

Abstract
Infection by human papillomavirus (HPV) is extremely common and associated with the development of benign warts or malignant lesions of the skin and mucosa. Infection by a high-risk (oncogenic) anogenital HPV type, most often through sexual contacts, is the starting point of virtually all cases of cervical cancers and the majority of anal cancers. The same viral types are also increasingly being linked with a subset of head-and-neck and non-melanoma skin cancers. Although prophylactic vaccines are now available to protect against the four types most commonly found in cervical and anal cancers (HPV16 and HPV18) and anogenital warts (HPV6 and HPV11), these neither protect against all genital HPVs nor are of therapeutic utility for already infected patients. Thus, the need for antiviral agents to treat HPV-associated diseases remains great, but none currently exist. This article reviews the recent progress made towards the development of antiviral agents to treat HPV infections, from target identification and validation to the discovery of lead compounds with therapeutic potential. Emphasis has been placed on novel low-molecular-weight compounds that antagonize HPV proteins or, alternatively, inhibit cellular proteins which have been usurped by papillomaviruses and are mediating their pathogenic effects.
Introduction
Human papillomaviruses (HPVs) are small DNA tumour viruses that infect keratinocytes of the differentiating epithelium of the skin and mucosa. Although most HPV infections remain asymptomatic, some result in the appearance of benign warts or more serious lesions that can progress to invasive cancer. It is now well established that infection by certain HPV types is a necessary event for the development of cervical cancer [1] and many anal cancers [2,3]. These viruses are also increasingly being associated with a subset of head-and-neck [4] and non-melanoma skin cancers [5]. More than 150 HPV types have been identified to date [6,7], each displaying a specific tropism for the epithelium of particular anatomical locations. For example, whereas HPV1 is found in plantar warts [8], HPV16 is found predominantly in the anogenital tract [9]. Approximately 30 HPV types infect the anogenital region. The oncogenic or high-risk types, such as HPV16, HPV18 and HPV31, are found in cancerous lesions and their precursors [10]. The low-risk types, including HPV6 and HPV11, cause benign genital warts (condylomas) or laryngeal papillomatosis [11], a rare but debilitating disease acquired by mother-to-child transmission of the virus during birth. Anogenital HPV infections are very common [12]. Of the 50 million Pap tests performed annually in the USA, about 3.5 million require some form of follow up and more than 250,000 reveal the presence of a high-grade cancer precursor lesion. Annually, approximately 4,000 US women die of cervical cancer and 10,000 are diagnosed with the disease [13]. Benign genital warts, although not life-threatening, are also very common, being clinically apparent in 1% of the sexually active population. A study by the US Centers for Disease Control and Prevention indicated that HPV is second to HIV in its contribution to the total medical costs associated with treating sexually transmitted diseases [14]. The burden of HPV-associated diseases is far worse in developing countries, where more than 200,000 women die of cervical cancer every year because of a lack of adequate screening programmes [15].
Treatment and prevention of HPV infection
Current therapies for HPV-associated lesions are mostly ablative and cytodestructive in nature (cryotherapy, surgical excision, topical application of cytotoxic agents, etc; [16,17]), although genital warts can be treated by topical application of the immunomodulator imiquimod [16] or the recently approved Polyphenon®E ointment made of catechins extracted from green tea [18]. As a prophylactic measure, a quadrivalent vaccine based on recombinant virus-like particles from HPV types 6, 11, 16 and 18 (Gardasil, Merck, Whitehouse Station, NJ, USA) was recently approved in several countries including the USA [19–21]. To date, this vaccine has been shown to be safe and highly effective in preventing the development of pre-cancerous cervical lesions associated with HPV16 and HPV18, and genital warts caused by HPV6 and HPV11 [21]. An analogous bivalent vaccine directed against HPV16 and HPV18 (Cervarix, GlaxoSmithKline, Brentford, Middlesex, UK) was expected to be submitted for US Food and Drug Administration approval in 2007 [22]. However, because immunological cross-reactivity between HPV types is limited, these vaccines will not protect against all anogenital HPV types nor eliminate the need for Pap screening. Nevertheless, because they are effective against the most prevalent low- and high-risk types, they are expected to significantly reduce the disease burden associated with anogenital HPV infections.
Surprisingly, there is no antiviral drug currently available for the treatment of HPV-associated diseases despite the high incidence of these viruses in the sexually active population (>50% of women [23]) and their well accepted aetiological role in the development of anogenital dysplasia and cancer. This article reviews the recent progress made towards the development of antiviral agents to treat HPV infections, from target identification and validation to the discovery of lead compounds with therapeutic potential. The emphasis has been placed on reports describing the identification of novel, low-molecular-weight (small molecule) compounds that bind and inhibit HPV proteins or, alternatively, that antagonize cellular proteins usurped by papillomaviruses and essential for their pathogenesis.
HPV proteins as candidate antiviral targets
Functions of E1 and E2 in replication of the viral episome
The life cycle of HPV is coupled to the cellular differentiation programme that keratinocytes undergo in the epithelium (Figure 1A). These viruses infect the basal cell layer where they establish their double-stranded DNA genome as a circular extrachromosomal element (episome) in the nucleus of infected cells (reviewed in [24]). Maintenance of the viral genome in 50–100 copies in basal cells is essential to the viral life cycle and its associated pathologies, thus making this process an attractive target for antiviral intervention. The HPV genome, slightly less than 8 kb in length, encodes eight well characterized proteins (Figure 1B). The genome also contains a regulatory locus termed the long control region (LCR) that encompasses the origin of viral DNA replication as well as the promoter and enhancer sequences necessary for transcription of the early and late genes [24]. Efficient maintenance of the papillomavirus episome in infected cells depends on its proper replication during S phase and segregation to daughter cells at mitosis. Replication of the genome is accomplished by the viral E1 and E2 proteins in concert with the cellular DNA replication machinery. E1 is an initiator protein and the only viral protein with enzymatic activities (ATPase and 3′-5′ helicase activities; Figure 2) [25]. E2 is a sequence-specific DNA-binding protein that can simultaneously bind to sites in the origin of replication (Figures 2, 3A and 3B) and to the E1 helicase. Through these interactions, E2 promotes the recruitment of E1 specifically at the origin and facilitates its assembly into replication-competent double hexamers needed for bidirectional DNA unwinding (Figure 3B) [26–29]. Assembly of E1 into double hexamers probably proceeds through double dimer and double trimer intermediates [30,31]. During replication, E1 unwinds the DNA ahead of the replication forks and interacts with essential replication factors including the host polymerase α-primase, the single-stranded DNA-binding protein replication protein A and topoisomerase I [32–37]. In addition to being necessary for replication, E2 also has essential roles in regulating transcription of the viral genes and segregation of the episome at mitosis [38,39]. Reverse genetic experiments have shown that E1 and E2 are both essential for maintenance of the viral episome in primary human keratinocyte cultures [40] and for pathogenesis (that is, papilloma induction) in the cottontail rabbit papillomavirus (CRPV) infection model [41]. As a consequence of their essential functions, E1 and E2 are considered attractive targets for the development of antiviral agents. Presumably, E1 and E2 are also required for the amplification of the viral genome to >1,000 copies, which occurs in infected cells that reach the upper layers of the epithelium (Figure 1A). Efficient amplification of the genome in organotypic raft cultures is also dependent on the action of E4 and E5 [24]. Although their exact molecular functions in this process remain unclear, it is believed that E4 and E5 promote amplification by modulating the activities of cell cycle regulatory proteins and growth factor receptors, respectively [24]. As for their roles in pathogenesis, reverse genetic experiments in the CRPV infection model have demonstrated that both are dispensable for papilloma induction [42,43], casting some doubts on the validity of inhibiting E4- and E5-mediated amplification of the viral episome as an antiviral strategy. Nevertheless, viral genome amplification is essential for expression of the late genes [44] encoding the capsid proteins L1 and L2, and thus for assembly of infectious virions (Figure 1A). It is therefore conceivable that amplification could be targeted for preventing transmission of the virus.
Figure 1. HPV life cycle and structure of the viral genome.

(A) Schematic representation of the human papillomavirus (HPV) life cycle within a differentiating epithelium. The different epithelial strata are indicated on the left. The diagram at the centre represents a prototypical infected keratinocyte undergoing terminal differentiation and harbouring viral episomes within its nucleus. Specific viral life cycle events occurring in each stratum are summarized on the right. (B) The genome of HPV16 is diagrammed in linear form. The coding regions of the early and late viral proteins are indicated by open boxes. Proteins that have been validated as potential antiviral targets are dark grey. The L1 protein, which has been shown to be a valid target for a microbicide, is light grey. The long-control region (LCR), which contains the transcriptional enhancer and promoter regions as well as the origin of DNA replication, is indicated by a two-way arrow.
Figure 2. HPV early proteins as antiviral targets.

The HPV early proteins E1, E2, E6 and E7 that have been validated as potential antiviral targets are represented by boxes. The locations of specific functional domains discussed in this review are indicated and their activities listed underneath. Because the length of each protein can vary slightly between viral types, an approximate length in amino acids (aa) is indicated at the right of each protein. DBD, DNA-binding domain; H, hinge region; HDAC, histone deacetylase; NES, nuclear export sequence; NLS, nuclear localization sequence; TAD, transactivation domain; Zn, zinc finger.
Figure 3. Initiation of HPV DNA replication and inhibitors thereof.

(A) Schematic representation of the minimal origin of viral DNA replication. Binding sites (BS) for E1 and E2 are indicated by white and black boxes, respectively. The location of an essential AT-rich region (AT) is indicated by a grey box. The nucleotide sequence of part of the HPV18 origin is given underneath. The left portion of this sequence contains a representative E2-binding site (ACCgN4cGGT) and is the target of E2 polyamides. The right portion of the sequence contains the four E1 binding sites, shown by arrows, which are the target of E1 polyamides. (B) Schematic representation of the initiation of papillomavirus DNA replication. DNA replication is initiated by the recruitment of E1, by E2, to the viral origin. This step is blocked by the indandione class of inhibitors which bind to the E2 transactivation domain and prevent its interaction with E1. Assembly of a replication-competent double hexamer proceeds through the initial assembly of two E1 dimers on the two pairs of inverted E1 binding sites present in the origin. Conversion of this tetrameric E1 intermediate to an active double hexameric helicase requires additional E1 molecules as well as ATP to promote their oligomerization. E1 also interacts with host cell replication factors such as the polymerase α-primase (Pol α) to promote viral DNA replication. Biphenylsulphonacetic acid and other E1 ATPase inhibitors abrogate the helicase activity of E1 responsible for unwinding the origin and the DNA ahead of the two replication forks. All inhibitors are indicated in boxes.
Role of E2 in transcriptional regulation and segregation of the viral episome
In addition to its role in viral DNA replication, E2 also serves as a transcription factor to regulate expression of the viral genes and as a segregation factor to ensure partitioning of the episome at mitosis [38,39]. E2 comprises two functional domains, an N-terminal transactivation domain (TAD) and a C-terminal DNA-binding/dimerization domain, separated by a hinge region. Both the TAD and DNA-binding domain (DBD) are necessary for E2 to modulate viral DNA replication, transcription and segregation [38,39,45,46]. As a transcription factor E2 can be either an activator or a repressor, depending on the promoter context, but is primarily a repressor of viral transcription initiated in the LCR [47–51]. As a segregation factor, E2 tethers the viral episome to mitotic chromatin [39,52–54], and/or the mitotic spindle [55,56]. The TAD of E2 binds to several cellular transcription factors including the long isoform of the bromodomain-containing protein 4, (Brd4) [57–59]. For some papillomavirus types, interaction with Brd4 was shown to be required for E2’s transcriptional and tethering activities [45,57,60–64]. It is currently unclear whether all papillomavirus types rely on Brd4 for segregation of their genomes or whether some are capable of using alternative mechanisms [65–67]. For HPV31, mutations in the E2 TAD that abrogate its binding to Brd4 were shown to have little effect on replication and maintenance of the viral episome in keratinocytes or on its amplification upon cellular differentiation [57,58,68]. By contrast, very similar mutations in CRPV E2 completely abolished the ability of the mutant genome to induce papillomas in the rabbit infection model [69]. This latter finding suggests that the interaction of E2 with Brd4 is essential for pathogenesis and thus warrants the search for small molecule inhibitors of the E2–Brd4 interaction as potential antiviral agents.
Finally, it is important to realize that inhibiting maintenance of the viral episome in infected cells might be a valuable antiviral strategy for treating lesions such as warts, in which the genome is maintained in episomal form, but not those in which the viral DNA has integrated into the host genome. Integration is detected in a majority of cervical and anal cancers and their high-grade precursors, despite the fact that it is not part of the normal viral life cycle [3,10]. Integration has often occurred in a way that disrupts the E2 open-reading frames, thus contributing to increased expression of the E6 and E7 oncogenes. In these cases, targeting E6 and E7 or the cellular proteins mediating their oncogenicity is clearly the only antiviral approach possible and is discussed below.
Activities of E6 and E7 needed for episomal maintenance and oncogenic transformation
The papillomavirus E6 and E7 proteins are small (<200 amino acids), zinc-finger-containing proteins (Figure 2) that target key cell-cycle-regulatory proteins to maintain infected keratinocytes in a proliferative state (reviewed in [7,24]). During the normal viral life cycle, these proteins act in concert to establish a cellular environment conducive to viral replication (Figure 4). Cell culture studies have shown that expression of E6 and E7 from high-risk HPV types is necessary and sufficient to immortalize primary keratinocytes [70,71], and that this immortalization process is dependent on the ability of E7 to stimulate quiescent cells to re-enter S phase and that of E6 to prevent the cellular growth arrest or apoptotic response brought about by this unscheduled DNA synthesis [24]. E7 from high-risk HPV types stimulates cells to undergo DNA synthesis by binding to Rb family members and promoting their degradation [72] whereas E6 prevents growth arrest and apoptosis by promoting degradation of the p53 tumour suppressor [73]. This capacity of high-risk E6 and E7 to antagonize the p53 and Rb pathways, respectively, is fundamental to their oncogenic properties. Importantly, continuous expression of E6 and E7 is needed for cervical carcinoma cells, such as HPV18-transformed HeLa cells, to maintain their transformed phenotypes [70]. Indeed, it has been shown that downregulation of E6 and E7 expression, by transfection of a functional E2 or by small interfering RNAs, results in restoration of the p53 and Rb pathways and the subsequent induction of cellular senescence [70,74–76]. These findings confirmed that antiviral approaches aimed at treating HPV-induced dysplasia should concentrate on inhibiting the oncogenic properties of E6 and E7 or those of cellular proteins mediating their effects.
