

Abstract
Adseverin is a member of the calcium-regulated gelsolin superfamily of actin-binding proteins. Here we report the crystal structure of the calcium-free N-terminal half of adseverin (iA1–A3) and the Ca2+-bound structure of A3, which reveal structural similarities and differences with gelsolin. Solution small-angle X-ray scattering combined with ensemble optimization revealed a dynamic Ca2+-dependent equilibrium between inactive, intermediate and active conformations. Increasing calcium concentrations progressively shift this equilibrium from a main population of inactive conformation to the active form. Molecular dynamics simulations of iA1–A3 provided insights into Ca2+-induced destabilization, implicating a critical role for the A2 type II calcium-binding site and the A2A3 linker in the activation process. Finally, mutations that disrupt the A1/A3 interface increase Ca2+-independent F-actin severing by A1–A3, albeit at a lower efficiency than observed for gelsolin domains G1–G3. Together, these data address the calcium dependency of A1–A3 activity in relation to the calcium-independent activity of G1–G3.
 The calcium-regulated gelsolin family of actin-binding proteins includes adseverin. Here, the authors report the X-ray crystal structure of the N-terminal portion of adseverin and use small-angle scattering and molecular dynamics simulations to examine the calcium-dependent function of this protein.
The calcium-regulated gelsolin family of actin-binding proteins includes adseverin. Here, the authors report the X-ray crystal structure of the N-terminal portion of adseverin and use small-angle scattering and molecular dynamics simulations to examine the calcium-dependent function of this protein.
Adseverin (also known as scinderin) is a member of the gelsolin superfamily of Ca2+-dependent actin-binding proteins, which regulate cytoskeletal dynamics by severing, bundling and capping of actin filaments1. The gelsolin superfamily groups a number of modular, evolutionary-related proteins that are composed of 3–6 globular gelsolin homology (GH) domains often fused to non-GH N- and/or C-terminal extensions2. In humans, family members include gelsolin, adseverin, villin, supervillin, advillin, villin-like protein, flightless I and CapG (reviewed in ref. 3). Adseverin was first identified in chromaffin cells of bovine adrenal medulla, where it acts as an actin depolymerizing agent that disassembles the cortical network of actin filaments in response to bursts of intracellular calcium, leading to exocytosis of secretary vesicles4,5,6. Adseverin has also been shown to be involved in the regulation of osteoclastogenesis7 and thrombocytic8, chondrocytic9 and odontoblastic differentiation10, as well as in cisplatin resistance in bladder cancer11. Recently, several studies revealed a role for adseverin in the proliferation of lung carcinoma12, and gastric13 and prostate cancer cell lines14.
Adseverin is composed of six GH domains (A1–A6) and shares 60% overall sequence identity with gelsolin (G1–G6). Adseverin and gelsolin have been shown to have two calcium-binding events, with Kds of 0.6 and 3 μM for adseverin4, and Kds of 0.3 and 1.2 μM for gelsolin15. Low-affinity calcium association occurs with Kds of 25 and 200 μM for gelsolin16. Crystallographic studies revealed that these binding events involve the association of eight calcium ions, six of which bind at a conserved site on each domain (type II), whereas the remaining two are also stabilized by contacts with actin (type I; refs 17, 18, 19, 20). Both proteins are able to sequester two monomers of actin, as well as sever and cap actin filaments through distinct Ca2+-induced domain rearrangements of the two halves of the molecule4,17,18,21,22,23. In addition, both proteins are inhibited by membrane lipids such as PIP and PIP2, although adseverin is additionally rendered inactive by phosphatidylinositol and phosphatidylserine24,25.
The crystal structure of calcium-free gelsolin26 revealed an inactive, globular state, where all the actin-binding sites are obscured by three Ca2+-dependent latches. In this state, the two halves of the molecule interact and adopt a pseudo-symmetric conformation in which the major actin-binding sites on domains G1 and G4 are, respectively, buried by interactions with G3 and G6, while the actin-binding site on G2 is masked by interactions with the C-terminal α-helical extension, which is absent in adseverin. In this conformation, the long α-helices of G3 and G6 adopt a kinked structure, which is straightened by the Ca2+-induced release of the latches27.
The structures of the N- and C-terminal halves of gelsolin in complex with actin17,18 have demonstrated that distinct Ca2+-induced domain rearrangements of each half of the molecule are crucial for actin binding. The N-terminal half adopts a conformation in which a new Ca2+-stabilized G2:G3 interface is formed while G1 dissociates from the rest of the molecule to bind in the groove between actin subdomains 1 and 3 (refs 18, 20). In contrast, the C-terminal half adopts a compact V-shaped structure where the G4/G6 latch is released by flipping over G6 to create a new G5:G6 interface17,22. Small-angle X-ray scattering (SAXS) studies showed a significant calcium-induced increase in the molecular dimensions of gelsolin, and ab initio reconstructions suggested an opening of the two halves and domain rearrangements consistent with the architecture of the active forms observed crystallographically28.
Despite extensive homology between adseverin and gelsolin, adseverin lacks the gelsolin C-terminal α-helix latch, a feature that likely explains a less complex relationship between severing rate and calcium concentration than observed for gelsolin21. In addition, the actin filament severing and capping activities of the N-terminal half of adseverin (A1–A3) are Ca2+ dependent29, whereas the same activities of G1–G3 are Ca2+ independent30. These differences, combined with similar activated structures and actin sequestering properties of the C-terminal halves, led to the proposal that the activation process of adseverin is dominated by calcium binding to its N-terminal half23. Recently, SAXS studies demonstrated that G1–G3 adopts an active conformation in the absence of calcium31, suggesting that the Ca2+-independent severing activity of this fragment results from instability of the G1/G3 latch in the absence of interaction with G4–G6 and the C-terminal α-helix latch.
Here we investigated the structure, dynamics and calcium activation of A1–A3 by using a combination of X-ray crystallography, SAXS, molecular dynamics simulations (MDS) and ensemble optimization. We crystallized A1–A3 in the presence of EGTA, revealing a compact auto-inhibited structure similar to the conformational state observed in inactive full-length gelsolin. Unexpectedly, attempts to crystallize the active form of A1–A3 in the presence of 5 mM calcium led to a structure of Ca2+-bound A3 following protein degradation within the crystallization drop. Using SAXS-based atomic ensemble optimization, we were able to draw a comprehensive picture of the conformational landscape of A1–A3 and track Ca2+-induced conformational changes. Classical MDS provided insights into the calcium-induced destabilization of inactive A1–A3, and revealed the location of potential low-affinity calcium-binding sites. Finally, actin depolymerization assays showed that mutations that destabilize the inactive A1:A3 interface increase A1–A3 severing activity in calcium-free conditions, demonstrating the importance of this latch.
Results
Calcium-free A1–A3 displays an inactive conformation
Purified recombinant A1–A3, supplemented with 5 mM EGTA, crystallized in space group C2 and its structure was solved by molecular replacement using the structure of G1–G3 excised from inactive full-length gelsolin as the search model and refined to a resolution of 2.9 Å (Fig. 1a and Table 1). Each domain adopts the classic GH fold, which is organized around a central five-stranded mixed β-sheet sandwiched between a long α-helix running approximately parallel to the strands, and a shorter α-helix running approximately perpendicular to the strands. The domains adopt the compact arrangement characteristic of inactive gelsolin, with A1 sandwiched between A2 and A3. The structure appears to be relatively rigid, with B-factors indicating that flexibility is limited mainly to the extended A2A3 linker and the short α-helices of A1 and A2 (Supplementary Fig. 1a). In this inactive conformation, the long α-helix of A3 is kinked to enable the formation of the A1/A3 latch, resulting in the masking of the major actin-binding interface on A1. The inactive forms of A1–A3 (iA1–A3) and G1–G3 (iG1–G3) are highly similar with an overall root mean squared deviation (r.m.s.d.) in Cα positions of 1.19 Å over 337 aligned residues, highlighting the structural conservation between gelsolin and adseverin (Fig. 1b). It is noteworthy that contrary to adseverin, iG1–G3 is unstable in the absence of G4–G6, leading to an open conformation of G1–G3 in the presence of EGTA31. The iA1–A3 crystal lattice is stabilized by a number of crystal contacts, including a relatively large packing interface of ∼1,518 Å2 (Supplementary Fig. 1b). Interestingly, comparisons with full-length gelsolin show that this interface largely buries the surfaces in iA1–A3 that bind the C-terminal half of the molecule, suggesting that the crystal environment partially mimics the inactive full-length protein (Supplementary Fig. 1c).
