

ABSTRACT
Gap junctions formed of connexin 36 (Cx36, also known as Gjd2) show tremendous functional plasticity on several time scales. Changes in connexin phosphorylation modify coupling in minutes through an order of magnitude, but recent studies also imply involvement of connexin turnover in regulating cell–cell communication. We utilized Cx36 with an internal HaloTag to study Cx36 turnover and trafficking in cultured cells. Irreversible, covalent pulse-chase labeling with fluorescent HaloTag ligands allowed clear discrimination of newly formed and pre-existing Cx36. Cx36 in junctional plaques turned over with a half-life of 3.1 h, and the turnover rate was unchanged by manipulations of protein kinase A (PKA) activity. In contrast, changes in PKA activity altered coupling within 20 min. New Cx36 in cargo vesicles was added directly to existing gap junctions and newly made Cx36 was not confined to points of addition, but diffused throughout existing gap junctions. Existing connexins also diffused into photobleached areas with a half-time of less than 2 s. In conclusion, studies of Cx36-HaloTag revealed novel features of connexin trafficking and demonstrated that phosphorylation-based changes in coupling occur on a different time scale than turnover.
KEY WORDS: Gap junction, Confocal microscopy, Connexin, Membrane trafficking, Pulse-chase, Tracer coupling
Summary: Fluorescent labeling of Cx36-HaloTag revealed that turnover occurs on a different time scale than phosphorylation-driven plasticity. Cx36-HaloTag is also partially mobile within gap junctions.
INTRODUCTION
Throughout the central nervous system, neurons are coupled to each other by electrical synapses, which consist of gap junctions that provide a direct intercellular pathway for current flow. They also allow transfer of ions and small molecules, including metabolites and second messengers (Saez et al., 2003). Electrical synapses are crucial in establishing network oscillations and play important roles in motor learning, memory consolidation, retinal signal processing and visual adaptation (Deans et al., 2001; Guldenagel et al., 2001; Hormuzdi et al., 2001; Van Der Giessen et al., 2008; Kothmann et al., 2009; Allen et al., 2011).
Connexins are the proteins comprising gap junctions in vertebrates, and they are diverse and ubiquitous. Connexin 36 (Gjd2, hereafter referred to as Cx36), the non-mammalian homolog of which is Cx35, is the predominant connexin that forms electrical synapses in neurons. It is widespread in the central nervous system, being found extensively in retina (O'Brien et al., 1998; Mills et al., 2001; Deans et al., 2002; O'Brien et al., 2004), olfactory bulb (Christie et al., 2005), neocortex (Deans et al., 2001; Blatow et al., 2003), hippocampus (Belluardo et al., 2000; Hormuzdi et al., 2001), inferior olive (Long et al., 2002; De Zeeuw et al., 2003) and cerebellum (Belluardo et al., 2000). Cx36 is also the primary connexin used in neuroendocrine cells of the pancreas (Serre-Beinier et al., 2000), pituitary and pineal organs (Belluardo et al., 2000), and is found in adrenal chromaffin cells (Martin et al., 2001).
Turnover and trafficking of ion channels and receptor proteins in chemical synapses have been well studied and have proven to be important regulators of synaptic strength and plasticity. Like chemical synapses, electrical synapses are dynamic and highly plastic. Cx36 gap junctions between rods and cones are strongly modulated by light adaptation and a circadian rhythm (Wang and Mangel, 1996; Ribelayga et al., 2002; Ribelayga et al., 2008). AII amacrine cells, also coupled by Cx36, also show significant plasticity with light adaptation state (Bloomfield and Völgyi, 2004). These changes in coupling are due to changes in the phosphorylation of Cx36 regulated by protein kinases and phosphatases (Kothmann et al., 2009; Li et al., 2009; Kothmann et al., 2012; Li et al., 2013). However, turnover of Cx36 could also contribute to plasticity by removal of functional phosphorylated connexins and changes in the total amount of connexins in electrical synapses. Turnover rates of connexins are exceptionally high. Non-neuronal connexin half-lives reported in the literature range from 1 to 10 h, with one exception in lens cells (Hervé et al., 2007). Changes in abundance of Cx36 have been proposed to contribute to plasticity of coupling in mouse photoreceptors (Katti et al., 2013). Furthermore, injection of a peptide that mimics the C-terminal tip of Cx35 into goldfish Mauthner cells, interfering with PDZ domain interactions, resulted in loss of Cx35 and reduced electrical coupling (Flores et al., 2012). This observation suggested that in the absence of the stabilizing influence of scaffolds, turnover of connexins quickly reduces coupling.
To date, very few studies have investigated the link between Cx36 turnover and functional plasticity. In this study, we developed procedures using HaloTag technology to study Cx36 turnover. We used a Cx36-HaloTag fusion construct to label Cx36 efficiently and irreversibly at narrowly defined time windows, allowing dual-color fluorescent pulse-chase labeling in transfected HeLa cells. Measurement of turnover rates revealed clear temporal and biochemical segregation of phosphorylation-based and turnover-based plasticity. The highly specific live fluorescent pulse-chase labeling also allowed us to examine aspects of the trafficking and turnover of connexins that have been difficult to study previously.
RESULTS
Cx36-HaloTag fusion protein forms functional, normally regulated gap junctions
In order to study Cx36 turnover we used a genetically encoded covalent labeling system, HaloTag, to permanently label pools of Cx36 at specific time points. The HaloTag is a mutated prokaryotic hydrolase that hydrolyzes chloroalkane ligands to form a covalent reaction intermediate that permanently labels the protein (Los and Wood, 2007). We used membrane-permeant fluorescent ligands for this study. The C-terminus of Cx36 is the site of a number of known protein–protein interactions and regulatory phosphorylation events (Li et al., 2004; Ouyang et al., 2005; Kothmann et al., 2007; Alev et al., 2008; del Corsso et al., 2012; Li et al., 2012). We chose to insert the HaloTag open reading frame between the two regulatory phosphorylation sites (S293 and S315) (Fig. 1) in order to avoid interfering with regulatory phosphorylation events and C-terminal protein–protein interactions. To determine if the Cx36-Halo construct forms gap junctions properly, we transfected HeLa cells with Cx36-Halo and labeled the cells with HaloTag tetramethylrhodamine (TMR) ligand. Live cell imaging revealed TMR-labeled gap junctions at cell–cell contacts, as well as TMR-labeled vesicles within cells (Fig. S1). Co-labeling fixed cells with an anti-Cx36 antibody showed that the HaloTag TMR labeling was co-extensive with structures labeled with Cx36 antibody (Fig. 2A), confirming that HaloTag TMR ligand efficiently labeled Cx36 gap junctions in transfected HeLa cells.
Fig. 1.
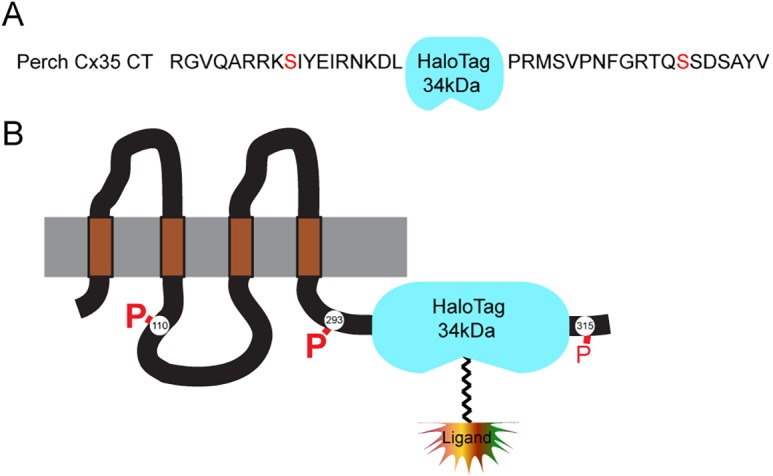
Construction of the Cx35-HaloTag fusion. (A) Sequence of perch Cx35 (non-mammalian homolog of Cx36) C-terminus indicating the insertion point of the HaloTag open reading frame. Regulatory serine phosphorylation sites (S276 and S298) are indicated in red type. (B) Ribbon structure of Cx36 showing the relative locations of HaloTag and regulatory phosphorylation sites. HaloTag ligands are covalently bound to the HaloTag protein when applied and become permanently attached to the Cx36-Halo protein.
Fig. 2.