Figure 4. Effects of E7 and E6 on the Rb and p53 pathways and inhibitors thereof.

Schematic diagram of how (A) E7 and (B) E6 promote cellular proliferation and inhibit apoptosis, respectively, by targeting the Rb and p53 pathways. The processes of cellular proliferation and apoptosis are indicated by downwards arrows on the left of the figure. E2F and Rb, two major effectors of cellular proliferation and apoptosis, are shown next to these arrows. (A) summarizes the key activities of E7 necessary for unscheduled E2F activation, namely its ability to promote hyperphosphorylation of Rb by inhibiting the cylcin (Cln)-dependent kinase (Cdk)2 inhibitors p21 and p27 and stimulating the activity of Cdk2, to promote the proteasomal degradation of pRb and to associate with histone deacetylases (HDAC). (B) summarizes how E6 of high-risk HPV types promotes the ubiquitination and proteasomal degradation of p53 by associating with the cellular ubiqiutin (Ub) ligase E6-associated protein (E6AP). Specific inhibitors of the Rb pathway and of the E6–E6AP protein interaction are indicated in boxes. P-PCI, purine-derived pharmacological Cdk inhibitors.
A few studies also investigated whether E6 and E7 are needed for maintenance of the viral episome in infected cells. For E7, two groups investigated whether its ability to bind Rb and related factors is necessary for episomal maintenance by mutating specific residues of the E7 LxCxE Rb-binding motif. One group observed for HPV31 that the mutant genome could still be maintained in primary HFK (human foreskin keratinocyte), albeit at a low copy number, even at early passages when most cells have presumably not begun to senesce [77]. In apparent contrast to this, the other group observed, for HPV16, that the mutant genome could still be maintained episomally, in this case in the spontaneously immortalized NIKS cell line [78]. Whether this apparent discrepancy is due to a difference in the HPV types used for these studies or the use of primary HFK versus an already immortalized cell line is currently unknown. Regardless of the explanation, both studies did agree that the mutant genomes could not become amplified upon epithelial differentiation, clearly supporting a role for the LxCxE motif in this process. In addition, mutation of the E7 LxCxE motif in the context of the CRPV genome did not abrogate its ability to induce papillomas in rabbits, suggesting that the E7–Rb interaction is not essential for pathogenesis and as would be expected if it were primarily needed for genome amplification in the upper layers of the epithelium [79]. There is ample evidence that E7 has additional functions: it can interact with several cellular proteins other than Rb and its family members [24]. Of relevance to this review is the interaction of E7 from high-risk types with histone deacetylases (HDACs) [77], a class of proteins that has been the subject of intense drug discovery efforts in recent years for different therapeutic indications. In the case of HPV infections, reverse genetic experiments with viral genomes encoding three E7 mutant proteins specifically defective in HDAC binding revealed that the E7–HDACs interaction is necessary for maintenance of the HPV31 episome in primary HFK [77], thus validating it as a potential antiviral target.
As for E6, two of its activities have been intensively studied and inferred by mutational analysis to be valid drug targets; these are its ability to promote the degradation of p53 and its binding to PDZ-domain-containing proteins (reviewed in [73]). Both of these activities are characteristics of the E6 proteins from high-risk HPV types and are not observed for those of the low-risk types. To promote degradation of p53, a key mediator of E7-induced apoptosis, E6 usurps the cellular ubiquitin ligase E6-associated proteins (E6AP) to form a complex that is capable of stimulating the poly-ubiquitination of p53 and its subsequent degradation by the proteasome [73]. Viral genomes harbouring mutations in E6 that reduce binding to E6AP or prevent degradation of p53 by other means are unable to immortalize primary keratinocytes and cannot be maintained in primary HFK [80]. These and other findings reviewed elsewhere [24] suggested that preventing E6-mediated degradation of p53 would be a useful therapeutic approach; a suggestion that has indeed been validated recently with small molecule inhibitors of the E6–E6AP interaction (see below).
E6 from high-risk HPV types also interacts with the PDZ-domain containing proteins hDlg, hScribb, MUPP1 and MAGI-1 to -3, to promote their proteasomal degradation [81–84]. These cellular proteins act as scaffolds for the assembly of multiprotein complexes at the plasma membrane and/or are involved in regulating various aspects of cell–cell contact and cell growth (hDlg and hScribb) [81,85–88]. Interestingly most of them have been shown to have tumour-suppressor activity in certain cellular contexts or in model organisms [81,86,89]. These proteins interact with E6 through a four amino acid motif, x-(T/S)-x-V (where x is any amino acid), located at its C-terminus. Deletion of this motif was shown to reduce, albeit not completely, maintenance of the HPV31 episome in primary HFK [90] and to abolish the ability of HPV16 E6 to induce epithelial hyper-plasia when expressed in the epidermis of transgenic mice from the keratin 14 promoter [91,92]. Collectively, these results suggest that down-regulation of PDZ-domain-containing proteins by E6 underlies part of its oncogenic activity. Interfering with this process may therefore be a valuable approach for the treatment of high-risk HPV infections, although to our knowledge this has not yet been attempted.
Inhibitors of E1- and E2-dependent viral DNA replication
E1–E2 protein interaction inhibitors
Papillomavirus DNA replication is initiated by the cooperative binding of E1 and E2 to the viral origin of replication (ori; Figure 3B). Formation of this ternary complex is an essential step towards the assembly of E1 double hexamers that have unwinding activity and are competent for replication [93,94]. Thus, preventing assembly of the initial E1–E2–ori complex represents an attractive antiviral strategy for the treatment of HPV lesions, in which the viral genome is maintained in episomal form, such as condylomas. Assembly of the E1–E2–ori complex depends on the interaction of E1 and E2 with DNA and on a critical protein–protein interaction between the TAD of E2 and the C-terminal helicase domain of E1 [27,40,93–98]. A small molecule inhibitor of the E1–E2 protein interaction has been identified by high-throughput screening of the Boehringer Ingelheim compound collection, using a scintillation proximity assay (SPA) that measures the cooperative binding of recombinant HPV11 E1 and E2 to radiolabelled origin DNA [99,100]. Medicinal chemistry efforts [99] led to the synthesis of more active analogues, such as inhibitors 1, 2 and 3 (Figure 5A), that antagonize assembly of the HPV11 E1–E2–ori complex with nanomolar potency in vitro. Structurally, these compounds comprise an indandione system spirofused onto an appropriately substituted tetrahydrofuran ring and hence have been termed indandione inhibitors. Structure–activity relationship (SAR) studies highlighted the benefit of the indandione system and of the carboxylate moiety for potency. Mechanistic studies, including the use of isothermal titration calorimetry, showed that the indandione inhibitors bind reversibly to the TAD of E2 with a 1:1 stoichiometry. Crystal structures of both the HPV11 TAD and of a complex between this domain and inhibitor 4 were obtained at 2.5 and 2.4 Å resolution, respectively (Figure 5B and C) [101]. Inhibitor binding did not alter the protein backbone, but caused the movement of several amino acid side chains at the binding site, in particular those of Tyr19, His32, Leu94 and Glu100, the net result being the formation of a deep hydrophobic pocket that surrounds the indandione system of the inhibitor (Figure 5B and 5C). Satisfyingly, the structure also revealed that the carboxylate moiety, shown by SAR to be important for potency, makes hydrogen bonds with amides from the protein backbone. These key features of the crystal structure were validated biochemically, by photo-affinity labelling with a reactive inhibitor analogue and by mutagenesis of several residues lining the inhibitor-binding pocket [102]. Of particular interest was the finding that substitution of Glu100 for alanine resulted in a mutant E2 protein displaying a 10-fold increased binding affinity for the inhibitors, perhaps because the entropic cost associated with the movement of the Glu100 side chain had been removed. Although the indandione inhibitors showed potent activity against the E2 proteins of the two most prevalent low-risk types, HPV6 and HPV11, they were unfortunately inactive against those of the high-risk types HPV16, HPV18 and HPV31 [100]. This lack of inhibition was correlated with variation of specific amino acids in or near the inhibitor-binding pocket, including His32 which is replaced by a tyrosine in the E2 of most high-risk types [101]. A recent crystal structure of the HPV18 E2 TAD in complex with the ATPase domain of E1 has revealed that both proteins interact through a surface area of close to 1,000 Å2, which includes the inhibitor-binding pocket [103]. Thus, the indandione inhibitors prevent the E1–E2 interaction by competing directly with E1 for binding to E2, rather than by an allosteric mechanism. The remarkable ability of the indandione inhibitors to antagonize a protein–protein interaction involving a relatively large contact area, a task usually considered unfeasible for a small molecule, provides a further incentive in the hunt for small molecule inhibitors of protein–protein interactions. Significantly, cell culture studies showed that the indandione inhibitors are also capable of antagonizing the E1–E2 interaction from HPV6 and HPV11 in vivo, in a two-hybrid-like system, and to inhibit HPV DNA replication in transiently transfected cells with a potency approaching 1 μM for the most potent inhibitor tested, compound 3 [100]. It remains an enigma why the indandione inhibitors have comparable potencies against HPV6 and HPV11 proteins in cell culture given that in vitro, in an E1–E2–ori-complex-formation assay, they are 10- to 30-fold more active against the HPV11 proteins than those of HPV6. It is conceivable that cellular proteins that interact with the E2 TAD, such as Brd4 [45], can influence the affinity of the inhibitor for E2 in vivo. Regardless of the explanation, the fact that these compounds can inhibit HPV6 and HPV11 genome replication in cells highlights, for the first time, the potential of E2 as a small molecule antiviral target for the treatment of genital warts and recurrent respiratory papillomatosis. It is hoped that the structural information available on the inhibitor-binding pocket will now stimulate the design of more active molecules [101].
Figure 5. E1–E2 interaction inhibitors.

(A) Structures of the indandione inhibitors 1, 2, 3 and 4, described in the text. These compounds inhibit the assembly of the HPV11 E1–E2–origin ternary complex in vitro with 50% inhibitory concentrations of 7.8, 0.35, 0.02 and 0.18 μm, respectively. (B) Surface representation of the crystal structure of the HPV11 E2 transactivation domain (TAD; Protein Data Bank accession number: 1R6K). (C) Portion of the crystal structure of the HPV11 E2 TAD in complex with compound 4 (PDB accession number: 1R6N) highlighting how the inhibitor (in red) binds on the surface of E2. In both (B) and (C), key residues of the inhibitor-binding pocket discussed in the text are coloured.
E1 and E2 DNA-binding inhibitors
The papillomavirus origin of DNA replication contains specific binding sites for the viral E1 and E2 proteins (Figure 3A). Binding of E2 to its cognate sites is essential for initiation of DNA replication, transcriptional repression of the viral oncogenes and segregation of the episome at mitosis [38,39,104]. As for E1, its specific binding to four sites in the origin, arranged as two pairs of inverted repeats, is needed for its correct assembly into a replication-competent double hexamer [104–108]. As a potential antiviral approach, hindering the binding of E1 or E2 to DNA has been attempted using polyamides (Figure 3B) [109,110]. Initially developed by Dervan and coworkers [111], polyamides are small amide polymers comprising organized N-methylpyrrole and N-methylimidazole amino acids, which can bind to the minor groove of DNA in a sequence-specific manner. One class of polyamides was designed to inhibit the interaction of E2 with one of its cognate binding sites in the LCR [109]. Although E2 interacts with the major grove of DNA, these minor-groove binding polyamides were shown to prevent the DNA bend that is induced by E2 and needed for stabilizing its association with its target site [109,112,113]. Optimization of this first generation of compounds through the resolution of co-crystal structures of polyamide–DNA and E2–DNA complexes, quantitative DNAse I footprinting and electromobility mobility shift assays culminated in the synthesis of an active analogue, PA1, which inhibited the binding of HPV18 E2 to DNA with a Ki of 2 nM in an in vitro binding assay [109]. Unfortunately, these compounds had no activity in cell culture assays, as they were unable to reach the nucleus of treated cells but rather accumulated primarily in the cytoplasm, a hurdle that will need to be overcome before these compounds can be developed further. An intriguing question of clinical importance is whether this class of compounds can also affect transcription of the viral oncogenes E6 and E7, in addition to interfering with viral DNA replication. In principle, these LCR-binding compounds could either increase transcription of E6/E7 by preventing E2 from binding to the LCR, or, alternatively, substitute for E2 and bring about repression of E6 and E7 expression. The future development of analogues that can reach the nucleus will be an important step towards addressing this question.
Specific polyamides have also been designed to inhibit the binding of E1 to origin DNA [110]. A brief report has described two specific imidazole–pyrrole polyamides which could significantly reduce the binding of HPV31 E1 to its cognate origin and lead to a more than 90%, dose-dependent loss of HPV31 episomes in cell cultures [110]. On the basis of these encouraging results with HPV31, similar polyamides are being developed against HPV16, the most prevalent type found in cervical cancers.
E1 ATPase and helicase inhibitors
E1 is one of the most conserved proteins among papillomaviruses and the only one with enzymatic activity [114,115]. This, coupled with the fact that viral DNA replication is absolutely dependent on E1, has contributed to making this protein an attractive target for the development of antiviral agents. During viral genome replication, E1 acts first as a DNA-binding protein to recognize the viral origin and subsequently as a helicase to unwind the DNA ahead of the replication fork [25,114,115]. The helicase activity of E1 is probably powered by ATP hydrolysis in vivo, although the enzyme can efficiently hydrolyse other nucleotides in vitro [116]. The helicase and ATPase activities of E1 are dependent on its assembly into hexamers and double hexamers and is encoded within its C-terminal half (Figure 2) [117–123]. The remaining portion of E1 contains the origin DNA-binding domain (DBD), located in the centre of the protein, and an N-terminal domain containing nuclear import and export sequences as well as phosphorylation sites for several regulatory kinases, including Cyclin A/E–Cdk2 (Figure 2). As expected from a domain with regulatory function, this N-terminal region is essential for E1 to support viral DNA replication in vivo but not in vitro [124].