Figure 1. Crystal structures of the N-terminal half of adseverin in calcium-free conditions (iA1–A3) and of calcium-bound domain A3 (aA3).

(a) The structure of iA1–A3 (residues 6σ349) is shown in cartoon representation and coloured from blue to red (N- to C terminus). (b) Comparison of the structure of iA1–A3 with domains iG1–G3 of gelsolin excised from the structure of full-length gelsolin in calcium-free conditions (PDB 1D0N)26, showing a similar compact arrangement of the domains that masks the conserved actin-binding interfaces on A1 and A2. G1–G3 is shown in grey cartoon representation. (c) The calcium-bound structure of adseverin domain A3 (aA3, residues 248–349), shown in cartoon representation and coloured from blue to red (N- to C terminus). The calcium ion is shown as a magenta sphere with coordinating residues as sticks. Inset: close up of the calcium-binding site, showing the coordination of the calcium ion by the Glu280 carbonyl, monodentate Glu281 and bidentate Glu305 side chains, as well as three water molecules. The 2Fo-Fc electron density map contoured at 1.5σ is shown as a mesh. (d) Comparison of the structure of aA3 (coloured as blue to red rainbow) and iA1–A3 (in grey). Superimposition highlights the conformational changes in the second β-strand and the long α-helix of A3 in response to calcium binding and dissociation from A1.
Table 1. Data collection and refinement statistics for iA1–A3 and aA3.
| iA1–A3 (PDB ID: 5A1K) | aA3 (PDB ID: 5A1M) | |
|---|---|---|
| Data collection | ||
| Space group | C2 | I422 |
| Cell dimensions | ||
| a, b, c (Å) | 135.4 , 65.3, 105.2 | 92.5, 92.5, 86.9 |
| α, β, γ (°) | 90, 118.3, 90 | 90, 90, 90 |
| Resolution (Å) | 59.6–2.9 (3.0–2.9) | 35.5–1.8 (1.9–1.8) |
| Rmerge, % | 15.3 (133.0) | 10.8 (88.9) |
| Average I/Io | 10.4 (1.5) | 21.6 (3.5) |
| Completeness, % | 98 (95) | 100 (97) |
| Redundancy | 5.5 (5.5) | 14.2 (14.1) |
| Refinement | ||
| Resolution (Å) | 59.6–2.9 (3.0–2.9) | 35.5–1.8 (1.9–1.8) |
| No. of reflections | 17,745 (2,818) | 13,831 (2,615) |
| Rwork/Rfree | 24.0 (30.2)/24.8 (33.3) | 18.2 (20.4)/18.9 (22.5) |
| No. of atoms | ||
| Protein | 5,402 | 815 |
| Ligand/ion | 0 | 1 |
| Water | 107 | 58 |
| B-factors | ||
| Protein | 61 | 20 |
| Ligand/ion | 0 | 17 |
| Water | 51 | 28 |
| r.m.s.d. | ||
| Bond lengths (Å) | 0.008 | 0.008 |
| Bond angles (°) | 1.120 | 0.980 |
r.m.s.d., root mean squared deviation.
Calcium-bound structure of A3
Attempts to crystallize the active form of A1–A3 (aA1–A3) using a 5-mM CaCl2 supplemented protein sample led to the appearance of a single crystal after ∼80 days, from which a 1.8-Å data set was collected. Cell content analysis indicated that the asymmetric unit was too small to accommodate more than a single GH domain, and the structure was solved by molecular replacement using gelsolin domain G3 excised from the structure of actin-bound G1–G3 as a search model (aG3) (PDB 1RGI). This finding was surprising given that homology with gelsolin suggests that in the active form, domains A2 and A3 interact through a Ca2+-stabilized interface, while A1 would be separated by the A1A2 linker18,20; it would thus have been expected that degradation would lead to a structure of isolated A1, or a compact, Ca2+-stabilized structure of A2–A3, rather than the Ca2+-bound A3 structure obtained here.
The conformation of aA3 is highly similar to aG3, with a r.m.s.d. in Cα positions of 0.82 Å over 93 Cα atoms, and differs from inactive iA3 and iG3 by the position of the second β-strand and the straightening of the long α-helix (Fig. 1c,d). The structure is stabilized by Ca2+ coordination at the type II binding site, which involves interactions with Glu280 carbonyl, the carboxylates of Glu281 and Glu305, and three water molecules with bond distances ranging between 2.3 and 2.5 Å (Fig. 1c, inset, and Supplementary Fig. 1d). As previously noted for gelsolin, this Ca2+-bound conformation is incompatible with the formation of the A1–A3 latch (Fig. 1d), suggesting that calcium binding at A3 moves A1–A3 away from the inactive conformation through straightening of the A3 long helix resulting in the disruption of the A1:A3 interface18,27.
Taken together, these results indicate that the Ca2+-induced conformational changes in adseverin mirror those observed in gelsolin, although in isolation iA1–A3 retains the inactive conformation that is lost by isolated Ca2+-free G1–G3. Furthermore, the structure of aA3 obtained by degradation of A1–A3 in the presence of calcium indicates that A3 may exist in a conformational state where it is distant from A1 and A2.
SAXS experiments
Next, we investigated the structure and calcium activation of A1–A3 using solution SAXS. Scattering profiles for A1–A3 were initially measured in the presence of 2 or 10 mM EGTA, and with increasing concentrations of CaCl2 (10–500 μM of free Ca2+), at protein concentrations of 1, 2, 4 and 8 mg ml−1 (Fig. 2a, curve series 1–4). The samples were aggregation free, as evidenced by the linearity of the Guinier region (Supplementary Fig. 2), with radii of gyration (Rg) ranging between 2.6 and 3.2 nm, and molecular weight estimates ranging from 35 to 42 kDa (Table 2), consistent with the theoretical value of 41.5 kDa. The Ca2+-induced changes in the scattering profiles were more pronounced at lower protein concentrations, which is to be expected as the Kds for the high-affinity calcium-binding sites are lower than the protein concentrations used in the experiment (Kd=0.6 and 3 μM versus c=24–192 μM). An additional calcium titration was performed at 4 mg ml−1 of protein using an increased range of calcium concentrations, in order to check for potential changes at very low (10 or 50 nM), as well as higher calcium concentrations (2 or 50 mM; Fig. 2a, curve series 5, and Table 2). These measurements, performed on a different synchrotron visit using a different protein preparation, showed Ca2+-induced changes that were similar to those observed in the previously acquired data (Fig. 2a, curve series 3, Supplementary Fig. 3a). All measured Rg were, however, smaller by 1–2 Å, suggesting mild degradation of the sample (Fig. 2b and Table 2). To gain qualitative insights into the Ca2+-induced conformational changes, the data were plotted using the Kratky representation (Fig. 2c). The analysis revealed that A1–A3 is predominantly globular in the absence of calcium, resulting in a bell-shaped curve, and that addition of calcium leads to a more complex profile suggesting modularity and significant interdomain flexibility32.
Figure 2. SAXS experiments.

(a) SAXS profiles of A1–A3 in the presence of 10 mM EGTA (black spheres), 2 mM EGTA (red spheres) and with increasing calcium concentrations (10, 220, 390 and 490 μM CaCl2, green, yellow, purple and pink spheres, respectively) measured at protein concentrations of 1, 2, 4 and 8 mg ml−1 (curve series 1, 2, 3 and 4, respectively). Curve series (5) corresponds to a second calcium titration at 4 mg ml−1 that spans a larger range of calcium concentrations (2 mM EGTA, 19 and 49 nM, 175 and 550 μM and 2.3 and 50 mM free Ca2+) and was measured on a separate synchrotron visit using a different protein preparation. (b) Changes in radius of gyration as a function of calcium concentration corresponding to the curve series in a: (1) black, (2) red, (3) green, (4) yellow and (5) purple line. (c) Model-free analysis of calcium-induced conformational changes. The data shown in a, curve series (5) is represented as Kratky plots, indicating a transition towards a less globular and more flexible structure.
