

Abstract
The synthesis, characterization and evaluation in solid-state devices of a series of 8 cationic iridium complexes bearing different numbers of methoxy groups on the cyclometallating ligands are reported. The optoelectronic characterization showed a dramatic red shift in the absorption and the emission and a reduction of the electrochemical gap of the complexes when a methoxy group was introduced para to the Ir-C bond. The addition of a second or third methoxy group did not lead to a significant further red shift in these spectra. Emission maxima over the series ranged from 595 to 730 nm. All complexes possessing a motif with a methoxy group at the 3-position of the cyclometalating ligands showed very short emission lifetimes and poor photoluminescence quantum yields whereas complexes having a methoxy group at the 4-position were slightly blue shifted compared to the unsubstituted parent complexes, resulting from the inductively electron withdrawing nature of this directing group on the Ir-C bond. Light-emitting electrochemical cells were fabricated and evaluated. These deep red emitters generally showed poor performance with electroluminescence mirroring photoluminescence. DFT calculations accurately modelled the observed photophysical and electrochemical behavior of the complexes and point to an emission from a mixed charge transfer state.
Light Emitting Electrochemical Cells (LEECs)1,2 are solid state lighting devices that typically incorporate a charged phosphorescent Ionic Transition Metal Complex (iTMC) as the luminophore. These devices offer some advantages over organic light-emitting diodes (OLEDs)2. LEECs have simple device architectures (single- or two-layer devices), use air-stable high work function electrodes (e.g., Al, Au) and are fabricated using solution printing processing with benign solvents such as acetonitrile (ACN), thus giving the potential to operate without encapsulation. They are thus cheaper to manufacture and can be driven by alternating current (AC). Employing LEEC technology, large-area artificial illumination is thus easier to attain3,4. In LEECs the iTMC accomplishes several functions including: lowering of the charge injection barrier by the displacement of ions; facilitating charge transport through iterative redox cycling; and generating light through phosphorescence. Based on the similarities in mechanism, LEECs should be able to attain similar efficiencies to OLEDs. Despite the advantages listed above, LEECs incorporating iTMCs have several weaknesses: (i) slow turn-on time compared to OLEDs; (ii) low EQEs; (iii) limited stability of the device; and (iv) color quality5. Red light emission is important as a component of white light emitting devices.
The principal strategies utilized to date to tune the emission energy into the red (λem > 630 nm) include the use of highly conjugated ancillary ligands6,7,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22 (N^N) such as biquinolines or the incorporation of electron-withdrawing groups23,24 onto the backbone of the ancillary ligand. Likewise, using conjugated cyclometallating ligands (C^N) such as phenylquinolines has also been exploited as a strategy for producing red-light emission10,16,20,25,26,27,28. Red-light emission was also obtained through energy transfer to a covalently tethered organic perylenediimide29. The use of pyridyl oxadiazoles30 and dipyridyl pyrazines31 as ancillary ligands has also resulted in deep red emission. LEECs incorporating these complexes emit with electroluminescent (EL) λmax between 630–665 nm and peak EQEs of between 0.02–7.4%32.
Oddly, the use of electron donating groups on the phenyl moiety of the C^N ligand has not be explored as a strategy to tune the emission energy to the red in cationic iridium(III) complexes despite the common use of electron withdrawing fluorine atoms to push the emission to the blue9,33,34,35,36,37,38,39. Herein we systematically explore the impact of methoxy substitution on the phenyl ring of the C^N ligands on the optoelectronic properties of a series of eight cationic iridium complexes40,41. DFT calculations rationalize the observed behavior. LEECs fabricated with these emitters demonstrate that this is a viable strategy for emission tuning in electroluminescent devices to the red, leading to a maximum emission wavelength of 650 nm and CIE(x,y) coordinates (0.61, 0.38) for the reddest complex. Unfortunately, the performance suffers in terms of brightness and device stability.