Small molecule inhibitors of HPV6 E1 have been identified by high-throughput screening of the Boehringer Ingelheim compound collection using an ATPase assay based on a novel scintillation proximity detection methodology [125]. Structurally, this class of compounds is characterized by the presence of a biphenyl group substituted with a sulphonacetic acid moiety (Figure 6A) [126]. Early SAR studies showed that both the biphenyl and sulphonacetic acid pharmacophores were important for potency and hence both groups were subjected to chemical modification for optimization. Of particular interest was the discovery that addition of a 3′-carboxamide group on the biphenyl moiety significantly improved potency, yielding compounds capable of inhibiting the ATPase activity of HPV6 E1 with a 50% inhibitory concentration (IC50) of 4 nM (optimized molecule, Figure 6) [126]. Studies on the mode of action of these compounds revealed that they act through a hyperbolic competitive mechanism, suggesting that they do not directly compete with ATP at the enzyme active site but rather affect substrate binding by allosteric means [127]. As anticipated, these inhibitors could also inhibit the helicase activity of HPV6 E1 in vitro. Surprisingly, they were 2- to 10-fold less potent against the ATPase activity of HPV11 E1 than against that of HPV6, despite the high degree of sequence similarity between both enzymes. Taking advantage of this high level of conservation, chimeric proteins of HPV6 and HPV11 E1 were constructed to narrow down a region important for inhibitor activity. These studies led to the identification of Tyr486 in HPV6 E1 as being critical for inhibitor potency. In HPV11 E1, a cysteine residue is found at this position. Satisfyingly, substitution of this cysteine by a tyrosine in HPV11 E1 dramatically increased its sensitivity to biphenyl-sulphonacetic acid inhibitors without affecting its basic kinetic parameters, thereby highlighting the requirement for a tyrosine at this position for inhibitor binding [127]. In the crystal structure of the helicase domain of HPV18 and bovine papillomavirus (BPV) E1, Tyr486 (Tyr492 in HPV18 E1 and Met439 in BPVE1) is located close to the conserved lysine of the Walker A motif, which binds the triphosphate tail of ATP (Figure 6B) [103]. It was speculated that interaction of the inhibitor with Tyr486 might indirectly reduce binding of ATP at the active site, a mechanism that would account for the allosteric mode of action of these inhibitors. Interestingly, this class of inhibitors was also found to be active against the E1 protein of a high-risk virus, HPV18, albeit at a reduced potency. Notwithstanding these encouraging characteristics, the biphenysulphonacetic acid inhibitors do have some undesired properties that will need to be addressed before their full potential can be realized. In particular, the stability of the sulphonylacetic acid moiety, which has a propensity to undergo decarboxylation in some conditions, needs to be increased. Perhaps of even greater importance is the fact that the current most potent compounds are not active in a cell-based assay of HPV DNA replication [126], activity being an obvious prerequisite for further development of these molecules. Given that the activity of these compounds varies as a function of the ATP concentration, which can be relatively high in vivo, achieving cellular activity will most certainly require the synthesis of more potent compounds. This task would benefit from structural studies aimed at identifying the precise inhibitor-binding pocket on E1 and how it relates to Tyr486. At this stage, the biphenysulphonacetic acid inhibitors should be viewed as an interesting starting point for further medicinal chemistry efforts as they have proven the concept that the E1 ATPase activity of low- and high-risk HPV types E1 is open to inhibition by small molecules.
Figure 6. E1 ATPase inhibitors.
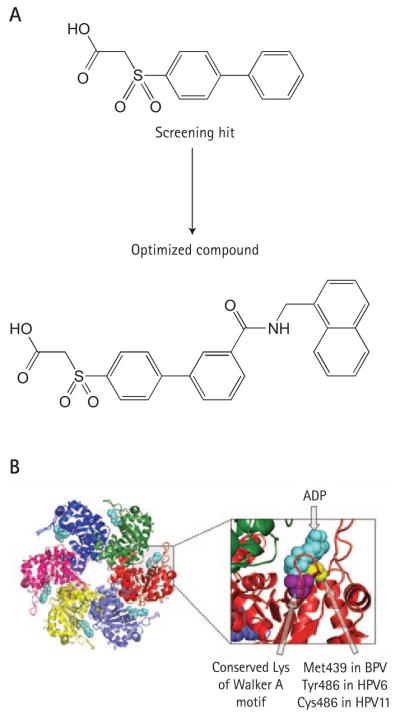
(A) Structures of the initial biphenylsulphonacetic acid inhibitor (screening hit) and of the most potent optimized compound. These compounds inhibit the ATPase activity of human papillomavirus 6 (HPV6) E1 in vitro with 50% inhibitory concentrations of 2 mM and 4 nM, respectively. (B) Side view of the crystal structure of the hexameric C-terminal helicase domain of bovine papillomavirus (BPV1) E1 bound to ADP (light blue; PDB accession number 2GXA). The monomers are differentially coloured. The right side of the panel shows an enlarged view of the ATP-binding pocket. The conserved lysine of the Walker A motif is coloured purple. The amino acid shown to be important for the binding of the biphenylsulphonic acid inhibitors is coloured yellow.
Inhibitors of the ATPase activity of HPV11 E1 have also been disclosed in a patent from Roche [128]. It is currently difficult to assess the true potential of these benzodiazepine derivatives as their activity against E1 either in vitro or in cell-based assays of HPV DNA replication has not been reported.
Inhibitors of E6-mediated p53 degradation
As mentioned above, one of the primary functions of high-risk E6 is to induce the degradation of the tumour-suppressor protein p53 as a means of preventing the growth arrest and/or apoptosis triggered by E7-induced, unscheduled DNA synthesis. To do so, E6 associates with the cellular E3 ubiquitin ligase E6AP to form a complex capable of binding p53 and promoting its ubiquitination and subsequent degradation by the proteasome (Figure 4; reviewed in [73]). From a therapeutic standpoint, it is expected that blocking the association of E6 with E6AP in high-risk HPV-infected cells will lead to an increase in p53 levels and trigger cell-cycle arrest or apoptosis in response to E7. Efforts to develop small molecule inhibitors of the E6–E6AP interaction have benefited from the knowledge that E6 interacts with E6AP, as well as other host cell proteins including E6BP and paxilin, through a short sequence motif present in these target proteins [129]. Sequence comparison led to the identification of the following consensus binding motif, LxxϕLsh, where xx is a dipeptide in which one of the residues is a aspartate, glutamate, asparagine or glutamine, ϕ is a hydrophobic residue, s is a small amino acid (alanine or glycine), and h is usually an aspartate, asparagine, glutamate or glutamine [130]. Mutagenesis of this motif revealed the critical importance of the three leucine residues (where ϕ is leucine) for E6 binding [129]. In addition, two charged residues are highly conserved amongst E6-interacting proteins that might be important for binding as they could potentially make hydrogen bonds with residues on E6. These features led to the suggestion that E6AP and other LxxϕLsh-containing proteins interact with E6 through a ‘charge leucine’ binding motif [129,130]. To date no precise binding interface has been mapped on E6 but mutagenesis experiments have indicated that both zinc fingers are required for interaction with E6AP. On that basis, it has been suggested that the binding pocket for E6AP on E6 lies, at least in part, at the junction of both metal-binding domains [130]. Significantly, an 18-residue peptide encompassing the LxxϕLsh motif of E6AP was found to inhibit the binding of full-length E6AP to HPV16 E6 with an IC50 of 10 μM in vitro, raising the possibility that a peptidomimetic approach might be feasible [131,132]. Unfortunately, more potent derivatives of this peptide have proven difficult to obtain [132]. Subsequent efforts were therefore focused on understanding the chemical nature of the E6–E6AP peptide interface. First, the structure of the E6-binding peptide lead was obtained by NMR [132]. This structure revealed that this peptide folds as an α-helix, with the three leucine residues (Leu9, Leu12 and Leu13) and the two negatively charged residues (Gln10 and Glu15) implicated in E6 binding being on opposite faces of the helix (Figure 7A) [129]. This solved structure, coupled with extensive mutagenesis of the E6AP peptide, allowed for a pharmacophore model to be created that was comprised of three hydrophobic centres representing the leucine residues, two hydrophilic centres representing the charged residues and an exclusion sphere accounting for the need for a small glycine residue at position 14 of the E6AP peptide [129]. This model was used for in silico screening of the National Cancer Institute (NCI) and Sigma-Aldrich chemical collections in order to identify small molecules that would fit these attributes and thus could possibly bind to E6 [130]. The most interesting virtual hits were then obtained and their potency assessed in an in vitro E6–E6AP binding assay as well as in in vitro and in vivo p53-degradation assays [130]. Five unrelated compounds were identified that showed significant activity in vitro. These compounds appeared to be specific as they were inactive in a counter assay based on the binding of HPV16 E7 to p107 [130]. Of these five compounds, two are of particular interest for differing reasons. One, shown in Figure 7B, is attractive because it is the most potent inhibitor identified in vitro (IC50 of ~17 μM in an E6–E6AP binding assay) and it conforms to Lipinski’s five rules of drug likeliness [130,133]. The other (Figure 7C), despite being less potent in vitro, is interesting because it showed activity in vivo, in a cell-based p53-degradation assay. Specifically, it led to an approximately fourfold increase in the levels of p53 when used at a concentration of 500 μM [130]. These encouraging findings prove the concept that E6-mediated p53 degradation can be inhibited by small molecules. Further optimization of these inhibitors would greatly benefit from structural studies aimed at understanding how these compounds bind to E6. The availability of one or more E6–inhibitor complex structures would not only help validating the pharmacophore model but also permit visualization of the inhibitor-binding pocket and pinpoint the surface of E6 involved in interacting with LxxϕLsh-motif-containing proteins. The recently reported structure of free E6 is one important step in this direction [134].
Figure 7. E6–E6AP interaction inhibitors.

(A) NMR structure of the E6-associated protein (E6AP; PDB accession number: IEQX). The positions of the three leucine residues important for binding to E6 as well as those of the two conserved charged residues are indicated by arrows. (B) Structure of the most potent E6–E6AP inhibitor (IC50 of ~17 μM in vitro). (C) Structure of the E6–E6AP inhibitor active in a cell-based p53-degradation assay. This compound leads to a more than 4-fold increase in the levels of p53 when used at a concentration of 500 μM.
Inhibitors of cellular proteins targeted by HPV gene products
HDAC inhibitors
The HPV E7 oncogene induces hyperproliferation of infected cells primarily by abrogating the functions of Rb and its family members p107 and p130, thereby causing an uncontrolled activation of E2F transcription factors and a concomitant increase in the transcription of proliferation-associated genes [24,135]. The hyperproliferative and immortalizing functions of E7 have also been linked to its capacity to associate with class I histone deacetylases (HDAC1 and HDAC2) (Figure 4) [77]. E7 binds indirectly to HDAC through Mi2β, a member of the nucleosome remodeling and histone deacetylation complex (NURD) [77,136,137]. This specific E7–Mi2β–HDAC interaction is mediated by the C-terminal zinc-binding domain of E7 and has been shown to be essential for several processes including the long-term maintenance of the viral episome in infected/transfected cells and extension of the cellular life span of undifferentiated keratinocytes [77]. Binding to HDAC is essential for E7 to modulate the transcription of E2F2 [77,136] and the immunomodulatory protein interferon regulatory factor (IRF)-1 [138]. HDACs generally inhibit gene transcription by decreasing the acetylation state of histones. To modulate the transcription of its target genes, E7 may therefore either sequester HDAC away from the promoter of E2F2 genes or recruit HDAC to the promoters of IRF-1 target genes such as IFN-β. Specific HDAC inhibitors could therefore be used to interfere with different cellular processes involved in HPV pathogenesis. HDAC inhibitors were originally found to induce the differentiation and/or growth arrest and apoptosis of various cancer cell lines (colon, prostate and ovarian) [139]. These inhibitors were also found to hinder the growth of HPV-immortalized cells by a number of mechanisms. These include restoring Cdk2 inhibitory function by increasing the transactivation of the p21 and p27 Cdk inhibitors (CDKI) [140,141] and restoring IRF-1 functions in immune responses [138]. More recently, HDAC inhibitors have also been found to induce apoptosis through an Rb–E2F–p73-mediated but p53-independent pathway [142].
Up to now, HDAC inhibitors have been grouped into five distinct classes on the basis of their chemical structure [143–145]. As listed in Table 1, these include short-chain fatty acids [146], hydroxamic acids [147–149], benzamide derivatives [150], epoxyketones and cyclic peptides [151] and hybrid molecules thereof [146,152]. Most of these compounds inhibit HDAC from both classes I and II to various degrees and with little specificity, by binding to their Zn-binding catalytic domain [146,152–154]. However, most are inactive against the class III enzymes which use nicotinamide adenine dinucleotide rather than zinc for catalysis [152]. Cell culture studies with some of these inhibitors demonstrated that butyrate (a short chain fatty acid) and trichostatin-A (a hydroxamic acid) can halt proliferation of cervical cancer cells [140,141] and that valproic acid reduces by 50% the growth of cervical and head-and-neck cancer cell lines at a concentration of 1 mM [155]. Phase I or II clinical trials with some HDAC inhibitors have now been initiated including some with valproic acid, which is already used clinically as an anticonvulsive drug [146,156]. Thus far, a Phase I clinical study has shown that oral administration of magnesium valproate reduced the tumour-associated deacetylase (HDAC) activity in 8 of 12 cervical cancer patients treated [157]. Derivatives of valproic acid that are 10 times more potent in vitro have now been identified but, unfortunately, these have not even been tested yet on HPV-containing cells [146]. In recent years, it has become evident that HDAC I and II affect many different cellular pathways, a fact that probably underlies the pleiotropic effects of current HDAC inhibitors [145]. In the future, it may become desirable to identify inhibitors that are selective for HDAC I over HDAC II or vice versa, to achieve a more targeted therapeutic effect. Although the biology clearly supports a role for HDAC in mediating the pathogenesis of HPV, it remains an open question as to whether and how HDAC inhibitors will be used for the treatment of HPV infections and HPV-induced cancers. It may be worthwhile to start addressing this question in animal models of HPV oncogenesis.