Table 2. SAXS-derived parameters.
| EGTA/free calcium concentration | c (mg ml−1) | MW (kDa) | Rg (nm) | Dmax (nm) | χexp |
|---|---|---|---|---|---|
| 10 mM EGTA | 1.00 | 40 | 2.64 | 9.0 | 0.915 |
| 10 mM EGTA | 2.00 | 42 | 2.62 | 9.1 | 1.123 |
| 10 mM EGTA | 4.00 | 39 | 2.65 | 9.1 | 1.524 |
| 10 mM EGTA | 8.00 | 40 | 2.67 | 9.3 | 1.674 |
| 2 mM EGTA | 1.00 | 39 | 2.61 | 9.1 | 0.832 |
| 2 mM EGTA | 2.00 | 39 | 2.61 | 9.3 | 1.098 |
| 2 mM EGTA | 4.00 | 39 | 2.69 | 9.1 | 1.198 |
| 2 mM EGTA | 8.00 | 40 | 2.70 | 9.2 | 1.461 |
| 10 μM Ca2+ | 1.00 | 36 | 3.05 | 10.0 | 0.803 |
| 10 μM Ca2+ | 2.00 | 39 | 3.05 | 10.6 | 0.865 |
| 10 μM Ca2+ | 4.00 | 38 | 2.95 | 10.3 | 0.939 |
| 10 μM Ca2+ | 8.00 | 39 | 2.90 | 10.1 | 1.291 |
| 220 μM Ca2+ | 1.00 | 33 | 3.07 | 10.4 | 0.786 |
| 220 μM Ca2+ | 2.00 | 37 | 3.02 | 10.4 | 0.862 |
| 220 μM Ca2+ | 4.00 | 38 | 2.96 | 10.3 | 0.947 |
| 220 μM Ca2+ | 8.00 | 40 | 2.98 | 10.4 | 1.361 |
| 380 μM Ca2+ | 1.00 | 36 | 3.03 | 10.5 | 0.827 |
| 380 μM Ca2+ | 2.00 | 36 | 3.03 | 11.5 | 0.862 |
| 380 μM Ca2+ | 4.00 | 38 | 3.02 | 10.0 | 1.162 |
| 380 μM Ca2+ | 8.00 | 40 | 2.97 | 9.9 | 1.281 |
| 490 μM Ca2+ | 1.00 | 35 | 3.10 | 10.5 | 0.830 |
| 490 μM Ca2+ | 2.00 | 36 | 3.10 | 10.4 | 0.874 |
| 490 μM Ca2+ | 4.00 | 39 | 3.19 | 10.2 | 0.914 |
| 490 μM Ca2+ | 8.00 | 40 | 3.19 | 10.5 | 1.131 |
| 2 mM EGTA | 4.00 | 38 | 2.56 | 8.8 | 1.386* |
| 19 nM Ca2+ | — | 38 | 2.57 | 8.9 | 1.323* |
| 49 nM Ca2+ | — | 38 | 2.60 | 8.7 | 1.343* |
| 175 μM Ca2+ | — | 38 | 2.91 | 9.7 | 1.006* |
| 550 μM Ca2+ | — | 39 | 3.16 | 10.9 | 0.947* |
| 2.3 mM Ca2+ | — | 43 | 3.61 | 12.3 | 1.262* |
| 50 mM Ca2+ | — | 43 | 3.67 | 13.5 | 2.053* |
SAXS, small-angle X-ray scattering.
*Optimal fitting of the data was obtained using ensembles that include a deletion of the last 15 C-terminal amino acids (residues 350–365) that belong to the A3–A4 linker and are intrinsically disordered in solution.
Ca2+-dependent activation of A1–A3
In order to analyse Ca2+-induced changes detected by SAXS, we combined atomistic coarse-grained MDS with ensemble optimization (Supplementary Note 1), demonstrating that A1–A3 transitions from the inactive state (I) to a gelsolin-like active state (A) through two distinct intermediates (Int1 and Int2) characterized by the release of A1/A3 latch and subsequent loss of the A1:A2 interface, respectively (Fig. 3a). All available data could be well fitted using this approach (Fig. 3b), with a goodness of fit χexp<2 for 0.08<Q<5 nm−1 (Table 2), except for data measured in the presence of 50 mM free calcium (χexp=2.05), which was not analysed further. Data measured at 4 mg ml−1 (Fig. 2a, curve series 5), which showed mild signs of degradation, could, however, only be optimally fitted using ensembles in which the last disordered 15 amino acids were truncated (residues 350–365—part of the A3A4 linker; Table 2 and Supplementary Fig. 4d,e). To understand the nature of Ca2+-induced conformational changes, we analysed the Rg distributions (Supplementary Fig. 4b,c), the relative populations of I, Int1, Int2 and A (Fig. 3c), and the distribution of A1/A2 and A2/A3 interdomain distances as a function of free calcium concentration (Fig. 3d–f and Supplementary Fig. 5).
Figure 3. Proposed mechanism of calcium activation of A1–A3 in solution.

(a) Model for A1–A3 transition between the inactive and active (actin-binding competent) states. The release of the A1/A3 latch from the inactive form (I) leads to a first intermediate (Int1), followed by the loss of the A1A2 interface (Int2). Finally, the calcium-stabilized interface between A2 and A3 is formed, resulting in the active conformation (a). (b) Fitted SAXS profiles. For clarity, only a subset of the data shown in Fig. 2a is represented. Experimental data are shown as black spheres and theoretical SAXS profiles for optimized ensembles of 50 models are drawn as red lines. (1) 490 μM CaCl2, 1 mg ml−1 of A1–A3; (2) 490 μM CaCl2, 2 mg ml−1 of A1–A3; (3) 490 μM CaCl2, 4 mg ml−1 of A1–A3; (4) 49 nM, 175 and 550 μM, and 2.3 mM CaCl2, 4 mg ml−1 of A1–A3; (5) 490 μM CaCl2, 8 mg ml−1 of A1–A3; (6) 2 mM EGTA, 8 mg ml−1 of A1–A3 mg ml−1. (c) Relative populations of inactive (I, black), intermediate (Int1, red and Int2, green) and active (A, blue) conformations of A1–A3, plotted as a function of free calcium concentration (x axis, bottom) and ratio of calcium ion over protein (x axis, top). All data measured at 4 mg ml−1 were used (Fig. 2 A, curve series 3 and 5). Error bars represent the s.d. of ensemble optimization results obtained from 10 independent ensembles of 6,000 models (see Methods section). (d–f) Two-dimensional (2D) histogram representations of the distribution of A1–A2 and A2–A3 interdomain distances (taken between their centres of mass) within the optimized ensembles, using data measured in the presence of 2 mM EGTA (d), 10 μM (e) and 550 μM free Ca2+ (f). The 2D histogram was calculated using a binning of 15 × 15 and coloured from red to blue (0–50 models).
In the presence of EGTA, the Rg distribution displays a major peak at 2.5 nm, and minor populations with Rg values broadly distributed ∼4.0–5.5 nm (Supplementary Fig. 4b,c), which correspond to 85–90% inactive conformation, 5% A and 9% Int1, respectively (Fig. 3c,d and Supplementary Fig. 5a). The open conformation Int2 is highly disfavoured with a population of 0–1%, suggesting that the transition to the active conformation following release of A3 is a fast process in these conditions. The presence of the minor populations is apparent in the significant improvement of χexp observed when using an optimized ensemble of 20 models (χexp=1.088) rather than a single best-fitting model of the inactive state to represent the data (χexp=1.919; Supplementary Fig. 6a). Accordingly, the minimal number of models required for optimal data fitting is constant at around seven models across the different calcium concentrations used, demonstrating that conformational heterogeneity exists regardless of the presence of calcium (Supplementary Fig. 6b).
The addition of 10 μM of free calcium stimulated the exchange process (Fig. 3e and Supplementary Fig. 5b), leading to significant changes in the conformational landscape of A1–A3: the proportion of inactive conformers fell to 50%, whereas Int1 and Int2 rose to ∼15% each, and active conformers increased to nearly 20%. Analysis of A2/A3 interdomain distances reveals a small population of Int1 conformers sampling distances very close to the inactive state as the A1/A3 latch opens up (Fig. 3e and Supplementary Fig. 5b,d), and the overall distribution of A2/A3 distances within the optimized ensemble is markedly different from that of the pool (Supplementary Fig. 5f). It is noteworthy that this significant increase in conformational dynamics and shift to the active conformation happens at a relatively low free calcium-to-protein ratio (0.1:1), which may be due to binding/unbinding of a single calcium ion to A1–A3 occurring much faster than the conformational changes it induces.