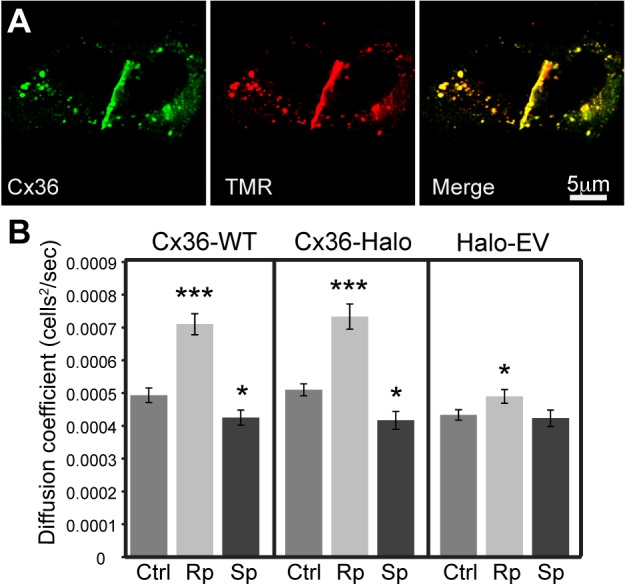
Cx36-HaloTag fusion protein forms functional gap junctions that are normally regulated by PKA activities. (A) Live cell labeling of HeLa cells transfected with Cx36-Halo with HaloTag TMR ligand (red) followed by fixation and labeling with Cx36 antibody (green). The labels are co-localized at cell–cell junctional plaques as well as in internal vesicles. Images shown are single confocal slices. Scale bar applies to all images. (B) Tracer coupling measurements in HeLa cells transiently transfected with Cx36-Halo, Cx36-WT and HaloTag empty vector (EV). Ctrl, control; Sp, 5 mM Sp-8-cpt-cAMPS (PKA activator); Rp, 5 mM Rp-8-cpt-cAMPS (PKA inhibitor). Data are means±s.e.m., n=3 experiments per condition; *P<0.05, ***P<0.001.
In previous studies, we established that Cx36 coupling is regulated by protein kinase A (PKA) activity and that the regulation of coupling observed in HeLa cells parallels that in retinal AII amacrine cells (O’Brien et al., 2004; Ouyang et al., 2005; Kothmann et al., 2009; Li et al., 2009). To determine whether inserting the HaloTag protein into Cx36 caused functional changes in Cx36 regulation, we performed scrape-loading experiments using HeLa cells transiently transfected with Cx36-Halo or wild-type Cx36. We observed similar regulation in both constructs (Fig. 2B): 10 min treatment with PKA inhibitor Rp-8-cpt-cAMPS (Rp) increased coupling significantly (mixed effects model: wt-Cx36, P<0.0001; Cx36-Halo, P<0.0001; n=3 per condition for each form), 10 min treatment with PKA activator Sp-8-cpt-cAMPS (Sp) slightly reduced coupling (wt-Cx36, P<0.05; Cx36-Halo, P<0.05; n=3 per condition for each form). Background tracer coupling in HeLa cells transfected with HaloTag vector alone (Halo-EV) was slightly elevated by Rp treatment (P<0.05, n=3 per condition), but this effect was significantly smaller than the effect on wild-type Cx36 or Cx36-Halo (P<0.0001 for both wt-Cx36 and Cx36-Halo; n=3 for each form).
Cx36 has a half-life of 3.1 h in HeLa cells, and the turnover rate is not affected by PKA activity
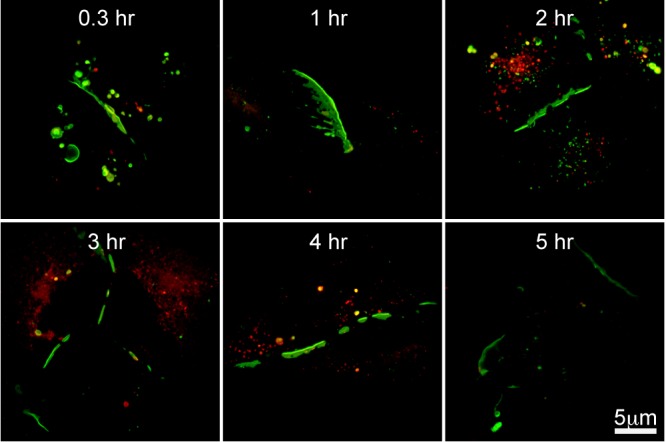
To study the turnover rate of Cx36 in HeLa cells, we performed pulse-chase studies using two different fluorescent ligands of the HaloTag protein. Cx36-Halo-transfected HeLa cells were labeled with HaloTag Oregon Green (OG) ligand at time zero and followed by HaloTag TMR labeling at hours 0.3, 1, 2, 3, 5 or 6. At 0.3 h, the majority of labeling in junctional plaques consisted of OG (Fig. 3). As time progressed, the amount of TMR labeling increased in the gap junctions and the amount of OG labeling decreased. At the last time point, the majority of labeling in the gap junctions consisted of TMR.
Fig. 3.

Pulse-chase labeling of HeLa cells transiently transfected with Cx36-Halo. Cells were labeled with OG (pulse; green label) at time 0 and TMR (chase; red label) at hours 0.3, 1, 2, 3, 5 and 6. Images are confocal stacks of 2.4–2.5 mm thickness. Scale bar applies to all images.
The TMR (chase) label was present in small vesicles throughout all time points, and was increasingly infused into the gap junctions (Fig. 3). This suggests a progressive removal of the old gap junction protein, which was labeled with OG, and replacement by new gap junction protein, which became labeled with TMR. The TMR label at early time points was patchy and at later time points was mixed throughout the whole gap junction (Fig. 3). The chase-labeled connexin did not appear to be added just on the outer edges of existing plaques, as previously reported (Gaietta et al., 2002; Lauf et al., 2002). This will be discussed further below.
Figure 4A shows a plot of the fraction of old gap junction protein in relation to the total amount of labeled gap junction protein present fitted with an exponential decay curve. The calculated half-life for replacement of old protein was 3.1 h, which is consistent with previous studies with other connexins (Hervé et al., 2007; Flores et al., 2012). To determine whether the turnover rate of Cx36 was affected by PKA activity we performed pulse-chase analyses in the presence of the PKA inhibitor Rp or the PKA activator Sp, which had significantly altered tracer coupling in 20 min. There were no significant differences in the half-life of Cx36 with either treatment (Fig. 4B; one-way ANOVA with Dunnett's post-hoc test: Sp, P=0.305; Rp, P=0.894; overall P=0.330; n=3 experiments per condition).
Fig. 4.
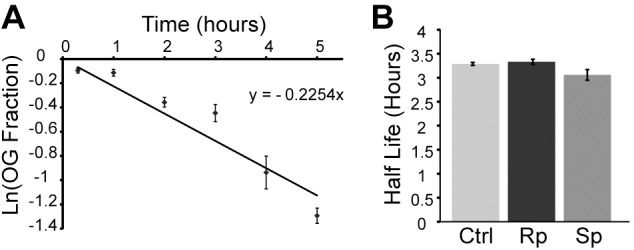
Half-life of Cx36 in HeLa cells. (A) Calculation of Cx36 half-life in HeLa cells. The x-axis represents time lapsed; the y-axis is the natural log of total green intensity divided by total intensity of green plus red in gap junctions (intracellular vesicles are excluded); n=10–20 gap junctions per time point. Half-life is calculated by the natural log of 2 divided by the gradient of the best-fitted straight line. (B) Half-lives of Cx36 gap junction plaques were not altered by the treatment with 5 mM Sp-8-cpt-cAMPS or Rp-8-cpt-cAMPS throughout the chase period; n=3 experiments with 10–20 gap junctions per time point in each. Data are means±s.e.m.
Cx36 gap junction assembly requires the Golgi apparatus
With the exception of Cx26 (Zhang et al., 1996), it is generally considered that connexins are modified in the endoplasmic reticulum (ER) (Ahmad et al., 1999; Zhang et al., 1996), transported to the Golgi complex, assembled in the trans-Golgi network (Musil and Goodenough, 1993; Koval et al., 1997), and finally inserted into the plasma membrane (Laird, 1996; Thomas et al., 2005). To determine if the trafficking of Cx36 to the plasma membrane involves the same pathway, we repeated the pulse chase analysis with treatment of Brefeldin A (BFA), which disassembles the Golgi apparatus. The original gap junctions were again labeled with OG, and chased with TMR. We saw that at time 0.3 h, all the gap junctions were labeled with OG, with a minimal amount of TMR labeling (Fig. 5). As time progressed, the amount of TMR labeling did not increase in the gap junctions. Most of the TMR label was present in small vesicles and some large vesicles; almost none integrated into the gap junctions, and OG remained the predominant label through the fifth hour (Fig. 5). The fraction of OG present in the gap junctions showed no significant change from hour 0 to hour 5 (one-way ANOVA: hour 0.3, 86.7±7.4%; hour 5, 91.2±6.5%; n=5 gap junctions per time point; P=0.33), the change of fraction of OG was significant when BFA was not added (one-way ANOVA: hour 0.3, 91.6±5.7%; hour 5, 31.4±6.2%; n=10–20 gap junctions per time point; P<0.0001). We conclude that the Golgi apparatus was essential for Cx36 gap junction assembly.
Fig. 5.
Pulse-chase analysis of Cx36-Halo in HeLa cells with Brefeldin A treatment throughout the chase period. Pulse (OG; green) and chase (TMR; red) ligands are the same as in Fig. 3. Treatment with BFA prevented insertion of newly made Cx36 into gap junctions. By 5 h, gap junctions were very rare and difficult to find. Images are confocal stacks 2.5 mm in thickness. Scale bar applies to all images.
