

Abstract
Chagas disease, which is caused by the parasite Trypanosoma cruzi, is an important cause of cardiomyopathy in Latin America. It is estimated that 10%–30% of all infected individuals will acquire chronic chagasic cardiomyopathy (CCC). The etiology of CCC is multifactorial and involves parasite genotype, host genetic polymorphisms, immune response, signaling pathways and autoimmune progression. Herein we verified the impact of the recombinant form of P21 (rP21), a secreted T. cruzi protein involved in host cell invasion, on progression of inflammatory process in a polyester sponge-induced inflammation model. Results indicated that rP21 can recruit immune cells induce myeloperoxidase and IL-4 production and decrease blood vessels formation compared to controls in vitro and in vivo. In conclusion, T. cruzi P21 may be a potential target for the development of P21 antagonist compounds to treat chagasic cardiomyopathy.
Chagas disease, which is caused by the parasite Trypanosoma cruzi, is an important cause of cardiomyopathy in Latin America. It is estimated that 10%–30% of all infected individuals will acquire chronic chagasic cardiomyopathy (CCC). This represents anywhere between 1.6 to 5.4 million CCC patients in Latin America1. CCC has been shown to cause social and economic burdens in endemic areas because of increased health care costs2. An estimated 20.000 deaths occur annually in endemic countries due to complications of CCC1. The prognosis for chagasic patients is rather bleak. In fact, CCC has been reported to be the main prognostic mortality factor among patients with heart failure of various etiologies1,2.
The etiology of CCC is multifactorial and involves parasite genotype3,4, host genetic polymorphisms5,6,7,8,9, immune response10,11,12,13, signaling pathways14 and autoimmunity15,16. An intriguing question remains unanswered: could trypanosome-derived components play an active role in CCC onset rather than serving as passive targets for the host immune response ? In this sense, trapped intracellular parasites may continue to secrete proteins that can enter the extracellular space after plasma membrane wounding and/or lysis and influence disease progression. Herein we verified the impact of the recombinant form of P21 (rP21), a secreted T. cruzi protein involved in host cell invasion17,18,19, on progression of chronic inflammatory processes in a polyester sponge-induced inflammation model.
Results and Discussion
rP21 treatment increased IL-4 expression in polyester sponge-induced inflammation
We observed that both rP21 and bacterial extract (B.E.) down-regulated interleukin-1β (IL-1β) and tumor necrosis factor-α (TNF-α) expression in in vivo polyester sponge-induced inflammation (Fig. 1A,B). However, while B.E. inhibited IL-4, rP21 treatment augmented this cytokine levels (Fig. 1C). Knowledge of the pathology and immune response to T. cruzi infection has been largely obtained from murine models. These models have shown that the innate and adaptive immune responses play an important role in parasite control, depending on the combined action of various cellular types including NK, CD4+ and CD8+ as well as on the production of antibodies by B cells. Resistance to T. cruzi infection has been associated with the production of the pro-inflammatory cytokines IL-12 and IFN-γ and with the local production of RANTES, MIP-1α, MIP-1β and MCP-1. These cytokines activate the production of nitric oxide by macrophages, which is responsible for elimination of the parasite. TNF-α has also been associated with macrophage activation as a secondary signal for nitric oxide production. In contrast, cytokines such as IL-4 and TGF-β are associated with parasite susceptibility (For review20). In vivo and in vitro studies have established a clear role for IL-4 in driving T-helper 2 (Th2) immune response21.
Figure 1. rP21 treatment increased IL-4 expression in polyester sponge-induced inflammation.
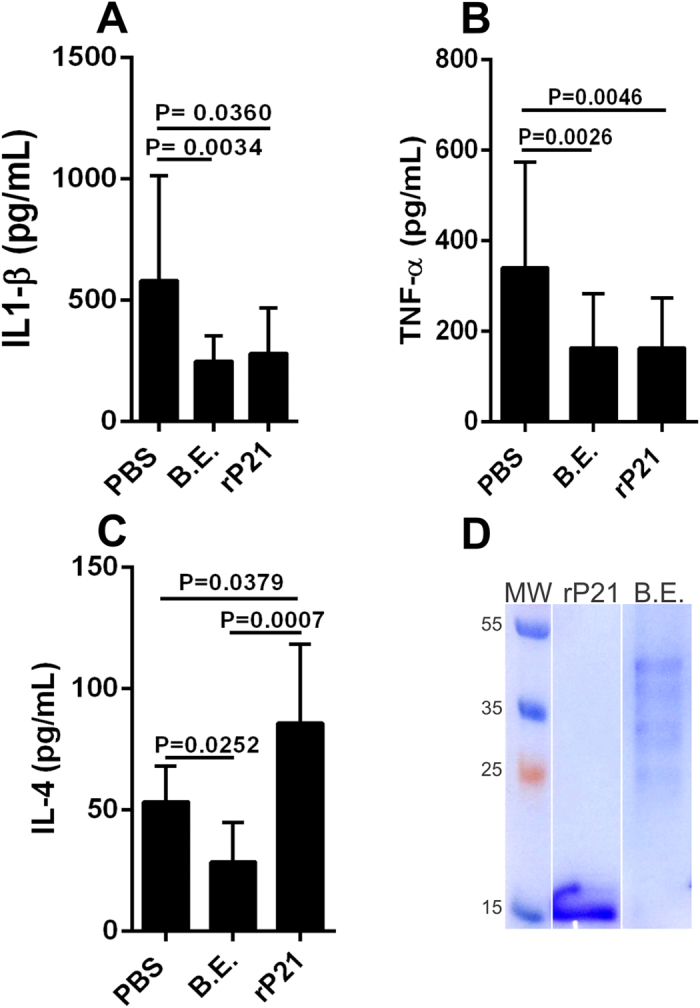
rP21 and bacterial extract (B.E.) down-regulated interleukin-1β (IL-1β) (A) and tumor necrosis factor-α (TNF-α) (B). B.E. inhibited IL-4 expression and rP21 treatment augmented this cytokine levels (C). 40 μg/mL of rP21 and B.E. were used. The experiment was performed twice using 10 animals/group. Samples were analyzed individually in triplicate. Data are expressed as mean ± standard deviation. Significant differences were determined by Dunn’s test (GraphPad Prism software, version 6.01). Differences were considered significant when p < 0.05. A 12% Coomassie Blue stained Sodium dodecyl sulfate-Polyacrylamide gel electrophoresis (SDS-PAGE) gel is shown to demonstrate de purity of purified rP21 and the bacterial protein pattern in B.E. (D). MW: molecular weight expressed in kilo Daltons (kDa). rP21 = 18 kDa.