Results and Discussion
Synthesis
The targeted methoxy-substituted C^N ligands were synthesized in two steps from the corresponding aryl bromide via arylboration and Suzuki coupling with 2-bromo-5-methyl pyridine (Figure S23). The non-optimized yields for boronic acid formation ranged from 36–65% while the cross-coupling reaction proceeded nearly quantitatively. The iridium dimers, [(C^N)2Ir(Cl)]2, were synthesized following the procedure first described by Nonoyama42. Heteroleptic cationic complexes 1a-4b were obtained in excellent yield through the cleavage of these dimers with 2.25 equiv. of ancillary diimine ligand (2,2’-bipyridine, bpy, or 4,4’-ditert-butyl-2,2’-bipyridine, dtBubpy) in refluxing DCM/MeOH (1:1 v/v) under N2 (Fig. 1)19. The complexes were isolated as their hexafluorophosphate salts following the dropwise addition of a methanolic solution of the corresponding chloride salts into an aqueous solution of NH4PF6. Each complex was purified by recrystallization from a 1:1 solution of DCM and diisopropyl ether. The structural identity and purity of each of the complexes were ascertained through 1H NMR and 13C NMR spectroscopy (Figures S1-S22), ESI-HRMS and melting point analyses.
Figure 1. Synthesis of complexes in study.

Reagents and conditions: a2-EtOC2H4OH/H2O (8:1 v/v), 110 °C, N2, 19 h. bi. CH2Cl2/MeOH (1:1 v/v), 55 °C, 19 h, N2; ii. Excess aq. NH4PF6.
The aromatic regions of the 1H NMR spectra of 1b, 2b, 3b and 4b obtained in CD3CN are shown in Fig. 2. From the overall pattern of the 1H NMR it is evident that each of 1b-4b possesses similar C2-symmetry. By contrast, Davies and co-workers reported the presence of two isomers for the related [Ir(3-MeOppz)2(bpy)]PF6 wherein a second minor isomer was also detected when cyclometallation occurred at C2 (3-MeOppzH = N-(3-methoxyphenyl)pyrazole)40. As previously observed by Davies, the H5 signal is the most upfield proton and is most strongly influenced by the adjacent 4-MeO substituent. The chemical shifts of the dtBubpy ligand (the three most downfield signals) are essentially invariant to methoxy substitution on the C^N ligands though are shifted slightly downfield in 1b compared to the other three complexes in the family. The 3-MeO group exhibits the strongest influence on the chemical shift of the H2 proton; in its absence, H2 in 2b is situated downfield at 7.71 ppm. The presence of two methoxy groups in 3b results in a downfield shift of the 3-MeO group and an upfield shift of the 4-MeO group compared to 1b and 2b, respectively. However, the additional incorporation of the 5-MeO group, positioned upfield at 2.87 ppm, in 4b promotes a significant 0.30 ppm downfield shift of the 4-MeO signal compared to that in 3b.
Figure 2. Aromatic region of the 1H NMR spectra of 1b-4b in CD3CN at 298 K.
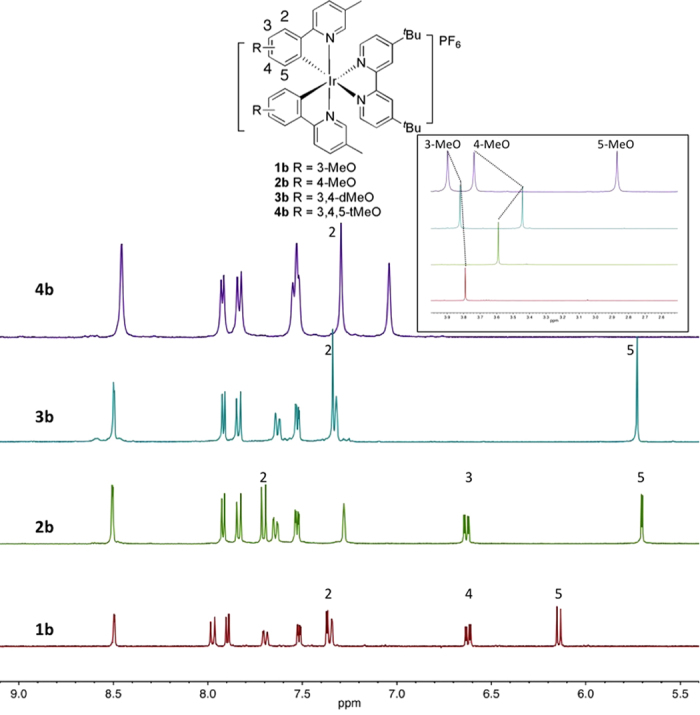
Inset shows MeO region of spectra.