Cx36 is trafficked to the plasma membrane in vesicles, and removed as annular gap junctions
It has been reported that new connexons are trafficked in vesicles as undocked hemichannels and removed as double membrane vesicles called annular junctions (Laird, 1996; Falk et al., 2009). In our confocal microscope images, we observed two different types of vesicles close to the gap junctions. These vesicles were present throughout all the time points.
The first type of vesicle was large and usually hollow in the middle. These vesicles could contain either pulse (OG) or chase (TMR) ligand, or both (Fig. 6A, arrows). When close to pre-existing gap junctions, these vesicles were mostly labeled with pulse ligand. We found some OG-labeled Cx36 budding off the pre-existing gap junctions near the ends (Fig. 6Aii, open arrowhead) as well as in the middle of plaques (Fig. 6Ai, asterisk). In the BFA-treated cells (Fig. 5), in which gap junctions were less abundant and contained very little chase label, we found that newly made Cx36 was added to the large internal vesicles throughout the chase period.
Fig. 6.
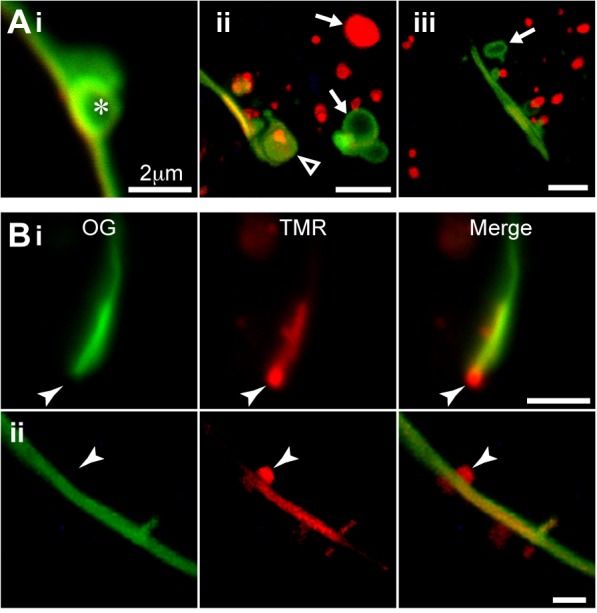
Intracellular vesicles containing Cx36-Halo are readily tracked in the pulse-chase paradigm. Pulse (OG; green) and chase (TMR; red) ligands are the same as in Fig. 3. (A) The removal of Cx36 from the gap junction. Large removal vesicles were usually a mixture of OG- and TMR-labeled, and were often circular (arrows). The vesicles can be removed from the edge (ii: open arrowhead) as well as center (i: asterisk) of a pre-existing gap junction. (B) Small insertion vesicles (arrowheads) containing new Cx36 were often found docked on gap junctions, and were often adjacent to bright patches of chase labeling within the gap junction. The insertion can happen at the edge (i) or the center (ii) of an existing gap junction. Images Aii and Aiii are 2.5 mm thick stacks; all other images are single confocal slices. Scale bars are 2 µm in each panel.
The second type of vesicle was smaller, solid and often very numerous. These vesicles were mostly labeled with the chase ligand (TMR), indicating that they were recently synthesized. These small, new vesicles were often observed on the periphery of the pre-existing gap junction plaques, either on the end (Fig. 6Bi) or in the middle (Fig. 6Bii). The small vesicles were often found adjacent to patches of chase-labeled gap junction, suggesting that they were supplying newly synthesized gap junction protein to existing plaques (Fig. 6Bi and ii). This addition of vesicles to existing gap junctions was not obviously symmetric, meaning that a vesicle could be added to a gap junction from either participating cell without concomitant addition of a vesicle from the other cell. This suggests that connexin hemichannels may be present in a gap junction prior to docking with hemichannels from the opposing cell.
Cx36-Halo diffuses laterally through gap junctions
The observation that recently synthesized Cx36 proteins spread throughout existing gap junction plaques differs from observations with Cx43 (Gaietta et al., 2002; Lauf et al., 2002; Falk et al., 2009) on which the common framework of understanding of connexin trafficking and turnover are based. Is this a result of the position of the tag within the connexin protein, of the type of connexin studied, or of the tag itself and observation methods? To address these questions we developed additional HaloTag constructs of Cx36 with the tag at the tip of the C-terminus, and of Cx43 with the tag positioned at the tip of the C-terminus or at either of two internal positions within the C-terminal domain (following amino acids 249 or 342). Cx43-HaloTag constructs did not form gap junctions when transfected into HeLa cells. However, all three Cx43 constructs tested formed gap junctions efficiently in HEK293 cells. We performed two types of experiments to determine whether these tagged connexins diffuse through gap junction plaques. First, we performed pulse-chase analysis of turnover with each of the constructs. Figure 7A and B show labeling of Cx36-Halo-C-IN (internal of C-terminus) and Cx36-Halo-C-END, respectively, at 2 h chase time in HEK293 cells. For both constructs, the TMR chase label was present throughout the gap junctions. Figure 7C and D show labeling for one of the Cx43 internal constructs (Cx43-Halo-C-249) and Cx43-Halo-C-END, respectively, at 2 h chase time. For both Cx43 constructs, the TMR chase label was present within the existing gap junction plaques. The Cx43 chase label was patchy in intensity, suggesting less mixing of the recently made connexin proteins with connexins present in the plaque.
Fig. 7.

Lateral diffusion of connexin protein is not dependent on tag position or cell type. (A) Cx36-C-IN and (B) Cx36-C-END transfected in HEK293 cells showed TMR chase label present throughout the gap junction. (C) Cx43-C-249 and (D) Cx43-C-END transfected in HEK293 cells showed TMR chase label present throughout the gap junction. TMR chase labeling was performed 2 h after the OG pulse labeling. All images are single confocal slices. Gap junctions are indicated by arrowheads at their ends. Scale bar applies to all images.
As a second test of the ability of connexin proteins to diffuse laterally through gap junction plaques, we performed live photobleaching studies of Cx36-Halo-C-IN in HeLa cells. Figure 8A shows a series of frames from a time sequence including one bleach of an OG-labeled Cx36 gap junction (boxed region) and an OG-labeled internal vesicle (circular region; see also Movie 1). Bleaching and imaging were done using a confocal microscope with a 20× objective and confocal zoom of 6. Under these conditions, the bleaching sweeps bleached more widely than the targeted band of interest, and the bleached area quickly recovered label in the OG channel. Figure 8B shows the intensity profiles of both bleached regions of interest through the entire experiment comprising four sequential bleaches (full sequence in Movie 2). Following each bleach, the OG label recovered to a new steady-state level with a first-order time course with a recovery half-time of 1.51±0.22 s (n=4 gap junctions, two to six bleaches per gap junction). The post-bleach steady state was below the original baseline, reflecting both bleaching of the total pool of OG-labeled Cx36 and a portion of the OG-labeled Cx36 pool that was effectively immobile within the time course of the experiment. The mobile fraction of Cx36 ranged from 56 to 41% and decreased with each bleach (Fig. S2). This decrease in mobile fraction with each subsequent bleach could be a result of light-induced damage to the gap junction or to preferential depletion of the mobile pool following the first bleach.
Fig. 8.

FRAP analysis of OG-labeled Cx36-Halo in HeLa cells. (A) Images of a gap junction prior to bleach (i) and at 0.5, 1.5 and 10.4 s post-bleach (ii–iv). The blue box is the bleached ROI and the red circle is an intracellular vesicle bleached at the same time. (B) Intensity profiles of the bleached ROIs: gap junction is the blue line; vesicle is the red line. (C) FRAP of an OG ligand droplet using the same bleaching parameters; images represent the time points indicated in A. (D) Intensity profile of droplet bleach.
Rapid recovery of photobleached connexin in gap junction plaques has not been previously reported and we questioned whether this might be an artifact resulting from reversible bleaching of the OG fluorochrome by the bleaching sweeps. To test this, we photobleached aqueous droplets of the OG HaloTag ligand suspended in mineral oil using the same bleaching paradigm. Figure 8C and D shows that each bleach destroyed a fraction of the fluorochrome with no recovery. This lack of recovery was replicated at several different laser power settings and confocal zoom settings. Thus, rapid recovery of OG-labeled Cx36 in the bleached area was not an artifact.
Finally, we also tested the ability of Cx43-Halo-C-249 to diffuse through existing gap junction plaques. Photobleaching of OG-labeled Cx43 resulted in partial recovery of fluorescence in the bleached area with an average time constant of 1.9 s and mobile fractions ranging from 78 to 46% (n=2 gap junctions, five to six bleaches per gap junction; Fig. S3). Thus, some Cx43-Halo was mobile within the gap junction.