Table 1.
Inhibitors of cellular proteins mediating HPV pathogenesis
| Target | Compound | Structure | Potency, IC50 | Reference |
|---|---|---|---|---|
| HDAC | ||||
| Benzamides |
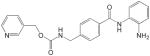
|
5 μM | [150] | |
| Short-chain fatty acids |
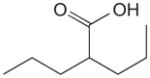
|
0.4 mM* | [146] | |
| Hydroxamic acids |
|
0.28 μM* | [147–149] | |
| Cyclic peptides and epoxyketones |
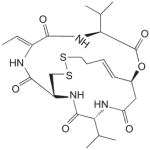
|
<5 nM | [151] | |
| Cdk2 | ||||
| Roscovitine |
|
0.7 μM* | [169,170] | |
| Indirubin-3-oxime |
|
0.25–0.44 μM* | [169,171] | |
| Sp1 | Tetra-O-methyl nordihydroguaiaretic acid (M4N) (R=OCH3) |
|
28 μM* | [182,186] |
|
|
|
|||
| Tetraacetyl nordihydroguaiaretic acid (R=O(C=O)CH3) | 11 μM | [182] | ||
For each class of inhibitors, only a single representative molecule is presented along with its structure and reported potency. For histone deacetylase (HDAC) inhibitors, the representative molecules of each chemical class are: benzanamide (MS-275), short-chain fatty acid (valproic acid), hydroxamic acid (suberoylanilide [SAHA]) and cyclic peptides (depsipeptide). The indicated potencies of these HDAC inhibitors are those measured with in vitro histone deacetylation assays. Potencies of the Cdk2 inhibitors are those reported using in vitro kinase assays. Potencies of the Sp1 inhibitors are those measured in reporter gene (luciferase) cell-based assays.
Molecules that have been tested on human papillomavirus (HPV)-infected cells or cervical cancer tissues.
Cdk2 inhibitors
Cdk2, in association with cyclin A or E, is an essential kinase for cellular proliferation, in particular for promoting the G1/S phase transition of the cell cycle. Cdk2 has therefore been considered an interesting target for the development of anti-proliferative drugs particularly in the area of oncology. The proliferation of HPV-immortalized cells, like that of most cancer cells, relies on Cdk2 activity. Two viral proteins, E7 and E1, have been shown to target Cdk2 to promote viral replication in infected cells. E7 increases Cdk2 activity by several mechanisms. It binds directly to cyclin E/A–Cdk2 to stimulate its activity [158]. It also upregulates Cdk2 indirectly by promoting the degradation of Rb and related family members, by inactivating the CDKIs p21 and p27 through direct protein–protein interactions and by increasing the expression of cyclin E and A through E2F transcription factor modulation (reviewed in [24]). These concerted actions on Cdk2 activity promote cellular proliferation and probably also underlie the ability of E7 to induce genomic instability by affecting centrosome copy numbers [159]. Indeed, centrosome abnormalities can be rapidly induced by E7 in transfected cells through a mechanism dependent on Cdk2 activity [160,161] and have also been observed in organotypic raft cultures of HPV-immortalized cells [161,162].
Cdk2 is also essential for the viral E1 helicase to support viral DNA replication [163,164]. E1 binds directly to cyclin A/E–Cdk2 though a cyclin-binding motif located in its N-terminal domain [164,165]. Phosphorylation of E1 by Cdk2 is essential for viral DNA replication, in part for E1 to accumulate in the nucleus. Indeed, recent studies have demonstrated that E1 shuttles in and out of the nucleus and that its phosphorylation at specific Cdk2 sites abrogates its nuclear export, thereby promoting its nuclear accumulation [166,167].
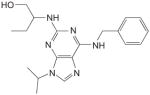
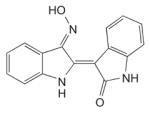
The findings presented above provide a good rationale for using small molecule Cdk2 inhibitors for the treatment of HPV infections and associated cancers [168,169]. In fact, roscovine, a Cdk2 inhibitor, has already been shown to delay proliferation of cervical carcinoma cells [170] and to inhibit the ability of E7 to cause centrosome abnormalities (see below). To date, two low-molecular-weight compounds belonging to the purine-derived pharmacological Cdk inhibitors family (P-PCI), indirubin-3′-oxime (IO) and roscovitine (Table 1), have demonstrated high potency against Cdk2. These small molecules inhibit the kinase activity of the enzyme by binding directly at its ATP-binding site [169]. IO has been shown to be a potent antagonist of several Cdks but especially of cyclin A–Cdk2 and cyclin E–Cdk2, which it can inhibit with an IC50 as low as 0.25 μM and 0.44 μM, respectively [169,171]. Moreover, IO has been reported to selectively inhibit HPV16 E7-induced centrosome abnormalities in human U2OS osteosarcoma and hTERT-immortalized human oral keratinocyte cell lines at a concentration of 0.1 μM [172]. Roscovitine, a less potent inhibitor of cyclin A/E–Cdk2 (IC50 of 0.7 μM), has also been found to block the proliferation of HPV16 E7-expressing cells and restore a normal number of centrosomes [169]. Interestingly, this inhibitor was also shown to be active in human cervical cancer tissues from different donors in vitro, where it leads to an average 61% reduction of DNA synthesis at a concentration of 100 μM [170]. The activity of more potent Cdk2 inhibitors such as flavopiridol has not yet been determined against HPV-infected cells [168]. Unfortunately, current Cdk2 inhibitors lack specificity and inhibit other cellular kinases including extracellular signal-regulated kinase (erk), protein kinase A (PKA) and casein kinase II (CKII), albeit at lower potencies (>5 μM) [169,173]. Further development of Cdk2 inhibitors will therefore need to address their relatively broad spectrum of activity against other kinases, either through the synthesis of more specific inhibitors or by demonstrating that this lack of specificity has no detrimental clinical consequences. In any case, because HPV-associated lesions appear to be particularly reliant on Cdk2 activity for proliferation and malignant progression, they should be prime candidates for treatment with effective and safe Cdk2 inhibitors.
Artemisinin
Dihydroartemisinin (DHA), the major metabolite of the antimalarial drug artemisin, was reported to be preferentially cytotoxic to HPV-containing cervical carcinoma cells, such as those from the HeLa cell line (IC50 of 7.5 μM), as well as to ectocervical cells transduced with both E6 and E7 [174,175]. Artemisinin, a natural product from the Chinese herb Artemisia annua, reacts with ferrous ions to generate reactive oxygen species capable of inducing apoptotic cell death [176,177]. Accordingly, iron was shown to be required for the cytotoxicity of DHA towards HPV-containing cells [175]. Moreover, cervical carcinoma cell lines and E6/E7-transduced ectocervical cells were found to express approximately twofold higher levels of the transferrin receptor, which for HeLa cells was associated with an increase in intracellular iron content [175]. Additional mechanistic studies indicated that DHA triggers apoptosis of carcinoma cells via the mitochondrial pathway in a p53- and E6/E7-independent manner. Remarkably, when tested topically in the canine papillomavirus infection model, DHA prevented papilloma formation in two out of three infected dogs and accelerated tumour regression in the third animal [175]. Furthermore, all three animals developed antibodies against L1, suggesting that DHA did not prevent infection or viral replication but rather inhibited tumour growth. These promising findings warrant the clinical evaluation of artemisinin and its derivatives for the treatment of HPV-induced lesions.
Sp1 inhibitors
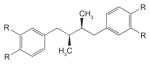
Transcription of the HPV genome is regulated by several cellular transcription factors, including Sp1, NFI-C, junB, transcription elongation factor 1 and AP1, which bind to specific sites in the viral LCR [178,179]. Among these, Sp1 is one of the best characterized [179,180]. Besides being involved in transcription of many cellular genes [181], Sp1 activates transcription of the viral E6 and E7 oncogenes by binding upstream of the early promoter p97 [182–184]. The presence of an Sp1 binding site in the viral LCR is a highly conserved feature of low- and high-risk HPV types [185]. These findings provided the rationale for inhibition of Sp1 as a potential therapy for the treatment of HPV-associated cancers. The natural product nordihydroguaiaretic acid (NDGA), isolated from the creosote bush Larrea tridentata, and its derivatives have been found to inhibit Sp1-mediated transcription by preventing binding of Sp1 to DNA [182]. In luciferase reporter gene assays, the two potent NDGA derivatives, tetra-O-methyl NDGA (M4N) and tetraacetyl NDGA (Table 1), have been shown to reduce Sp1-activated transcription from the HPV16 p97 at IC50s of 28 and 11 μM, respectively [182]. M4N was also shown to inhibit the growth of the C33A (HPV-negative) and C3 (HPV-16/Ras-transformed) cervical carcinoma cell lines, albeit after 3 days of incubation with 50 μM of inhibitor [186]. In animal studies, daily intratumoral injection of 20 mg of M4N led to reduction in the size of C3-induced tumours in mice after 2 weeks of treatment [186]. In addition to their direct effect on the HPV promoter, Sp1 inhibitors also promote apoptosis by downregulating expression of the Cdc2 kinase and survivin genes [187] at concentrations that apparently do not affect Sp1-modulated housekeeping genes [188]. Despite these interesting observations, the fact that Sp1 is involved in the transcription of many cellular genes raises important questions about the safety of these inhibitors. The demonstration that this class of inhibitors can provide therapeutic benefit at doses that do not compromise their safety profile has yet to be achieved.
Carrageenan: an attachment inhibitor and potential microbicide
All of the inhibitors described above are aimed at treating HPV-induced lesions by targeting viral and cellular proteins required for pathogenesis after infection. A complementary approach is to prevent HPV infections through the use of microbicides. The recent development of methods to produce pseudoviruses (PSVs) has greatly facilitated the study of how papillomaviruses attach to and enter keratinocytes, and has opened the door for the identification of inhibitors of these processes as candidate microbiocidal compounds. Using a high-throughput screening assay based on the PSV-induced delivery of a fluorescent protein into HeLa cells, Buck et al. (2006) recently identified carrageenan as a potent HPV cellular attachment inhibitor [189]. Carrageenan is a sulphated polysaccharide (Figure 8) extracted from seaweed (red algae) that is closely related to heparin, a known inhibitor of HPV cell entry [190,191]. Although structurally related, carrageenan is 1,000-fold more potent than heparin and both are believed to exert their inhibitory effect by mimicking heparin sulphate, an attachment factor for HPV virions on the cell surface [191]. Like heparin, carrageenan was shown to bind specifically to the L1 capsid protein. Accordingly, order-of-addition studies showed that carrageenan acts primarily by inhibiting attachment of PSV to the cell surface although it did also show a post-attachment, heparin-sulphate-independent inhibitory activity at higher doses [189]. To date, carrageenan has showed high activity against PSV from anogenital HPV types 16, 18, 31, 45 and 6, a 100-fold lower potency against those from BPV and CRPV, and, somewhat surprisingly, no activity against HPV5 [189]. In in vitro-focal transformation-based assays, carrageenan inhibited native BPV1 virions at an IC50 varying between 1 and 10 μg/ml, consistent with its activity against BPV1 PSV [189]. Importantly, these inhibitors remained highly active at the lower pH of 4.5, characteristic of the human vaginal region [189]. What makes carrageenan an especially attractive potential microbicide is the fact that it has already proven to be safe. In fact, it is currently widely used as a thickener in the food and cosmetic industries as well as in some sexual lubricants. Therefore clinical trials could, in principle, be initiated relatively soon to evaluate the efficacy of carrageenan as a topical microbicide for the prevention of anogenital HPV infections.
Figure 8. Structure of carrageenan.

Carrageenan is a polymer of the indicated structure. The average number of repeated units is ~ 500 [189, 192].
Conclusions
There are currently no antiviral agents to treat HPV-associated diseases despite the fact that infections by these viruses are extremely common, are the cause of a significant proportion of dermatological and oncological disorders, and are directly responsible for more than 200,000 deaths annually due to cervical cancer [15]. HPV has been considered a difficult virus to tackle by antiviral therapy, in part because the pharmaceutical industry has historically preferred enzymes as antiviral drug targets and HPV encodes only a single enzyme, the E1 helicase. Fortunately, this situation has begun to change in recent years as our greater understanding of the molecular biology of these viruses has permitted the discovery of new potential antiviral targets and approaches. Among the HPV early proteins, E1, E2, E6 and E7 have now been validated through mutations as being essential for pathogenesis and hence are considered the most attractive targets against which to develop drugs. It is exciting that small molecules that can inhibit specific functions of these early proteins are now emerging. Maintenance of the viral episome in infected cells can now be antagonized with indandione, polyamides and biphenylsulphonacetic inhibitors that respectively inhibit the E1–E2 protein interaction, the binding of E1 and E2 to origin DNA and the ATPase and heli-case activities of E1. In addition to being the foundations for drug discovery programmes, these inhibitors have proven that hindering viral DNA replication by pharmacological means is feasible. Because these compounds and their targets are relevant to infections in which the viral genome is maintained in episomal form, rather than integrated, they represent attractive entry points for the development of drugs to treat anogenital and cutaneous warts, and perhaps even low-grade squamous intraepithelial lesions in which the viral DNA is most often found not integrated. Clearly, once integration has occurred, E6 and E7 are the prime targets for therapeutic intervention. Our enhanced understanding of how E6 from high-risk HPV types targets p53 for degradation has permitted the design of E6–E6AP interaction inhibitors and established that restoration of p53 levels in HPV-infected cells can be achieved with small molecules. Although the discoveries of these E1, E2 and E6 inhibitors are exciting, they need to be viewed with caution as none of these compounds have yet all of the attributes of a real drug. In fact, for most of these inhibitors, potency and drug-like properties still need to be improved to reach nanomolar levels of activity in cellular assays prior to further development.
