constants for the reactions of hydrogen sulfide toward mixed disulfides formed between Cys-34 of HSA and LMW thiols in 100 mm phosphate buffer at 25 °C
Reaction is as follows: HS− + HSA-SSR → HSA-SS− + RSH or HSA-SH + RSS−.
| Disulfide | pKa of the thiol | k pH 7.4 | k pH-independent |
|---|---|---|---|
| m−1 s−1 | m−1 s−1 | ||
| HSA-TNB | 4.4a | (3.5 ± 0.8) × 102 | (5 ± 1) × 102 |
| HSA-CysOMe | 6.45b | (5.5 ± 1.3) × 101 | (8 ± 2) × 101 |
| HSA-Cys | 8.29c | 3.0 ± 1.1 | 4 ± 2 |
| HSA-GSH | 8.94c | 1.7 ± 1.1 | 2 ± 1 |
| HSA-Hcy | 9.1c | (2.3 ± 0.8) × 10−1 | (3 ± 1) × 10−1 |
| HSA-β-mercaptoethanol | 9.6c | (5.1 ± 0.4) × 10−1 | (7.2 ± 0.6) × 10−1 |
FIGURE 2.
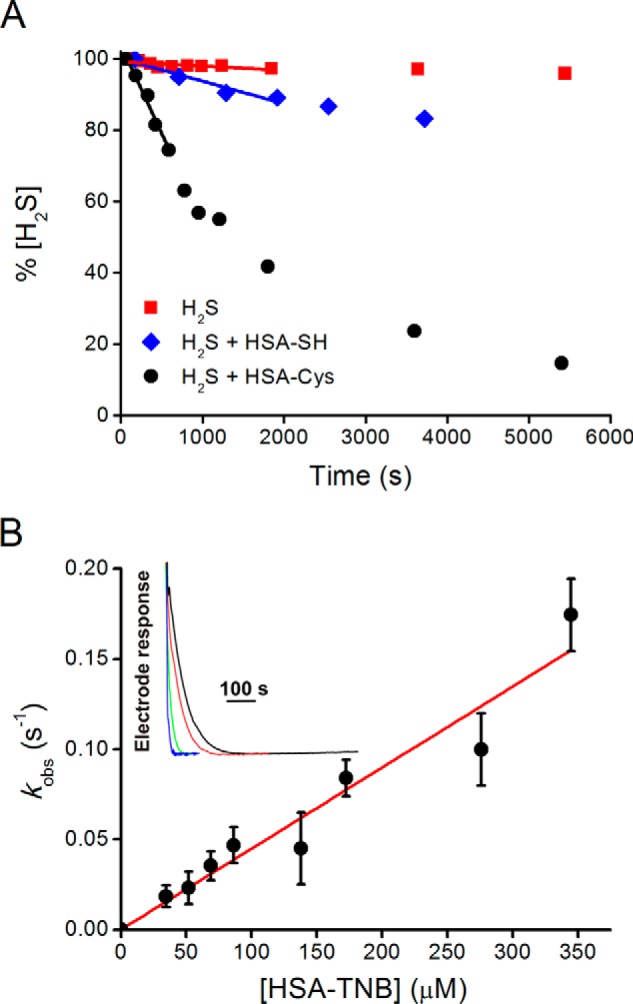
Kinetics of reaction of hydrogen sulfide with HSA mixed disulfides. A, Na2S (476 μm) was incubated with HSA-Cys (470 μm, black circles). Controls were prepared using Na2S alone (476 μm, red squares) or Na2S (118 μm) with HSA-SH (439 μm, blue diamonds) in tris buffer (0.2 m, pH 7.4, 0.1 mm DTPA, 25 °C). Aliquots were analyzed for H2S content with the methylene blue method. B, kinetics of H2S decay in the presence of HSA-TNB, measured by selective H2S electrode. Inset, original traces of H2S decay upon addition of 34.5 μm (black line), 51.7 μm (red line), 172.4 μm (green line), and 344.7 μm (blue line) HSA-TNB into 10 μm Na2S in phosphate buffer (50 mm, pH 7.4, 25 °C).
Mechanism of the Reaction of Hydrogen Sulfide toward Disulfides
The data obtained for H2S can be rationalized in the context of reported data for thiol-disulfide interchange reactions. These interchange reactions occur by single concerted SN2 mechanisms where the reactive species involved is the thiolate anion (45). When the pH-independent rate constants are plotted against the pKa values of a variety of attacking thiols and target disulfides, Brønsted correlations are obtained according to Equation 3,
where βnuc, βc, and βlg are the Brønsted coefficients for the nucleophilic sulfur, central sulfur, and leaving group sulfur, respectively. Analogously, for the reaction studied herein of HS− with symmetrical disulfides producing a thiolate (leaving group) and a persulfide, the logarithm of the pH-independent rate constants obtained showed a negative linear correlation with the pKa of the leaving thiol (Fig. 3A), supporting a simple concerted SN2 mechanism and indicating that acidic thiols, with lower pKa values, are better leaving groups. The slope of this curve represents the addition of the Brønsted coefficients of central and leaving sulfurs (βc + βlg) and has a value of −0.75, consistent with the values reported for the reaction with thiolates (45). In the case of mixed disulfides of HSA and LMW thiols, the slope of the Brønsted plot was −0.6, reflecting βc or βlg (Fig. 3B). The leaving group can be either HSA-S− or RS−. In the absence of steric effects, the most acidic groups will be the preferred leaving groups.
FIGURE 3.
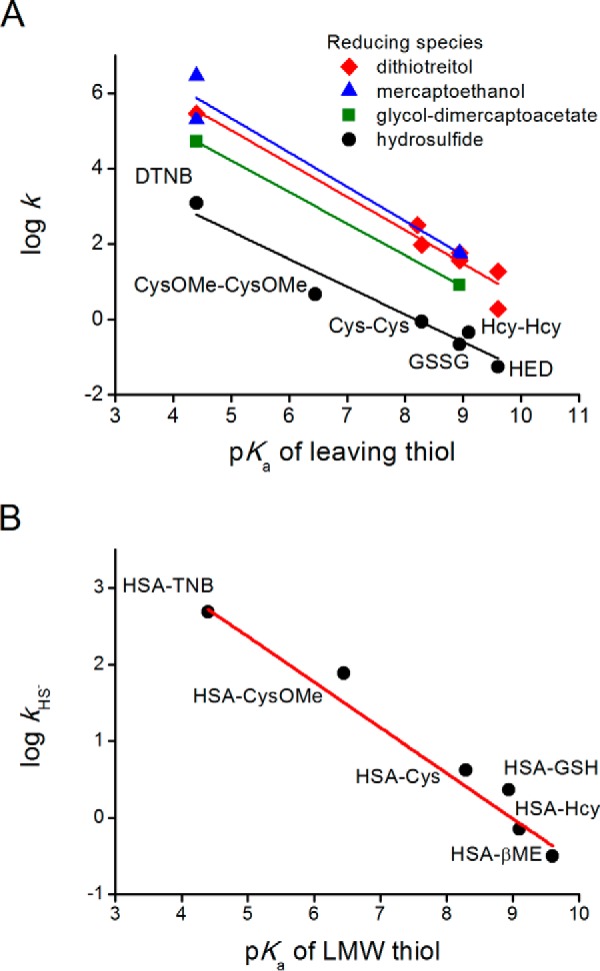
Comparison of the reactivities of HS− and thiolates toward disulfides depending on the pKa value of the leaving thiol. A, pH-independent rate constants of the reactions of LMW disulfides with HS− (this work) or with LMW thiolates (data reported in Refs. 45, 47, 61, 62). B, pH-independent rate constants of the reactions of HS− with mixed HSA disulfides.