DISCUSSION
Validation of Connexin-HaloTag fusion constructs
To study the turnover rate of gap junction protein, it is important to develop a means to label and track gap junction protein efficiently and specifically. In this study, we used the HaloTag technology to label Cx36 and Cx43. The HaloTag offers many advantages, including covalent labeling with non-toxic fluorescent ligands and monomeric structure (Encell et al., 2013). However, at 34 kDa, the HaloTag is about 25% larger than enhanced green fluorescent protein (EGFP) and almost as large as Cx36 itself. For reference, this tag is a little bit less than two-thirds the size of tdTomato. In spite of the large tag size, the protein produced by the Cx36-Halo construct successfully trafficked through the Golgi apparatus to the plasma membrane and formed gap junctions, even when bound to HaloTag ligands. Furthermore, the Cx36-Halo gap junctions were functional and were regulated by PKA activity in the same manner as wild-type Cx36 gap junctions as assessed by Neurobiotin tracer coupling.
The position of the HaloTag within the connexin did result in minor differences in gap junction formation that we did not assess quantitatively. Cx36 with an internal tag produced more gap junctions and fewer large internal vesicles than Cx36 with a C-terminal tag. Likewise, Cx43 with an internal tag after amino acid 249 produced more gap junctions and fewer large internal vesicles than Cx43 with an internal tag after amino acid 342 or with a C-terminal tag. Thus, there may be aspects of the HaloTag that result in context-specific anomalies. Nonetheless, the labeling system was very effective for studying Cx36.
Novel features of Connexin-HaloTag trafficking
Previous studies have reported that connexons are trafficked to the plasma membrane as undocked hemichannels packaged in vesicles, accreting at the edge of gap junctions (Gaietta et al., 2002; Lauf et al., 2002), and removed as paired channels that form double membrane vesicles known as annular junctions (Laird, 1996; Gaietta et al., 2002; Lauf et al., 2002). These annular gap junctions are subsequently transported to lysosomes, where they are degraded (Laird, 1996; Piehl et al., 2007). Our microscopic evidence is consistent with these observations by showing two different classes of vesicles: small newly formed vesicles presumably for exocytosis, and large ring-shaped vesicles presumably resulting from endocytosis. In the BFA-treated cells, newly synthesized Cx36 did not integrate into gap junctions, but still formed large hollow vesicles that morphologically resembled annular junctions. These are unlikely to be the conventional annular junctions since the newly synthesized Cx36 never reached the plasma membrane and docked with hemichannels from the adjacent cells. Kumar and Gilula showed that when connnexins are overexpressed, double-membraned gap junction-like sheets form within the intracellular compartment (Kumar and Gilula, 1992). Many of the TMR-labeled Cx36 annular junctions in our system are probably artifacts of overexpression of Cx36 in HeLa cells. These annular junctions are transported to lysosomes subsequently where the gap junction proteins are degraded (Laird, 1996; Thomas et al., 2002).
A consistent observation in our imaging studies is that the small vesicles carrying newly made Cx36 were often found attached to existing gap junction plaques, and patches of chase label could be seen in the gap junction adjacent to the vesicles. This strongly implies that these vesicles dock at existing gap junctions and their cargo diffuses into the gap junction plaque. These findings agree with those of Shaw et al., who found that Cx43 cargo vesicles were targeted directly to adherens junctions adjacent to gap junction plaques through microtubule plus-end tracking proteins (Shaw et al., 2007).
Several previous studies have found that Cx43 present as undocked hemichannels in the plasma membrane accreted to the outside edges of existing gap junctions and mixed very little with existing gap junction channels (Gaietta et al., 2002; Lauf et al., 2002; Falk et al., 2009). Our observations did not show such apparent accretion at the edges of gap junctions, but instead chase-labeled Cx36 was found diffusely throughout the gap junction plaques. Furthermore, our photobleaching studies showed that about half of the labeled Cx36 was extraordinarily mobile within the gap junction plaque, suggesting that we would not be able to detect accretion of new connexins on plaque edges. It is not clear why these results differ so dramatically from those using other connexins with other tags. Our results with Cx43-HaloTag constructs showed some mixing throughout gap junctions, although to a lesser extent than Cx36. Cx43 also recovered partially from photobleaching, again with about half of the labeled protein being mobile. Thus, whereas the properties of individual connexin types do differ, the presence of the HaloTag may impart greater mobility to connexins than a fluorescent protein or tetracysteine tag. We should note that the fluorescently labeled HaloTag connexins are much brighter than fluorescent-protein-labeled connexins, so our observations of the chase label may have higher sensitivity. Additional studies will be required to determine if the HaloTag specifically imparts higher mobility to connexins or other proteins.
Gap junction protein turnover
Pulse-chase analysis is the most commonly used method to study the turnover rate of a protein in cell cultures. With the Cx36-Halo construct, we were able to label Cx36 at two different time points efficiently and specifically. Our results demonstrate that Cx36 protein in gap junction plaques is replaced with a half-life of 3.1 h, which is consistent with prior studies of turnover rates of other connexins. Furthermore, this turnover rate was not affected by manipulating the activity of PKA, whereas tracer coupling of Cx36 gap junctions is regulated by the same perturbations of PKA activity within minutes (O’Brien et al., 2004; Ouyang et al., 2005). Thus regulation of functional plasticity and turnover are at least partially separate. It is important to note that this is the turnover rate for Cx36 in gap junction plaques; the measurement is not confounded by the presence of Cx36 protein in other cellular compartments. In this sense, the pulse-chase imaging approach has been able to provide information that has not been available with traditional pulse-chase methodologies.
Although our reported half-life of Cx36 in HeLa cells is consistent with the studies of other connexins in cell cultures and whole organs (Hervé et al., 2007), there are still considerable factors that may influence the measurement of half-lives. Turnover rates of plasma membrane proteins have been reported to be very different in different cell types. For example, Cx32 showed a half-life of 4–6 h in rat hepatocytes (Hidaka et al., 1989), but only 2.5–3 h in mouse embryo hepatocytes (Traub et al., 1987). In contrast, pulse-chase analyses showed that Cx43 had a similar half-life in metabolically labeled rat heart (Beardslee et al., 1998) and cultured myocytes (Laird et al., 1991; Darrow et al., 1995; Laing et al., 1998). HeLa cells are a transformed epithelial cell line in which Cx36 may turn over at a different rate than in neurons.
The reasons for the relatively fast turnover rates of connexins have remained elusive. It is possible that connexin proteins, like many other integral membrane proteins, have to respond constantly to physiological changes. For example, degradation of Cx43 in bovine retinal endothelial cells is enhanced by hyperglycemia, reducing the half-life of Cx43 from 2.3 to 1.9 h (Fernandes et al., 2004). Gap junction intercellular communication was concomitantly reduced, suggesting that natural stress can modify coupling by altering the turnover rate of connexins.
Gap junctions consisting of Cx36 also undergo substantial plasticity that can be related to connexin turnover. In brain injuries, including ischemic events, epilepsy and compression injury, transient increases in neuronal coupling and Cx36 expression are observed (Oguro et al., 2001; Ohsumi et al., 2006; Wang et al., 2012). This coupling has negative consequences for neuron survival as the extent of cell death is greatly reduced in Cx36 knockout animals or when gap junction inhibitors are applied (Talhouk et al., 2008; Paschon et al., 2012; Belousov and Fontes, 2013). The increase in Cx36 expression is driven by type II metabotropic glutamate receptors and involves post-transcriptional mechanisms (Wang et al., 2012). In insulin-secreting β-cells of pancreatic islets, glucose suppresses Cx36 expression through a cAMP-cAMP response element signaling pathway (Allagnat et al., 2005). Changes in coupling of these cells have a strong influence on insulin secretion (Ravier et al., 2005). In each of these examples, control of turnover could play a significant role in plasticity.
Contribution of Cx36 turnover to electrical synaptic plasticity
Electrical synapses display a great deal of functional plasticity, being subject to alterations in coupling strength due to biophysical properties of the coupled cells, activity-dependent modification of the synapses, and modification driven by neurotransmitters (Pereda et al., 2013; O'Brien, 2014). The mechanisms responsible for these forms of plasticity are of great interest. In previous studies, profound dopamine-driven short-term plasticity of coupling in Cx36 gap junctions on AII amacrine cell dendrites in the rabbit retina was found to be directly correlated with phosphorylation of regulatory sites on Cx36, with no changes in number or size of the gap junctions (Kothmann et al., 2009). Similarly, Li et al. also showed a direct relation of dramatic changes in photoreceptor coupling during light or dark adaptation in the mouse retina to Cx36 phosphorylation (Li et al., 2013), whereas the number of Cx36 plaques per unit area in the photoreceptor synaptic layer was not affected. However, Katti et al., found that Cx36 protein level was regulated by diurnal and circadian rhythms in mouse retina (Katti et al., 2013). The level of Cx36 transcript peaked in the late night and immunolabeling showed higher Cx36 expression level in the night than in the day in the photoreceptor synaptic layer. This suggests that Cx36 gap junctions in the photoreceptor layer may contain more protein at night than during the day, reflecting the fluctuation in Cx36 protein synthesis rate and potentially contributing to the elevated coupling at night.