An important issue is whether development of a pan-HPV antiviral is possible. As can be appreciated from this review, compounds that are active against one HPV type are often less active or inactive against others, sometimes because of a single amino acid difference in the target protein. Although HPV proteins are highly conserved structurally between types, they are not as similar at the primary amino acid sequence level, a fact that may well prevent the development of pan-HPV antivirals. Thus, treatment of HPV-associated diseases might require the development of several classes of drugs that would each target a subset of types associated with a given medical indication. One potential way circumvent this type-specificity issue would be to develop inhibitors of cellular proteins that mediate the pathogenesis of most, if not all, HPV types. Examples of this approach are the HDAC and Cdk inhibitors, which were found to be effective at inhibiting the ability of E7 to promote cellular immortalization and induce centrosomal abnormalities, respectively. Obviously a major hurdle in using these inhibitors is that they will also affect normal cell functions, and thus might be more likely to exhibit mechanism-based toxicity. If so, topical application rather than systemic administration of these drugs might partly alleviate these undesirable effects.
However, it is important to realize that none of the liabilities associated with current inhibitors of the HPV early proteins or of cellular factors mediating their pathogenic effects are insurmountable and, in fact, they are typical concerns for most drug discovery programmes. Thus we can be cautiously optimistic that some of these compounds will be further developed and that we may witness in the relatively near future the first clinical trials with candidate antiviral drugs to treat papillomavirus infections and associated cancers. As resistance to such drugs is not expected to be an issue given that the HPV genome is replicated by cellular DNA polymerases with high fidelity, these novel HPV antiviral agents could rapidly become the gold-standard first-line therapy to treat HPV infections in diseased patients.
Acknowledgments
We thank Dr Simon Joubert and Dr Keith Schappert for helpful comments on this manuscript. Work in the authors’ laboratory is supported by grants from the Canadian Institutes of Health Research and The Cancer Research Society Inc. JA is a senior scholar from the Fonds de la Recherche en Santé du Québec (FRSQ).
References
- 1.Bosch FX, Lorincz A, Munoz N, Meijer CJ, Shah KV. The causal relation between human papillomavirus and cervical cancer. J Clin Pathol. 2002;55:244–265. doi: 10.1136/jcp.55.4.244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Daling JR, Madeleine MM, Johnson LG, et al. Human papillomavirus, smoking, and sexual practices in the etiology of anal cancer. Cancer. 2004;101:270–280. doi: 10.1002/cncr.20365. [DOI] [PubMed] [Google Scholar]
- 3.Frisch M, Fenger C, van den Brule AJ, et al. Variants of squamous cell carcinoma of the anal canal and perianal skin and their relation to human papillomaviruses. Cancer Res. 1999;59:753–757. [PubMed] [Google Scholar]
- 4.Syrjanen S. Human papillomavirus (HPV) in head and neck cancer. J Clin Virol. 2005;32(Suppl 1):S59–S66. doi: 10.1016/j.jcv.2004.11.017. [DOI] [PubMed] [Google Scholar]
- 5.Akgul B, Cooke JC, Storey A. HPV-associated skin disease. J Pathol. 2006;208:165–175. doi: 10.1002/path.1893. [DOI] [PubMed] [Google Scholar]
- 6.de Villiers EM, Fauquet C, Broker TR, Bernard HU, zur Hausen H. Classification of papillomaviruses. Virology. 2004;324:17–27. doi: 10.1016/j.virol.2004.03.033. [DOI] [PubMed] [Google Scholar]
- 7.Munger K, Baldwin A, Edwards KM, et al. Mechanisms of human papillomavirus-induced oncogenesis. J Virol. 2004;78:11451–11460. doi: 10.1128/JVI.78.21.11451-11460.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Clad A, Gissmann L, Meier B, Freese UK, Schwarz E. Molecular cloning and partial nucleotide sequence of human papillomavirus type 1a DNA. Virology. 1982;118:254–259. doi: 10.1016/0042-6822(82)90341-5. [DOI] [PubMed] [Google Scholar]
- 9.Chan SY, Delius H, Halpern AL, Bernard HU. Analysis of genomic sequences of 95 papillomavirus types: uniting typing, phylogeny, and taxonomy. J Virol. 1995;69:3074–3083. doi: 10.1128/jvi.69.5.3074-3083.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Woodman CB, Collins SI, Young LS. The natural history of cervical HPV infection: unresolved issues. Nat Rev Cancer. 2007;7:11–22. doi: 10.1038/nrc2050. [DOI] [PubMed] [Google Scholar]
- 11.Gissmann L, Wolnik L, Ikenberg H, Koldovsky U, Schnurch HG, zur Hausen H. Human papillomavirus types 6 and 11 DNA sequences in genital and laryngeal papillomas and in some cervical cancers. Proc Natl Acad Sci U S A. 1983;80:560–563. doi: 10.1073/pnas.80.2.560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Koutsky L. Epidemiology of genital human papillomavirus infection. Am J Med. 1997;102:3–8. doi: 10.1016/s0002-9343(97)00177-0. [DOI] [PubMed] [Google Scholar]
- 13.American Cancer Society [homepage on the internet] Cancer Facts and Figures 2006. Atlanta: American Cancer Society, Inc; 2006. Available from http://www.cancer.org/downloads/STT/CAFF2006PWSecured.pdf. [Google Scholar]
- 14.Chesson HW, Blandford JM, Gift TL, Tao G, Irwin KL. The estimated direct medical cost of sexually transmitted diseases among American youth, 2000. Perspect Sex Reprod Health. 2004;36:11–19. doi: 10.1363/psrh.36.11.04. [DOI] [PubMed] [Google Scholar]
- 15.Batson A, Meheus F, Brooke S. Chapter 26: Innovative financing mechanisms to accelerate the introduction of HPV vaccines in developing countries. Vaccine. 2006;24(Suppl 3):S219–S225. doi: 10.1016/j.vaccine.2006.05.042. [DOI] [PubMed] [Google Scholar]
- 16.Scheinfeld N, Lehman DS. An evidence-based review of medical and surgical treatments of genital warts. Dermatol Online J. 2006;12:5. [PubMed] [Google Scholar]
- 17.Gross G. Therapy of human papillomavirus infection and associated epithelial tumors. Intervirology. 1997;40:368–377. doi: 10.1159/000150569. [DOI] [PubMed] [Google Scholar]
- 18.US Food and Drugs Administration. VEREGEN™ (Kunecatechins) Ointment, 15%. Bradley Pharmaceuticals, Inc. under license from MediGene AG. Rockville: US Food and Drug Administration; 2006. Application No 021902. Available from http://www.fda.gov/cder/foi/label/2006/021902lbl.pdf. [Google Scholar]
- 19.US Food and Drug Administration [homepage on the internet] Rockville: US Food and Drug Administration; Merck & Co., Inc; 2006. GARDASIL®; Product Approval Information – Licensing Action. [updated 2006 June 8, cited 8th June 2006] Available from http://www.fda.gov/cber/products/hpvmer060806.htm. [Google Scholar]
- 20.Frazer IH. Prevention of cervical cancer through papillomavirus vaccination. Nat Rev Immunol. 2004;4:46–54. doi: 10.1038/nri1260. [DOI] [PubMed] [Google Scholar]
- 21.Villa LL, Costa RL, Petta CA, et al. High sustained efficacy of a prophylactic quadrivalent human papillomavirus types 6/11/16/18 L1 virus-like particle vaccine through 5 years of follow-up. Br J Cancer. 2006;95:1459–1466. doi: 10.1038/sj.bjc.6603469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.GlaxoSmithKline. New data show cervarixTM, GSK’s HPV 16/18 cervical cancer candidate vaccine is highly immunogenic and well-tolerated in women over 25 years of age. Mississauga: GlaxoSmithKline; 2006. [updated 2006 June 5, cited 5th June 2006] Available from http://www.gsk.ca/en/media_room/news/20060605.pdf. [Google Scholar]
- 23.Moscicki AB. Impact of HPV infection in adolescent populations. J Adolesc Health. 2005;37:S3–S9. doi: 10.1016/j.jadohealth.2005.09.011. [DOI] [PubMed] [Google Scholar]
- 24.Hebner CM, Laimins LA. Human papillomaviruses: basic mechanisms of pathogenesis and oncogenicity. Rev Med Virol. 2006;16:83–97. doi: 10.1002/rmv.488. [DOI] [PubMed] [Google Scholar]
- 25.Sverdrup F, Myers G. The E1 proteins. In: Myers G, Baker C, Münger K, Sverdruo F, McBride A, Bernard HU, editors. Human papillomavirus. Los Alamos: Theoretical Biology and Biophysics, Los Alamos National Laboratory; 1997. pp. 37–53. [Google Scholar]
- 26.Frattini MG, Laimins LA. Binding of the human papillomavirus E1 origin-recognition protein is regulated through complex formation with the E2 enhancer-binding protein. Proc Natl Acad Sci U S A. 1994;91:12398–12402. doi: 10.1073/pnas.91.26.12398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mohr IJ, Clark R, Sun S, Androphy EJ, MacPherson P, Botchan MR. Targeting the E1 replication protein to the papillomavirus origin of replication by complex formation with the E2 transactivator. Science. 1990;250:1694–1699. doi: 10.1126/science.2176744. [DOI] [PubMed] [Google Scholar]
- 28.Lusky M, Hurwitz J, Seo YS. The bovine papillomavirus E2 protein modulates the assembly of but is not stably maintained in a replication-competent multimeric E1-replication origin complex. Proc Natl Acad Sci U S A. 1994;91:8895–8899. doi: 10.1073/pnas.91.19.8895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sanders CM, Stenlund A. Mechanism and requirements for bovine papillomavirus, type 1, E1 initiator complex assembly promoted by the E2 transcription factor bound to distal sites. J Biol Chem. 2001;276:23689–23699. doi: 10.1074/jbc.M101861200. [DOI] [PubMed] [Google Scholar]
- 30.Schuck S, Stenlund A. Assembly of a double hexameric helicase. Mol Cell. 2005;20:377–389. doi: 10.1016/j.molcel.2005.09.020. [DOI] [PubMed] [Google Scholar]
- 31.Schuck S, Stenlund A. Role of papillomavirus E1 initiator dimerization in DNA replication. J Virol. 2005;79:8661–8664. doi: 10.1128/JVI.79.13.8661-8664.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Conger KL, Liu JS, Kuo SR, Chow LT, Wang TS. Human papillomavirus DNA replication. Interactions between the viral E1 protein and two subunits of human DNA polymerase α/primase. J Biol Chem. 1999;274:2696–2705. doi: 10.1074/jbc.274.5.2696. [DOI] [PubMed] [Google Scholar]
- 33.Clower RV, Fisk JC, Melendy T. Papillomavirus E1 protein binds to and stimulates human topoisomerase I. J Virol. 2006;80:1584–1587. doi: 10.1128/JVI.80.3.1584-1587.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Han Y, Loo YM, Militello KT, Melendy T. Interactions of the papovavirus DNA replication initiator proteins, bovine papillomavirus type 1 E1 and simian virus 40 large T antigen, with human replication protein A. J Virol. 1999;73:4899–4907. doi: 10.1128/jvi.73.6.4899-4907.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Loo YM, Melendy T. Recruitment of replication protein A by the papillomavirus E1 protein and modulation by single-stranded DNA. J Virol. 2004;78:1605–1615. doi: 10.1128/JVI.78.4.1605-1615.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Masterson PJ, Stanley MA, Lewis AP, Romanos MA. A C-terminal helicase domain of the human papillomavirus E1 protein binds E2 and the DNA polymerase α-primase p68 subunit. J Virol. 1998;72:7407–7419. doi: 10.1128/jvi.72.9.7407-7419.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Park P, Copeland W, Yang L, Wang T, Botchan MR, Mohr IJ. The cellular DNA polymerase α-primase is required for papillomavirus DNA replication and associates with the viral E1 helicase. Proc Natl Acad Sci U S A. 1994;91:8700–8704. doi: 10.1073/pnas.91.18.8700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hegde RS. The papillomavirus E2 proteins: structure, function, and biology. Annu Rev Biophys Biomol Struct. 2002;31:343–360. doi: 10.1146/annurev.biophys.31.100901.142129. [DOI] [PubMed] [Google Scholar]
- 39.Ilves I, Kivi S, Ustav M. Long-term episomal maintenance of bovine papillomavirus type 1 plasmids is determined by attachment to host chromosomes, which is mediated by the viral E2 protein and its binding sites. J Virol. 1999;73:4404–4412. doi: 10.1128/jvi.73.5.4404-4412.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Frattini MG, Laimins LA. The role of the E1 and E2 proteins in the replication of human papillomavirus type 31b. Virology. 1994;204:799–804. doi: 10.1006/viro.1994.1596. [DOI] [PubMed] [Google Scholar]