At ∼500 μM of free calcium (calcium-to-protein ratio 5:1), which is enough to saturate the three conserved type II calcium-binding sites, the population of active conformers reaches a peak at ∼50%, while the inactive form drops to 20%. Int1 and Int2 represent, respectively, 10 and 20% (Fig. 3c,f). This shift towards a main population of active conformers is apparent in Rg distributions (Supplementary Fig. 4b,c), with the appearance of a broad peak centred at 3 nm. Interestingly, the selected active conformers preferentially sample relatively short distances between A1 and A2 compared with the pool distribution (similar to the G1–G2 distance in gelsolin-bound actin; Fig. 3f and Supplementary Fig. 5c,d,h), which may reflect a conformational optimization of the A1/A2 linker for actin binding. At even higher free calcium concentrations (2 mM, calcium-to-protein ratio 20:1), the open state (Int2) becomes favoured over the active conformation (55 versus 5%), while the inactive conformation is reduced to 5% (Fig. 3c and Supplementary Fig. 4c). These observations are in line with the increase in the measured Rg (3.6 nm; Table 2), and the predominance of the open state may explain the degradation of A1–A3 that led to crystallization of A3 in the presence of 5 mM calcium.
MDS of calcium binding to inactive A1–A3
Ensemble optimization demonstrated a complex calcium-induced remodelling of A1–A3 conformational landscape. To gain insights into the initial events that shift the equilibrium towards the active state, we performed explicit solvent MDS of iA1–A3 in the presence of 50 mM CaCl2 or 50 mM NaCl (Fig. 4). Although well above the physiological range, such high calcium concentrations enable the detection of regions that display conformational sensitivity to calcium on fast timescales, as well as potential low-affinity sites, within reasonable simulation times (for example, ref. 33). For each system, we ran two independent simulations of 1 μs and 0.1 μs in the presence and absence of calcium, respectively. The protein structure was stable over the course of the simulations, with the r.m.s.d. from the starting structure stabilizing ∼3–4 Å after an initial rise during the first 20 ns (Fig. 4a,b). Analysis of the root mean square fluctuations indicated flexibility mainly in the linker regions, which was enhanced by calcium (Fig. 4c).
Figure 4. MDS of A1–A3 in the inactive state.

(a,b) r.m.s.d. of Cα atoms from the starting structure in the presence of 50 mM CaCl2 (a) or NaCl (b). The results from two independent simulations are shown as black and red lines, respectively. (c) Root mean square fluctuations (r.m.s.f.) of Cα atoms along the protein sequence. The averages of the two independent simulations are shown as black and red lines for systems simulated in the absence and presence of calcium, respectively. (d) Analysis of calcium binding to inactive iA1–A3 by MDS. Calcium ions displaying the lowest time-averaged distance to protein atoms were extracted from eight snapshots taken at 100 ns of interval and are represented as magenta spheres. Four overlaid snapshots of the protein are shown in two views rotated by 90° in cartoon representation with the protein backbone coloured from blue to red (N- to C terminus). The protein backbone from four additional snapshots, for which the calcium ions are shown, have been hidden for clarity. Close ups of the calcium-binding sites of interest are shown as insets.
We identified bound calcium by analysing time-averaged distances between Ca2+ and protein atoms (Fig. 4d). Ca2+ clusters mainly around the conserved type II binding sites (Fig. 4d, inset 1, 2 and 5), as well as the A2A3 linker and the C-terminal disordered extension belonging to the A3A4 linker, in a manner that is consistent with the distribution of negative charges on the protein surface (Supplementary Fig. 7). Additional Ca2+ is observed between the short α-helices of A1 and A2, and their second and fourth β-strands, respectively (Fig. 4d, inset 3 and 4). These sites involve calcium coordination by Glu9 and Glu28 carboxylates, and the backbone oxygen of Leu29 in A1, and Glu219 and Glu173 carboxylates and the hydroxyl of Tyr175 in A2. These interactions create additional contacts between the short α-helices and β-strands of A1 and A2, and may represent low-affinity-binding sites that stabilize the GH domains at high calcium concentrations.
Examination of Ca2+ dynamics at the high-affinity sites indicates that only the type II binding site at A2 can bind Ca2+ in the inactive conformation (Fig. 4d, inset 5), showing stable coordination of Ca2+ by Asp164 and Glu186 side chains, as well as Gly163 carbonyl oxygen. In contrast, binding at A1 is hindered by the interaction of Asp44 with Arg23 side chain and Val122 backbone nitrogen, resulting from the interaction with A2 (Fig. 4d, inset 1). Similarly, formation of the type II binding site in A3 is prevented by the kink of the long α-helix that interacts with A2, resulting in distances between Glu305, Asp256 and Glu281 that are too large to coordinate calcium tightly (Fig. 4d, inset 2).
Next, we analysed the effect of calcium on the dynamics of the A2A3 linker, which is located close to the type II binding site at A2 (Fig. 5), and in actin-bound G1–G3 contacts calcium through Asp259 side chain (Asp238 in adseverin), thus providing additional stability to the active state20. In the absence of calcium, the extended A2A3 linker samples the conformational space in the vicinity of the crystal structure (Fig. 5a). In contrast, calcium perturbs the linker dynamics, leading to an initial increase in flexibility within the first 100-ns of simulation (Fig. 5b) followed by sampling of calcium-induced, non-native conformational states of the linker associated with calcium binding to the type II binding site in A2 and to the acidic patch of the A2A3 linker (Fig. 5a,d). The observation of calcium-induced changes in A2A3 linker dynamics suggests that this linker acts as a calcium-sensitive conformational switch that modulates the stability of the inactive, intermediate and active states.
Figure 5. Effect of calcium on A2A3 linker dynamics as observed in MDS.

In each panel, the crystal structure of iA1–A3 is shown in grey cartoons for comparison with the A2A3 linker represented using a larger cartoon loop radius. (a) Linker dynamics in the absence of calcium. Ten overlaid snapshots of the protein taken at 10 ns of interval (10–100 ns) are shown in cartoon representation with the protein backbone coloured from blue to red (N- to C terminus). (b–d) Linker dynamics in the presence of calcium, illustrating the increased flexibility and deviation from the crystal structure. Ten overlaid snapshots of the protein taken at 10 ns of interval were extracted from one of the two 1 μs MDS of iA1–A3 in the presence of calcium and are shown as rainbow-coloured cartoons (0–90, 400–490 and 900–990 ns in b–d respectively). In each case, bound calcium ions are represented in magenta spheres for one of the snapshots, with coordinating side chains as sticks (100 ns in b, 500 ns in c and 1,000 ns in d).
Impact of A1/A3 interface mutations on F-actin severing
The detailed picture of the Ca2+-dependent activation mechanism of A1–A3 brought by the combination of X-ray crystallography, SAXS and MDS explains its reported Ca2+-dependent severing activity, which contrasts with the open conformation and Ca2+-independent severing of actin filaments described for the equivalent fragment of gelsolin (G1–G3; refs 30, 31). These differences prompted us to investigate the contribution of the stability of the A1/A3 latch to severing activity. To this end, two mutants of A1–A3 (Met310Asp and Glu314Ser) were designed that were aimed at disrupting hydrophobic interactions between Met310 and Phe64, and a salt bridge involving Glu314 and Arg97 at the A1:A3 interface (Fig. 6a). Interestingly, structural comparisons show that in gelsolin, the equivalent residue to Glu314 is a serine, resulting in loss of the salt bridge, while the Met310/Phe64 pair is replaced by Leu332/Tyr87, suggesting a potentially less stable G1:G3 interface (Fig. 6b).
Figure 6. Calcium-independent actin-severing activity of A1–A3 is increased by mutations that disrupt the A1/A3 interface.

(a) Details of the A1/A3 interface. The mutated residues Met310 and Glu314 are shown as grey sticks. (b) Similar view of the G1/G3 interface. (c,d) Actin depolymerization assays. A total of 6 μM of each protein (actin control, black; A1–A3wt, red; E314S mutant, green; M310D mutant, yellow; and G1–G3, purple) was added to 6 μM of F-actin in the presence of 1 mM EGTA (c) or 0.5 mM CaCl2 buffer (d).
F-actin depolymerization by A1–A3 and its mutants, as well as by G1–G3 was monitored by the loss of fluorescence of pyrene-labelled F-actin in the presence of 1 mM EGTA (Fig. 6c) or 500 μM CaCl2 (Fig. 6d). In the presence of Ca2+, all proteins showed a similar actin filament depolymerization pattern, with rapid loss of fluorescence. In contrast, in the presence of EGTA, A1–A3 and the M310D and E314S mutants showed reduced severing activity, while only G1–G3 severing activity remained robust, as expected from previous reports29,30. Interestingly, A1–A3 severing of F-actin was roughly 10 times slower than in the presence of Ca2+ (Fig. 6c,d). Although the M310D and E314S mutants showed increased activity compared with wild-type A1–A3, they remained much less active than G1–G3 in Ca2+-free conditions, suggesting that the destabilization of the A1/A3 latch in the mutants may only partially shift the equilibrium towards the active state. In addition, M310D was slightly more active than E314S, indicating a larger contribution to destabilizing the interface, as may be expected based on the nature of the mutation (hydrophobic to charged versus charged to hydrophilic).