Several studies22,23,24 demonstrated the importance of antibodies for survival and parasite clearance. Brener25 proposed that trypanolytic antibodies elicited by an active infection are the major and possibly the sole immune effector mechanism controlling murine and human T. cruzi infection. Although parasite-specific antibodies are essential for controlling T. cruzi infection, it has been described that B cells from acutely T. cruzi-infected mice showed a reduced reactivity to parasite antigens in vitro26. Minoprio et al.27 proposed that the humoral immunosuppression to parasite antigens observed during the acute phase of the infection might be due to the fact that parasite-specific B cells are outnumbered by polyclonally activated cells which produce non-specific antibodies that fail to bind parasite antigens. In addition, Hayes & Kierszenbaum28 described that B cells isolated only at the acute phase of the infection show an impaired reactivity to polyclonal activators despite the significant number of B lymphocytes, indicating a B cell alteration, a suppressive phenomenon, or a combination of both. In this context, it has been reported that stimulated B lymphocytes display reduced IL-2R expression in the presence of T. cruzi trypomastigotes29. Other researchers have suggested that a membrane antigen of T. cruzi is involved in immunosuppression30. Also, it has been demonstrated that polyclonal B cell activation during acute experimental infection is not a generalized response31. Altogether, these findings provide evidence for the coexistence of B cell immunosuppression and polyclonal activation during acute T. cruzi infection. The coexistence of these apparently contradictory immunological phenomena is consistent with the hypothesis that a continuous stimulation of lymphocytes leads them to an immune dysfunction: a loss of ability to respond to an antigen and/or an increase in the levels of apoptosis. In this sense, B cell apoptosis and cell cycle arrest could be the mechanisms that control intense B cell expansion, but at the same time could be delaying the emergence of a specific immune response against the parasite32. Moreover, authors have shown that IL-4 reduces B cells apoptosis in T. cruzi infected mice, blocks the differentiation of these cells to plasma cells, favoring the development of a memory B cell phenotype in a galectin-3-dependent pathway. This phenotype results in reduced immunoglobulin production and parasite clearance33. In this context, P21-driven IL-4 secretion may have impact on promoting a suitable environment for T. cruzi infection.
A 12% Coomassie Blue stained Sodium dodecyl sulfate-Polyacrylamide gel electrophoresis (SDS-PAGE) gel is shown to demonstrate de purity of purified rP21 and the bacterial protein pattern in B.E. (Fig. 1D).
rP21 promoted myeloperoxidase expression and leukocytes chemotaxis
rP21 treatment enhanced the levels of myeloperoxidase (MPO) but not of N-acetyl-beta-D-glucosaminidase (NAG) compared to control treatments in polyester sponge-induced inflammation (Fig. 2A,B). Moreover, the inflammatory score analysis showed that polyester sponges treated with rP21 displayed moderate inflammatory infiltrate, mostly composed of macrophages. B.E. treated sponges exhibited intense inflammatory infiltrated mainly composed of neutrophil and lymphocytes with a moderate level of necrotic tissue (Table 1). MPO is a lysosomal enzyme produced in high amounts by neutrophils, especially during their early maturation phase. MPO catalyzes the production of hypochlorous acid from hydrogen peroxide and chloride anion and oxidizes tyrosine to tyrosyl radicals. These radicals are cytotoxic to a variety of microorganisms. However, MPO released into extracellular milieu also contributes to tissue injury and disease pathology34. The lack of correlation between MPO levels and neutrophil infiltrate in rP21 treated sponges may be in part due to the expression of this enzyme by macrophages and B-lymphocytes35. Moreover, authors have identified granulocyte macrophage colony-stimulating factor as an endogenous regulator of macrophage myeloperoxidase expression in human atherosclerosis36.
Figure 2. rP21 promoted myeloperoxidase expression and leukocytes chemotaxis.

rP21 treatment enhanced the levels of myeloperoxidase (MPO) (A) but not of N-acetyl-beta-D-glucosaminidase (NAG) (B). rP21 but not B.E. induced macrophage migration in a trans-well system. Negative control (-control): serum-free medium; Positive control (+control): stromal cell-derived factor 1 (SDF-1α – CXCL12) (C). rP21 promoted neutrophil chemotaxis in Boyden chamber. This activity was completely abolished by previous cell treatment with pertussis toxin (PTX; negative control). Positive control: macrophage inflammatory protein (MIP-2) Experiments were performed four times in triplicate (D). rP21 treatment induced the recruitment of higher number of total leukocytes to mice peritoneal cavity (E). Phenotypic cell identification showed that at two hours rP21 promoted higher recruitment of granulocytes (F), lymphocytes (G) and monocytes (H). The ability of rP21 to recruit higher number of monocytes extended up to six hours post-inoculation. Higher number of mastocytes was observed in rP21-treated sponges compared to PBS and B.E. treatments. Experiments were performed twice using 6 animals/group. Samples were analyzed individually (I). 40 μg/mL of rP21 and B.E. were used. Data are expressed as mean ± standard deviation of experiments performed in triplicate. Significant differences were determined by one-way (A,B,C,D,I) or two-way ANOVA (E,F,G,H) and Turkey’s multiple comparisons test (GraphPad Prism software, version 6.01). Differences were considered significant when p < 0.05.