- 41.Wu X, Xiao W, Brandsma JL. Papilloma formation by cottontail rabbit papillomavirus requires E1 and E2 regulatory genes in addition to E6 and E7 transforming genes. J Virol. 1994;68:6097–6102. doi: 10.1128/jvi.68.9.6097-6102.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Peh WL, Brandsma JL, Christensen ND, Cladel NM, Wu X, Doorbar J. The viral E4 protein is required for the completion of the cottontail rabbit papillomavirus productive cycle in vivo. J Virol. 2004;78:2142–2151. doi: 10.1128/JVI.78.4.2142-2151.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Brandsma JL, Yang ZH, DiMaio D, Barthold SW, Johnson E, Xiao W. The putative E5 open reading frame of cottontail rabbit papillomavirus is dispensable for papilloma formation in domestic rabbits. J Virol. 1992;66:6204–6207. doi: 10.1128/jvi.66.10.6204-6207.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ruesch MN, Stubenrauch F, Laimins LA. Activation of papillomavirus late gene transcription and genome amplification upon differentiation in semisolid medium is coincident with expression of involucrin and transglutaminase but not keratin-10. J Virol. 1998;72:5016–5024. doi: 10.1128/jvi.72.6.5016-5024.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.McBride AA, McPhillips MG, Oliveira JG. Brd4: tethering, segregation and beyond. Trends Microbiol. 2004;12:527–529. doi: 10.1016/j.tim.2004.10.002. [DOI] [PubMed] [Google Scholar]
- 46.Howley PM, Lowy DR. Papillomaviruses and their replication. In: Fields BN, Knipe DM, Howley PM, editors. Fields Virology. 4. Vol. 2. Philadelphia: Lippincott Williams & Wilkins; 2001. pp. 2197–2229. [Google Scholar]
- 47.Kovelman R, Bilter GK, Glezer E, Tsou AY, Barbosa MS. Enhanced transcriptional activation by E2 proteins from the oncogenic human papillomaviruses. J Virol. 1996;70:7549–7560. doi: 10.1128/jvi.70.11.7549-7560.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Demeret C, Desaintes C, Yaniv M, Thierry F. Different mechanisms contribute to the E2-mediated transcriptional repression of human papillomavirus type 18 viral oncogenes. J Virol. 1997;71:9343–9349. doi: 10.1128/jvi.71.12.9343-9349.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Thierry F, Yaniv M. The BPV1-E2 trans-acting protein can be either an activator or a repressor of the HPV18 regulatory region. EMBO J. 1987;6:3391–3397. doi: 10.1002/j.1460-2075.1987.tb02662.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bernard BA, Bailly C, Lenoir MC, Darmon M, Thierry F, Yaniv M. The human papillomavirus type 18 (HPV18) E2 gene product is a repressor of the HPV18 regulatory region in human keratinocytes. J Virol. 1989;63:4317–4324. doi: 10.1128/jvi.63.10.4317-4324.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Soeda E, Ferran MC, Baker CC, McBride AA. Repression of HPV16 early region transcription by the E2 protein. Virology. 2006;351:29–41. doi: 10.1016/j.virol.2006.03.016. [DOI] [PubMed] [Google Scholar]
- 52.Lehman CW, Botchan MR. Segregation of viral plasmids depends on tethering to chromosomes and is regulated by phosphorylation. Proc Natl Acad Sci U S A. 1998;95:4338–4343. doi: 10.1073/pnas.95.8.4338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Skiadopoulos MH, McBride AA. Bovine papillomavirus type 1 genomes and the E2 transactivator protein are closely associated with mitotic chromatin. J Virol. 1998;72:2079–2088. doi: 10.1128/jvi.72.3.2079-2088.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bastien N, McBride AA. Interaction of the papillomavirus E2 protein with mitotic chromosomes. Virology. 2000;270:124–134. doi: 10.1006/viro.2000.0265. [DOI] [PubMed] [Google Scholar]
- 55.Dao LD, Duffy A, Van Tine BA, et al. Dynamic localization of the human papillomavirus type 11 origin-binding protein E2 through mitosis while in association with the spindle apparatus. J Virol. 2006;80:4792–4800. doi: 10.1128/JVI.80.10.4792-4800.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Van Tine BA, Dao LD, Wu SY, et al. Human papillomavirus (HPV) origin-binding protein associates with mitotic spindles to enable viral DNA partitioning. Proc Natl Acad Sci U S A. 2004;101:4030–4035. doi: 10.1073/pnas.0306848101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schweiger MR, You J, Howley PM. Bromodomain protein 4 mediates the papillomavirus E2 transcriptional activation function. J Virol. 2006;80:4276–4285. doi: 10.1128/JVI.80.9.4276-4285.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Senechal H, Poirier GG, Coulombe B, Laimins LA, Archambault J. Amino acid substitutions that specifically impair the transcriptional activity of papillomavirus E2 affect binding to the long isoform of Brd4. Virology. 2007;358:10–17. doi: 10.1016/j.virol.2006.08.035. [DOI] [PubMed] [Google Scholar]
- 59.Abbate EA, Voitenleitner C, Botchan MR. Structure of the papillomavirus DNA-tethering complex E2:Brd4 and a peptide that ablates HPV chromosomal association. Mol Cell. 2006;24:877–889. doi: 10.1016/j.molcel.2006.11.002. [DOI] [PubMed] [Google Scholar]
- 60.Wu SY, Lee AY, Hou SY, et al. Brd4 links chromatin targeting to HPV transcriptional silencing. Genes Dev. 2006;20:2383–2396. doi: 10.1101/gad.1448206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.You J, Schweiger MR, Howley PM. Inhibition of E2 binding to Brd4 enhances viral genome loss and phenotypic reversion of bovine papillomavirus-transformed cells. J Virol. 2005;79:14956–14961. doi: 10.1128/JVI.79.23.14956-14961.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ilves I, Maemets K, Silla T, Janikson K, Ustav M. Brd4 is involved in multiple processes of the bovine papillomavirus type 1 life cycle. J Virol. 2006;80:3660–3665. doi: 10.1128/JVI.80.7.3660-3665.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Brannon AR, Maresca JA, Boeke JD, Basrai MA, McBride AA. Reconstitution of papillomavirus E2-mediated plasmid maintenance in Saccharomyces cerevisiae by the Brd4 bromodomain protein. Proc Natl Acad Sci U S A. 2005;102:2998–3003. doi: 10.1073/pnas.0407818102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.You J, Croyle JL, Nishimura A, Ozato K, Howley PM. Interaction of the bovine papillomavirus E2 protein with Brd4 tethers the viral DNA to host mitotic chromosomes. Cell. 2004;117:349–360. doi: 10.1016/s0092-8674(04)00402-7. [DOI] [PubMed] [Google Scholar]
- 65.McPhillips MG, Oliveira JG, Spindler JE, Mitra R, McBride AA. Brd4 is required for E2-mediated transcriptional activation but not genome partitioning of all papillomaviruses. J Virol. 2006;80:9530–9543. doi: 10.1128/JVI.01105-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.McPhillips MG, Ozato K, McBride AA. Interaction of bovine papillomavirus E2 protein with Brd4 stabilizes its association with chromatin. J Virol. 2005;79:8920–8932. doi: 10.1128/JVI.79.14.8920-8932.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Baxter MK, McPhillips MG, Ozato K, McBride AA. The mitotic chromosome binding activity of the papillomavirus E2 protein correlates with interaction with the cellular chromosomal protein, Brd4. J Virol. 2005;79:4806–4818. doi: 10.1128/JVI.79.8.4806-4818.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Stubenrauch F, Colbert AM, Laimins LA. Transactivation by the E2 protein of oncogenic human papillomavirus type 31 is not essential for early and late viral functions. J Virol. 1998;72:8115–8123. doi: 10.1128/jvi.72.10.8115-8123.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jeckel S, Huber E, Stubenrauch F, Iftner T. A transactivator function of cottontail rabbit papillomavirus E2 is essential for tumor induction in rabbits. J Virol. 2002;76:11209–11215. doi: 10.1128/JVI.76.22.11209-11215.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.DeFilippis RA, Goodwin EC, Wu L, DiMaio D. Endogenous human papillomavirus E6 and E7 proteins differentially regulate proliferation, senescence, and apoptosis in HeLa cervical carcinoma cells. J Virol. 2003;77:1551–1563. doi: 10.1128/JVI.77.2.1551-1563.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hawley-Nelson P, Vousden KH, Hubbert NL, Lowy DR, Schiller JT. HPV16 E6 and E7 proteins cooperate to immortalize human foreskin keratinocytes. EMBO J. 1989;8:3905–3910. doi: 10.1002/j.1460-2075.1989.tb08570.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gonzalez SL, Stremlau M, He X, Basile JR, Munger K. Degradation of the retinoblastoma tumor suppressor by the human papillomavirus type 16 E7 oncoprotein is important for functional inactivation and is separable from proteasomal degradation of E7. J Virol. 2001;75:7583–7591. doi: 10.1128/JVI.75.16.7583-7591.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Scheffner M, Whitaker NJ. Human papillomavirus-induced carcinogenesis and the ubiquitin-proteasome system. Semin Cancer Biol. 2003;13:59–67. doi: 10.1016/s1044-579x(02)00100-1. [DOI] [PubMed] [Google Scholar]
- 74.Moon MS, Lee CJ, Um SJ, Park JS, Yang JM, Hwang ES. Effect of BPV1 E2-mediated inhibition of E6/E7 expression in HPV16-positive cervical carcinoma cells. Gynecol Oncol. 2001;80:168–175. doi: 10.1006/gyno.2000.6053. [DOI] [PubMed] [Google Scholar]
- 75.Nishimura A, Nakahara T, Ueno T, et al. Requirement of E7 oncoprotein for viability of HeLa cells. Microbes Infect. 2006;8:984–993. doi: 10.1016/j.micinf.2005.10.015. [DOI] [PubMed] [Google Scholar]
- 76.Yamato K, Fen J, Kobuchi H, et al. Induction of cell death in human papillomavirus 18-positive cervical cancer cells by E6 siRNA. Cancer Gene Ther. 2006;13:234–241. doi: 10.1038/sj.cgt.7700891. [DOI] [PubMed] [Google Scholar]
- 77.Longworth MS, Laimins LA. The binding of histone deacetylases and the integrity of zinc finger-like motifs of the E7 protein are essential for the life cycle of human papillomavirus type 31. J Virol. 2004;78:3533–3541. doi: 10.1128/JVI.78.7.3533-3541.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Collins AS, Nakahara T, Do A, Lambert PF. Interactions with pocket proteins contribute to the role of human papillomavirus type 16 E7 in the papillomavirus life cycle. J Virol. 2005;79:14769–14780. doi: 10.1128/JVI.79.23.14769-14780.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Defeo-Jones D, Vuocolo GA, Haskell KM, et al. Papillomavirus E7 protein binding to the retinoblastoma protein is not required for viral induction of warts. J Virol. 1993;67:716–725. doi: 10.1128/jvi.67.2.716-725.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Thomas JT, Hubert WG, Ruesch MN, Laimins LA. Human papillomavirus type 31 oncoproteins E6 and E7 are required for the maintenance of episomes during the viral life cycle in normal human keratinocytes. Proc Natl Acad Sci U S A. 1999;96:8449–8454. doi: 10.1073/pnas.96.15.8449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Massimi P, Gammoh N, Thomas M, Banks L. HPV E6 specifically targets different cellular pools of its PDZ domain-containing tumour suppressor substrates for proteasome-mediated degradation. Oncogene. 2004;23:8033–8039. doi: 10.1038/sj.onc.1207977. [DOI] [PubMed] [Google Scholar]
- 82.Glaunsinger BA, Lee SS, Thomas M, Banks L, Javier R. Interactions of the PDZ-protein MAGI-1 with adenovirus E4-ORF1 and high-risk papillomavirus E6 oncoproteins. Oncogene. 2000;19:5270–5280. doi: 10.1038/sj.onc.1203906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Thomas M, Laura R, Hepner K, et al. Oncogenic human papillomavirus E6 proteins target the MAGI-2 and MAGI-3 proteins for degradation. Oncogene. 2002;21:5088–5096. doi: 10.1038/sj.onc.1205668. [DOI] [PubMed] [Google Scholar]
- 84.Mantovani F, Banks L. The human papillomavirus E6 protein and its contribution to malignant progression. Oncogene. 2001;20:7874–7887. doi: 10.1038/sj.onc.1204869. [DOI] [PubMed] [Google Scholar]
- 85.Woods DF, Hough C, Peel D, Callaini G, Bryant PJ. Dlg protein is required for junction structure, cell polarity, and proliferation control in Drosophila epithelia. J Cell Biol. 1996;134:1469–1482. doi: 10.1083/jcb.134.6.1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bilder D, Li M, Perrimon N. Cooperative regulation of cell polarity and growth by Drosophila tumor suppressors. Science. 2000;289:113–116. doi: 10.1126/science.289.5476.113. [DOI] [PubMed] [Google Scholar]
- 87.Bilder D, Perrimon N. Localization of apical epithelial determinants by the basolateral PDZ protein Scribble. Nature. 2000;403:676–680. doi: 10.1038/35001108. [DOI] [PubMed] [Google Scholar]
- 88.Fanning AS, Anderson JM. PDZ domains: fundamental building blocks in the organization of protein complexes at the plasma membrane. J Clin Invest. 1999;103:767–772. doi: 10.1172/JCI6509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Borg JP. hScrib: un nouveau suppresseur de tumeur à l’horizon [hScrib: a potential novel tumor suppressor] Pathol Biol (Paris) 2004;52:328–331. doi: 10.1016/j.patbio.2003.09.015. French. [DOI] [PubMed] [Google Scholar]