Computational Modeling
The structures of the reactant complex, transition state, and product complex for the reaction mechanism of HS− with cystine as a representative LMW disulfide are shown in Fig. 4. The reaction exhibits a concerted linear transition state (with an associated imaginary frequency of 233i cm−1) consistent with a typical Sn2 mechanism. The explicit water molecules present in the model are not directly involved in the reaction coordinate but play an ancillary role stabilizing the participant moieties through hydrogen bonding. The Gibbs free energy of reaction is predicted to be of 13.4 kcal mol−1. Although formation of the products would thus appear to be thermodynamically disfavored, fast acid base equilibration of the products is expected to drive the outcome of the process. Precisely, because CysSH has a pKa of 8.29 and the predicted pKa for CysSSH as obtained from Equation 2 is 4.34 (ΔpKa of −3.96, see Table 3), the products at physiological pH become CysSS− and CysSH, preventing the reverse reaction. The activation Gibbs free energy is calculated to be of 19.9 kcal mol−1. This value is comparable with the experimental one of 17.5 kcal mol−1 that can be derived from the rate constant by applying the Eyring-Polanyi equation, confirming the consistency between theory and experiment in the description of the reaction.
FIGURE 4.

Computational modeling of the reaction of hydrosulfide toward cystine. DFT optimized structures in water of the reactant complex, transition state, and product complex for the reaction between cystine and HS−. Values in parentheses correspond to relative Gibbs free energies at 298 K calculated with respect to the reactant complex.
TABLE 3.
Comparison of basicity, nucleophilicity (KS-HOMO energies), and chemical hardness (η) values for thiolates, HS−, and CysSS− calculated at the DFT level in aqueous solution
| Species | pKa | EHOMO | η |
|---|---|---|---|
| eV | eV | ||
| CysOMe− | 6.45a | −7.63 | 4.53 |
| HS− | 7.00b | −7.84 | 4.94 |
| CysGly− | 7.95c | −7.57 | 4.67 |
| Cys− | 8.29c | −7.50 | 4.72 |
| Hcy− | 9.1c | −7.33 | 4.58 |
| β-Mercaptoethanol− | 9.60c | −7.39 | 4.75 |
| CysSS− | (4.34)d | −6.97 | 4.16 |
a Data are from this work.
b Data are from Ref. 2.
c Data are from Ref. 64.
d Data are estimated here from the predicted shift with respect to Cys of −3.96 (see Equation 2).
Comparison of Nucleophilicities of Hydrosulfide and Thiolates
The pH-independent rate constants obtained for HS− reactions with the six symmetric disulfides studied are about 1 order of magnitude lower than those reported for alkyl thiolates of comparable pKa values (Fig. 3A). This decreased nucleophilic character of HS− could be due to the lack of the inductive effect of the adjacent methylene of the sulfur, changes in the polarizability of the sulfur, or solvation effects (8, 46).
The effect of the pKa of the nucleophile on the reaction with a disulfide is evident from Fig. 5, where the pH-independent rate constants of thiol disulfide interchange reactions for different thiolates with DTNB or GSSG, taken from the literature (45, 47), are compared with the value obtained for HS−. Again, the intrinsic reactivity of HS− is lower than that of thiolates, by factors of 20–25.
FIGURE 5.
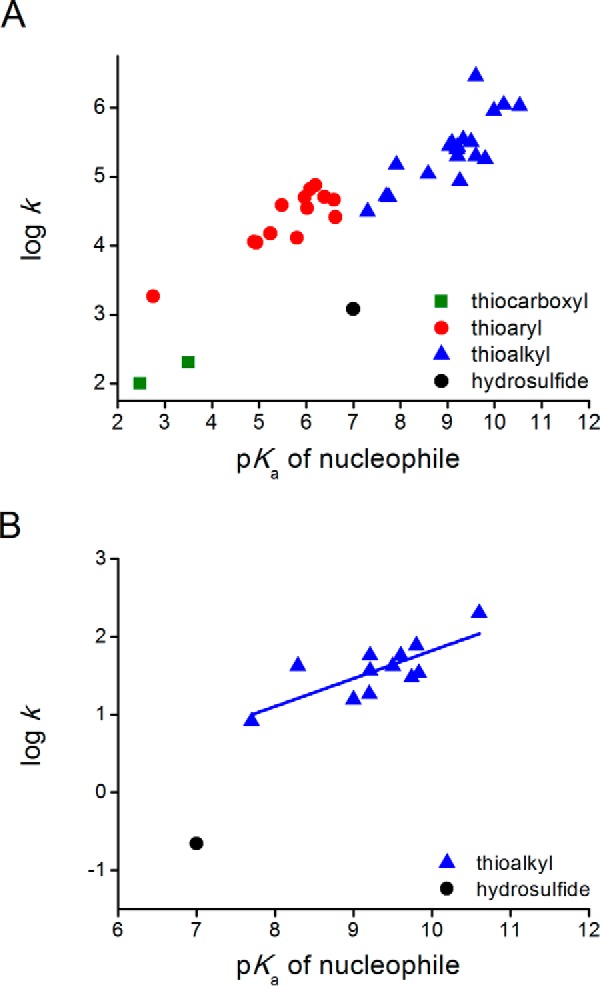
Comparison of the reactivities of HS− and thiolates toward disulfides depending on the pKa of the attacking thiol. A, DTNB as electrophile. pH-independent rate constants for the reactions of HS− (this work) or thiolates (45, 47, 62) with DTNB. Data are grouped for thiocarboxyls (green squares), aryl thiols (red circles), alkyl thiols (blue triangles), and H2S (black circle). Slopes (βnuc) are 0.30, 0.38, and 0.46 for attacking thiocarboxyls, thioaryls, and thioalkyls, respectively. B, GSSG as electrophile. pH-independent rate constants for alkyl thiols (blue triangles (45, 61, 63)) and H2S (black circle, this work). Slope (βnuc) is 0.36.
As shown by inspection of the calculated descriptors of global reactivity and nucleophilicity collected in Table 3, HS− is harder and a weaker nucleophile than the thiolates. The nucleophilicities (estimated through the KS-HOMO energies, EHOMO, as a measure of the facility of each species to donate electrons) of the five thiolates linearly correlate with the corresponding experimental pKa (Fig. 6) mirroring the trend emerging from the experimental data and showing that more basic thiolates are better nucleophiles (Fig. 5). In contrast, the nucleophilicity of HS− falls below that of a thiolate of comparable basicity.
FIGURE 6.
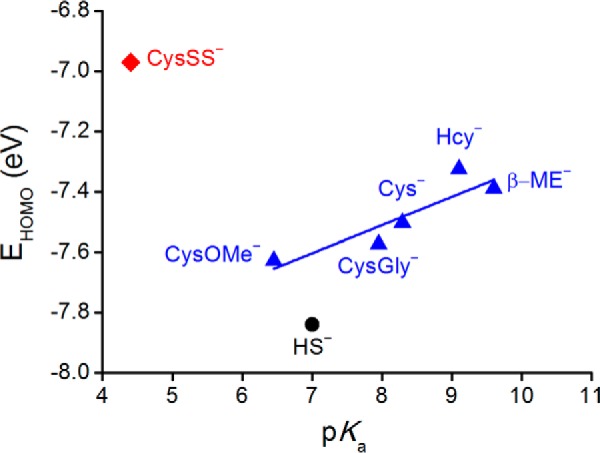
KS-HOMO energies for thiolates, HS−, and CysSS− calculated at the IEFPCM/ωB97X-D/6-31+G(d) level of theory as a function of pKa of conjugated acid.
Although the intrinsic reactivity of HS− is lower than that of thiolates, at pH 7.4 its availability is higher. Indeed, the ratio HS−/H2S is 2.51 at pH 7.4 due to the low pKa of H2S, and the ratios thiolate/thiol for cysteine and glutathione are 0.13 and 0.03, respectively. The Brønsted correlation can be used to obtain a theoretical optimum pKa value for maximizing the observed rate for reactions at a certain pH (43) as shown in Equation 4.