In all electrical synapses, the continuous insertion and removal of connexons through turnover has the potential to alter electrical synaptic strength. In studies of goldfish Mauthner cell mixed synapses, Flores et al. found that introduction of synthetic peptides that disrupt endocytosis or formation of SNARE complexes enhanced or reduced electrical coupling, respectively (Flores et al., 2012). Glutamatergic transmission was also altered in parallel. Peptides that mimicked the C-terminus of Cx36, interfering with Cx36 interactions with scaffolding proteins, also reduced electrical coupling. These experiments indicate that steady-state maintenance of gap junctions through turnover is important for maintenance of electrical coupling. They further demonstrate that the C-terminus of Cx36 is important in stabilizing the gap junctions.
Our studies in HeLa cells indicate that turnover of Cx36 and functional plasticity driven by changes in connexin phosphorylation occur on different time scales. This is likely also to be the case in neurons, in which short-term changes in coupling of neurons driven by transmitters such as dopamine, which take minutes to achieve, do not appear to be accompanied by a change in Cx36 abundance. However, it is certainly reasonable to expect regulation of insertion and removal/degradation rates to contribute to changes in coupling strength in longer time frames such as through a circadian cycle. Changes in Cx36 expression and neuronal coupling occur on an hours to weeks time scale during the course of central nervous system development and following a variety of types of injury. These changes are regulated by neurotransmitters and involve both transcriptional and post-transcriptional mechanisms (Belousov and Fontes, 2013). Factors that control turnover must be important in establishing the steady-state level of Cx36 expression, and it is apparent that these can be modified under certain circumstances.
MATERIALS AND METHODS
Connexin-HaloTag constructs
The HaloTag open reading frame was inserted into an internal site in the C-terminus of perch Cx35 (non-mammalian homolog of Cx36) in pcDNA 3.1 Zeo (O’Brien et al., 2004). The location of the insertion was chosen so as not to disrupt the regulatory C-terminal phosphorylation sites nor block the C-terminal PDZ interaction motif (see Fig. 1). Cx35 pcDNA was split in its C-terminus using whole plasmid polymerase chain reaction (PCR) amplification with primers 5′-TTCAGAGCCCGAGGATGAGTGTGCC-3′ (forward) and 5′-TCTGCCATCAAGTCCTTATTTCTGATCTC-3′ (reverse). The HaloTag open reading frame was amplified by PCR from the HaloTag-N vector (Promega, Madison, WI) with primers 5′-AGGACTTGATGGCAGAAATCGGTACTGGC-3′ (forward) and 5′-ATCCTCGGGCTCTGAAAGTACAGATCCTCAGTG-3′ (reverse). Phusion high-fidelity polymerase (New England BioLabs, Ipswich, MA) was used for both PCR reactions. The purified PCR products were cloned using cold fusion cloning (System Biosciences, Mountain View, CA). This clone is called Cx36-Halo-C-IN, and referred to as Cx36-Halo throughout the manuscript.
A conventional C-terminal fusion of the HaloTag open reading frame to perch Cx35 was made using Cx35 in the EGFP-N1 vector (Clontech, Mountain View, CA). Enhanced green fluorescent protein (EGFP) was deleted with SmaI and NotI. The HaloTag open reading frame was cut out from the HaloTag-C vector (Promega) with XhoI, filled in with Klenow polymerase, and NotI, and cloned into the Cx35-containing vector. This clone is called Cx36-Halo-C-END.
A human Cx43 cDNA clone (NM_000165.3) in pReceiver-M02 was purchased from Genecopoeia (Rockville, MD). Two constructs containing the HaloTag at internal sites within the C-terminus were created using native XcmI and EcoRI restriction sites. These constructs were named with reference to the amino acid position following which the HaloTag was inserted: Cx43-Halo-Cinternal-249 and Cx43-Halo-Cinternal-342, respectively. HaloTag was amplified from HaloTag-N (Promega) with primer pairs 5′-TTCCCCGATGATAACCAGATGGCAGAAATCGGTACTGGCT-3′ (forward) and 5′-AGCAGCTAGTTTTTTAGAATTCTCGCCGGAAATCTCGAGC-3′ (reverse) for EcoRI and 5′-AGCGACCCTTACCATGCGATGGCAGAAATCGGTACTGGCT-3′ (forward) and 5′-GGCTCAGCGCACCACTGGTCTCGCCGGAAATCTCGAGC-3′ (reverse) for XcmI using Q5 polymerase (New England BioLabs), and cloned into the respective restriction sites of Cx43 using Gibson Assembly (New England BioLabs). A C-terminal fusion of Cx43 was produced by amplifying Cx43 with primer pair 5′-GCAAAGCGATCGCTTCCGGAGTTCGAACCATGGGTGACTG-3′ (forward) and 5′-GATCCTCAGTGGTTGGCTCGAGGATCTCCAGGTCATCAGGCCG-3′ (reverse) using Q5 polymerase and cloning into EcoRI and XhoI sites of pHaloTag-N using Gibson Assembly. All clones were fully sequenced through the connexin-HaloTag fusion open reading frames.
Cell culture and reagents
All media, fetal bovine serum and cell culture reagents were obtained from Invitrogen (Grand Island, NY). HeLa cells (catalog number CCL-2) and HEK293 cells (catalog number CRL-1573) were obtained from American Type Culture Collection (Rockville, MD). Cells were grown in complete minimum essential medium (MEM) supplemented with 10% fetal bovine serum and 1% antibiotic-antimycotic (penicillin/streptomycin/amphotericin B). Cells were plated on 12 mm cover glasses, grown to 75% confluence overnight in 35 mm culture dishes, and transiently transfected with 2 µg of plasmid DNA per 35 mm culture dish using GenePORTER® 2 transfection reagent (Genlantis, San Diego, CA). Plasmids transfected included the connexin-HaloTag constructs described above, untagged Cx36 in pcDNA (Cx36 wild type) or HaloTag-N empty vector (control). Experiments were conducted 24 h after transfection.
General laboratory chemicals were obtained from Sigma (St Louis, MO). PKA activator, Sp-8-cpt-cAMPS and PKA inhibitor, Rp-8-cpt-cAMPS, were obtained from Alexis (San Diego, CA). HaloTag ligands Oregon Green (OG) and tetramethylrhodamine (TMR) were purchased from Promega, and Brefeldin A was purchased from Cell Signaling Technology (Danvers, MA).
Tracer coupling
Transfected HeLa cell cover glasses were maintained in oxygenated Ringer medium at 35°C. The medium was supplemented with 0.05% Neurobiotin and cells were scraped with a 25-gauge needle. Incubation with Neurobiotin was continued for 10 min to allow loading and diffusion. Cells were then washed twice with 0.1 M phosphate buffer to remove excess Neurobiotin and fixed with 4% paraformaldehyde (Electron Microscopy Sciences, Hatfield, PA) in 0.1 M phosphate buffer. When used, the PKA activator or inhibitor was added to the oxygenated incubation medium 10 min before the scrape for pre-incubation and replaced with fresh drug upon Neurobiotin addition. Drugs were present throughout the 10 min tracer diffusion period. Following fixation, cells were probed with streptavidin-Cy3 (Jackson ImmunoResearch, West Grove, PA) and photographed on a Zeiss (Thornwood, NY) fluorescence microscope (Axiovert 200 with 40×, 0.5 NA Hoffman Modulation Contrast objective) with a Hamamatsu C4742-95 digital camera using HCImage software (Hamamatsu, Sewickley, PA). Five images were taken of different patches of loaded cells for each experiment and treated as replicates in the data analysis.
The diffusion coefficient of Neurobiotin through the coupled network of HeLa cells was determined from fluorescence intensity data using a compartmental diffusion model (Zimmerman and Rose, 1985). The analysis uses a linear 25-compartment diffusion model to fit Neurobiotin concentration and diffusion distance measurements (O'Brien et al., 2004). This model has been applied to neural networks to assess gap junction coupling in the retina (Mills and Massey, 1998; O'Brien et al., 2004; Li et al., 2009). The movement of tracer between adjacent compartments is described by a series of 25 differential equations that are solved for tracer flux given the total amount of diffusion time and a diffusion coefficient, k. The diffusion coefficient k represents the proportion of tracer that diffuses from the first compartment to the next per second. Optimal fitting of intensity data to the model was determined in MATLAB (MathWorks, Natick, MA) by varying the diffusion coefficient k and another parameter, bo, the bolus loading rate. The parameter bo is defined as the rate of addition of tracer to the initial compartment for the loading period, which was assumed to be 1 min in the scrape-loading experiments, and was set to zero thereafter. Data fits were determined by plotting cell intensities on a log intensity axis and determining the diffusion coefficient k that best fitted the rate of decline of tracer intensity with distance from the cell of origin, and the rate of delivery, bo, that fitted the overall tracer concentration (Mills and Massey, 1998; O'Brien et al., 2004; Ouyang et al., 2005; Li et al., 2009). Replicate measurements were averaged to yield a single value for each treatment condition in each experiment. Diffusion coefficients were compared under different drug treatment conditions using a mixed effects model.