- 90.Lee C, Laimins LA. Role of the PDZ domain-binding motif of the oncoprotein E6 in the pathogenesis of human papillomavirus type 31. J Virol. 2004;78:12366–12377. doi: 10.1128/JVI.78.22.12366-12377.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Nguyen ML, Nguyen MM, Lee D, Griep AE, Lambert PF. The PDZ ligand domain of the human papillomavirus type 16 E6 protein is required for E6’s induction of epithelial hyperplasia in vivo. J Virol. 2003;77:6957–6964. doi: 10.1128/JVI.77.12.6957-6964.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Nguyen MM, Nguyen ML, Caruana G, Bernstein A, Lambert PF, Griep AE. Requirement of PDZ-containing proteins for cell cycle regulation and differentiation in the mouse lens epithelium. Mol Cell Biol. 2003;23:8970–8981. doi: 10.1128/MCB.23.24.8970-8981.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Berg M, Stenlund A. Functional interactions between papillomavirus E1 and E2 proteins. J Virol. 1997;71:3853–3863. doi: 10.1128/jvi.71.5.3853-3863.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Sanders CM, Stenlund A. Recruitment and loading of the E1 initiator protein: an ATP-dependent process catalysed by a transcription factor. EMBO J. 1998;17:7044–7055. doi: 10.1093/emboj/17.23.7044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Blitz IL, Laimins LA. The 68-kilodalton E1 protein of bovine papillomavirus is a DNA binding phosphoprotein which associates with the E2 transcriptional activator in vitro. J Virol. 1991;65:649–656. doi: 10.1128/jvi.65.2.649-656.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Bonne-Andrea C, Tillier F, McShan GD, Wilson VG, Clertant P. Bovine papillomavirus type 1 DNA replication: the transcriptional activator E2 acts in vitro as a specificity factor. J Virol. 1997;71:6805–6815. doi: 10.1128/jvi.71.9.6805-6815.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Seo YS, Muller F, Lusky M, et al. Bovine papilloma virus (BPV)-encoded E2 protein enhances binding of E1 protein to the BPV replication origin. Proc Natl Acad Sci U S A. 1993;90:2865–2869. doi: 10.1073/pnas.90.7.2865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Yang L, Li R, Mohr IJ, Clark R, Botchan MR. Activation of BPV-1 replication in vitro by the transcription factor E2. Nature. 1991;353:628–632. doi: 10.1038/353628a0. [DOI] [PubMed] [Google Scholar]
- 99.Yoakim C, Ogilvie WW, Goudreau N, et al. Discovery of the first series of inhibitors of human papillomavirus type 11: inhibition of the assembly of the E1-E2-Origin DNA complex. Bioorg Med Chem Lett. 2003;13:2539–2541. doi: 10.1016/s0960-894x(03)00510-9. [DOI] [PubMed] [Google Scholar]
- 100.White PW, Titolo S, Brault K, et al. Inhibition of human papillomavirus DNA replication by small molecule antagonists of the E1-E2 protein interaction. J Biol Chem. 2003;278:26765–26772. doi: 10.1074/jbc.M303608200. [DOI] [PubMed] [Google Scholar]
- 101.Wang Y, Coulombe R, Cameron DR, et al. Crystal structure of the E2 transactivation domain of human papillomavirus type 11 bound to a protein interaction inhibitor. J Biol Chem. 2004;279:6976–6985. doi: 10.1074/jbc.M311376200. [DOI] [PubMed] [Google Scholar]
- 102.Davidson W, McGibbon GA, White PW, et al. Characterization of the binding site for inhibitors of the HPV11 E1-E2 protein interaction on the E2 transactivation domain by photoaffinity labeling and mass spectrometry. Anal Chem. 2004;76:2095–2102. doi: 10.1021/ac035335o. [DOI] [PubMed] [Google Scholar]
- 103.Abbate EA, Berger JM, Botchan MR. The X-ray structure of the papillomavirus helicase in complex with its molecular matchmaker E2. Genes Dev. 2004;18:1981–1996. doi: 10.1101/gad.1220104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Sedman T, Sedman J, Stenlund A. Binding of the E1 and E2 proteins to the origin of replication of bovine papillomavirus. J Virol. 1997;71:2887–2896. doi: 10.1128/jvi.71.4.2887-2896.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Chen G, Stenlund A. Characterization of the DNA-binding domain of the bovine papillomavirus replication initiator E1. J Virol. 1998;72:2567–2576. doi: 10.1128/jvi.72.4.2567-2576.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Sun YN, Lu JZ, McCance DJ. Mapping of HPV-11 E1 binding site and determination of other important cis elements for replication of the origin. Virology. 1996;216:219–222. doi: 10.1006/viro.1996.0050. [DOI] [PubMed] [Google Scholar]
- 107.Holt SE, Wilson VG. Mutational analysis of the 18-base-pair inverted repeat element at the bovine papillomavirus origin of replication: identification of critical sequences for E1 binding and in vivo replication. J Virol. 1995;69:6525–6532. doi: 10.1128/jvi.69.10.6525-6532.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Titolo S, Brault K, Majewski J, White PW, Archambault J. Characterization of the minimal DNA binding domain of the human papillomavirus e1 helicase: fluorescence anisotropy studies and characterization of a dimerization-defective mutant protein. J Virol. 2003;77:5178–5191. doi: 10.1128/JVI.77.9.5178-5191.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Schaal TD, Mallet WG, McMinn DL, et al. Inhibition of human papilloma virus E2 DNA binding protein by covalently linked polyamides. Nucleic Acids Res. 2003;31:1282–1291. doi: 10.1093/nar/gkg206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Bashkin J, Fisher C. Potential new treatment for human papillomavirus: Scientists from UM-St. Louis, NanoVir make discovery. 19th International Conference for Antiviral Research; 7–11 May 2006; San Juan, Puerto Rico. p. Abstract 35. Available from http://m114.phrm.cf.ac.uk/ISAR2/2006.sanjuan.icar.program.pdf. [Google Scholar]
- 111.Dervan PB, Burli RW. Sequence-specific DNA recognition by polyamides. Curr Opin Chem Biol. 1999;3:688–693. doi: 10.1016/s1367-5931(99)00027-7. [DOI] [PubMed] [Google Scholar]
- 112.Zimmerman JM, Maher LJ., III Solution measurement of DNA curvature in papillomavirus E2 binding sites. Nucleic Acids Res. 2003;31:5134–5139. doi: 10.1093/nar/gkg697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Bedrosian CL, Bastia D. The DNA-binding domain of HPV-16 E2 protein interaction with the viral enhancer: protein-induced DNA bending and role of the nonconserved core sequence in binding site affinity. Virology. 1990;174:557–575. doi: 10.1016/0042-6822(90)90109-5. [DOI] [PubMed] [Google Scholar]
- 114.Wilson VG, West M, Woytek K, Rangasamy D. Papillomavirus E1 proteins: form, function, and features. Virus Genes. 2002;24:275–290. doi: 10.1023/a:1015336817836. [DOI] [PubMed] [Google Scholar]
- 115.Stenlund A. Initiation of DNA replication: lessons from viral initiator proteins. Nat Rev Mol Cell Biol. 2003;4:777–785. doi: 10.1038/nrm1226. [DOI] [PubMed] [Google Scholar]
- 116.Rocque WJ, Porter DJ, Barnes JA, et al. Replication-associated activities of purified human papillomavirus type 11 E1 helicase. Protein Expr Purif. 2000;18:148–159. doi: 10.1006/prep.1999.1182. [DOI] [PubMed] [Google Scholar]
- 117.Titolo S, Pelletier A, Sauve F, et al. Role of the ATP-binding domain of the human papillomavirus type 11 E1 helicase in E2-dependent binding to the origin. J Virol. 1999;73:5282–5293. doi: 10.1128/jvi.73.7.5282-5293.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Titolo S, Pelletier A, Pulichino AM, et al. Identification of domains of the human papillomavirus type 11 E1 helicase involved in oligomerization and binding to the viral origin. J Virol. 2000;74:7349–7361. doi: 10.1128/jvi.74.16.7349-7361.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.White PW, Pelletier A, Brault K, et al. Characterization of recombinant HPV6 and 11 E1 helicases: effect of ATP on the interaction of E1 with E2 and mapping of a minimal helicase domain. J Biol Chem. 2001;276:22426–22438. doi: 10.1074/jbc.M101932200. [DOI] [PubMed] [Google Scholar]
- 120.Sedman J, Stenlund A. The papillomavirus E1 protein forms a DNA-dependent hexameric complex with ATPase and DNA helicase activities. J Virol. 1998;72:6893–6897. doi: 10.1128/jvi.72.8.6893-6897.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Hughes FJ, Romanos MA. E1 protein of human papillomavirus is a DNA helicase/ATPase. Nucleic Acids Res. 1993;21:5817–5823. doi: 10.1093/nar/21.25.5817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Jenkins O, Earnshaw D, Sarginson G, et al. Characterization of the helicase and ATPase activity of human papillomavirus type 6b E1 protein. J Gen Virol. 1996;77:1805–1809. doi: 10.1099/0022-1317-77-8-1805. [DOI] [PubMed] [Google Scholar]
- 123.Santucci S, Bonne-Andrea C, Clertant P. Bovine papillomavirus type 1 E1 ATPase activity does not depend on binding to DNA nor to viral E2 protein. J Gen Virol. 1995;76:1129–1140. doi: 10.1099/0022-1317-76-5-1129. [DOI] [PubMed] [Google Scholar]
- 124.Amin AA, Titolo S, Pelletier A, Fink D, Cordingley MG, Archambault J. Identification of domains of the HPV11 E1 protein required for DNA replication in vitro. Virology. 2000;272:137–150. doi: 10.1006/viro.2000.0328. [DOI] [PubMed] [Google Scholar]
- 125.Jeffery JA, Sharom JR, Fazekas M, et al. An ATPase assay using scintillation proximity beads for high-throughput screening or kinetic analysis. Anal Biochem. 2002;304:55–62. doi: 10.1006/abio.2002.5632. [DOI] [PubMed] [Google Scholar]
- 126.Faucher AM, White PW, Brochu C, Grand-Maitre C, Rancourt J, Fazal G. Discovery of small-molecule inhibitors of the ATPase activity of human papillomavirus E1 helicase. J Med Chem. 2004;47:18–21. doi: 10.1021/jm034206x. [DOI] [PubMed] [Google Scholar]
- 127.White PW, Faucher AM, Massariol MJ, et al. Biphenylsulfonacetic acid inhibitors of the human papillomavirus type 6 E1 helicase inhibit ATP hydrolysis by an allosteric mechanism involving tyrosine 486. Antimicrob Agents Chemother. 2005;49:4834–4842. doi: 10.1128/AAC.49.12.4834-4842.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Hurst DN, Jones PS, Parkes KEB, Parratt MJ, Wilson FX, inventors. Hoffmann–La Roche Inc, assignee. United State patent US 6703387. Inhibitors of HPV E1 helicase enzyme. 2004 Mar 9;
- 129.Be X, Hong Y, Wei J, Androphy EJ, Chen JJ, Baleja JD. Solution structure determination and mutational analysis of the papillomavirus E6 interacting peptide of E6AP. Biochemistry. 2001;40:1293–1299. doi: 10.1021/bi0019592. [DOI] [PubMed] [Google Scholar]
- 130.Baleja JD, Cherry JJ, Liu Z, et al. Identification of inhibitors to papillomavirus type 16 E6 protein based on three-dimensional structures of interacting proteins. Antivir Res. 2006;72:49–59. doi: 10.1016/j.antiviral.2006.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Sterlinko GH, Weber M, Elston R, et al. Inhibition of E6-induced degradation of its cellular substrates by novel blocking peptides. J Mol Biol. 2004;335:971–985. doi: 10.1016/j.jmb.2003.10.079. [DOI] [PubMed] [Google Scholar]
- 132.Liu Y, Liu Z, Androphy E, Chen J, Baleja JD. Design and characterization of helical peptides that inhibit the E6 protein of papillomavirus. Biochemistry. 2004;43:7421–7431. doi: 10.1021/bi049552a. [DOI] [PubMed] [Google Scholar]
- 133.Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 2001;46:3–26. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 134.Nomine Y, Masson M, Charbonnier S, et al. Structural and functional analysis of E6 oncoprotein: insights in the molecular pathways of human papillomavirus-mediated pathogenesis. Mol Cell. 2006;21:665–678. doi: 10.1016/j.molcel.2006.01.024. [DOI] [PubMed] [Google Scholar]
- 135.Zhang B, Chen W, Roman A. The E7 proteins of low- and high-risk human papillomaviruses share the ability to target the pRB family member p130 for degradation. Proc Natl Acad Sci U S A. 2006;103:437–442. doi: 10.1073/pnas.0510012103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Longworth MS, Wilson R, Laimins LA. HPV31 E7 facilitates replication by activating E2F2 transcription through its interaction with HDACs. EMBO J. 2005;24:1821–1830. doi: 10.1038/sj.emboj.7600651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Brehm A, Nielsen SJ, Miska EA, et al. The E7 oncoprotein associates with Mi2 and histone deacetylase activity to promote cell growth. EMBO J. 1999;18:2449–2458. doi: 10.1093/emboj/18.9.2449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Park JS, Kim EJ, Kwon HJ, Hwang ES, Namkoong SE, Um SJ. Inactivation of interferon regulatory factor-1 tumor suppressor protein by HPV E7 oncoprotein. Implication for the E7-mediated immune evasion mechanism in cervical carcinogenesis. J Biol Chem. 2000;275:6764–6769. doi: 10.1074/jbc.275.10.6764. [DOI] [PubMed] [Google Scholar]
- 139.Marks P, Rifkind RA, Richon VM, Breslow R, Miller T, Kelly WK. Histone deacetylases and cancer: causes and therapies. Nat Rev Cancer. 2001;1:194–202. doi: 10.1038/35106079. [DOI] [PubMed] [Google Scholar]
- 140.Finzer P, Ventz R, Kuntzen C, Seibert N, Soto U, Rosl F. Growth arrest of HPV-positive cells after histone deacetylase inhibition is independent of E6/E7 oncogene expression. Virology. 2002;304:265–273. doi: 10.1006/viro.2002.1667. [DOI] [PubMed] [Google Scholar]
- 141.Finzer P, Kuntzen C, Soto U, zur Hausen H, Rosl F. Inhibitors of histone deacetylase arrest cell cycle and induce apoptosis in cervical carcinoma cells circumventing human papillomavirus oncogene expression. Oncogene. 2001;20:4768–4776. doi: 10.1038/sj.onc.1204652. [DOI] [PubMed] [Google Scholar]