Considering a βnuc value of 0.38 (Fig. 5), the optimal pKa for the reaction at pH 7.4 is 7.2. This means that at physiological pH the nucleophilic capacity of H2S is maximally exploited. In other words, although H2S is a relatively weak nucleophile, its reactivity is maximized at physiological pH values.
Detection of HSA Persulfide
The formation of a persulfide in HSA could not only represent a potential physiological product, but it could also be a useful model to study its features, as in the example of sulfenic acids (26). Using the method of cold cyanolysis, the formation of sulfane sulfur, a group of compounds that includes persulfide, was evaluated. Sulfane sulfur was detected in reaction mixtures of asymmetric HSA disulfides with Na2S and coeluted with protein in gel filtration separations (Fig. 7A).
FIGURE 7.
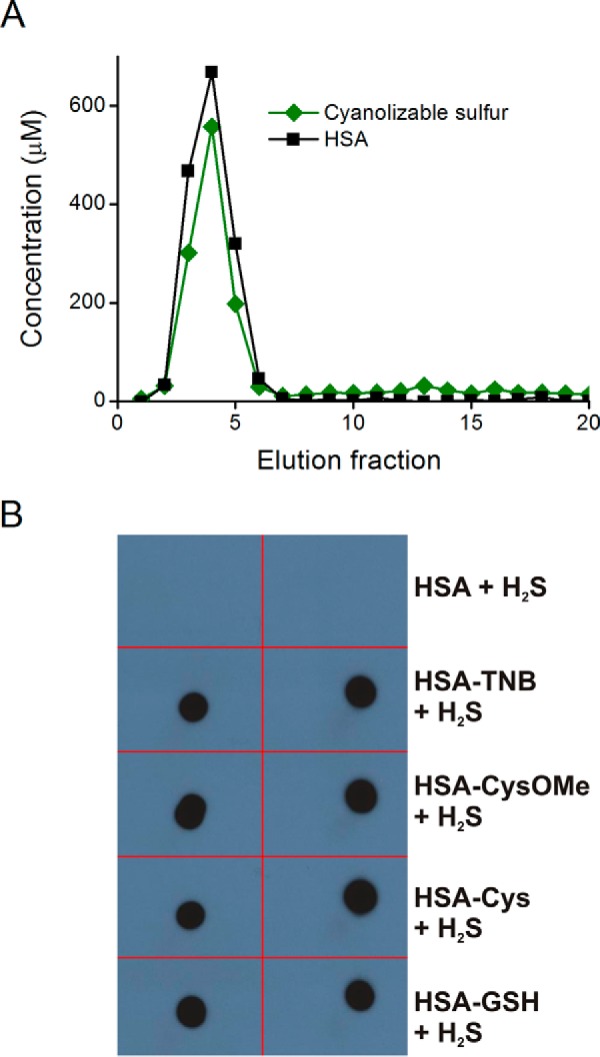
Product characterization. A, detection of sulfane sulfur. HSA-Hcy (350 μm) was incubated with Na2S (350 μm) during 3 h. The reaction mixture was gel-filtrated using a PD-10 column, and sulfanes were quantified in elution fractions by cold cyanolysis. B, dot-blot detection of HSA-SSH using the tag-switch assay. Different mixed disulfides of HSA were treated with Na2S to produce persulfides, which were then labeled by the tag-switch method. Duplicates of aliquots (5 μl) from the samples (30 μm) were added to a nitrocellulose membrane, and biotinylation was visualized on radiographic films using chemiluminescent reagents.
In addition, a selective method for persulfide detection has been recently developed, the tag-switch assay, which labels protein persulfides with biotin (19, 30). HSA-SSH formed through the reaction of several mixed disulfides with Na2S was analyzed for the level of biotin labeling by dot-blot assay to confirm that indeed, in all tested cases, HSA-SSH is formed (Fig. 7B). Control experiments performed with the mixed disulfides in the absence of Na2S gave negative results, with the only exception being HSA-TNB, which showed a weak signal (data not shown).
Sulfane sulfur reacts with triphenylphosphine and its derivatives to give the corresponding sulfide. This reaction has been recently used as a basis for a new method for sulfane sulfur quantification (48). We used a water-soluble phosphine, TDMSP, to prove persulfide formation in HSA. HSA-SSH prepared from HSA-TNB incubation with Na2S was treated with TDMSP and analyzed by negative ion mode ultra-high resolution ESI-TOF MS. The spectrum of the reaction mixture (Fig. 8, A and B, for two different m/z ranges) suggested formation of TDMSP sulfide (TDMSP=S). Although the strongest signals were assigned to unreacted TDMSP, detected as [TDMSP + Na]2− (Fig. 8C) and [TDMSP + 2Na]− (Fig. 8D), the presence of TDMSP=S with one sodium (Fig. 8E) or two sodium cations (Fig. 8F) was also observed. The peak intensity was ∼4.5% matching nicely the assumed TDMSP/persulfide ratio.
FIGURE 8.

Detection of HSA persulfide with TDMSP. A and B, mass spectrum of the reaction mixture obtained by mixing HSA-SSH (20 μm) with TDMSP (400 μm) during 30 min at 25 °C. Two different m/z ranges are shown from the same spectrum, m/z 200–500 (A) and m/z 500–800 (B). The peak assignment (C–F) reveals that the most intense peaks correspond to unreacted TDMSP detected in a cluster with one or two sodium cations (C and D). However, formation of TDMSP sulfide (TDMSP=S) could also be detected (E and F). Observed peaks (blue) match perfectly the predicted isotopic distribution (red). For TDMSP, the observed and predicted masses are 303.0111 and 303.0109, respectively, for the monosodium species, and the corresponding values for the disodium species are 629.0111 and 629.0116. For TDMSP=S, the observed and predicted masses are 318.9972 and 318.9970, respectively, for the monosodium or 660.9831 and 660.9837 for the disodium species.
HSA-SSH was also analyzed by MS before and after the equimolar addition of Na2S to HSA-TNB. An obvious shift toward lower m/z could be observed in the sample of HSA-TNB treated with H2S (Fig. 9, A and B) suggesting the loss of the TNB moiety. Spectral deconvolution confirmed this as shown in Fig. 9C. The appearance of a new peak corresponded well to the loss of the TNB moiety and the addition of sulfur, i.e. formation of HSA-SSH. In addition, a mass change consistent with the formation of HSA-SSO3− was observed, in accordance with previous observations on bovine serum albumin and glutathione peroxidase (18, 19). The mass of the protein was higher than expected, probably because of the presence of sodium ions. Nevertheless, the mass differences are in agreement with the changes expected.
FIGURE 9.

Mass spectrometry of HSA persulfide. A, mass spectrum of HSA-TNB (black) and HSA-TNB mixed with equimolar amounts of Na2S (red). B, zoomed overlay of the MS spectra showing difference. C, deconvoluted mass spectra of HSA-TNB (black) and HSA-TNB + Na2S (red). The spectrum shows the mass difference of 166 ± 1 suggesting the formation of protein persulfide. In addition, a peak assigned to HSA-SSO3H could be also observed. D, methodological approach to label and elute the peptide containing persulfidated cysteine. E, observed (black) and simulated (red) isotopic pattern of a peptide eluted following the protocol depicted in D. The mass is consistent with the doubly charged ALVLIAFAQYLQQCPFEDHVK peptide (where underline indicates Cys-34). F, MS/MS spectrum confirms the sequence.