Table 1. Qualitative analyses of cell infiltrate in sponges treated with PBS, B.E or rP21.
| Histological criteria | PBS | B.E. | rP21 |
|---|---|---|---|
| Neutrophils | + | +++ | + |
| Macrophages | ++ | ++ | +++ |
| Lymphocytes | +++ | +++ | ++ |
| Plasma cells | ++ | + | + |
| Necrotic tissue | − | ++ | + |
| Inflammatory response | ++ | +++ | ++ |
Our previous studies showed that rP21 enhanced macrophage phagocytosis and actin polymerization by binding to the CXCR4 receptor18. The ability of rP21 to bind to CXCR4 indicated a potential chemotaxis activity driven by this protein. We performed different in vitro and in vivo experiments in order to gain insight into the direct effect of rP21 over leukocyte migration. First, we addressed the ability of rP21 to induce migration of peritoneal macrophages in a trans-well system through a polycarbonate membrane over a thin layer of extracellular matrix. Our results showed that rP21 but not B.E. induced macrophage migration (Fig. 2C). After, we verified that rP21 promoted neutrophil chemotaxis in Boyden chamber. This activity was completely abolished by previous cell treatment with pertussis toxin (PTX) (Fig. 2D). Finally, to assess whether rP21 has chemo-attractive activity in vivo, it was injected intra-peritoneally into mice and we observed cell recruitment at different time point compared to injection of PBS or B.E. The results showed that at two and six hours post-inoculation, rP21 treatment induced the recruitment of higher number of total leukocytes compared to the control treatments (Fig. 2E). Phenotypic cell identification showed that at two hours rP21 promoted higher recruitment of granulocytes, lymphocytes and monocytes (Fig. 2F,G). Leukocytes recovered from peritoneal cavity previous to PBS, B.E. or rP21 inoculation was similar to the result obtained at 0.5 hour post-inoculation (data not shown). The ability of rP21 to recruit higher number of monocytes extended up to six hours post-inoculation (Fig. 2H).
The literature has shown that the inflammatory process that characterizes CCC is accentuated during the acute phase of the disease. Although it may be clinically silent after the acute phase, inflammation is continuously present in patients with indeterminate and chronic forms of the disease37,38. Moreover, trypanosome antigens and/or its genomic material are found in inflammatory foci39,40,41,42,43. These observations provide a basis to propose a role for P21 regarding the onset and maintenance of chagasic heart inflammation.
Also, we observed higher number of mastocytes in rP21-treated sponges compared to PBS and B.E. treatments (Fig. 2I). This result may be in part explained by the higher levels of IL-4 detected in rP21-treated sponges. IL-4 is known to enhance mastocytes maturation, survival, proliferation and migration44. Mastocytes have been implicated in cardiovascular dysfunctions, such as ischemic heart disease, experimental myocardial infarction, myocarditis, heart failure, transplant-related fibrosis, and hypertensive heart disease45,46,47,48. In vitro and in vivo studies have shown the presence of mastocytes associated with cardiac T. cruzi infection49,50,51.
rP21 inhibited angiogenesis in vivo and in vitro
We observed a significant decrease in hemoglobin content in rP21 treated sponges (Fig. 3A). This result suggested a potential anti-angiogenic role of rP21. To confirm this hypothesis, we determined the number of blood vessels on histological preparations. Sponges treated with rP21 showed a decreased number of blood vessels compared to the control treatments (Fig. 3B). We also treated a murine endothelial cell line derived from thymus hemangioma (tEnd) with different concentrations of rP21 or B.E. and observed that rP21 did not alter cell viability and adhesion to a thin layer of extracellular matrix (Fig. 3C,D) but strongly inhibited vessel formation (Fig. 3E). B.E. showed cell toxicity and inhibition of adhesion at the concentrations of 40 and 80 μg/mL (Fig. 3C–E). The toxicity and inhibition of cell adhesion observed in B.E. treated cells may explain the lack of vessel formation. Representative images highlighting cell and vessels morphologies are also shown (Fig. 3F). The anti-angiogenic activity of rP21 may be due to a cascade of events triggered by direct interaction of the protein to endothelial cells. This possibility will be addressed in novel studies from our research group. However, another non excluding hypothesis is that rP21-induced IL-4 drives macrophage to produce soluble fms-like tyrosine kinase 1 (sFlt-1) and express an Arginase-1(+) phenotype resulting in the inhibition of angiogenesis52.
Figure 3. rP21 inhibited angiogenesis in vivo and in vitro.

Decrease in hemoglobin content was observed in rP21 treated sponges (A). Sponges treated with rP21 showed a decreased number of blood vessels compared to the control treatments (B). Representative HE-stained sponges are shown (panel on the up-right position) and yellow asterisks indicate blood vessels. 40 μg/mL of rP21 and B.E. were used. rP21 did not alter tEnd cell viability (C), adhesion (D) to extracellular matrix and inhibited vessel formation (E). B.E. showed cell toxicity and inhibition of adhesion at the concentrations of 40 and 80 μg/mL (representative results of four independent experiments performed in triplicate). Representative images highlighting vessels morphology (yellow dotted line) are also shown (F). Data are expressed as mean ± standard deviation of experiments performed in triplicate. Significant differences were determined by one-way ANOVA and Turkey’s multiple comparisons test (GraphPad Prism software, version 6.01). Differences were considered significant when p < 0.05.
Functional and structural micro-vascular abnormalities occur in CCC53,54. Consequently, vasospasms, decreased blood flow, focal ischemia, platelet thrombi, increased platelet aggregation, and elevated levels of thromboxane A-2 and endothelin-1 are frequently observed55,56,57,58. As such, P21 could impact on micro-vascular ischemia, which is believed to amplify chronic inflammatory aggression toward myocardial tissue59.