Cyclic voltammetry
The electrochemical behavior of 1a-4b was investigated through cyclic voltammetry (CV) studies in deaerated ACN solution containing n-NBu4PF6 as the supporting electrolyte and using Fc/Fc+ as an internal standard at 298 K. All potentials are referenced to SCE (Fc/Fc+ = 0.38 V in ACN)43. The electrochemistry data, obtained at a scan rate of 50 mV s−1, are summarized in Table S1. The CV behavior was reproducible at the faster scan rate of 200 mV s−1. The CV traces for 1a-4b are shown in Fig. 3. All complexes exhibit reversible first reduction waves and all complexes save 2a and 2b exhibit reversible first oxidation waves.
Figure 3. Cyclic voltammograms for 1a-4b recorded at 298 K at 50 mVs−1 in deaerated ACN with 0.1 M (n-Bu4N)PF6.

The first oxidation wave, corresponding to the HOMO, is attributed to the IrIII/IrIV couple with contribution from the C^N ligands. For all the complexes in the study this wave is significantly cathodically shifted compared to both [Ir(ppy)2(bpy)]PF6 (E1/2,ox = 1.25 V) and [Ir(ppy)2(dtBubpy)]PF6 (E1/2,ox = 1.29 V) owing to the mesomeric electron donating character of the methoxy group44. The incorporation of electron-donating tert-butyl groups generally promote a further cathodic shift of between 0.02–0.06 V. Methoxy groups are Janus-like, being electron donating mesomerically but electron withdrawing inductively, evident from an evaluation of Hammet sigma parameters (σm = 0.12, σp = −0.27). Complexes 2a and 2b, with 4-MeOppy C^N ligands, are shifted cathodically the least in the series compared to their unsubstituted analogs. Moving the MeO group to the 3-position results in a 200 mV shift to lower positive potentials for 1a vs 2a and 230 mV for 1b vs 2b. The presence of two MeO groups results in a synergistic destabilization of the oxidation wave by a further 110 mV for 3a vs 1a and 120 mV for 3b vs 1b. The addition of a third MeO group in 4a and 4b does not result in any further appreciable tuning of the oxidation wave. This first oxidation wave is irreversible for 2a and 2b but is a pseudo-reversible one-electron process for the other complexes in the series.
A second irreversible oxidation wave was found between 1.30–1.57 V in all eight complexes. This oxidation wave is the least cathodically shifted in 2a and 2b with peak potentials of 1.55 and 1.57 V, respectively. By contrast, for 1a and 1b the second oxidation waves at 1.33 and 1.32 V, respectively, are significantly cathodically shifted compared to those in 2a and 2b. These second oxidation waves are pushed to lower positive potentials with the presence of two methoxy groups with irreversible oxidation waves for 3a and 3b of 1.13 and 1.25 V, respectively. With three methoxy groups present in the cases of 4a and 4b these waves remain at 1.21 V. From the similar electrochemical behavior between the first and second oxidation waves, we can conclude that this second wave is also localized on the C^N ligand.
The reduction potential for 1a-4a is quite invariant at between −1.40 and −1.42 V; similarly that for 1b-4b resides between −1.49 and −1.52 V. The clustering of reduction potentials points to a reversible one-electron first reduction wave, and thus the LUMO, that is localized on the ancillary diimine ligand. The presence of the MeO groups though located on the C^N ligands act to destabilize the reduction wave compared to both [Ir(ppy)2(bpy)]PF6 (E1/2,red = −1.36 V) and [Ir(ppy)2(dtBubpy)]PF6 (E1/2,red = −1.38 V). Surprisingly, there is a significantly larger influence for the family of complexes containing a dtBubpy N^N ligand than the congeners 1a-4a.
The electrochemical gaps for 1a-4b range from 2.27 for 3a to 2.63 V for 2b. Thus, the use of a 4-MeOppy C^N ligand has a negligible effect on the tuning of the HOMO-LUMO gap compared to the parent complexes [Ir(ppy)2(bpy)]PF6 (ΔEredox = 2.65 V) and [Ir(ppy)2(dtBubpy)]PF6 (ΔEredox = 2.71 V). The use of dtBubpy as the ancillary ligand promotes a widening of the gap by between 0.08–0.11 V except for 2a and 2b where there is little influence observed. Within each family of complexes 3a and 3b possess the smallest electrochemical gaps of 2.27 and 2.35 V, respectively.