Finally, we also proved that the actual site of HSA persulfidation is Cys-34 (Fig. 9, D–F). To do so, we designed an approach, adapted from Ref. 5, in which HSA-SSH, generated in the reaction between HSA-TNB and H2S, was mixed with biotin-maleimide. This alkylated free thiols and also reacted with persulfides forming a mixed disulfide. Protein was then trypsinized, and streptavidin beads were added into the peptide mixture. Only labeled peptides were expected to bind to the streptavidin beads. After careful washout, the peptide that originally contained persulfide was eluted with DTT, which reduced the mixed disulfide formed in the reaction with biotin-maleimide (Fig. 9D). The peptide was then analyzed by HPLC-MS. The positive-ion mode analysis revealed the presence of the Cys-34 peptide ALVLIAFAQYLQQCPFEDHVK as the doubly charged species (Fig. 9E). Subsequent collision-induced dissociation analysis further confirmed it (Fig. 9F).
Kinetics of Reaction of Hydrogen Sulfide with the Sulfenic Acid of HSA
The reaction of H2S toward sulfenic acid provides an alternative way to form persulfide. To kinetically evaluate this reaction in HSA, an experimental design involving competition between H2S and TNB for HSA-SOH was used. The kobs of TNB consumption increased and the amplitude decreased with Na2S concentration (Fig. 10, A and B). From the slope obtained (Equation 5), we could calculate a rate constant of (2.7 ± 0.8) × 102 m−1 s−1 for the reaction of H2S with HSA-SOH at pH 7.4 and a pH-independent rate constant of (4 ± 1) × 102 m−1 s−1 (assuming that all HSA-SOH was protonated).
This is to our knowledge the first kinetic report regarding the reactivity of H2S toward a sulfenic acid. In contrast to the trend observed for LMW disulfides (Figs. 3 and 5), the pH-independent rate constant of HS− reaction with HSA-SOH is higher than those of Cys, GSH, and TNB (1.1, 1.0, and 1.9 × 102 m−1 s−1, respectively (24), and similar to Hcy (4.8 × 102 m−1 s−1), in which pKa is 2 units higher. This difference can be explained by steric hindrance, because Cys-34 is partially buried in the protein surface, and HS− could have easier access than thiolates.
FIGURE 10.
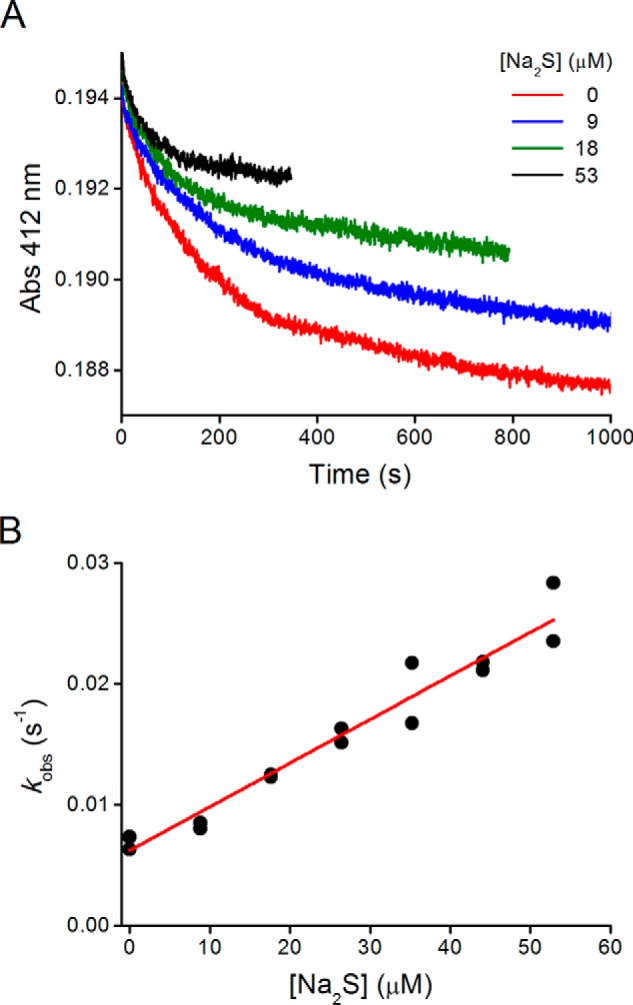
Competition kinetics for the reaction of HSA sulfenic acid with hydrogen sulfide. HSA-SOH (0.5 μm) was incubated with TNB (66 μm) and increasing concentrations of Na2S in phosphate buffer (0.1 m, pH 7.4, 0.1 mm DTPA, 25 °C). A, time courses of TNB consumption at 412 nm. B, correlation of kobs versus concentration of Na2S.
Reactivity of Hydrogen Sulfide toward Disulfides and Sulfenic Acids in Cellular Context
Oxidized thiols that could constitute molecular targets for H2S (i.e. disulfides and sulfenic acids) are scarce in the cytoplasmic environment. Accordingly, the majority of protein persulfides has been found to be located in the endoplasmic reticulum, an organelle rich in protein-oxidized thiols (19). The influence of oxidative conditions could increase the probability of inducing the formation of persulfides in cells. We addressed this possibility by treating cells in culture with H2O2, with or without the prior inhibition of the intracellular production of H2S. We used the tag-switch assay to detect persulfide levels in HUVEC and SH-SY5Y cells (Fig. 11). In both cell lines the treatment with H2O2 led to a significant increase in the total persulfide levels as evidenced by fluorescence microscopy. More importantly, we assessed the effect of the inhibition of the endogenous production of H2S. For HUVEC, propargylglycine was used, a relatively specific inhibitor of cystathionine γ-lyase. For SH-SY5Y cells, aminooxyacetic acid was used, which inhibits both cystathionine β-synthase and cystathionine γ-lyase, as shown recently (49). Inhibition of endogenous H2S production abolished H2O2-induced effects suggesting that the reaction of H2S with oxidized thiols could be an important source of endogenous persulfides (Fig. 11).
FIGURE 11.

Effect of H2O2 on protein persulfidation in SH-SY5Y and HUVEC cells. A, SH-SY5Y cells were preincubated or not with 2 mm aminooxyacetic acid (inhibitor of cystathionine β-synthase and cystathionine γ-lyase) and treated with 300 μm H2O2 for 30 min. Cells were fixed with methanol and labeled for persulfide detection using the tag-switch assay. The biotinylation of the proteins was visualized by fluorescence microscopy using DyLight 488 streptavidin. Nuclei were stained with DAPI. B, change in the fluorescence was semi-quantified using ImageJ, n = 100, from the experiment done in triplicate. Columns represent the mean ± S.E. *, p < 0.01 versus control by analysis of variance (simple paired Student's t test). C, HUVEC were preincubated or not with 2 mm propargylglycine (PG, inhibitor of cystathionine γ-lyase) for 1.5 h and treated with 500 μm H2O2 for 30 min. Cells were fixed, labeled, and visualized as in A. D, change in the fluorescence was semi-quantified using ImageJ, n = 20, from the experiment done in triplicate. Columns represent the mean ± S.E. *, p < 0.05 versus control, or **, p = 0.053 versus H2O2, by analysis of variance (simple paired Student's t test).
Kinetics of Reaction of the Persulfide of HSA toward Electrophiles
Both reactions of H2S with disulfides and with sulfenic acids yield persulfides. These products are likely to have enhanced nucleophilic reactivity with respect to thiolates. Several publications suggest that persulfides are better nucleophiles than thiols but so far there are no quantitative kinetic analyses reported (13, 17).