Envisaged mechanism triggered by rP21 in inflammation
Taken our results together with the current literature we propose that rP21 induces recruitment of leukocytes to the site of inflammation and up-regulates the expression of IL-4. IL-4 is produced by lymphocytes, basophils and mastocytes and promotes a Th2 immune response. Also, IL-4 induces macrophages to acquire an Arginase-1(+) phenotype and to increase production of sFlt-1 that culminates in the inhibition of angiogenesis. Moreover, IL-4 promotes survival and differentiation of B-lymphocytes in memory cells negatively impacting on immunoglobulin production and secretion. Mastocytes in the inflammatory foci will liberate several bioactive molecules that may participate in tissue damage, recruitment of neutrophils and amplification of IL-4-driven pathways (Fig. 4).
Figure 4. Envisaged mechanism triggered by rP21 in inflammation.
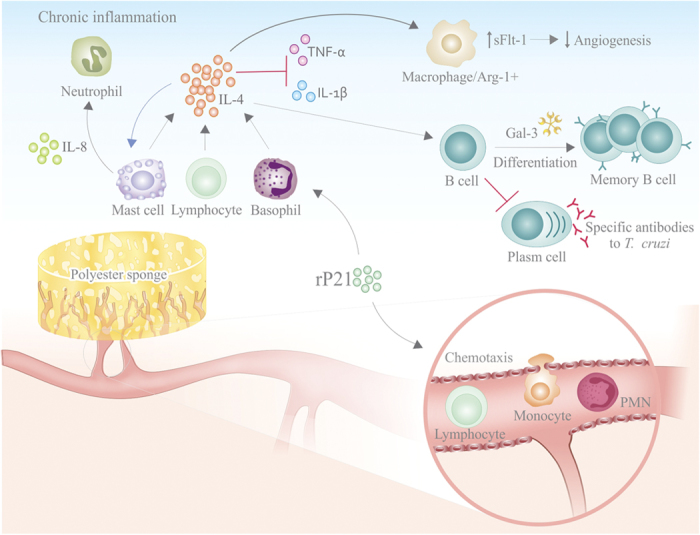
rP21 induces recruitment of leukocytes to the site of inflammation and up-regulates the expression of IL-4. IL-4 is produced by lymphocytes, basophils and mastocytes and promotes a Th2 immune response. Also, IL-4 induces macrophages to acquire an Arginase-1(+) phenotype and to increase production of sFlt-1 that culminates in the inhibition of angiogenesis. Moreover, IL-4 promotes survival and differentiation of B-lymphocytes in memory cells negatively impacting on immunoglobulin production and secretion. Mastocytes in the inflammatory foci will liberate several bioactive molecules that may participate in tissue damage, recruitment of neutrophils and amplification of IL-4-driven pathways.
This putative mechanism suggests a potential role of native P21 during T. cruzi pathogenesis and highlights P21 as a novel target for the development of innovative therapeutics agents against CCC.
Experimental procedures
Animals and ethics
Six-to-eight-week-old male C57BL/6 mice were maintained under standard conditions on a 12-h light-dark cycle in a temperature controlled setting (25 °C) with food and water ad libitum. Maintenance and care of animals complied with the guidelines of the Laboratory Animal Ethics Committee from the Universidade Federal de Uberlândia. Animal euthanasia was performed based on international welfare grounds according to the American Veterinary Medical Association Guidelines on Euthanasia. This study was approved by the ethics committee for animal research at Universidade Federal de Uberlândia.
Cell culture
Murine endothelial cell lines derived from thymus hemangioma (tEnd)60 were cultivated in RPMI 1640 medium supplemented with 10% bovine fetal serum, 2 mM l-glutamine, 2 mM sodium pyruvate, 1 mM non-essential amino acids, 100 U/mL penicillin and 100 μg/mL streptomycin and incubated at 37 °C and 5% CO2.
rP21 purification
rP21 (GenBank: EU004210.1) was purified as previously described17,19. B.E was obtained under the same conditions without inducing rP21 expression. The quality of purification was demonstrated by SDS-PAGE. Similar concentration of rP21 and B.E. was used in the experimental procedures.
Polyester inflammation induction and treatment scheme
A polyester sponge with 1.2 cm diameter (Vitafoam Ltd.) was stored in 70% ethanol v/v for at least 24 hours and subsequently boiled in distilled water for 30 minutes61,62. C57BL/6 mice were divided into groups of 10 animals for biochemical and 6 animals for histological analysis, treated with PBS, 40 μg/mL of rP2117,18,19 or B.E. The animals were anesthetized via intra-peritoneal with ketamine-xylazine (Syntec) (60 mg–4 mg/kg) and subjected to trichotomy and antisepsis in the dorsal region with alcohol 70% v/v. The sponge was introduced by midline incision in the inter-scapular region. Incisions were closed with a Donati suture using nylon 3.0. After recovery from anesthesia, animals were placed in individual cages with food and water ad libitum50. Treatments were injected into the sponge every 72 hours and analyzed during the chronic (9 days) phase of inflammation.
Hemoglobin content
Determination of the intra-implant hemoglobin content was performed using Drabkin’s reagent61,62. Implants were removed and mass determined 9 days post-implantation. Samples that showed macroscopic bleeding or infection were excluded from analysis. Each implant was homogenized (Tekmar TR-10, Ohio, USA) in 2 mL of a hemoglobin-specific chromogen reagent (Drabkin kit, Labtest) and added to 2 mL tubes. Samples were centrifuged at 4 °C for 30 minutes at 12,000 × g and homogenates were filtered through 0.22 μm filters (Millipore). Spectrophotometric analysis was performed at 540 nm using 96-well plates. The hemoglobin concentration of each sample was calculated based on a standard curve and the results were expressed as hemoglobin concentration (micrograms) per milligram wet weight of the implant.