UV-Visible absorption spectroscopy
The UV-Visible absorption spectra for 1a-4b were recorded in aerated ACN at 298 K, the spectra of which are shown in Fig. 4. The calculated molar absorptivities, ε, are collected in Table S2. As with many cationic heteroleptic iridium complexes the absorption spectra exhibit certain characteristic features. There are intense bands between 244 and 278 nm, which are assigned to the spin-allowed ligand centered (1LC) π-π* transitions for cyclometallating (C^N) ppy and ancillary (N^N) bpy ligands. Lower intensity absorption bands below 300 nm are assigned to spin-allowed mixed charge transfer (CT) bands while bands visible past 450 nm are assigned to spin-forbidden mixed CT bands.
Figure 4. UV-vis absorption spectra for 1a-4b in ACN at 298 K. Inset: Zoomed UV-vis spectra for the low energy region between 350–550 nm.
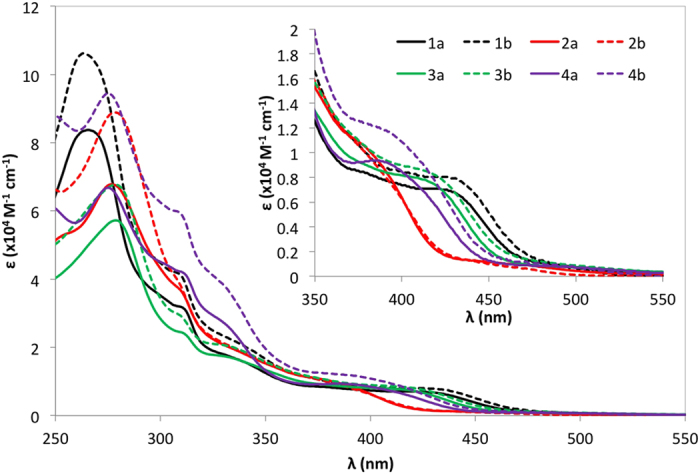
The molar absorptivities for each of the absorption bands are on the order of 104 M−1 cm−1. The profile and intensity of these bands are similar to other bis(heteroleptic) cationic iridium complexes reported elsewhere in the literature45,46,47,48,49. The 1LC bands for the dtBubpy family of complexes are generally about 20% more intense than those of the bpy family of complexes. The 1LC bands for 1a and 1b, found at ca. 265 nm, are hypsochromically shifted by 1635 and 2052 cm−1, respectively, compared to those of 2a and 2b found at 277 nm. The absorption spectra for the latter two complexes also show the presence of an addition 1LC band at 255 and 250 nm, respectively. This absorption profile is mirrored for 3a, 3b, 4a and 4b wherein a high intensity band for each is located between 275–279 nm and the lower intensity higher energy band is also observable in 4a and 4b (244 and 248 nm, respectively); this latter band appears only as a shoulder in 3a and 3b. All complexes possess an absorption band at ca. 308–312 nm. Similar to other heteroleptic iridium (III) cationic complexes50,51, hypochromic bands between 330–425 nm result from mixed (1CT) transitions resulting from a combination of metal-to-ligand and ligand-to-ligand CT transitions (1MLCT and 1LLCT, respectively). The low energy bands for 1a and 1b are located at 425 nm whereas 2a and 2b these are found at 390 nm. The red shifted nature of these transitions in 1a and 1b are in line with their electrochemistry and smaller electrochemical gaps. Additionally, owing to strong spin-orbit coupling on iridium there is the presence of very low intensity bands in the region of 400–500 nm that correspond to spin-forbidden 3CT transitions.
From the UV-Vis spectroscopy we can conclude that placing a methoxy group in the 3-position, para to the C-Ir bond, invokes a larger red-shift in the absorption compared to substitution at the 4-position. The incorporation of two methoxy groups does not lead to a further red shift in the absorption profile. In fact, 3a and 3b possess bands at 419 and 424 nm, respectively that are slightly blue shifted compared to those of 1a and 1b. With three methoxy groups present on the C^N ligand, a further blue shift is imparted in 4a and 4b where these transitions are found at 385 and 395 nm, respectively.