We first studied the reactivity of HSA-SSH toward peroxynitrite (ONOO−/ONOOH), the product of the reaction of nitric oxide and superoxide radicals. For these experiments, the persulfide concentration was judged based on the yield of TNB released from HSA-TNB after it reacted with Na2S and prior to desalting. An initial rate approach was used to analyze the kinetic traces obtained (50), because a pseudo-first order excess of HSA-SSH could not be reached. The initial rate increased linearly with the assumed concentration of HSA-SSH, leading to a rate constant of (1.2 ± 0.4) × 104 m−1 s−1 at 20 °C (Fig. 12). This value is likely an underestimation due to HSA-SSH instability and to the difficulty in exactly knowing its concentration. For comparison, HSA-SH reacted more slowly with peroxynitrite, (2.7 ± 0.3) × 103 m−1 s−1, under the same conditions, confirming the higher reactivity of the persulfide with respect to the thiol. It is worth mentioning that along with the increased kinetics for the reaction with peroxynitrite, we observed decreased nitrotyrosine levels (0.041 versus 0.071 mol of nitrotyrosine/mol of protein for HSA-SSH versus HSA-SH, respectively).
FIGURE 12.
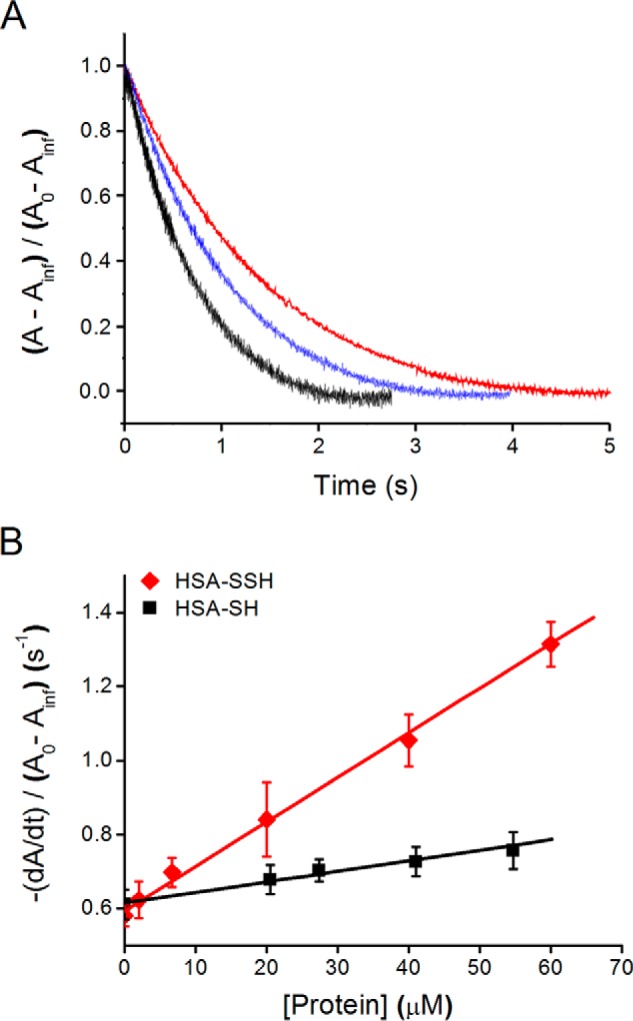
Kinetics of the reaction of HSA persulfide with peroxynitrite. A, kinetic traces of peroxynitrite decay (9 μm) in the presence of 7 μm (red), 20 μm (blue), and 40 μm (black) HSA-SSH. B, analysis of kinetic traces using an initial rate approach for HSA-SSH (red diamonds) and HSA-SH (black squares). Error bars represent the standard deviation (n = 5).
To improve the quantitative assessment of the nucleophilicity of HSA-SSH, we then evaluated its reactivity toward DTDPy. The use of the disulfide DTDPy as target presents advantages as follows. It has high intrinsic reactivity; the reaction can be followed through thiopyridone absorbance, and it can be used in pseudo-first order excess, eliminating the need for accurately knowing the concentration of the persulfide. When equimolar amounts of HSA-SOH and Na2S were incubated for a certain time and aliquots were mixed with excess DTDPy in a stopped-flow device, an initial fast increment in thiopyridone absorbance was detected (Fig. 13A). Controls using HSA-SH and HSA-SOH preparations in the absence of H2S also showed an absorbance increase but lacked the first fast phase. For the latter, the absorbance increase likely represented remnant thiols. The observed rate constants were linearly dependent on DTDPy concentration, and a second-order rate constant of (1.7 ± 0.1) × 104 m−1 s−1 for HSA-SSH was determined (pH 7.4, 25 °C), higher than those observed for HSA-SH or HSA-SOH preparations as follows: (7.6 ± 0.4) × 102 m−1 s−1 and (6.4 ± 0.1) × 102 m−1 s−1, respectively (Fig. 13B). In addition, an increase in the amplitude of the initial fast phase with time was observed, indicative of the time course of HSA-SSH formation (Fig. 13, C and D). The rate constant of the reaction between HSA-SOH and H2S was estimated from the fit of the amplitude versus incubation time plot using a Levenberg-Marquardt algorithm included in Gepasi, considering also the spontaneous decay of HSA-SOH (5.6 × 10−4 s−1 at 25 °C (23)) (Fig. 13D). A value of (4.7 ± 0.2) × 102 m−1 s−1 was obtained, consistent with that determined by competition with TNB.
FIGURE 13.

Kinetics of the reaction of HSA persulfide with DTDPy. A, kinetic traces of thiopyridone formation at 324 nm during the incubation of ∼3 μm HSA-SSH (red) with 32 μm DTDPy are compared with those produced by 18 μm HSA-SH (black) and 3 μm HSA-SOH (blue) preparations. B, dependence of kobs on the concentration of DTDPy for HSA-SSH (red diamonds), HSA-SH (black squares), and HSA-SOH (blue triangles) preparations. C, kinetic traces when HSA-SOH (6 μm, initial) was incubated with Na2S (6 μm, initial) during 50 s and mixed with 32 μm DTDPy every 3 min. D, red points, amplitudes obtained from fits of the first phase produced by incubation of DTDPy (32–207 μm) with the mixture of HSA-SOH and Na2S (final concentration: 3 μm each) at different incubation times. Black line, numerical determination of the second order rate constant between HSA-SOH and H2S. The spontaneous decay of HSA-SOH was also considered. Rate constant estimated: (4.7 ± 0.2) × 102 m−1 s−1.
The evaluation of nucleophilicity by means of DTDPy demonstrates 20-fold increased reactivity of the persulfide at pH 7.4 in comparison with thiol. The pKa of HSA-SSH is likely to be several units lower than the original HSA-SH (Table 3) resulting in an estimated pH-independent rate constant close to the value determined at pH 7.4, ∼2 × 104 m−1 s−1. In contrast, the pH-independent rate constant for HSA-SH can be calculated considering the pKa of 8.1 as ∼7 × 103 m−1 s−1. This value is lower than that determined for HSA-SSH by a factor of ∼3. The enhanced reactivity of a nucleophilic atom when it is adjacent to an atom containing one or more unshared pairs of electrons is a classic observation, referred to as the α effect (7, 8). For another reaction, that of different nucleophiles with the ester p-nitrophenyl acetate, the α effect is associated with increases in reactivity by factors of 10–100 (7, 51). By comparison, the extent of the effect in our case is more modest. The higher reactivity of HSA-SSH with respect to HSA-SH is consistent with the computational calculations performed for cysteine persulfide, which show a higher intrinsic nucleophilicity than the corresponding thiol (increased KS-HOMO energy) as well as decreased chemical hardness (Table 3 and Fig. 6). The latter could confer preferential reactivity toward soft electrophiles such as heavy metal cations (52), alkyl halides, and Michael electrophiles (18). Our study is, to our knowledge, the first evaluation of the magnitude of the α effect for a persulfide.
Biological Implications
Consistent with the limited intrinsic nucleophilicity of H2S, the rate constants of the reactions with LMW and HSA disulfides are relatively low. To extrapolate how these reactions could contribute to H2S decay, factors of rate constant × concentration together with competing pathways and compartmentalization aspects need to be taken into account.