Myeloperoxidase (MPO) activity
After determining the hemoglobin content, the supernatant was stored in protease inhibitor cocktail (Sigma Aldrich) at −80 °C. The pellet was resuspended in 2 mL sodium phosphate buffer (pH 5.4). 300 μL was transferred to tubes and supplemented with 600 μL of 0.5% w/v HTAB (hexadecyltrimethylammonium bromide, Sigma) diluted in phosphate buffer (pH 5.4). After, samples were centrifuged at 10,000 × g for 10 minutes at 4 °C and the supernatant was used. Finally, 100 μL of 0.003% hydrogen peroxide and 100 μL TMB (3,3′,5,5′-tetramethylbenzidine, Sigma) at 4 mM diluted in DMSO (dimethyl sulfoxide, Merck) were added in a novel tube followed by 200 μL of the sample supernatant and incubated for 1 minute. The reaction was stopped by adding 100 μL of 4 M H2SO4 (sulfuric acid, Merck). 200 μL of the final solution was added to 96-well plate and spectrophotometric analysis was performed at 450 nm. Results are expressed as the MPO index (absorbance/mg wet weight of the implant).
N-acetyl-beta-D-glucosaminidase (NAG) activity
After determining the hemoglobin content, the supernatant was stored in protease inhibitor cocktail (Sigma Aldrich) at −80 °C. The pellet was resuspended in 2.0 mL 0.9% saline with 0.1% Triton X-100 (Promega) and centrifuged at 3000 × g for 10 minutes at 4 °C. 100 μL of the supernatant was added to a 96-well plate in duplicate. After, 100 μL of substrate (p-nitrophenyl-N-acetyl-beta-D-glucosaminide, Sigma) diluted in citrate /phosphate buffer pH 4.5 was added to the samples and incubated at 37 °C for 30 minutes. Finally, 100 μL of 0.2 M glycine buffer pH 10.6 was added and absorbance was measured in spectrophotometer at 400 nm. NAG activity was calculated using a standard curve of p-nitrophenol. Results are expressed as nmol mL-1/mg wet weight of the implant.
Cytokines detection
The supernatant obtained after determining the hemoglobin content was used to measure the levels of IL-1β, TNF-α and IL-4 by Enzyme-Linked Immuno Sorbent Assay (ELISA) according to the manufacturer (BD Biosciences).
Histological analysis
Sponges designated for histological analysis were fixed in Metacarn buffer (60% methanol, 30% chloroform, 10% acetic acid) for 3 hours at 4 °C. Sponges were prepared for inclusion in ethyl alcohol PA for 30 minutes with 4 washes in xylene, followed by additional 30 minutes of incubation, three washes and finally embedded in paraffin. The blocks were processed for hematoxylin and eosin (HE) and toluidine blue staining. Images were obtained in a Leica DM500 microscope coupled to Las Ez camera and software. The type and severity of the inflammatory infiltrate were determined by the presence or absence of (i) neutrophils, (ii) macrophages, (iii) lymphocytes, (iv) plasmocytes, (v) necrotic tissue, (vi) intensity of inflammatory response. These features were scored for intensity: (−) absent, (+) mild, (++) moderate, (+++) intense.
Cell viability and adhesion
The viability of tEnd cells treated with rP21 was evaluated using the MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide) assay. Cells were seeded at 1.5 × 104 cells/well in 96-well micro-plates. After adhesion, cells were treated with different concentrations of rP21, Bacterial Extract (B.E.) or culture medium for 24 hours at 37 °C and 5% CO2. After 24 hours, cells were incubated with 5 mg/mL MTT for 3 hours at 37 °C. Formosan crystals resulting from MTT reduction were dissolved by addition of 100 μL of PBS containing 10% SDS and 0.01 M HCl (18 h, 37 °C and 5% CO2). Absorbance (570 nm) was measured on a multi-well scanning spectrophotometer (Thermo Scientific).
For adhesion assay, tEnd cells (2 × 104 cells/well) were pre-incubated for 30 min at 37 °C with different concentration of rP21, B.E. or culture medium. After, cells were added to 96-well plate at 37 °C and 5% CO2. After 2 h, detached cells were removed by two washes using PBS. Finally, the plate was incubated with 5 mg/mL MTT for 3 h at 37 °C, as described above.
Angiogenesis assay
The influence of rP21 on endothelial cell tube formation was evaluated by Matrigel tube formation assays. Experiments were performed as described63. tEnds cells (2 × 105 cells/well) were pre-incubated with 10, 40 and 80 μg/mL of rP21, B.E. or only culture medium for 30 minutes at room temperature and plated on 24-well plates previously coated with 250 μL of 5.25 mg/mL Matrigel (BD Bioscience) and supplemented with basic Fibroblast Growth Factor (bFGF) (30 ng/mL). After 18 hours of incubation at 37 °C and 5% CO2, images were acquired at×20 magnifications in bright light microscope.
In vitro and in vivo chemotaxis assays
The in vitro chemotaxis assay for peritoneal macrophages was performed in a trans-well system through a polycarbonate membrane with a defined pore size of 8 μm (Chemicon Cell Invasion Assay Kit, EMD Millipore Corporation, Darmstadt, Alemanha). Peritoneal macrophages from C57BL/6 mice were seeded at 106 in top of the insert in serum-free medium. Serum-free medium contained 40 μg/mL of rP21 or B.E. or stromal cell-derived factor 1 (SDF-1α–CXCL12–50 ng/mL–Sigma Aldrich) or medium only was added in the bottom well. Cells were incubated for 72 hours. Migratory cells were stained and counted using an optical microscope.
For neutrophil chemotaxis, a Boyden chamber assay was used, consisting of upper and lower chambers separated by a polycarbonate membrane with an 8-μm pore. Neutrophils were extracted from C57BL/6 bone marrow using Ficoll-Paque (Sigma Aldrich) to separate them from other cells. Some of the cells were pre-treated with pertussis toxin (PTX – 100 ng/mL–Sigma Aldrich) (G-protein coupled receptor inhibitor) for 1 hour. Treated and non-treated cells were placed in the upper chamber. In the lower chamber, only media, rP21 or macrophage inflammatory protein (MIP-2–30 ng/mL–Life Technologies), a chemotaxis positive control, were added. After 1 hour, the membrane was removed, fixed and colored with panoptic dye. Cells that migrated were counted using an optical microscope.