Discussion
The structural data presented here have revealed that isolated A1–A3 can adopt, in the crystal and in solution, an inactive conformation (I) characterized by the formation of the A1/A3 latch as observed in calcium-free full-length gelsolin. Using SAXS and ensemble optimization, we have demonstrated that the conformational landscape of A1–A3 is composed of a heterogeneous mixture of dynamically exchanging conformers, where the inactive conformation coexists with activation intermediates and gelsolin-like active conformers in a 9:1 ratio in the absence of calcium. The addition of moderate (10 μM)-to-high (500 μM) calcium concentrations increased conformational exchange, resulting in a larger proportion of intermediate forms (30%), and progressively shifted the equilibrium towards the active form, up to a maximum of ∼55%. These calcium concentrations correlate well with the ∼ 50 μM peaks in subplasmalemmal calcium observed in adrenal chromaffin cells34, which enable adseverin to depolymerize the cortical network of actin filaments. Interestingly, we found that higher calcium concentrations (2 mM, close to blood plasma calcium concentration of 1.0–1.5 mM) induced further expansion leading to a main population of open conformers (Int2). Because of its intrinsic disorder, this open conformation may result in faster actin binding and severing through a fly casting mechanism35,36, and increase the activity of the protein when present in the extracellular environment, for example, following injury as adseverin is highly expressed in soft tissue3,37. The predominance of Int2 at high calcium concentration may explain the slow degradation of A1–A3 and crystallization of A3 in the presence of 5 mM calcium owing to the increased accessibility of the A1A2 and A2A3 linkers to proteolytic cleavage. This crystal structure revealed the presence of a single calcium ion bound at the type II binding site, associated with the straightened long α-helix that is characteristic of the active forms of gelsolin domains G3 and G6, highlighting the similarity of Ca2+-induced conformational changes in both proteins.
Classical MDS provided detailed insights into the binding of calcium to inactive A1–A3, indicating that binding at A2 can occur in the inactive state while binding at the other type II calcium sites on A1 and A3 is hindered by interdomain contacts (Fig. 4). This bound calcium likely plays a major role in shifting the equilibrium towards the active state at low calcium concentrations by perturbing the conformation of the A2A3 linker (Fig. 5), which undergoes significant refolding during the inactive-to-active transition (Fig. 7a). However, an additional, perhaps more important, mechanism by which calcium binding at A2 may shift the conformational landscape is through stabilization of the active A2:A3 interface (Fig. 7b), likely resulting in an increased lifetime of the active conformation. The inaccessibility of the A1 and A3 type II calcium-binding sites in the inactive conformation implies that the increase in the population of active conformers observed at calcium-to-protein ratios >1 is a consequence of calcium binding at A1 and A3 in the Int1, Int2 and A conformers, which, in combination with the straightening of the long α-helix of A3, prevents the molecule from transitioning back to the inactive form. Finally, we show that very high calcium concentrations destabilize the active A2:A3 interface, leading to a majority of open conformers. Based on the MDS results, we propose that additional calcium binding in the A2A3 linker region, and in particular at the highly acidic stretch 232-DGGDDDD-238, may cause the observed destabilization. Interestingly, potential additional low-affinity sites that might play a role in stabilizing the A1 and A2 domain structure were also identified in MDS (Fig. 4), although they have not been verified experimentally. The A2A3 linker appears to be optimized for calcium-induced conformational change through allosteric regulation by association with calcium (Fig. 5). Furthermore, 232-DGGDDDD-238 in the A2A3 linker may offer sequential aspartic acids to the occupied type II calcium-binding site on A2. Thus, as A3 approaches A2, a pathway of calcium/aspartic acid interactions may lead to the docking of the A2:A3 interface, which is stabilized by the final aspartic acid in the sequence (Asp238) coordinating the type II calcium bound to A2 (Fig. 7). Such functionality in linker regions is beginning to be recognized across a range of multidomain proteins38.
Figure 7. Role of the A2A3 linker in activation and comparison with gelsolin.

(a) Conformation of the A2A3 linker in the inactive structure (grey cartoons) and active model (rainbow-coloured cartoons) of A1–A3. The crystal structure of iA1–A3 was superimposed onto a homology model of the gelsolin-like active conformation through structural alignment of A2. (b,c) Calcium coordination at the type II binding site of domain 2 in the active A1–A3 model (b) and in the crystal structure of actin-bound G1–G3 (PDB 3FFK) (c), highlighting sequence divergence in this region with a more acidic linker in adseverin.
Actin depolymerization assays have shown that A1–A3 is able to slowly sever actin filaments in EGTA conditions, although this activity was roughly 10% of the severing rate observed in the presence of 500 μM calcium. Interestingly, this correlates well with the proportion of active conformers detected by SAXS in similar conditions, which are, respectively, 5 and 50% in the presence of 2 mM EGTA and 500 μM calcium. This suggests that F-actin binding by A1–A3 is dominated by conformational selection of the active form, and that the inactive-to-active transition may be relatively slow compared to F-actin binding and severing, preventing population shift from occurring. Alternatively, the large concentration of G-actin present in the assay might hinder population shift through sequestering of G-actin by A1–A3 following an initial binding event to the inactive conformation.
The M310D and E314S mutants of A1–A3 showed increased severing activity in the absence of calcium when compared with wild-type A1–A3, most probably due to a partial shift towards the active conformation, which highlighted the importance of the A1/A3 contacts in stabilizing the inactive state. However, the activity of the mutants in calcium-free conditions remained much lower than for wild-type A1–A3 in the presence of calcium, or G1–G3 that displays fast depolymerization activity regardless of the presence of calcium30. The fact that these mutations were unable to completely turn A1–A3 into a Ca2+-independent severer suggests that the high level of activity of G1–G3 does not primarily result from the instability of the inactive state G1–G3. In support of this, comparison of predicted ΔiG for the interfaces between domains 1–2 and 1–3 in iA1–A3 and iG1–G3 using the Pisa Web server39 revealed no major difference in stability, and a ∼150-ns MDS of iG1–G3 suggested that the G1:G3 interface is stable on such timescales (Supplementary Fig. 8), also indicating that the domain rearrangements required for activation are slow.
A second structural difference between the two proteins may reside in the stability of the active form, which is tightly influenced by the sequence of the A2A3 linker. Indeed, the concentration of negative charges in the highly acidic stretch 232-DGGDDDD-238 of A1–A3, as well as the loss of ionic interactions between Lys166 and Glu263, may lead to an active structure that is less stable than aG1–G3 (Fig. 7b,c). Consistent with this, SAXS studies of G1–G3 and fragments G1–G2 and G2–G3 showed that G1–G3 mainly adopts an active/open conformation in solution regardless of calcium concentration, explaining its calcium-independent activity31. Although ensemble analysis was not performed on this data, the construct used (gelsolin residues 1–371) has the same length as A1–A3 (residues 1–365+5 residues remaining from the cleaved purification tag), which enables direct comparisons between measured Rg. Interestingly, a mild compaction of G1–G3 from 3.4 to 3.2 nm was observed upon addition of 1 mM calcium. This Rg of 3.2 nm fits well with the Rg of A1–A3 in the presence of 500 μM calcium, which is dominated by active conformers. It is thus tempting to speculate that this slight calcium-induced compaction of G1–G3 may result from a shift between open (Int2) and active conformations through stabilization of the G2:G3 interface by calcium binding at the type II site of G2. In addition, the same study showed that mutations in the G2G3 linker could induce Ca2+-dependent severing activity in G1–G3. The mutants were found to adopt a collapsed state in the absence of calcium (Rg=2.6 nm similar to inactive A1–A3) and expand in the presence of 1 mM calcium to Rg=3.8 nm. These dimensions are too large to represent exclusively active conformers and suggest the presence of fully expanded conformers, possibly resulting from destabilization of the linker structure. These results, together with the predominance of the open state of A1–A3 at high calcium concentrations, the changes in dynamics associated with the binding of calcium ions to the A2A3 linker observed in MDS, and sequence divergence in this region between adseverin and gelsolin indicate that the linker between domains 2 and 3 plays a critical role in controlling protein dynamics by tuning the relative stability of the inactive, intermediate and active conformations.