Labeling, immunostaining and imaging
Transfected HeLa cell cover glasses were incubated in Ringer medium containing 5 µM HaloTag TMR fluorescent ligand for 15 min. Cover glasses were then washed with ligand-free medium to remove unbound ligand and transferred to a microscope to capture live images.
After live cell imaging, cells were fixed with 4% paraformaldehyde in 0.1 M phosphate buffer for 10 min. Cover glasses were then incubated in immunolabeling buffer (PBS with 0.5% Triton X-100 and 0.1% NaN3, pH 7.4) with 10% donkey serum (Jackson ImmunoResearch, West Grove, PA) to block non-specific binding. Cover glasses were incubated overnight at 4°C with monoclonal mouse anti-Cx36 (mCx36, MAB3045, 1:1000 dilution; Millipore, Billerica, MA) primary antibody in immunolabeling buffer with 10% donkey serum, followed by Cy5 conjugated donkey anti-mouse secondary antibody (1:500 dilution, Jackson ImmunoResearch) in immunolabeling buffer with 5% donkey serum for 3 h. Cover glasses were then washed, mounted and transferred to a confocal microscope to capture images. HaloTag TMR was visualized with the TRITC filter set, and the Cx36 was visualized with the Cy5 filter set. Images of HeLa cells were digitally captured using a Zeiss LSM 510 Meta confocal microscope with a 63×, 1.4 NA Plan-Apochromat oil immersion objective using the PMT detectors and similar settings of pinhole, contrast and brightness parameters. Images were exported unaltered in TIFF format with Zeiss LSM Image Browser.
Pulse-chase analysis
Transfected HeLa cell cover glasses were incubated in Ringer medium containing 5 µM pulse labeling ligand OG for 15 min in a 24-well plate in a 37°C incubator. Cover glasses were then washed to remove unbound ligand and incubated with Ringer medium at 37°C. To analyze whether PKA activity altered Cx36 turnover rate, transfected HeLa cells were treated with Rp-8-cpt-cAMPS (5 µM) or Sp-8-cpt-cAMPS (5 µM) during the pulse-chase analysis. Rp or Sp was added to HeLa cells after 15 min of pulse label with OG, and was replaced every hour when necessary until the time for chase label. Cells were then labeled with tetramethylrhodamine (TMR) HaloTag ligand at various times (0.3, 1, 2, 3, 5 and 6 h after pulse labeling) for 15 min. Some experiments with Cx43-Halo and Cx36-Halo constructs used transfected human embryonic kidney (HEK) cell cover glasses with an abbreviated set of chase times. All cover glasses were fixed with 4% paraformaldehyde and transferred to a confocal microscope for image capturing. HaloTag TMR was visualized with the TRITC filter set, and HaloTag OG was visualized with the FITC filter set.
Images were captured with Zeiss LSM 510 or LSM 780 confocal microscopes as a series of confocal slices at 0.3 to 0.5 µm intervals. Acquisition parameters were initially set on control samples so that the brightest regions just reached saturation in a few pixels, while the background just reached zero. Subsequent images were collected with the same settings. Post-imaging processing was limited to making maximum intensity projections of stacks of images. Images were analyzed with the same settings using SimplePCI software (Hamamatsu, Sewickley, PA). Regions of interest (ROI) were selected by setting an intensity threshold and applying a minimum size threshold. ROIs were defined as contiguous pixels with intensity threshold greater than 20% of the total intensity range and which covered a minimum area of 200 pixels. Each image was then manually scanned for individual ROIs that fitted the criteria but were not part of the gap junction plaques. These were manually removed from the analysis. Mean and total fluorescent intensity was measured in each channel for each ROI. Ten to 20 gap junctions were analyzed at each time point in each experiment. At any given time point, the fraction of pulse label remaining was defined as the total pulse label divided by the total labeling (pulse+chase). The half-life of Cx36 was calculated by fitting an exponential function to the fraction of pulse label remaining over time.
Drug treatment: Brefeldin A
To analyze the transport of Cx36 from ER to the plasma membrane, transfected HeLa cells were treated with 2 µg/ml Brefeldin A in Ringer medium with 0.1% dimethyl sulfoxide (DMSO) during the pulse-chase analysis. BFA was added to the cells after 15 min of pulse label OG incubation and washing, and was left in the wells before the chase label TMR was added. Cells were incubated in TMR for 15 min. In the control experiment, cover glasses were treated with DMSO (0.1%) and the treatment was identical to that of BFA.
FRAP experiments
Fluorescence recovery after photobleaching (FRAP) experiments were performed using Zeiss LSM 780 and 880 confocal microscopes using 20× objectives and gallium arsenide phosphide (GaAsP) detectors employing the FRAP routines in Zeiss Zen software. Immediately after HaloTag OG labeling and 15 min of wash period, coverslips were mounted in Ringer medium and gap junctions imaged at a single focal plane at 0.5 to 2 s intervals using a low laser intensity to ensure gap junction stability. An ROI was selected away from edges of a gap junction plaque and a series of laser sweeps at 100% laser power from 0.4 to 9 s was used to bleach the selected area. Immediately after bleaching, the sample was imaged at regular time intervals using the same low intensity as before the bleaching until the intensity of the bleached area had reached a plateau. Bleachings were repeated three more times after the intensity plateaued to study the effect of repeated exposure. An internal vesicle was selected as an ROI as a negative control and was bleached and imaged in the same manner as the ROI in the gap junction plaque.
The droplet control was performed by suspending 5 µl of 1× working solution of the HaloTag OG ligand (Ringer medium containing 5 µM OG) in 1 ml mineral oil by vortexing. Drops of the emulsion on a coverslip were bleached and individual HaloTag OG ligand droplets were imaged in the same manner as connexin gap junctions.
Acknowledgements
We would like to thank Dr Alice Z. Chuang for assistance with statistical analyses.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
H.Y.W. designed the research, performed experiments, collected and analyzed data, and wrote the paper. C.K.M. and Y.-P.L. provided experimental support. S.R. provided assistance with FRAP experiments on the Zeiss LSM 880 confocal microscope. J.O.’B. designed the research, analyzed data and wrote the paper.
Funding
This research was supported by National Institutes of Health [grant numbers EY12857 and core grant EY10608], and by a challenge grant to the Department of Ophthalmology & Visual Science from Research to Prevent Blindness. Additional support was provided by the Vale-Asche Foundation through the Frederic B. Asche Endowment. H.Y.W. has support from the University of Texas Graduate School of Biomedical Sciences at Houston. Deposited in PMC for release after 12 months.