In vivo, C57BL/6 mice received three different intra-peritoneal treatments: PBS, 40 μg/mL of rP2117,18,19 or B.E. The animals were euthanized at intervals of 0.5, 02 and 06 hours. Peritoneal lavage was obtained by inoculating 4 mL PBS into the peritoneum cavity. Total leukocyte recruitment was quantified in a Neubauer chamber. Cells were phenotypically identified by size and granularity in flow cytometry (BD FacsCanto II).
Statistical analysis
Data are expressed as mean ± standard deviation of experiments performed in triplicate. Significant differences were determined by one-way or two-way ANOVA and Turkey’s multiple comparisons test (parametric) or Dunn’s test (nonparametric), according to the experimental design (GraphPad Prism software, version 6.01). Differences were considered significant when p < 0.05.
Additional Information
How to cite this article: Teixeira, T. L. et al. Trypanosoma cruzi P21: a potential novel target for chagasic cardiomyopathy therapy. Sci. Rep. 5, 16877; doi: 10.1038/srep16877 (2015).
Acknowledgments
This study was supported by grants from Fundação de Amparo à Pesquisa do Estado de Minas Gerais (FAPEMIG - Processes numbers: APQ-00305-12; APQ-01855-13); Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES) (Process number: 23038.005295/2011-40) and Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq – Process number: 444862/2014-5). This manuscript has been edited by native English-speaking experts from BioMed Proofreading LLC.
Footnotes
Author Contributions T.L.T., F.C.M., A.A.S. and S.C.T. contributed to the experimental manipulations, analyzed and interpreted data, wrote the first drafts of the manuscript. B.C.B., M.A.S., F.A.M., P.C.B., A.A.R., A.F.O.N., B.A.F., J.P.S.S., S.R.D. and D.S.L. performed experiments. V.M.R.A., F.A.A., T.C.T. and M.J.B.S. designed the biological experiments, analyzed and interpreted data. C.V.S. coordinated, designed the biological experiments, analyzed, interpreted the data, edited the manuscript and got grants. All authors reviewed the manuscript.
References
- Carvalho A. C. C. et al. Cell therapy in Chagas Disease. Interd. Perspc. Infect. Dis. 2009, 1–6 (2009). [Google Scholar]
- Abuhad A., Segre C. A. W., Demarchi L. M. M. F. & Aiello V. Case 06/2005 –progressive heart failure in a 65-year-old man with permanent cardiac pacemaker. Arq. Bras.Ccardiol. 85, 437–443 (2005). [DOI] [PubMed] [Google Scholar]
- Ramírez J. D. et al. Chagas Cardiomyopathy Manifestations and Trypanosoma cruzi Genotypes Circulating in Chronic Chagasic Patients. PLoS. Negl. Trop. Dis. 4, e899, doi: 10.1371/journal.pntd.0000899 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrer M. F., Pascuale C. A. & Gomez R. M. DTU I isolates of Trypanosoma cruzi induce upregulation of Galectin-3 in murine myocarditis and fibrosis. Parasitol. 141, 849–858 (2014). [DOI] [PubMed] [Google Scholar]
- Cruz-Robles D. et al. Association between IL-1B and IL-1RN gene polymorphisms and Chagas’ disease development susceptibility. Immunol. Invest. 38, 231–239 (2009). [DOI] [PubMed] [Google Scholar]
- Boldt A. B., Luz P. R. & Messias-Reason I. J. MASP2 haplotypes are associated with high risk of cardiomyopathy in chronic Chagas disease. Clin. Immunol. 140, 63–70 (2011). [DOI] [PubMed] [Google Scholar]
- Henao-Martínez A. F., Schwartz D. A. & Yang I. V. Chagasic cardiomyopathy, from acute to chronic: is this mediated by host susceptibility factors ? Trans. R. Soc. Trop. Med. Hyg. 106, 521–527 (2012). [DOI] [PubMed] [Google Scholar]
- Pissetti C. W. et al. Association between the lymphotoxin-alpha gene polymorphism and chagasic cardiopathy. J. Interf. Cytok. Res. 33, 130–135 (2013). [DOI] [PubMed] [Google Scholar]
- Frade A. F. et al. Genetic susceptibility to Chagas disease cardiomyopathy: involvement of several genes of the innate immunity and chemokine-dependent migration pathways. BMC Infect. Dis. 13, 587, doi: 10.1186/1471-2334-13-587 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lula J. F. et al. Plasma concentrations of tumor necrosis factor-alpha, tumor necrosis factor-related apoptosis-inducing ligand, and Fas Ligand/CD95L in patients with Chagas cardiomyopathy correlate with left ventricular dysfunction. Eur. J. Heart Fail. 11, 825–831 (2009). [DOI] [PubMed] [Google Scholar]
- Silverio J. C. et al. Perforin-expressing cytotoxic cells contribute to chronic cardiomyopathy in Trypanosoma cruzi infection. Int. J. Exp. Pathol. 91, 72–86 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cutrullis R. A. et al. Elevated serum levels of macrophage migration inhibitory factor are associated with progressive chronic cardiomyopathy in patients with Chagas disease. PLoS ONE. 8, e57181, doi: 10.1371/journal.pone.0057181 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sousa G. R. et al. Plasma Cytokine Expression Is Associated with Cardiac Morbidity in Chagas Disease. PLoS ONE 9, e87082, doi: 10.1371/journal.pone.0087082 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roman-Campos D. et al. Novel insights into the development of chagasic cardiomyopathy: role of PI3Kinase/NO axis. Int. J. Cardiol. 167, 3011–3020 (2013). [DOI] [PubMed] [Google Scholar]