In conclusion, we have drawn a quantitative picture of the conformational landscape of A1–A3 and provided structural insights into how calcium binding induces large conformational changes by affecting the relative stability of the accessible conformational states of A1–A3. Our biophysical characterization of A1–A3 in solution helps in explaining the apparent structural similarity yet different calcium-regulated dynamics that characterize the N-terminal halves of adseverin and gelsolin. The origin of these differences is best understood in the context of the full-length proteins. In the absence of a gelsolin C-terminal α-helix latch, adseverin requires tighter interactions than gelsolin in the A1/A3 latch to maintain calcium control of activation.
Methods
Cloning, expression and purification
The cloning and Escherichia coli expression of the N-terminal halves of adseverin (A1–A3, residues 1–365) and gelsolin (G1–G3, residues 25–372) have been described previously in refs 23, 20. Briefly, the constructs were engineered into a modified pET-21d(+) (Novagen) plasmid, pSY5, to encode an 8-histidine tag, followed by a PreScission protease cleavage site, ahead of the N terminus of the proteins. The A1–A3 M310D and E314S mutants were generated using the QuikChange site-directed mutagenesis kit (Stratagene) and the following oligonucleotide pairs: 5′- CCCAAGAGAGGAAGGCTGCAGATAAGACAGCTGAAGAATTTC -3′; 5′- GAAATTCTTCAGCTGTCTTATCTGCAGCCTTCCTCTCTTGGG -3′ for M310D and 5′- GGCTGCAATGAAGACAGCTTCAGAATTTCTACAGCAAATG -3′; 5′ CATTTGCTGTAGAAATTCTGAAGCTGTCTTCATTGCAGCC -3′ for E314S. Constructs were transformed into E. coli Rosetta 2 (DE3) cells (Novagen) for expression according to the manufacturer's protocol. Overnight starter cultures were prepared by growing the cells in LB liquid media supplemented with 100 μg ml−1 ampicillin and 34 μg ml−1 chloramphenicol at 37 °C with shaking at 250 r.p.m. for 16 h. Fresh LB liquid media for expression of each construct was inoculated with 50 ml of overnight starter culture per liter, and incubated with shaking at 250 r.p.m. at 37 °C until cell density reached OD600=0.6. Proteins were expressed by induction with 1 mM isopropyl β-D-1-thiogalactopyranoside at 23 °C overnight. Cells were harvested by centrifugation at 3,700g for 30 min. The pellets were subsequently resuspended in 100 ml binding buffer (50 mM Tris-HCl (pH 8.0), 500 mM, NaCl and 20 mM imidazole), and disrupted by 2 mg ml−1 lysozyme treatment for 1 h at 4 °C followed by sonication.The suspensions were clarified by centrifugation at 32,000g for 45 min in an SS-34 rotor in a RC 5C Plus centrifuge (Sorvall) at 4 °C. Nickel-nitrilotriacetic acid (Ni-NTA) beads (2 ml) were added to the supernatant and mixed continuously for 30 min at 4 °C. The beads were pelleted at 1,000g at 4 °C for 15 min, and resuspended in buffer containing 300 mM NaCl, 20 mM imidazole, 10 mM Tris-HCl (pH 7.5). After washing the beads, proteins were eluted by raising the imidazole concentration to 250 mM. The His-tag was cleaved by addition of human rhinovirus 3C protease followed by overnight dialysis against 150 mM NaCl and 20 mM Tris-HCl (pH 7.5). Proteins were further purified by size-exclusion chromatography on a HiLoad 16/60 Superdex 200 prep grade column (GE Healthcare), first equilibrated with 125 ml of 100 mM EGTA, 20 mM Tris-HCl (pH 7.2), 150 mM NaCl, re-equilibrated with 250 ml gel-filtration buffer (0.2 mM EGTA, 20 mM Tris-HCl (pH 7.2) and 150 mM NaCl). Proteins were concentrated to 10 mg ml−1 by Ultra-15, MWCO 10 kDa, Amicon Ultra centrifugal filter units.
Crystallization and data collection
Crystallization was carried out by vapour diffusion using a Cartesian Technologies pipetting system40. Solutions containing 10 mg ml−1 of A1–A3 in 50 mM NaCl and 10 mM Tris-HCl (pH 7.5) supplemented with 5 mM of either EDTA or CaCl2 crystallized in 20% polyethylene glycol 3,350, 100 mM Bis-Tris propane (pH 7.5) or 30% polyethylene glycol 6,000, 100 mM HEPES (pH 7.0), respectively, which led to the crystal structures of iA1–A3 and aA3. Crystals were frozen in liquid nitrogen after being soaked in a mother liquor solution supplemented with 25% glycerol. Diffraction data were recorded on the ID29 and ID14-2 beamlines at the European Synchrotron Radiation Facility, Grenoble, France. Wavelength was 1.033 and 0.933 Å for iA1–A3 and aA3, respectively. All data were automatically processed by xia2 (ref. 41).
Structure determination and refinement
Calcium-free iA1–A3 and calcium-bound aA3 structures were solved by molecular replacement using the calcium-free N-terminal half of gelsolin, iG1–G3, excised from PDB 1D0N (ref. 26) or the calcium-bound gelsolin domain 3, aG3 excised from PDB 1RGI (ref. 18) as search models in PHASER42. The molecular replacement solutions were subjected to repetitive rounds of restrained refinement in PHENIX43 and Autobuster44, and manual building in COOT45. Translation/Libration/Screw (TLS) parameters were included in the final round of refinement. The CCP4 programme suite46 was used for coordinate manipulations. The structures were validated with Molprobity47. 97 and 99% of residues were in the Ramachandran-favoured region for iA1–A3 and aA3, respectively. Refinement statistics are given in Table 1 and a portion of the electron density map for each structure is shown in Supplementary Fig. 9.
SAXS experiments
Protein samples (10 mg ml−1 in 0.2 mM EGTA, 20 mM Tris-HCl (pH 7.2) and 150 mM NaCl) were mixed with 10 × calcium or EGTA stock solutions to achieve the required free calcium concentrations, which were calculated using the Ca-EGTA Calculator v1.3 webserver (http://maxchelator.stanford.edu/CaEGTA-TS.htm)48. SAXS data were collected at the European Synchrotron Radiation Facility on beamline ID14-3. The sample-to-detector distance was 1 m and the wavelength of the X-rays was 0.0995, nm. Data acquisition was performed at 20 °C. Protein concentrations ranged from 1 to 8 mg ml−1. Data reduction was performed using the established procedure available at ID14-3, and buffer background runs were subtracted from sample runs. The radius of gyration was determined with the programme PRIMUS49 according to the Guinier approximation at low Q values, in a Q.Rg range up to 1.3:
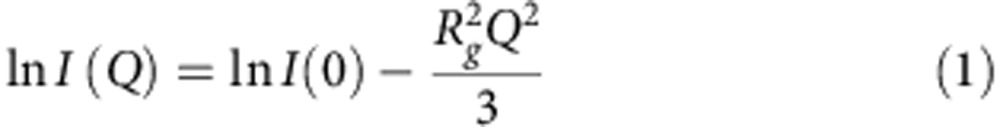 |
The pairwise distance distribution functions, P(r), were calculated with the programme GNOM50. Molecular weights were estimated based on ref. 51.
Ensemble generation
An ensemble approach was used to model the N-terminal half of adseverin (A1–A3). We employed all-atom coarse-grained MDS52 to generate candidate structures based on available X-ray crystallographic data. Starting coordinates for the inactive state (I) were obtained from the crystal structure of calcium-free N-terminal half of adseverin or from a homology model based on PDB 1RGI for the active structure (A)18. Complete models including the missing disordered termini were generated in Modeller53. An additional homology model based on the structure of the gain-of-function mutant of the actin capping protein CapG (PDB 2J72; ref. 54), which differs from the active structure by having a different interface between domains 2 and 3, was also generated and used as a qualitative control of the accuracy of the ensemble optimization approach. Starting conformers for intermediates 1 and 2 (Int1 and Int2) were obtained by dissociating domain 3 and then domain 1 from the iA1–A3 model. This was performed by defining and altering backbone torsions within the interdomain linker regions in VEGA ZZ55. An additional model for Int2 was generated by dissociating domain 3 from domain 2 in the active structure, leading to an alternative conformer of Int2 where the alpha helix in the A2A3 linker is folded. Models of A1–A3 (I, Int1, Int2, A and the CapG-derived model) were simulated in GROMACS56 using an atomistic coarse-grained structure-based model57,58. A timestep of 0.0005 time units was used and the simulation was coupled to a temperature bath via Langevin dynamics. 10,000 Snapshots were extracted from each simulation, and a 1,000 models from each simulation were randomly dispatched into 10 independent ensembles of 6,000 models.