In the intravascular space, a compartment where disulfides are abundant, factors of rate constant × concentration can be evaluated considering the total concentration levels of LMW and HSA disulfides (∼ 250 μm (23)), and the rate constants now known (Tables 1 and 2). Thus, a global half-life of 26 min can be estimated for this process in plasma. However, from previous work (53, 54) it is known that H2S can traverse membranes easily and that methemoglobin in red blood cells can participate in its clearance. Recently, consumption rates for H2S by methemoglobin consistent with a half-life of 4 min in blood were reported (55). This value is within 1 order of magnitude of that ascribed to disulfides. Thus, variations in the levels of methemoglobin or disulfides could produce changes in the relative contributions of disulfides and red blood cells to H2S decay rate.
Targets in the extravascular milieu can also be involved in H2S clearance. The high permeability of membranes allows mitochondria to become a sink for H2S, limiting its half-life in the cell. Formation of persulfides by direct reaction of H2S with disulfides and sulfenic acids is probably under kinetic competition with mitochondrial consumption. In this regard, an interesting scenario could be set in the nervous system, where H2S scavenging by mitochondria is inefficient due to the lack of sulfide:quinone oxidoreductase activity (56), the enzyme responsible for the first step of the mitochondrial catabolism of H2S, and the effects of endogenous H2S on non-mitochondrial persulfide generation are probably maximized.
In the cytoplasmic environment, the reaction of H2S with LMW and typical protein disulfides or sulfenic acids is kinetically challenged due to the low concentration of oxidized thiols and the relatively low rate constants, in the context of high concentrations of reduced thiols that might compete with H2S. Thus, situations that lead to increased steady state levels of oxidized thiols increase persulfide formation. Based on our experiments with cells in cultures treated with H2O2, the reaction of H2S with oxidized thiols is a plausible pathway for persulfide production in cytoplasmic and reticulum environments in response to oxidative signals. In addition to the direct reaction of H2S with oxidized thiols, alternative pathways for persulfide formation in vivo can involve the consumption of cystine by cystathionine β-synthase or cystathionine γ-lyase (13). Furthermore, proteins that participate in H2S oxidation pathways can generate intermediate persulfides that can subsequently be transferred to other targets (57).
It is important to bear in mind that the rate constants of H2S reactions can be accelerated in the case of particular protein contexts. Proof of concept for the potential acceleration of the reaction of H2S with a disulfide by the protein environment is provided by sulfide:quinone oxidoreductase. This flavoprotein catalyzes the oxidation of H2S to sulfane sulfur at the expense of ubiquinone. The process starts with the reaction of H2S with a critical protein disulfide, and values of kcat/Km, sulfide of 107 m−1 s−1 (58, 59) demonstrate an extraordinary acceleration with respect to free cystine (0.6 m−1 s−1). Likewise, wide variations in rate constants are expected for the reactions of H2S with different protein sulfenic acids. To achieve specificity, one aspect that H2S could invoke, as suggested by the case of HSA-SOH, is a steric advantage for reaction with constrained disulfides and sulfenic acids due to its small size in comparison with other cellular nucleophiles. In fact, from the point of view of biological function, the low rate constants of the uncatalyzed processes constitute actually an asset, opening the way to kinetic control.
Persulfides are promising candidates to constitute intermediate species in the modulation of cellular functions by H2S. The levels of persulfides are so far hard to evaluate in biological samples due to the instability of these compounds and to the lack of rapid and selective analytical methods, which are still under development. Persulfides have been proposed to protect the original thiol from irreversible oxidative modifications (60). They have been reported to participate in the synthesis of sulfur-containing molecules (11) and to be involved in enzymatic catalysis and regulation (6). The 20-fold increased reactivity of the HSA persulfide relative to the HSA thiol at pH 7.4 underscores the improved nucleophilicity of these derivatives with respect to the parent thiol and to H2S. As functions for persulfides in biological systems continue to be unraveled, this increased nucleophilicity is likely to play a key role in explaining the underlying molecular mechanisms.
Conclusions
Our study reveals several aspects relative to the reactivity of H2S, summarized as follows. 1) Hydrogen sulfide reacts with LMW and mixed protein disulfides to produce persulfides. 2) The reaction occurs through a concerted mechanism, similar to the thiol-disulfide interchange reaction. 3) The intrinsic reactivity observed for HS− toward disulfides is lower than that reported for thiolates, probably due to the absence of the inductive effect of the methylene adjacent to the sulfur atom, increased chemical hardness, or solvation. 4) Hydrogen sulfide is able to react with the sulfenic acid in HSA, proving another pathway for persulfide formation. 5) Experiments with cells in culture indicate that persulfide formation increases upon exposure to H2O2, consistent with the proposed processes. 6) Kinetic determinations indicate that the persulfide has increased nucleophilic reactivity than the parent thiol, as expected from the α effect. Overall, our study sets the stage for understanding the reactivity of H2S toward oxidized thiol derivatives and the possible contribution of persulfides to the biological effects of this novel endogenous modulator.
Author Contributions
E. C. designed, performed, and analyzed experiments and wrote the manuscript. J. B. and E. L. C. designed, performed and analyzed the computational experiments and wrote the corresponding parts of the manuscript. G. F. S. helped design and analyze experiments and edited the manuscript. M. R. F. designed, performed, and analyzed mass spectrometry, tag-switch, amperometric kinetics, peroxynitrite and cell experiments with the assistance of M. L., and co-wrote the manuscript. B. A. conceived, designed, and analyzed the experiments and wrote the manuscript. All authors approved the final version.
Acknowledgments
We thank Matías N. Möller, Laura Antmann, Lucía Turell, and Martina Steglich (Universidad de la República, Uruguay) and Jan Miljkovic (University of Erlangen-Nuremberg, Germany) for helpful discussions and technical assistance. We are grateful to Professor Ivana Ivanovic-Burmazovic (Friedrich-Alexander University) for the use of the equipment.
This work was supported in part by grants from Comisión Sectorial de Investigación Científica (Universidad de la República) and L'Oréal-UNESCO, Uruguay (to B. A.), and Emerging Field Initiative intramural grant from Friedrich-Alexander University of Erlangen-Nuremberg (MRIC) (to M. R. F.). The authors declare that they have no conflicts of interest with the contents of this article.
This article was selected as a Paper of the Week.
The mixture of H2S and HS− present in aqueous solution according to the working pH is referred as “H2S,” unless otherwise specified. The IUPAC-recommended names are sulfane and hydrogen sulfide for H2S and sulfanide and hydrogen(sulfide)(1−) for hydrosulfide anion, HS−.
The term “persulfides” is used in this text for the mixture of RSSH (hydrodisulfide derivative) and RSS− (disulfanidyl derivative) in solution according to the working pH, unless otherwise specified. The IUPAC-recommended names for RSSH are disulfanyl and dithiohydroperoxide.
- HSA
- human serum albumin
- LMW
- low molecular weight
- DTNB
- 5,5′-dithiobis-(2-nitrobenzoic acid)
- DTPA
- diethylenetriaminepentaacetic acid
- TNB
- 5-thio-2-nitrobenzoic acid
- Cys-Cys
- cystine
- CysOMe-CysOMe
- cystine dimethyl ester
- HED
- hydroxyethyldisulfide
- Hcy-Hcy
- homocystine
- Hcy
- homocysteine
- DTDPy
- 4,4′-dithiodipyridine
- TDMSP
- tris(2,4-dimethyl-5-sulfophenyl)phosphine
- ESI
- electrospray ionization
- HUVEC
- human umbilical vein endothelial cell
- DFT
- density functional theory.