- Macedo L. G. R. et al. Rationale and Design of a Prospective Study to Assess the Effect of Microcirculatory Antiplatelet and Vasodilation Therapy in Patients with Chronic Chagas Disease and Coronary Microvascular Disease. Rev. Bras. Cardiol. Invasiva. 20, 82–88 (2012). [Google Scholar]
- Nunes M. C. et al. Chagas disease: an overview of clinical and epidemiological aspects. J. Am. Coll. Cardiol. 62, 767–776 (2013). [DOI] [PubMed] [Google Scholar]
- Silva C. V. et al. Characterization of a 21 kDa protein from Trypanosoma cruzi associated with mammalian cell invasion. Microbes Infect. 11, 563–570 (2009). [DOI] [PubMed] [Google Scholar]
- Rodrigues A. A. et al. A Recombinant Protein Based on Trypanosoma cruzi P21 Enhances Phagocytosis. Plos ONE. 7, e51384, doi: 10.1371/journal.pone.0051384 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dos Santos M. A. et al. A successful strategy for the recovering of active P21, an insoluble recombinant protein of Trypanosoma cruzi. Sci. Rep. 4, 4259, doi: 10.1038/srep04259 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodrigues A. A. et al. IFN-γ Plays a unique role in protection against low virulent Trypanosoma cruzi strain. PLoS Negl. Trop. Dis. 6, e1598, doi: 10.1371/journal.pntd.0001598 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi P. & Reiser H. IL-4: role in disease and regulation of production. Clin. Exp. Immunol. 113, 317–319 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krettli A. U. & Brener Z. Protective effects of specific antibodies in Trypanosoma cruziinfection. J Immunol. 116, 755–761 (1976). [PubMed] [Google Scholar]
- Rodriguez A. M. et al. Trypanosoma cruzi infection in B cell deficient rats. Infect. Immun. 31, 524–530 (1981). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kierszenbaum F. & Howard J. G. Mechanisms of resistance against Trypanosoma cruzi infection: the importance of antibodies and antibody forming capacity in biozzi high and low responder mice. J. Immunol. 116, 1208–1214 (1976). [PubMed] [Google Scholar]
- Brener Z. Why vaccines do not work in Chagas’ disease ? Parasitol. Today. 2, 196–202 (1986). [DOI] [PubMed] [Google Scholar]
- Tarleton R. L. & Kuhn R. E. Measurement of parasite-specific immune responses in vitro: evidence for suppression of the antibody response to Trypanosoma cruzi. Eur. J. Immunol. 15, 845–850 (1985). [DOI] [PubMed] [Google Scholar]
- Hayes M. M. & Kierszenbaum F. Experimental Chagas’ disease: kinetics of lymphocyte responses and immunological control of transition from acute to chronic T. cruzi infection. Infect. Immun. 31, 1117–1124 (1981). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kierszenbaum F., Moretti E. & Sztein M. B. Molecular basis of Trypanosoma cruzi-induced immunosuppression. Altered expression by activated human lymphocytes of molecules which regulate antigen recognition and progression through the cell cycle. Biol. Res. 26, 197–207 (1993). [PubMed] [Google Scholar]
- Hernandez-Munain C. et al. GP 50/55, a membrane antigen of Trypanosoma cruzi involved in autoimmunity and immunosuppression. Biol. Res. 26, 209–218 (1993). [PubMed] [Google Scholar]
- Bryan M. A., Guyach S. E. & Norris K. A. Specific Humoral Immunity versus Polyclonal B Cell Activation in Trypanosoma cruzi Infection of Susceptible and Resistant Mice. PLoS Negl. Trop. Dis. 4, e733, doi: 10.1371/journal.pntd.0000733 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuñiga E. et al. Trypanosoma cruzi-induced immunosuppression: B cells undergo spontaneous apoptosis and lipopolysaccharide (LPS) arrests their proliferation during acute infection. Clin. Exp. Immunol. 119, 507–515, doi: 10.1046/j.1365-2249.2000.01150.x (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acosta-Rodríguez E. V. et al. Galectin-3 mediates IL-4-induced survival and differentiation of B cells: functional cross-talk and implications during Trypanosoma cruzi infection. J. Immunol. 172, 493–502 (2004). [DOI] [PubMed] [Google Scholar]
- Klebanoff S. J. Myeloperoxidase: friend and foe. J. Leukoc. Biol. 77, 598–625 (2005). [DOI] [PubMed] [Google Scholar]
- de Araujo T. H. et al. Intracellular localization of myeloperoxidase in murine peritoneal B-lymphocytes and macrophages. Cell. Immunol. 281, 27–30 (2013). [DOI] [PubMed] [Google Scholar]
- Sugiyama S. et al. Macrophage Myeloperoxidase Regulation by Granulocyte Macrophage Colony-Stimulating Factor in Human Atherosclerosis and Implications in Acute Coronary Syndromes. Am. J. Pathol. 158, 879–891 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira-Barretto A. C. et al. Right ventricular endomyocardial biopsy in chronic Chagas’ disease. Am. Heart J. 111, 307–312 (1986). [DOI] [PubMed] [Google Scholar]
- Carrasco-Guerra H. A. et al. Clinical, histochemical, and ultrastructural correlation in septal endomyocardial biopsies from chronic chagasic patients: detection of early myocardial damage. Am. Heart J. 113, 716–724 (1987). [DOI] [PubMed] [Google Scholar]