Ensemble optimization
For each model from each ensemble, theoretical SAXS patterns were calculated with the programme CRYSOL59, and ensemble optimization fitting was performed for each of the 10 independent ensembles with GAJOE60. GAJOE uses a genetic algorithm to select from a large pool of conformers optimized sub-ensembles that minimize the discrepancy between the experimental and calculated curves χexp according to the following equation:
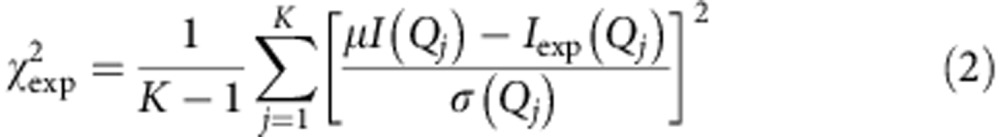 |
where K is the number of points in the experimental curve, σ is the s.d. and μ is a scaling factor. The ensemble size was varied from 1 to 50 models and variations in the quality of the fit to the experimental data χexp were monitored in order to analyse protein flexibility (Supplementary Fig. 6). Additional validation of the ensemble calculations was also performed by fitting the experimental data to pool ensembles including only I, A or I+A models, which yielded fits of poorer quality than when using ensembles that included the intermediate states (I+Int1+Int2+A; Supplementary Table 1 and Supplementary Fig. 3). In order to build conformational landscapes, 50 models were selected per ensemble, resulting in 50 × 10=500 optimized models. Interdomain distances were extracted from the models using GROMACS routines and then used to calculate two-dimensional histograms (using a binning of 15 × 15). The relative populations of conformers were counted in each optimized ensemble and averaged for each experimental SAXS profile to gain insight into the mechanism of calcium activation.
Explicit solvent MDS
Classical MD simulations of iA1–A3 in the presence and absence of calcium were performed using GROMACS 4 (ref. 56) and the AMBER99SB-ILDN force field61. At the beginning of each simulation, the protein was immersed in a box of SPC/E water, with a minimum distance of 1.0 nm between protein atoms and the edges of the box. An amount of 50 mM of either CaCl2 or NaCl were then added using genion. Long-range electrostatics were treated with the particle-mesh Ewald summation62. Bond lengths were constrained using the P-LINCS algorithm. The integration timestep was 5 fs. The v-rescale thermostat and the Parrinello–Rahman barostat were used to maintain a temperature of 300 K and a pressure of 1 a.t.m. Each system was energy minimized using 1,000 steps of steepest descent and equilibrated for 200 ps with restrained protein heavy atoms. For each system, two independent production simulations were obtained by using different initial velocities. The aggregated simulation time was 200 and 2,000 ns for the simulations in the absence and presence of calcium, respectively. r.m.s.d. and root mean square fluctuations were calculated using GROMACS routines.
Actin depolymerization assays
Polymerization buffer (final concentration, 50 mM KCl, 0.2 mM ATP, 2 mM MgCl2, 0.5 mM dithiothreitol, 1 mM EGTA and 50 mM HEPES, pH 7.5) was added to 10% pyrene-labelled G-actin (6 μM) in 96-well, flat-bottomed plates (Corning), and was incubated for 30 min to allow the formation of F-actin. Calcium was then added to obtain the required free calcium concentration. Reactions were equilibrated for 1 h before 6 μM of the respective proteins (A1–A3, A1–A3-M310D, A1–A3-E314S or G1–G3) were added. The final volume in each well was 100 μl. Fluorescence intensity was measured kinetically at excitation and emission wavelengths of 365 and 407 nm, respectively, using a Safire2 fluorimeter (Tecan).
Additional information
How to cite this article: Chumnarnsilpa, S. et al. Calcium-controlled conformational choreography in the N-terminal half of adseverin. Nat. Commun. 6:8254 doi: 10.1038/ncomms9254 (2015).
Supplementary Material
Supplementary Figures 1-9, Supplementary Table 1, Supplementary Note 1 and Supplementary References.
Acknowledgments
We thank the European Synchrotron Radiation Facility (Grenoble, France) for beamtime, and the staff of beamlines ID29, ID14-2 and ID14-3 for assistance with crystallography and SAXS data collection.
Footnotes
Author contributions S.C., R.C.R., J.M.G. and C.L. designed the research; S.C., J.M.G. and C.L. performed experiments; S.C., R.C.R., J.M.G. and C.L. analysed data; and S.C., R.C.R., J.M.G. and C.L. wrote the paper.
References
- Silacci P. et al. Gelsolin superfamily proteins: key regulators of cellular functions. Cell. Mol. Life Sci. 61, 2614–2623 (2004). [DOI] [PubMed] [Google Scholar]
- Ghoshdastider U., Popp D., Burtnick L. D. & Robinson R. C. The expanding superfamily of gelsolin homology domain proteins. Cytoskeleton (Hoboken) 70, 775–795 (2013). [DOI] [PubMed] [Google Scholar]
- Nag S., Larsson M. r., Robinson R. C. & Burtnick L. D. Gelsolin: the tail of a molecular gymnast. Cytoskeleton 70, 360–384 (2013). [DOI] [PubMed] [Google Scholar]
- Rodriguez Del Castillo A. et al. Chromaffin cell scinderin, a novel calcium-dependent actin filament-severing protein. EMBO J. 9, 43–52 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitale M. L., Rodriguez Del Castillo A., Tchakarov L. & Trifaro J. M. Cortical filamentous actin disassembly and scinderin redistribution during chromaffin cell stimulation precede exocytosis, a phenomenon not exhibited by gelsolin. J. Cell Biol. 113, 1057–1067 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumitrescu Pene T., Rose S. D., Lejen T., Marcu M. G. & Trifaro J. M. Expression of various scinderin domains in chromaffin cells indicates that this protein acts as a molecular switch in the control of actin filament dynamics and exocytosis. J. Neurochem. 92, 780–789 (2005). [DOI] [PubMed] [Google Scholar]
- Hassanpour S., Jiang H., Wang Y., Kuiper J. W. & Glogauer M. The actin binding protein adseverin regulates osteoclastogenesis. PLoS ONE 9, e109078 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zunino R. et al. Expression of scinderin in megakaryoblastic leukemia cells induces differentiation, maturation, and apoptosis with release of plateletlike particles and inhibits proliferation and tumorigenesis. Blood 98, 2210–2219 (2001). [DOI] [PubMed] [Google Scholar]
- Nurminsky D., Magee C., Faverman L. & Nurminskaya M. Regulation of chondrocyte differentiation by actin-severing protein adseverin. Dev. Biol. 302, 427–437 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X. et al. Expression and function of the actin-severing protein adseverin in the proliferation, migration, and differentiation of dental pulp cells. J Endod. 41, 493–500 (2015). [DOI] [PubMed] [Google Scholar]
- Miura N. et al. Adseverin: a novel cisplatin-resistant marker in the human bladder cancer cell line HT1376 identified by quantitative proteomic analysis. Mol. Oncol. 6, 311–322 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H., Shi D., Liu T., Yu Z. & Zhou C. Lentivirus-mediated silencing of SCIN inhibits proliferation of human lung carcinoma cells. Gene 554, 32–39 (2014). [DOI] [PubMed] [Google Scholar]
- Chen X. M. et al. Suppression of scinderin modulates epithelialmesenchymal transition markers in highly metastatic gastric cancer cell line SGC7901. Mol. Med. Rep. 10, 2327–2333 (2014). [DOI] [PubMed] [Google Scholar]
- Wang D. et al. Suppression of SCIN inhibits human prostate cancer cell proliferation and induces G0/G1 phase arrest. Int. J. Oncol. 44, 161–166 (2013). [DOI] [PubMed] [Google Scholar]
- Lin K. M., Mejillano M. & Yin H. L. Ca2+ regulation of gelsolin by its C-terminal tail. J. Biol. Chem. 275, 27746–27752 (2000). [DOI] [PubMed] [Google Scholar]
- Ditsch A. & Wegner A. Two low-affinity Ca(2+)-binding sites of gelsolin that regulate association with actin. Eur. J. Biochem. 229, 512–516 (1995). [DOI] [PubMed] [Google Scholar]
- Robinson R. C. et al. Domain movement in gelsolin: a calcium-activated switch. Science 286, 1939–1942 (1999). [DOI] [PubMed] [Google Scholar]
- Burtnick L. D., Urosev D., Irobi E., Narayan K. & Robinson R. C. Structure of the N-terminal half of gelsolin bound to actin: roles in severing, apoptosis and FAF. EMBO J. 23, 2713–2722 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chumnarnsilpa S. et al. Calcium ion exchange in crystalline gelsolin. J. Mol. Biol. 357, 773–782 (2006). [DOI] [PubMed] [Google Scholar]