Supplementary information
Supplementary information available online at http://jcs.biologists.org/lookup/suppl/doi:10.1242/jcs.162586/-/DC1
References
- Ahmad S., Diez J. A., George C. H. and Evans W. H. (1999). Synthesis and assembly of connexins in vitro into homomeric and heteromeric functional gap junction hemichannels. Biochem. J. 339, 247-253. 10.1042/bj3390247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alev C., Urschel S., Sonntag S., Zoidl G., Fort A. G., Hoher T., Matsubara M., Willecke K., Spray D. C. and Dermietzel R. (2008). The neuronal connexin36 interacts with and is phosphorylated by CaMKII in a way similar to CaMKII interaction with glutamate receptors. Proc. Natl. Acad. Sci. USA 105, 20964-20969. 10.1073/pnas.0805408105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allagnat F., Martin D., Condorelli D. F., Waeber G. and Haefliger J.-A. (2005). Glucose represses connexin36 in insulin-secreting cells. J. Cell Sci. 118, 5335-5344. 10.1242/jcs.02600 [DOI] [PubMed] [Google Scholar]
- Allen K., Fuchs E. C., Jaschonek H., Bannerman D. M. and Monyer H. (2011). Gap junctions between interneurons are required for normal spatial coding in the hippocampus and short-term spatial memory. J. Neurosci. 31, 6542-6552. 10.1523/JNEUROSCI.6512-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beardslee M. A., Laing J. G., Beyer E. C. and Saffitz J. E. (1998). Rapid turnover of connexin43 in the adult rat heart. Circ. Res. 83, 629-635. 10.1161/01.RES.83.6.629 [DOI] [PubMed] [Google Scholar]
- Belluardo N., Mudò G., Trovato-Salinaro A., Le Gurun S., Charollais A., Serre-Beinier V., Amato G., Haefliger J.-A., Meda P. and Condorelli D. F. (2000). Expression of connexin36 in the adult and developing rat brain. Brain Res. 865, 121-138. 10.1016/S0006-8993(00)02300-3 [DOI] [PubMed] [Google Scholar]
- Belousov A. B. and Fontes J. D. (2013). Neuronal gap junctions: making and breaking connections during development and injury. Trends Neurosci. 36, 227-236. 10.1016/j.tins.2012.11.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blatow M., Rozov A., Katona I., Hormuzdi S. G., Meyer A. H., Whittington M. A., Caputi A. and Monyer H. (2003). A novel network of multipolar bursting interneurons generates theta frequency oscillations in neocortex. Neuron 38, 805-817. 10.1016/S0896-6273(03)00300-3 [DOI] [PubMed] [Google Scholar]
- Bloomfield S. A. and Völgyi B. (2004). Function and plasticity of homologous coupling between AII amacrine cells. Vis. Res. 44, 3297-3306. 10.1016/j.visres.2004.07.012 [DOI] [PubMed] [Google Scholar]
- Christie J. M., Bark C., Hormuzdi S. G., Helbig I., Monyer H. and Westbrook G. L. (2005). Connexin36 mediates spike synchrony in olfactory bulb glomeruli. Neuron 46, 761-772. 10.1016/j.neuron.2005.04.030 [DOI] [PubMed] [Google Scholar]
- Darrow B. J., Laing J. G., Lampe P. D., Saffitz J. E. and Beyer E. C. (1995). Expression of multiple connexins in cultured neonatal rat ventricular myocytes. Circ. Res. 76, 381-387. 10.1161/01.RES.76.3.381 [DOI] [PubMed] [Google Scholar]
- De Zeeuw C. I., Chorev E., Devor A., Manor Y., Van Der Giessen R. S., De Jeu M. T., Hoogenraad C. C., Bijman J., Ruigrok T. J., French P. et al. (2003). Deformation of network connectivity in the inferior olive of connexin 36-deficient mice is compensated by morphological and electrophysiological changes at the single neuron level. J. Neurosci. 23, 4700-4711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deans M. R., Gibson J. R., Sellitto C., Connors B. W. and Paul D. L. (2001). Synchronous activity of inhibitory networks in neocortex requires electrical synapses containing connexin36. Neuron 31, 477-485. 10.1016/S0896-6273(01)00373-7 [DOI] [PubMed] [Google Scholar]
- Deans M. R., Volgyi B., Goodenough D. A., Bloomfield S. A. and Paul D. L. (2002). Connexin36 is essential for transmission of rod-mediated visual signals in the mammalian retina. Neuron 36, 703-712. 10.1016/S0896-6273(02)01046-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- del Corsso C., Iglesias R., Zoidl G., Dermietzel R. and Spray D. C. (2012). Calmodulin dependent protein kinase increases conductance at gap junctions formed by the neuronal gap junction protein connexin36. Brain Res. 1487, 69-77. 10.1016/j.brainres.2012.06.058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Encell L. P., Friedman Ohana R., Zimmerman K., Otto P., Vidugiris G., Wood M. G., Los G. V., McDougall M. G., Zimprich C., Karassina N. et al. (2013). Development of a dehalogenase-based protein fusion tag capable of rapid, selective and covalent attachment to customizable ligands. Curr. Chem. Genomics 6, 55-71. 10.2174/1875397301206010055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falk M. M., Baker S. M., Gumpert A. M., Segretain D. and Buckheit R. W. III (2009). Gap junction turnover is achieved by the internalization of small endocytic double-membrane vesicles. Mol. Biol. Cell 20, 3342-3352. 10.1091/mbc.E09-04-0288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandes R., Girao H. and Pereira P. (2004). High glucose down-regulates intercellular communication in retinal endothelial cells by enhancing degradation of connexin 43 by a proteasome-dependent mechanism. J. Biol. Chem. 279, 27219-27224. 10.1074/jbc.M400446200 [DOI] [PubMed] [Google Scholar]
- Flores C. E., Nannapaneni S., Davidson K. G. V., Yasumura T., Bennett M. V. L., Rash J. E. and Pereda A. E. (2012). Trafficking of gap junction channels at a vertebrate electrical synapse in vivo. Proc. Natl. Acad. Sci. USA 109, E573-E582. 10.1073/pnas.1121557109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaietta G., Deerinck T. J., Adams S. R., Bouwer J., Tour O., Laird D. W., Sosinsky G. E., Tsien R. Y. and Ellisman M. H. (2002). Multicolor and electron microscopic imaging of connexin trafficking. Science 296, 503-507. 10.1126/science.1068793 [DOI] [PubMed] [Google Scholar]
- Guldenagel M., Ammermuller J., Feigenspan A., Teubner B., Degen J., Sohl G., Willecke K. and Weiler R. (2001). Visual transmission deficits in mice with targeted disruption of the gap junction gene connexin36. J. Neurosci. 21, 6036-6044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hervé J.-C., Derangeon M., Bahbouhi B., Mesnil M. and Sarrouilhe D. (2007). The connexin turnover, an important modulating factor of the level of cell-to-cell junctional communication: comparison with other integral membrane proteins. J. Membr. Biol. 217, 21-33. 10.1007/s00232-007-9054-8 [DOI] [PubMed] [Google Scholar]
- Hidaka S., Shingai R., Dowling J. E. and Naka K. (1989). Junctions form between catfish horizontal cells in culture. Brain Res. 498, 53-63. 10.1016/0006-8993(89)90398-3 [DOI] [PubMed] [Google Scholar]
- Hormuzdi S. G., Pais I., LeBeau F. E. N., Towers S. K., Rozov A., Buhl E. H., Whittington M. A. and Monyer H. (2001). Impaired electrical signaling disrupts gamma frequency oscillations in connexin 36-deficient mice. Neuron 31, 487-495. 10.1016/S0896-6273(01)00387-7 [DOI] [PubMed] [Google Scholar]
- Katti C., Butler R. and Sekaran S. (2013). Diurnal and circadian regulation of connexin 36 transcript and protein in the mammalian retina. Invest. Ophthalmol. Vis. Sci. 54, 821-829. 10.1167/iovs.12-10375 [DOI] [PubMed] [Google Scholar]
- Kothmann W. W., Li X., Burr G. S. and O'Brien J. (2007). Connexin 35/36 is phosphorylated at regulatory sites in the retina. Vis. Neurosci. 24, 363-375. 10.1017/S095252380707037X [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kothmann W. W., Massey S. C. and O'Brien J. (2009). Dopamine-stimulated dephosphorylation of connexin 36 mediates AII amacrine cell uncoupling. J. Neurosci. 29, 14903-14911. 10.1523/JNEUROSCI.3436-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kothmann W. W., Trexler E. B., Whitaker C. M., Li W., Massey S. C. and O'Brien J. (2012). Non-synaptic NMDA receptors mediate activity-dependent plasticity of gap junctional coupling in the AII amacrine cell network. J. Neurosci. 32, 6747-6759. 10.1523/JNEUROSCI.5087-11.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koval M., Harley J. E., Hick E. and Steinberg T. H. (1997). Connexin46 is retained as monomers in a trans-Golgi compartment of osteoblastic cells. J. Cell Biol. 137, 847-857. 10.1083/jcb.137.4.847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar N. M. and Gilula N. B. (1992). Molecular biology and genetics of gap junction channels. Semin. Cell Biol. 3, 3-16. 10.1016/S1043-4682(10)80003-0 [DOI] [PubMed] [Google Scholar]
- Laing J. G., Tadros P. N., Green K., Saffitz J. E. and Beyer E. C. (1998). Proteolysis of connexin43-containing gap junctions in normal and heat-stressed cardiac myocytes. Cardiovasc. Res. 38, 711-718. 10.1016/S0008-6363(98)00060-1 [DOI] [PubMed] [Google Scholar]
- Laird D. W. (1996). The life cycle of a connexin: gap junction formation, removal, and degradation. J. Bioenerg. Biomembr. 28, 311-318. 10.1007/BF02110107 [DOI] [PubMed] [Google Scholar]