Solution state photophysical behavior
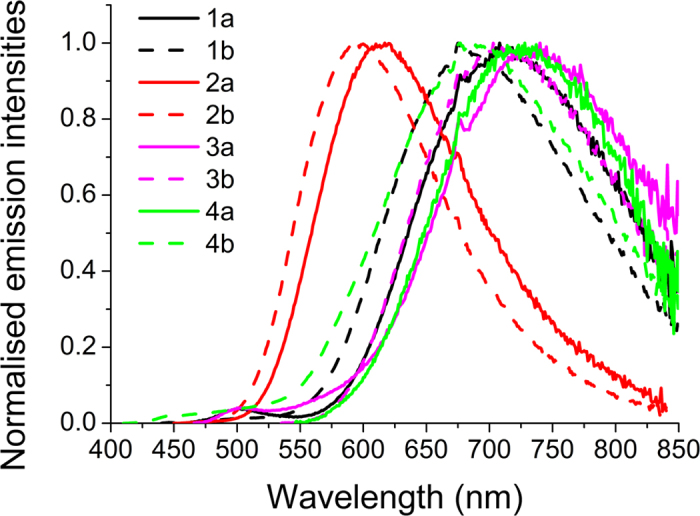
The steady state emission spectra for complexes 1a-4b were recorded in degassed ACN at 298 K (Fig. 5). The emission profiles are broad and featureless with emission maxima ranging from 580–730 nm. The yellow-to-near infrared emission is assigned to a mixed metal-to-ligand and ligand-to-ligand charge transfer transitions (3MLCT and 3LLCT), commonly observed for heteroleptic cationic iridium complexes9,49. Excepting 2a and 2b, the emission bands of these complexes are red shifted compared to [Ir(ppy)2 (bpy)]PF6 (λem = 610 nm), which had been previously reported by us49 and others52,53,54. The emission maxima for 3a, 3b, 4a and 4b are further red-shifted compared to complexes 1a and 1b. The reddest emission occurs in 3a. The trends observed in the emission spectra mirror those in the absorption spectra and the CV thus demonstrating that the T1 state is modulated similarly to the S0 ground state. Not surprisingly, the use of the inductively electron-donating dtBubpy N^N ligands in 1b-4b blue shifts the emission compared to their respective analogs in 1a-4a.
Figure 5. Steady state emission spectra for 1a-4b in degassed ACN at 298 K.
The photoluminescence quantum yields (ΦPL) for 1a-4b were determined in degassed ACN at 298 K using by comparing with a reference standard [Ru(bpy)3](PF6)2 (ФPL = 9.5%)55 under optically dilute conditions as described by Demas and Crosby56 (Table 1). With the exception of 2a (5.7%) and 2b (15.4%), the ΦPL values are exceedingly low, especially compared to other iridium complexes emitting in the same spectral region57. Further information about the photophysics of the compounds were obtained through time-resolved phosphorescence (PL) measurements by exciting the deaerated samples at room temperature at 390 nm. The results are shown in Fig. 6. Compound 1a shows monoexponential decay with a lifetime of 4.7 ns with no presence of long lived emission.
Table 1. Relevant photophysical data for complex 1a-4b.
| Complex | λem (nm)a | ФPL (%)b | τ1 (ns)a | Α1c | τ2 (ns)a | Α2c |
|---|---|---|---|---|---|---|
| 1a | 710 | 0.2 | 4.72 | 1 | – | – |
| 1b | 680 | 0.3 | 10.2 | 0.84 | 174 | 0.16 |
| 2a | 618 | 5.7 | 194 | 0.97 | 1505 | 0.03 |
| 2b | 595 | 15.4 | 388 | 0.96 | 2411 | 0.04 |
| 3a | 730 | 0.3 | 1.6 | 0.95 | 29.3 | 0.05 |
| 3b | 700 | 0.1 | 3.2 | 0.82 | 100 | 0.18 |
| 4a | 720 | 0.2 | 1.66 | 0.91 | 23.3 | 0.09 |
| 4b | 685 | 0.2 | 3.2 | 0.78 | 70 | 0.22 |
aMeasured in deaerated ACN at 298 K at excitation wavelength 390 nm.
bUsing [Ru(bpy)3](PF6)2 as the standard (ФPL = 9.5% in deaerated ACN at 298 K).
cA1 and A2 are the pre-exponential factors for the phosphorescence lifetime.