- Nag S. et al. Ca2+ binding by domain 2 plays a critical role in the activation and stabilization of gelsolin. Proc. Natl Acad. Sci. USA 106, 13713–13718 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lueck A., Yin H. L., Kwiatkowski D. J. & Allen P. G. Calcium regulation of gelsolin and adseverin: a natural test of the helix latch hypothesis. Biochemistry 39, 5274–5279 (2000). [DOI] [PubMed] [Google Scholar]
- Choe H. et al. The calcium activation of gelsolin: insights from the 3A structure of the G4-G6/actin complex. J. Mol. Biol. 324, 691–702 (2002). [DOI] [PubMed] [Google Scholar]
- Chumnarnsilpa S. et al. The crystal structure of the C-terminus of adseverin reveals the actin-binding interface. Proc. Natl Acad. Sci. USA 106, 13719–13724 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maekawa S. & Sakai H. Inhibition of actin regulatory activity of the 74-kDa protein from bovine adrenal medulla (adseverin) by some phospholipids. J. Biol. Chem. 265, 10940–10942 (1990). [PubMed] [Google Scholar]
- Janmey P. A. & Stossel T. P. Modulation of gelsolin function by phosphatidylinositol 4,5-bisphosphate. Nature 325, 362–364 (1987). [DOI] [PubMed] [Google Scholar]
- Burtnick L. D. et al. The crystal structure of plasma gelsolin: implications for actin severing, capping, and nucleation. Cell 90, 661–670 (1997). [DOI] [PubMed] [Google Scholar]
- Wang H. et al. Helix straightening as an activation mechanism in the gelsolin superfamily of actin regulatory proteins. J. Biol. Chem. 284, 21265–21269 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashish, et al. Global structure changes associated with Ca2+ activation of full-length human plasma gelsolin. J. Biol. Chem. 282, 25884–25892 (2007). [DOI] [PubMed] [Google Scholar]
- Sakurai T., Kurokawa H. & Nonomura Y. The Ca2(+)-dependent actin filament-severing activity of 74-kDa protein (adseverin) resides in its NH2-terminal half. J. Biol. Chem. 266, 4581–4585 (1991). [PubMed] [Google Scholar]
- Chaponnier C., Janmey P. A. & Yin H. L. The actin filament-severing domain of plasma gelsolin. J. Cell Biol. 103, 1473–1481 (1986). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peddada N. et al. Global shapes of F-actin depolymerization-competent minimal gelsolins: insight into the role of g2-g3 linker in pH/Ca2+ insensitivity of the first half. J. Biol. Chem. 288, 28266–28282 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernado P. Effect of interdomain dynamics on the structure determination of modular proteins by small-angle scattering. Eur. Biophys. J. 39, 769–780 (2009). [DOI] [PubMed] [Google Scholar]
- Leyrat C., Renner M., Harlos K., Huiskonen J. T. & Grimes J. M. Structure and self-assembly of the calcium binding matrix protein of human metapneumovirus. Structure 22, 136–148 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia A. G., Garcia-De-Diego A. M., Gandia L., Borges R. & Garcia-Sancho J. Calcium signaling and exocytosis in adrenal chromaffin cells. Physiol. Rev. 86, 1093–1131 (2006). [DOI] [PubMed] [Google Scholar]
- Huang Y. & Liu Z. Kinetic advantage of intrinsically disordered proteins in coupled folding-binding process: a critical assessment of the "fly-casting" mechanism. J. Mol. Biol. 393, 1143–1159 (2009). [DOI] [PubMed] [Google Scholar]
- Shoemaker B. A., Portman J. J. & Wolynes P. G. Speeding molecular recognition by using the folding funnel: the fly-casting mechanism. Proc. Natl Acad. Sci. USA 97, 8868–8873 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tchakarov L., Vitale M. L., Jeyapragasan M., Rodriguez Del Castillo A. & Trifaro J. M. Expression of scinderin, an actin filament-severing protein, in different tissues. FEBS Lett. 268, 209–212 (1990). [DOI] [PubMed] [Google Scholar]
- Ma B., Tsai C. J., Haliloglu T. & Nussinov R. Dynamic allostery: linkers are not merely flexible. Structure 19, 907–917 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krissinel E. & Henrick K. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372, 774–797 (2007). [DOI] [PubMed] [Google Scholar]
- Walter T. S. et al. A procedure for setting up high-throughput nanolitre crystallization experiments. Crystallization workflow for initial screening, automated storage, imaging and optimization. Acta Crystallogr. D Biol. Crystallogr. 61, 651–657 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter G., Lobley C. M. & Prince S. M. Decision making in xia2. Acta Crystallogr. D Biol. Crystallogr. 69, 1260–1273 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy A. J. et al. Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams P. D. et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanc E. et al. Refinement of severely incomplete structures with maximum likelihood in BUSTER-TNT. Acta Crystallogr. D Biol. Crystallogr. 60, 2210–2221 (2004). [DOI] [PubMed] [Google Scholar]
- Emsley P., Lohkamp B., Scott W. G. & Cowtan K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winn M. D. et al. Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 67, 235–242 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen V. B. et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoenmakers T. J., Visser G. J., Flik G. & Theuvenet A. P. CHELATOR: an improved method for computing metal ion concentrations in physiological solutions. Biotechniques 12, 870–874 876-9 (1992). [PubMed] [Google Scholar]
- Svergun D. I., Konarev P. V., Volkov V. V., Sokolova A. V. & Koch M. H. J. PRIMUS: a Windows PC-based system for small-angle scattering data analysis. J. Appl. Crystallogr. 36, 1277–1282 (2003). [Google Scholar]
- Semenyuk A. V. & Svergun D. I. Gnom - a Program package for small-angle scattering data-processing. J. Appl. Crystallogr. 24, 537–540 (1991). [Google Scholar]
- Rambo R. P. & Tainer J. A. Accurate assessment of mass, models and resolution by small-angle scattering. Nature 496, 477–481 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onuchic J. N. et al. An all-atom structure-based potential for proteins: Bridging minimal models with all-atom empirical forcefields. Proteins 75, 430–441 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eswar N., Eramian D., Webb B., Shen M. Y. & Sali A. Protein structure modeling with MODELLER. Methods Mol. Biol. 426, 145–159 (2008). [DOI] [PubMed] [Google Scholar]
- Zhang Y. et al. A CapG gain-of-function mutant reveals critical structural and functional determinants for actin filament severing. EMBO J. 25, 4458–4467 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vistoli G., Pedretti A. & Villa L. VEGA - An open platform to develop chemo-bio-informatics applications, using plug-in architecture and script programming. Journal of Computer-Aided Molecular Design 18, 167–173 (2004). [DOI] [PubMed] [Google Scholar]
- Hess B., Kutzner C., van der Spoel D. & Lindahl E. GROMACS 4: algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 4, 435–447 (2008). [DOI] [PubMed] [Google Scholar]
- Noel J. K., Whitford P. C., Sanbonmatsu K. Y. & Onuchic J. N. SMOG@ctbp: simplified deployment of structure-based models in GROMACS. Nucleic Acids Res. 38, W657–W661 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitford P. C. et al. An all-atom structure-based potential for proteins: bridging minimal models with all-atom empirical forcefields. Proteins 75, 430–441 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svergun D., Barberato C. & Koch M. H. J. CRYSOL-a program to evaluate X-ray solution scattering of biological macromolecules from atomic coordinates. J. Appl. Crystallogr. 28, 768–773 (1995). [Google Scholar]
- Bernado P., Mylonas E., Petoukhov M. V., Blackledge M. & Svergun D. I. Structural characterization of flexible proteins using small-angle X-ray scattering. J. Am. Chem. Soc. 129, 5656–5664 (2007). [DOI] [PubMed] [Google Scholar]
- Lindorff-Larsen K. et al. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins 78, 1950–1958 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Essmann U. et al. A smooth particle mesh Ewald method. J. Chem. Phys. 103, 8577–8593 (1995). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figures 1-9, Supplementary Table 1, Supplementary Note 1 and Supplementary References.