References
- 1.Kabil O., and Banerjee R. (2010) Redox biochemistry of hydrogen sulfide. J. Biol. Chem. 285, 21903–21907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cotton F., and Wilkinson G. (1988) Advanced Inorganic Chemistry, John Wiley & Sons, Inc., New York [Google Scholar]
- 3.Paulsen C. E., and Carroll K. S. (2013) Cysteine-mediated redox signaling: chemistry, biology, and tools for discovery. Chem. Rev. 113, 4633–4679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mustafa A. K., Gadalla M. M., Sen N., Kim S., Mu W., Gazi S. K., Barrow R. K., Yang G., Wang R., and Snyder S. H. (2009) H2S signals through protein S-sulfhydration. Sci. Signal. 2, ra72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sen N., Paul B. D., Gadalla M. M., Mustafa A. K., Sen T., Xu R., Kim S., and Snyder S. H. (2012) Hydrogen sulfide-linked sulfhydration of NF-κB mediates its antiapoptotic actions. Mol. Cell 45, 13–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Toohey J. I. (1989) Sulphane sulphur in biological systems: a possible regulatory role. Biochem. J. 264, 625–632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jencks W. P., and Carriuolo J. (1960) Reactivity of nucleophilic reagents toward esters. J. Am. Chem. Soc. 82, 1778–1786 [Google Scholar]
- 8.Edwards J. O., and Pearson R. G. (1962) The factors determining nucleophilic reactivities. J. Am. Chem. Soc. 84, 16–24 [Google Scholar]
- 9.Toohey J. I. (2011) Sulfur signaling: is the agent sulfide or sulfane? Anal. Biochem. 413, 1–7 [DOI] [PubMed] [Google Scholar]
- 10.Olson K. R. (2009) Is hydrogen sulfide a circulating “gasotransmitter” in vertebrate blood? Biochim. Biophys. Acta 1787, 856–863 [DOI] [PubMed] [Google Scholar]
- 11.Wright C. M., Christman G. D., Snellinger A. M., Johnston M. V., and Mueller E. G. (2006) Direct evidence for enzyme persulfide and disulfide intermediates during 4-thiouridine biosynthesis. Chem. Commun. 29, 3104–3106 [DOI] [PubMed] [Google Scholar]
- 12.Mueller E. G. (2006) Trafficking in persulfides: delivering sulfur in biosynthetic pathways. Nat. Chem. Biol. 2, 185–194 [DOI] [PubMed] [Google Scholar]
- 13.Ida T., Sawa T., Ihara H., Tsuchiya Y., Watanabe Y., Kumagai Y., Suematsu M., Motohashi H., Fujii S., Matsunaga T., Yamamoto M., Ono K., Devarie-Baez N. O., Xian M., Fukuto J. M., and Akaike T. (2014) Reactive cysteine persulfides and S-polythiolation regulate oxidative stress and redox signaling. Proc. Natl. Acad. Sci. U.S.A. 111, 7606–7611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hildebrandt T. M., and Grieshaber M. K. (2008) Three enzymatic activities catalyze the oxidation of sulfide to thiosulfate in mammalian and invertebrate mitochondria. FEBS J. 275, 3352–3361 [DOI] [PubMed] [Google Scholar]
- 15.Kabil O., and Banerjee R. (2012) Characterization of patient mutations in human persulfide dioxygenase (ETHE1) involved in H2S catabolism. J. Biol. Chem. 287, 44561–44567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ubuka T. (2002) Assay methods and biological roles of labile sulfur in animal tissues. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 781, 227–249 [DOI] [PubMed] [Google Scholar]
- 17.Francoleon N. E., Carrington S. J., and Fukuto J. M. (2011) The reaction of H(2)S with oxidized thiols: generation of persulfides and implications to H(2)S biology. Arch. Biochem. Biophys. 516, 146–153 [DOI] [PubMed] [Google Scholar]
- 18.Pan J., and Carroll K. S. (2013) Persulfide reactivity in the detection of protein S-sulfhydration. ACS Chem. Biol. 8, 1110–1116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang D., Macinkovic I., Devarie-Baez N. O., Pan J., Park C.-M., Carroll K. S., Filipovic M. R., and Xian M. (2014) Detection of protein S-sulfhydration by a tag-switch technique. Angew. Chem. Int. Ed. Engl. 53, 575–581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nagahara N., Nirasawa T., Yoshii T., and Niimura Y. (2012) Is novel signal transducer sulfur oxide involved in the redox cycle of persulfide at the catalytic site cysteine in a stable reaction intermediate of mercaptopyruvate sulfurtransferase? Antioxid. Redox Signal. 16, 747–753 [DOI] [PubMed] [Google Scholar]
- 21.Chatterji T., Keerthi K., and Gates K. S. (2005) Generation of reactive oxygen species by a persulfide (BnSSH). Bioorg. Med. Chem. Lett. 15, 3921–3924 [DOI] [PubMed] [Google Scholar]
- 22.Bailey T. S., Zakharov L. N., and Pluth M. D. (2014) Understanding hydrogen sulfide storage: probing conditions for sulfide release from hydrodisulfides. J. Am. Chem. Soc. 136, 10573–10576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Turell L., Radi R., and Alvarez B. (2013) The thiol pool in human plasma: the central contribution of albumin to redox processes. Free Radic. Biol. Med. 65, 244–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Turell L., Botti H., Carballal S., Ferrer-Sueta G., Souza J. M., Durán R., Freeman B. A., Radi R., and Alvarez B. (2008) Reactivity of sulfenic acid in human serum albumin. Biochemistry. 47, 358–367 [DOI] [PubMed] [Google Scholar]
- 25.Torres M. J., Turell L., Botti H., Antmann L., Carballal S., Ferrer-Sueta G., Radi R., and Alvarez B. (2012) Modulation of the reactivity of the thiol of human serum albumin and its sulfenic derivative by fatty acids. Arch. Biochem. Biophys. 521, 102–110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Alvarez B., Carballal S., Turell L., and Radi R. (2010) Formation and reactions of sulfenic acid in human serum albumin. Methods Enzymol. 473, 117–136 [DOI] [PubMed] [Google Scholar]
- 27.Iversen R., Andersen P. A., Jensen K. S., Winther J. R., and Sigurskjold B. W. (2010) Thiol-disulfide exchange between glutaredoxin and glutathione. Biochemistry. 49, 810–820 [DOI] [PubMed] [Google Scholar]
- 28.Wood J. L. (1987) Sulfane sulfur. Methods Enzymol. 143, 25–29 [DOI] [PubMed] [Google Scholar]
- 29.Carballal S., Trujillo M., Cuevasanta E., Bartesaghi S., Möller M. N., Folkes L. K., García-Bereguiaín M. A., Gutiérrez-Merino C., Wardman P., Denicola A., Radi R., and Alvarez B. (2011) Reactivity of hydrogen sulfide with peroxynitrite and other oxidants of biological interest. Free Radic. Biol. Med. 50, 196–205 [DOI] [PubMed] [Google Scholar]
- 30.Park C.-M., Macinkovic I., Filipovic M. R., and Xian M. (2015) Use of the “tag-switch” method for the detection of protein S-sulfhydration. Methods Enzymol. 555, 39–56 [DOI] [PubMed] [Google Scholar]
- 31.Saha A., Goldstein S., Cabelli D., and Czapski G. (1998) Determination of optimal conditions for synthesis of peroxynitrite by mixing acidified hydrogen peroxide with nitrite. Free Radic. Biol. Med. 24, 653–659 [DOI] [PubMed] [Google Scholar]
- 32.Chai J.-D., and Head-Gordon M. (2008) Long-range corrected hybrid density functionals with damped atom-atom dispersion corrections. Phys. Chem. Chem. Phys. 10, 6615–6620 [DOI] [PubMed] [Google Scholar]