- 142.Finzer P, Krueger A, Stohr M, et al. HDAC inhibitors trigger apoptosis in HPV-positive cells by inducing the E2F-p73 pathway. Oncogene. 2004;23:4807–4817. doi: 10.1038/sj.onc.1207620. [DOI] [PubMed] [Google Scholar]
- 143.Duenas–Gonzalez A, Lizano M, Candelaria M, Cetina L, Arce C, Cervera E. Epigenetics of cervical cancer. An overview and therapeutic perspectives. Mol Cancer. 2005;4:38. doi: 10.1186/1476-4598-4-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Monneret C. Histone deacetylase inhibitors. Eur J Med Chem. 2005;40:1–13. doi: 10.1016/j.ejmech.2004.10.001. [DOI] [PubMed] [Google Scholar]
- 145.Drummond DC, Noble CO, Kirpotin DB, Guo Z, Scott GK, Benz CC. Clinical development of histone deacetylase inhibitors as anticancer agents. Annu Rev Pharmacol Toxicol. 2005;45:495–528. doi: 10.1146/annurev.pharmtox.45.120403.095825. [DOI] [PubMed] [Google Scholar]
- 146.Lu Q, Yang YT, Chen CS, et al. Zn2+-chelating motif-tethered short-chain fatty acids as a novel class of histone deacetylase inhibitors. J Med Chem. 2004;47:467–474. doi: 10.1021/jm0303655. [DOI] [PubMed] [Google Scholar]
- 147.Marks PA, Richon VM, Rifkind RA. Cell cycle regulatory proteins are targets for induced differentiation of transformed cells: Molecular and clinical studies employing hybrid polar compounds. Int J Hematol. 1996;63:1–17. doi: 10.1016/0925-5710(95)00428-9. [DOI] [PubMed] [Google Scholar]
- 148.Suzuki T, Matsuura A, Kouketsu A, Nakagawa H, Miyata N. Identification of a potent non-hydroxamate histone deacetylase inhibitor by mechanism-based drug design. Bioorg Med Chem Lett. 2005;15:331–335. doi: 10.1016/j.bmcl.2004.10.074. [DOI] [PubMed] [Google Scholar]
- 149.Suzuki T, Nagano Y, Kouketsu A, et al. Novel inhibitors of human histone deacetylases: design, synthesis, enzyme inhibition, and cancer cell growth inhibition of SAHA-based non-hydroxamates. J Med Chem. 2005;48:1019–1032. doi: 10.1021/jm049207j. [DOI] [PubMed] [Google Scholar]
- 150.Suzuki T, Ando T, Tsuchiya K, et al. Synthesis and histone deacetylase inhibitory activity of new benzamide derivatives. J Med Chem. 1999;42:3001–3003. doi: 10.1021/jm980565u. [DOI] [PubMed] [Google Scholar]
- 151.Nakajima H, Kim YB, Terano H, Yoshida M, Horinouchi S. FR901228, a potent antitumor antibiotic, is a novel histone deacetylase inhibitor. Exp Cell Res. 1998;241:126–133. doi: 10.1006/excr.1998.4027. [DOI] [PubMed] [Google Scholar]
- 152.Lin HY, Chen CS, Lin SP, Weng JR, Chen CS. Targeting histone deacetylase in cancer therapy. Med Res Rev. 2006;26:397–413. doi: 10.1002/med.20056. [DOI] [PubMed] [Google Scholar]
- 153.de Ruijter AJ, van Gennip AH, Caron HN, Kemp S, van Kuilenburg AB. Histone deacetylases (HDACs): characterization of the classical HDAC family. Biochem J. 2003;370:737–749. doi: 10.1042/BJ20021321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Hildmann C, Wegener D, Riester D, et al. Substrate and inhibitor specificity of class 1 and class 2 histone deacetylases. J Biotechnol. 2006;124:258–270. doi: 10.1016/j.jbiotec.2006.01.030. [DOI] [PubMed] [Google Scholar]
- 155.Chavez-Blanco A, Perez-Plasencia C, Perez-Cardenas E, et al. Antineoplastic effects of the DNA methylation inhibitor hydralazine and the histone deacetylase inhibitor valproic acid in cancer cell lines. Cancer Cell Int. 2006;6:2. doi: 10.1186/1475-2867-6-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Drummond DC, Noble CO, Kirpotin DB, Guo Z, Scott GK, Benz CC. Clinical development of histone deacetylase inhibitors as anticancer agents. Annu Rev Pharmacol Toxicol. 2005;45:495–528. doi: 10.1146/annurev.pharmtox.45.120403.095825. [DOI] [PubMed] [Google Scholar]
- 157.Chavez-Blanco A, Segura-Pacheco B, Perez-Cardenas E, et al. Histone acetylation and histone deacetylase activity of magnesium valproate in tumor and peripheral blood of patients with cervical cancer. A Phase I study. Mol Cancer. 2005;4:22. doi: 10.1186/1476-4598-4-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.He W, Staples D, Smith C, Fisher C. Direct activation of cyclin-dependent kinase 2 by human papillomavirus E7. J Virol. 2003;77:10566–10574. doi: 10.1128/JVI.77.19.10566-10574.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Duensing S, Munger K. Centrosome abnormalities and genomic instability induced by human papillomavirus oncoproteins. Prog Cell Cycle Res. 2003;5:383–391. [PubMed] [Google Scholar]
- 160.Duensing S, Munger K. Human papillomaviruses and centrosome duplication errors: modeling the origins of genomic instability. Oncogene. 2002;21:6241–6248. doi: 10.1038/sj.onc.1205709. [DOI] [PubMed] [Google Scholar]
- 161.Duensing S, Duensing A, Flores ER, Do A, Lambert PF, Munger K. Centrosome abnormalities and genomic instability by episomal expression of human papillomavirus type 16 in raft cultures of human keratinocytes. J Virol. 2001;75:7712–7716. doi: 10.1128/JVI.75.16.7712-7716.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Duensing S, Lee LY, Duensing A, et al. The human papillomavirus type 16 E6 and E7 oncoproteins cooperate to induce mitotic defects and genomic instability by uncoupling centrosome duplication from the cell division cycle. Proc Natl Acad Sci U S A. 2000;97:10002–10007. doi: 10.1073/pnas.170093297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Lin BY, Ma T, Liu JS, et al. HeLa cells are phenotypically limiting in cyclin E/CDK2 for efficient human papillomavirus DNA replication. J Biol Chem. 2000;275:6167–6174. doi: 10.1074/jbc.275.9.6167. [DOI] [PubMed] [Google Scholar]
- 164.Ma T, Zou N, Lin BY, Chow LT, Harper JW. Interaction between cyclin-dependent kinases and human papillomavirus replication-initiation protein E1 is required for efficient viral replication. Proc Natl Acad Sci U S A. 1999;96:382–387. doi: 10.1073/pnas.96.2.382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Cueille N, Nougarede R, Mechali F, Philippe M, Bonne-Andrea C. Functional interaction between the bovine papillomavirus virus type 1 replicative helicase E1 and cyclin E-Cdk2. J Virol. 1998;72:7255–7262. doi: 10.1128/jvi.72.9.7255-7262.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Hsu CY, Mechali F, Bonne-Andrea C. Nucleocytoplasmic shuttling of bovine papillomavirus E1 helicase downregulates viral DNA replication in S phase. J Virol. 2007;81:384–394. doi: 10.1128/JVI.01170-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Deng W, Lin BY, Jin G, et al. Cyclin/CDK regulates the nucleocytoplasmic localization of the human papillomavirus E1 DNA helicase. J Virol. 2004;78:13954–13965. doi: 10.1128/JVI.78.24.13954-13965.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Schang LM. Effects of pharmacological cyclin-dependent kinase inhibitors on viral transcription and replication. Biochim Biophys Acta. 2004;1697:197–209. doi: 10.1016/j.bbapap.2003.11.024. [DOI] [PubMed] [Google Scholar]
- 169.Schang LM. Cyclin-dependent kinases as cellular targets for antiviral drugs. J Antimicrob Chemother. 2002;50:779–792. doi: 10.1093/jac/dkf227. [DOI] [PubMed] [Google Scholar]
- 170.Vitali L, Yakisich JS, Vita MF, et al. Roscovitine inhibits ongoing DNA synthesis in human cervical cancer. Cancer Lett. 2002;180:7–12. doi: 10.1016/s0304-3835(01)00827-8. [DOI] [PubMed] [Google Scholar]
- 171.Hoessel R, Leclerc S, Endicott JA, et al. Indirubin, the active constituent of a Chinese antileukaemia medicine, inhibits cyclin-dependent kinases. Nat Cell Biol. 1999;1:60–67. doi: 10.1038/9035. [DOI] [PubMed] [Google Scholar]
- 172.Duensing S, Duensing A, Lee DC, et al. Cyclin-dependent kinase inhibitor indirubin-3′-oxime selectively inhibits human papillomavirus type 16 E7-induced numerical centrosome anomalies. Oncogene. 2004;23:8206–8215. doi: 10.1038/sj.onc.1208012. [DOI] [PubMed] [Google Scholar]
- 173.Meijer L, Borgne A, Mulner O, et al. Biochemical and cellular effects of roscovitine, a potent and selective inhibitor of the cyclin-dependent kinases cdc2, cdk2 and cdk5. Eur J Biochem. 1997;243:527–536. doi: 10.1111/j.1432-1033.1997.t01-2-00527.x. [DOI] [PubMed] [Google Scholar]
- 174.Golenser J, Waknine JH, Krugliak M, Hunt NH, Grau GE. Current perspectives on the mechanism of action of artemisinins. Int J Parasitol. 2006;36:1427–1441. doi: 10.1016/j.ijpara.2006.07.011. [DOI] [PubMed] [Google Scholar]
- 175.Disbrow GL, Baege AC, Kierpiec KA, et al. Dihydroartemisinin is cytotoxic to papillomavirus-expressing epithelial cells in vitro and in vivo. Cancer Res. 2005;65:10854–10861. doi: 10.1158/0008-5472.CAN-05-1216. [DOI] [PubMed] [Google Scholar]
- 176.Meshnick SR. Artemisinin: mechanisms of action, resistance and toxicity. Int J Parasitol. 2002;32:1655–1660. doi: 10.1016/s0020-7519(02)00194-7. [DOI] [PubMed] [Google Scholar]
- 177.O’Neill PM, Posner GH. A medicinal chemistry perspective on artemisinin and related endoperoxides. J Med Chem. 2004;47:2945–2964. doi: 10.1021/jm030571c. [DOI] [PubMed] [Google Scholar]
- 178.Bernard HU. Gene expression of genital human papillomaviruses and considerations on potential antiviral approaches. Antivir Ther. 2002;7:219–237. [PubMed] [Google Scholar]
- 179.Demeret C, Le Moal M, Yaniv M, Thierry F. Control of HPV 18 DNA replication by cellular and viral transcription factors. Nucleic Acids Res. 1995;23:4777–4784. doi: 10.1093/nar/23.23.4777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Gloss B, Bernard HU. The E6/E7 promoter of human papillomavirus type 16 is activated in the absence of E2 proteins by a sequence-aberrant Sp1 distal element. J Virol. 1990;64:5577–5584. doi: 10.1128/jvi.64.11.5577-5584.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Black AR, Black JD, Azizkhan-Clifford J. Sp1 and kruppel-like factor family of transcription factors in cell growth regulation and cancer. J Cell Physiol. 2001;188:143–160. doi: 10.1002/jcp.1111. [DOI] [PubMed] [Google Scholar]
- 182.Craigo J, Callahan M, Huang RC, DeLucia AL. Inhibition of human papillomavirus type 16 gene expression by nordihydroguaiaretic acid plant lignan derivatives. Antivir Res. 2000;47:19–28. doi: 10.1016/s0166-3542(00)00089-9. [DOI] [PubMed] [Google Scholar]
- 183.Gloss B, Bernard HU. The E6/E7 promoter of human papillomavirus type 16 is activated in the absence of E2 proteins by a sequence-aberrant Sp1 distal element. J Virol. 1990;64:5577–5584. doi: 10.1128/jvi.64.11.5577-5584.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Butz K, Hoppe-Seyler F. Transcriptional control of human papillomavirus (HPV) oncogene expression: composition of the HPV type 18 upstream regulatory region. J Virol. 1993;67:6476–6486. doi: 10.1128/jvi.67.11.6476-6486.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.O’Connor M, Chan SY, Bernard HU. Transcription factor binding sites in the long control region of genital HPVs. In: Myers G, Bernard HU, Delius H, Baker C, Icenogle J, Halpern A, Wheeler C, editors. Human Papillomaviruses 1995 Compendium, part III-A. Los Alamos: Los Alamos National Laboratory; 1995. pp. 21–40. [Google Scholar]
- 186.Heller JD, Kuo J, Wu TC, Kast WM, Huang RC. Tetra-O-methyl nordihydroguaiaretic acid induces G2 arrest in mammalian cells and exhibits tumoricidal activity in vivo. Cancer Res. 2001;61:5499–5504. [PubMed] [Google Scholar]
- 187.Chang CC, Heller JD, Kuo J, Huang RC. Tetra-O-methyl nordihydroguaiaretic acid induces growth arrest and cellular apoptosis by inhibiting Cdc2 and survivin expression. Proc Natl Acad Sci U S A. 2004;101:13239–13244. doi: 10.1073/pnas.0405407101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Huang RC, Li Y, Giza PE, et al. Novel antiviral agent tetraglycylated nordihydroguaiaretic acid hydrochloride as a host-dependent viral inhibitor. Antivir Res. 2003;58:57–64. doi: 10.1016/s0166-3542(02)00189-4. [DOI] [PubMed] [Google Scholar]
- 189.Buck CB, Thompson CD, Roberts JN, Muller M, Lowy DR, Schiller JT. Carrageenan is a potent inhibitor of papillomavirus infection. PLoS Pathog. 2006;2:e69. doi: 10.1371/journal.ppat.0020069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Giroglou T, Sapp M, Lane C, et al. Immunological analyses of human papillomavirus capsids. Vaccine. 2001;19:1783–1793. doi: 10.1016/s0264-410x(00)00370-4. [DOI] [PubMed] [Google Scholar]
- 191.Joyce JG, Tung JS, Przysiecki CT, et al. The L1 major capsid protein of human papillomavirus type 11 recombinant virus-like particles interacts with heparin and cell-surface glycosaminoglycans on human keratinocytes. J Biol Chem. 1999;274:5810–5822. doi: 10.1074/jbc.274.9.5810. [DOI] [PubMed] [Google Scholar]
- 192.Tobacman JK. Review of harmful gastrointestinal effects of carrageenan in animal experiments. Environ Health Perspect. 2001;109:983–994. doi: 10.1289/ehp.01109983. [DOI] [PMC free article] [PubMed] [Google Scholar]