- Laird D. W., Puranam K. L. and Revel J. P. (1991). Turnover and phosphorylation dynamics of connexin43 gap junction protein in cultured cardiac myocytes. Biochem. J. 273, 67-72. 10.1042/bj2730067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauf U., Giepmans B. N. G., Lopez P., Braconnot S., Chen S.-C. and Falk M. M. (2002). Dynamic trafficking and delivery of connexons to the plasma membrane and accretion to gap junctions in living cells. Proc. Natl. Acad. Sci. USA 99, 10446-10451. 10.1073/pnas.162055899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X., Olson C., Lu S., Kamasawa N., Yasumura T., Rash J. E. and Nagy J. I. (2004). Neuronal connexin36 association with zonula occludens-1 protein (ZO-1) in mouse brain and interaction with the first PDZ domain of ZO-1. Eur. J. Neurosci. 19, 2132-2146. 10.1111/j.0953-816X.2004.03283.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Chuang A. Z. and O'Brien J. (2009). Photoreceptor coupling is controlled by connexin 35 phosphorylation in zebrafish retina. J. Neurosci. 29, 15178-15186. 10.1523/JNEUROSCI.3517-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X., Lynn B. D. and Nagy J. I. (2012). The effector and scaffolding proteins AF6 and MUPP1 interact with connexin36 and localize at gap junctions that form electrical synapses in rodent brain. Eur. J. Neurosci. 35, 166-181. 10.1111/j.1460-9568.2011.07947.x [DOI] [PubMed] [Google Scholar]
- Li H., Zhang Z., Blackburn M. R., Wang S. W., Ribelayga C. P. and O'Brien J. (2013). Adenosine and dopamine receptors coregulate photoreceptor coupling via gap junction phosphorylation in mouse retina. J. Neurosci. 33, 3135-3150. 10.1523/JNEUROSCI.2807-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long M. A., Deans M. R., Paul D. L. and Connors B. W. (2002). Rhythmicity without synchrony in the electrically uncoupled inferior olive. J. Neurosci. 22, 10898-10905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Los G. V. and Wood K. (2007). The HaloTag: a novel technology for cell imaging and protein analysis. Methods Mol. Biol. 356, 195-208. [DOI] [PubMed] [Google Scholar]
- Martin A. O., Mathieu M. N., Chevillard C. and Guerineau N. C. (2001). Gap junctions mediate electrical signaling and ensuing cytosolic Ca2+ increases between chromaffin cells in adrenal slices: a role in catecholamine release. J. Neurosci. 21, 5397-5405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills S. L. and Massey S. C. (1998). The kinetics of tracer movement through homologous gap junctions in the rabbit retina. Vis. Neurosci. 15, 765-777. 10.1017/S0952523898154159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills S. L., O'Brien J. J., Li W., O'Brien J. and Massey S. C. (2001). Rod pathways in the mammalian retina use connexin 36. J. Comp. Neurol. 436, 336-350. 10.1002/cne.1071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musil L. S. and Goodenough D. A. (1993). Multisubunit assembly of an integral plasma membrane channel protein, gap junction connexin43, occurs after exit from the ER. Cell 74, 1065-1077. 10.1016/0092-8674(93)90728-9 [DOI] [PubMed] [Google Scholar]
- O'Brien J. (2014). The ever-changing electrical synapse. Curr. Opin. Neurobiol. 29, 64-72. 10.1016/j.conb.2014.05.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Brien J., Bruzzone R., White T. W., Al-Ubaidi M. R. and Ripps H. (1998). Cloning and expression of two related connexins from the perch retina define a distinct subgroup of the connexin family. J. Neurosci. 18, 7625-7637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Brien J., Nguyen H. B. and Mills S. L. (2004). Cone photoreceptors in bass retina use two connexins to mediate electrical coupling. J. Neurosci. 24, 5632-5642. 10.1523/JNEUROSCI.1248-04.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oguro K., Jover T., Tanaka H., Lin Y., Kojima T., Oguro N., Grooms S. Y., Bennett M. V. and Zukin R. S. (2001). Global ischemia-induced increases in the gap junctional proteins connexin 32 (Cx32) and Cx36 in hippocampus and enhanced vulnerability of Cx32 knock-out mice. J. Neurosci. 21, 7534-7542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohsumi A., Nawashiro H., Otani N., Ooigawa H., Toyooka T., Yano A., Nomura N. and Shima K. (2006). Alteration of gap junction proteins (connexins) following lateral fluid percussion injury in rats. Acta Neurochir. Suppl. 96, 148-150. 10.1007/3-211-30714-1_33 [DOI] [PubMed] [Google Scholar]
- Ouyang X., Winbow V. M., Patel L. S., Burr G. S., Mitchell C. K. and O'Brien J. (2005). Protein kinase A mediates regulation of gap junctions containing connexin35 through a complex pathway. Mol. Brain Res. 135, 1-11. 10.1016/j.molbrainres.2004.10.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paschon V., Higa G. S. V., Resende R. R., Britto L. R. G. and Kihara A. H. (2012). Blocking of connexin-mediated communication promotes neuroprotection during acute degeneration induced by mechanical trauma. PLoS ONE 7, e45449 10.1371/journal.pone.0045449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereda A. E., Curti S., Hoge G., Cachope R., Flores C. E. and Rash J. E. (2013). Gap junction-mediated electrical transmission: regulatory mechanisms and plasticity. Biochim. Biophys. Acta 1828, 134-146. 10.1016/j.bbamem.2012.05.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piehl M., Lehmann C., Gumpert A., Denizot J.-P., Segretain D. and Falk M. M. (2007). Internalization of large double-membrane intercellular vesicles by a clathrin-dependent endocytic process. Mol. Biol. Cell 18, 337-347. 10.1091/mbc.E06-06-0487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravier M. A., Guldenagel M., Charollais A., Gjinovci A., Caille D., Sohl G., Wollheim C. B., Willecke K., Henquin J.-C. and Meda P. (2005). Loss of connexin36 channels alters beta-cell coupling, islet synchronization of glucose-induced Ca2+ and insulin oscillations, and basal insulin release. Diabetes 54, 1798-1807. 10.2337/diabetes.54.6.1798 [DOI] [PubMed] [Google Scholar]
- Ribelayga C., Wang Y. and Mangel S. C. (2002). Dopamine mediates circadian clock regulation of rod and cone input to fish retinal horizontal cells. J. Physiol. 544, 801-816. 10.1113/jphysiol.2002.023671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribelayga C., Cao Y. and Mangel S. C. (2008). The circadian clock in the retina controls rod-cone coupling. Neuron 59, 790-801. 10.1016/j.neuron.2008.07.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saez J. C., Berthoud V. M., Branes M. C., Martinez A. D. and Beyer E. C. (2003). Plasma membrane channels formed by connexins: their regulation and functions. Physiol. Rev. 83, 1359-1400. 10.1152/physrev.00007.2003 [DOI] [PubMed] [Google Scholar]
- Serre-Beinier V., Le Gurun S., Belluardo N., Trovato-Salinaro A., Charollais A., Haefliger J. A., Condorelli D. F. and Meda P. (2000). Cx36 preferentially connects beta-cells within pancreatic islets. Diabetes 49, 727-734. 10.2337/diabetes.49.5.727 [DOI] [PubMed] [Google Scholar]
- Shaw R. M., Fay A. J., Puthenveedu M. A., von Zastrow M., Jan Y.-N. and Jan L. Y. (2007). Microtubule plus-end-tracking proteins target gap junctions directly from the cell interior to adherens junctions. Cell 128, 547-560. 10.1016/j.cell.2006.12.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talhouk R. S., Zeinieh M. P., Mikati M. A. and El-Sabban M. E. (2008). Gap junctional intercellular communication in hypoxia–ischemia-induced neuronal injury. Prog. Neurobiol. 84, 57-76. 10.1016/j.pneurobio.2007.10.001 [DOI] [PubMed] [Google Scholar]
- Thomas M. A., Huang S., Cokoja A., Riccio O., Staub O., Suter S. and Chanson M. (2002). Interaction of connexins with protein partners in the control of channel turnover and gating. Biol. Cell 94, 445-456. 10.1016/S0248-4900(02)00015-1 [DOI] [PubMed] [Google Scholar]
- Thomas T., Jordan K., Simek J., Shao Q., Jedeszko C., Walton P. and Laird D. W. (2005). Mechanisms of Cx43 and Cx26 transport to the plasma membrane and gap junction regeneration. J. Cell Sci. 118, 4451-4462. 10.1242/jcs.02569 [DOI] [PubMed] [Google Scholar]
- Traub O., Look J., Paul D. and Willecke K. (1987). Cyclic adenosine monophosphate stimulates biosynthesis and phosphorylation of the 26 kDa gap junction protein in cultured mouse hepatocytes. Eur. J. Cell Biol. 43, 48-54. [PubMed] [Google Scholar]
- Van Der Giessen R. S., Koekkoek S. K., van Dorp S., De Gruijl J. R., Cupido A., Khosrovani S., Dortland B., Wellershaus K., Degen J., Deuchars J. et al. (2008). Role of olivary electrical coupling in cerebellar motor learning. Neuron 58, 599-612. 10.1016/j.neuron.2008.03.016 [DOI] [PubMed] [Google Scholar]
- Wang Y. and Mangel S. C. (1996). A circadian clock regulates rod and cone input to fish retinal cone horizontal cells. Proc. Natl. Acad. Sci. USA 93, 4655-4660. 10.1073/pnas.93.10.4655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., Song J.-H., Denisova J. V., Park W.-M., Fontes J. D. and Belousov A. B. (2012). Neuronal gap junction coupling is regulated by glutamate and plays critical role in cell death during neuronal injury. J. Neurosci. 32, 713-725. 10.1523/JNEUROSCI.3872-11.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J. T., Chen M., Foote C. I. and Nicholson B. J. (1996). Membrane integration of in vitro-translated gap junctional proteins: co- and post-translational mechanisms. Mol. Biol. Cell 7, 471-482. 10.1091/mbc.7.3.471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmerman A. L. and Rose B. (1985). Permeability properties of cell-to-cell channels: kinetics of fluorescent tracer diffusion through a cell junction. J. Membr. Biol. 84, 269-283. 10.1007/BF01871390 [DOI] [PubMed] [Google Scholar]