- 33.Tomasi J., Mennucci B., and Cancès E. (1999) The IEF version of the PCM solvation method: an overview of a new method addressed to study molecular solutes at the QM ab initio level. J. Mol. Struct. Theochem. 464, 211–226 [Google Scholar]
- 34.Ditchfield R., Hehre W. J., and Pople J. A. (1971) Self-consistent molecular-orbital methods. IX. An extended Gaussian-type basis for molecular-orbital studies of organic molecules. J. Chem. Phys. 54, 724–728 [Google Scholar]
- 35.Frisch M. J., Trucks G. W., Schlegel H. B., Scuseria G., Robb M. A., Cheeseman J. R., Scalmani G., Barone V., Mennucci B., Petersson G. A., Nakatsuji H., Caricato M., Li X., Hratchian H. P., Izmaylov A. F., et al. (2013) Gaussian 09, Revision D.01 ed., Gaussian Inc., Wallingford, CT [Google Scholar]
- 36.Bondi A. (1964) van der Waals volumes and radii. J. Phys. Chem. 68, 441–451 [Google Scholar]
- 37.Fukui K. (1981) The path of chemical reactions–the IRC approach. Acc. Chem. Res. 14, 363–368 [Google Scholar]
- 38.Hratchian H. P., and Schlegel H. B. (2005) Using Hessian updating to increase the efficiency of a Hessian based predictor-corrector reaction path following method. J. Chem. Theory Comput. 1, 61–69 [DOI] [PubMed] [Google Scholar]
- 39.Geerlings P., Fias S., Boisdenghien Z., and De Proft F. (2014) Conceptual DFT: chemistry from the linear response function. Chem. Soc. Rev. 43, 4989–5008 [DOI] [PubMed] [Google Scholar]
- 40.Yu H.-Z., Yang Y.-M., Zhang L., Dang Z.-M., and Hu G.-H. (2014) Quantum-chemical predictions of pKa's of thiols in DMSO. J. Phys. Chem. A 118, 606–622 [DOI] [PubMed] [Google Scholar]
- 41.Mendes P. (1993) GEPASI: a software package for modelling the dynamics, steady states and control of biochemical and other systems. Comput. Appl. Biosci. 9, 563–571 [DOI] [PubMed] [Google Scholar]
- 42.Liu D. K., and Chang S. G. (1987) Kinetic study of the reaction between cystine and sulfide in alkaline solutions. Can. J. Chem. 65, 770–774 [Google Scholar]
- 43.Singh R., and Whitesides G. M. (1993) in Sulphur-Containing Functional Groups (Patai S., and Rappoport Z., eds) pp. 633–658, John Wiley & Sons, Inc. New York [Google Scholar]
- 44.Vasas A., Dóka É., Fábián I., and Nagy P. (2015) Kinetic and thermodynamic studies on the disulfide-bond reducing potential of hydrogen sulfide. Nitric Oxide 46, 93–101 [DOI] [PubMed] [Google Scholar]
- 45.Szajewski R. P., and Whitesides G. M. (1980) Rate constants and equilibrium constants for thiol-disulfide interchange reactions involving oxidized glutathione. J. Am. Chem. Soc. 102, 2011–2026 [Google Scholar]
- 46.Bunnett J. F. (1963) Nucleophilic reactivity. Annu. Rev. Phys. Chem. 14, 271–290 [Google Scholar]
- 47.Wilson J. M., Bayer R. J., and Hupe D. J. (1977) Structure-reactivity correlations for the thiol-disulfide interchange reaction. J. Am. Chem. Soc. 99, 7922–7926 [Google Scholar]
- 48.Liu C., Zhang F., Munske G., Zhang H., and Xian M. (2014) Isotope dilution mass spectrometry for the quantification of sulfane sulfurs. Free Radic. Biol. Med. 76, 200–207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Asimakopoulou A., Panopoulos P., Chasapis C. T., Coletta C., Zhou Z., Cirino G., Giannis A., Szabo C., Spyroulias G. A., and Papapetropoulos A. (2013) Selectivity of commonly used pharmacological inhibitors for cystathionine β synthase (CBS) and cystathionine γ lyase (CSE). Br. J. Pharmacol. 169, 922–932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Alvarez B., Ferrer-Sueta G., Freeman B. A., and Radi R. (1999) Kinetics of peroxynitrite reaction with amino acids and human serum albumin. J. Biol. Chem. 274, 842–848 [DOI] [PubMed] [Google Scholar]
- 51.Frey P. A., and Hegeman A. D. (2007) Enzymatic Reaction Mechanisms, pp. 16–21, Oxford University Press, New York [Google Scholar]
- 52.Abiko Y., Yoshida E., Ishii I., Fukuto J. M., Akaike T., and Kumagai Y. (2015) Involvement of reactive persulfides in biological bismethylmercury sulfide formation. Chem. Res. Toxicol. 28, 1301–1306 [DOI] [PubMed] [Google Scholar]
- 53.Cuevasanta E., Denicola A., Alvarez B., and Möller M. N. (2012) Solubility and permeation of hydrogen sulfide in lipid membranes. PLoS ONE. 7, e34562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mathai J. C., Missner A., Kügler P., Saparov S. M., Zeidel M. L., Lee J. K., and Pohl P. (2009) No facilitator required for membrane transport of hydrogen sulfide. Proc. Natl. Acad. Sci. U.S.A. 106, 16633–16638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Vitvitsky V., Yadav P. K., Kurthen A., and Banerjee R. (2015) Sulfide oxidation by a noncanonical pathway in red blood cells generates thiosulfate and polysulfides. J. Biol. Chem. 290, 8310–8320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lagoutte E., Mimoun S., Andriamihaja M., Chaumontet C., Blachier F., and Bouillaud F. (2010) Oxidation of hydrogen sulfide remains a priority in mammalian cells and causes reverse electron transfer in colonocytes. Biochim. Biophys. Acta 1797, 1500–1511 [DOI] [PubMed] [Google Scholar]
- 57.Mishanina T. V., Libiad M., and Banerjee R. (2015) Biogenesis of reactive sulfur species for signaling by hydrogen sulfide oxidation pathways. Nat. Chem. Biol. 11, 457–464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jackson M. R., Melideo S. L., and Jorns M. S. (2012) Human sulfide:quinone oxidoreductase catalyzes the first step in hydrogen sulfide metabolism and produces a sulfane sulfur metabolite. Biochemistry 51, 6804–6815 [DOI] [PubMed] [Google Scholar]
- 59.Libiad M., Yadav P. K., Vitvitsky V., Martinov M., and Banerjee R. (2014) Organization of the human mitochondrial hydrogen sulfide oxidation pathway. J. Biol. Chem. 289, 30901–30910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ono K., Akaike T., Sawa T., Kumagai Y., Wink D. A., Tantillo D. J., Hobbs A. J., Nagy P., Xian M., Lin J., and Fukuto J. M. (2014) Redox chemistry and chemical biology of H2S, hydropersulfides, and derived species: implications of their possible biological activity and utility. Free Radic. Biol. Med. 77, 82–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Creighton T. E. (1975) Interactions between cysteine residues as probes of protein conformation: the disulphide bond between Cys-14 and Cys-38 of the pancreatic trypsin inhibitor. J. Mol. Biol. 96, 767–776 [DOI] [PubMed] [Google Scholar]
- 62.Whitesides G. M., Lilburn J. E., and Szajewski R. P. (1977) Rates of thiol-disulfide interchange reactions between mono- and dithiols and Ellman's reagent. J. Org. Chem. 42, 332–338 [Google Scholar]
- 63.Keire D. A., Strauss E., Guo W., Noszal B., and Rabenstein D. L. (1992) Kinetics and equilibria of thiol/disulfide interchange reactions of selected biological thiols and related molecules with oxidized glutathione. J. Org. Chem. 57, 123–127 [Google Scholar]
- 64.Portillo-Ledesma S., Sardi F., Manta B., Tourn M. V., Clippe A., Knoops B., Alvarez B., Coitiño E. L., and Ferrer-Sueta G. (2014) Deconstructing the catalytic efficiency of peroxiredoxin-5 peroxidatic cysteine. Biochemistry 53, 6113–6125 [DOI] [PubMed] [Google Scholar]