- Jones E. M. et al. Amplification of a Trypanosoma cruzi DNA sequence from inflammatory lesions in human chagasic cardiomyopathy. Am. J. Trop. Med. Hyg. 48, 348–357 (1993). [DOI] [PubMed] [Google Scholar]
- Bellotti G. et al. In vivo detection of Trypanosoma cruzi antigens in hearts of patients with chronic Chagas’ heart disease. Am. Heart J. 131, 301–307 (1996). [DOI] [PubMed] [Google Scholar]
- Zhang L. & Tarleton R. L. Parasite persistence correlates with disease severity and localization in chronic Chagas’ disease. J. Infect. Dis. 180, 480–486 (1999). [DOI] [PubMed] [Google Scholar]
- Palomino S. A., Aiello V. D. & Higuchi M. L. Systematic mapping of hearts from chronic chagasic patients: the association between the occurrence of histopathological lesions and Trypanosoma cruzi antigens. Ann. Trop. Med. Parasitol. 94, 571–579 (2000). [DOI] [PubMed] [Google Scholar]
- Tarleton R. L. Parasite persistence in the etiology of Chagas disease. Int. J. Parasitol. 31, 550–554 (2001). [DOI] [PubMed] [Google Scholar]
- Olsson N., Taub D. D. & Nilsson G. Regulation of mast cell migration by T and T cytokines: identification of tumor necrosis factor-alpha and interleukin-4 as mast cell chemotaxins. Scand. J. Immunol. 59, 267–272 (2004). [DOI] [PubMed] [Google Scholar]
- Kalesnikoff J. & Galli S. J. New developments in mast cell biology. Nat. Immunol. 9, 1215–1223 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palladini G., Tozzi R. & Perlini S. Cardiac mast cells in the transition to heart failure: innocent bystanders or key actors? J. Hypertens. 21, 1823–1825 (2003). [DOI] [PubMed] [Google Scholar]
- Francis G. S. & Tang W. H. Histamine, mast cells, and heart failure: is there a connection? J. Am. Coll. Cardiol. 48, 1385–1386 (2006). [DOI] [PubMed] [Google Scholar]
- Martin T. R. et al. Mast cells contribute to the changes in heart rate, but not hypotension or death, associated with active anaphylaxis in mice. J. Immunol. 151, 367–376 (1993). [PubMed] [Google Scholar]
- Brower G. L. & Janicki J. S. Pharmacologic inhibition of mast cell degranulation prevents left ventricular remodeling induced by chronic volume overload in rats. J. Card. Fail. 11, 548–556 (2005). [DOI] [PubMed] [Google Scholar]
- Almeida H. O., Pereira F. E. & Tafuri W. L. Mast cells in Chagas’ chronic cardiopathy. Rev. Inst. Med. Trop. Sao Paulo. 17, 5–9 (1975). [PubMed] [Google Scholar]
- Postan M., Correa R., Ferrans V. J. & Tarleton R. L. In vitro culture of cardiac mast cells from mice experimentally infected with Trypanosoma cruzi. Int. Arch. Allergy Immunol. 105, 251–257 (1994). [DOI] [PubMed] [Google Scholar]
- Meuser-Batista M., Correa J. R., Soares M. J. & Henriques-Pons A. Isolation of cardiac mast cells in experimental Trypanosoma cruzi infection. Tissue Cell. 40, 309–316 (2008). [DOI] [PubMed] [Google Scholar]
- Wu W.-K. et al. IL-4 Regulates Specific Arg-1(+) Macrophage sFlt-1-Mediated Inhibition of Angiogenesis. Am. J. Pathol. 185, 2324–2335 (2015). [DOI] [PubMed] [Google Scholar]
- Punukollu G. et al. Clinical aspects of the Chagas’ heart disease. Int. J. Cardiol. 115, 279–283 (2007). [DOI] [PubMed] [Google Scholar]
- Marin-Neto J. A. et al. Pathogenesis of chronic Chagas heart disease. Circulation. 115, 1109–1123 (2007). [DOI] [PubMed] [Google Scholar]
- Rossi M. A., Goncalves S. & Ribeiro-dos-Santos R. Experimental Trypanosoma cruzi cardiomyopathy in BALB/c mice. The potential role of intravascular platelet aggregation in its genesis. Am. J. Pathol. 114, 209–216 (1994). [PMC free article] [PubMed] [Google Scholar]
- Rossi M. A. Microvascular changes as a cause of chronic cardiomyopathy in Chagas’ disease. Am. Heart J. 120, 233–236 (1990). [DOI] [PubMed] [Google Scholar]
- Tanowitz H. B. et al. Enhanced platelet adherence and aggregation in Chagas’ disease: a potential pathogenic mechanism for cardiomyopathy. Am. J. Trop. Med. Hyg. 43, 274–281 (1990). [DOI] [PubMed] [Google Scholar]
- Tanowitz H. B. et al. Compromised microcirculation in acute murine Trypanosoma cruzi infection. J. Parasitol. 82, 124–130 (1996). [PubMed] [Google Scholar]
- Biolo A., Ribeiro A. L. & Clausell N. Chagas Cardiomyopathy–Where Do We Stand After a Hundred Years ? Prog. Cardiovac. Dis. 52, 300–316 (2010). [DOI] [PubMed] [Google Scholar]
- Achê D. C. et al. Biochemical properties of a new PI SVMP from Bothrops pauloensis: Inhibition of cell adhesion and angiogenesis. Int. J. Biol. Macromol. 72, 445–453 (2014). [DOI] [PubMed] [Google Scholar]
- Andrade S. P., Fan T. P. D. & Lewis G. P. Quantitative in vivo studies on angiogenesis in a rat sponge model. Br. J. Exp. Pathol. 68, 755–766 (1987). [PMC free article] [PubMed] [Google Scholar]
- Plunkett M. L. & Hailey J. A. An in vivo quantitative angiogenesis model using tumor cells entrapped in alginate. Lab. Invest. 62, 510–517 (1990). [PubMed] [Google Scholar]
- Hu D. E., Hiley C. R., Smither R. L., Gresham G. A. & Fan T. P. D. Correlation of 133Xe clearance, blood flow and histology in the rat sponge model for angiogenesis. Lab. Invest. 72, 601–610 (1995). [PubMed] [Google Scholar]