Figure 6. Time-resolved photoluminescence spectra for 1a-4b in degassed ACN at 298 K.

The samples were excited at 390 nm. The emission is collected in each case at the peak of the PL spectra. The red dotted sprectrum represent the instrument response function.
The presence of the bulky dtBubpy N^N ligand in compound 1b enhances the lifetime corresponding to phosphorescence emission but still nonradiative decay dominates as evident from poor PLQY and the short-lived decay seen in Fig. 6a. For compounds 2a and 2b, Fig. 6b, emission lifetimes are much longer and in the sub-microsecond regime, 194 and 388 ns, respectively, with pre-exponential factors of up to 97%. So the position of the methoxy group strongly influences the photophysical properties of the compounds as we saw with PLQY results. The addition of a further MeO moiety in 3a drastically alters the PL lifetime and essentially all the emission is quenched nonradiatively. The addition of dtBubpy N^N ligand helps in improving the metal-ligand coupling but still most of the photons decay nonraditively as shown in Fig. 6c. Further addition of methoxy groups in 4a and 4b does not contribute to enhancing radiative decay. The PL decay characteristics are summerised in Table 1. The presence of iridium and the large observed stokes shifts, ranging from 7806–11606 cm−1, clearly indicate phosphorescence despite the short emission state lifetimes.
The markedly low quantum yields are accompanied by very short, nanosecond emission lifetimes. The structural commonality in these poorly emissive complexes is the presence of the 3-MeO groups on the C^N ligands. Similarly low luminescence had been previously reported for red-emitting fac-Ir(atpy)3, where atpy = 2-(5′-amino-4′-tolyl)pyridine (λmax = 613 nm in DMSO, ΦPL = 9.4 × 10−3)58. Thus, though substitution at this position red shifts the emission energy the most, electron donation into the Ir-CC^N bond leads to rapid decay of the excited state. Generally, the τe’s of 1b-4b are nearly twofold longer than those of 1a-4a due to the presence of the bulky tert-butyl groups.
The photoluminescence spectra in thin films for 1a-4b were obtained by spin-coating an acetonitrile solution of each complex mixed with the ionic liquid (IL) 1-butyl-3-methylimidazolium hexafluorophosphate [BMIM+:PF6−], in a similar configuration to the light-emitting layer in LEECs (vide infra). The films were prepared on quartz substrates and recorded with λexc = 350 nm at room temperature (see Fig. 7).
Figure 7. Photoluminescence spectra of complexes 1a-4b in thin solid films mixed with [BMIM+ :PF6–] in a molar ratio 4:1.
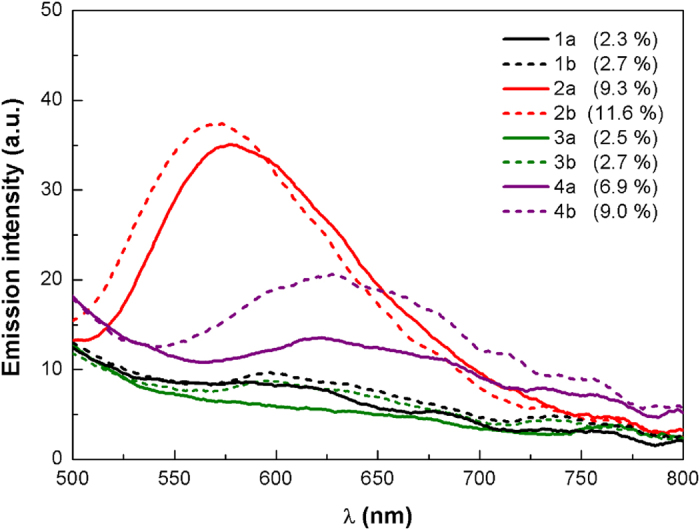
The photoluminescence quantum yield (ΦPL) for the solid films are indicated in the legend (numbers in brackets).
In thin solid-films, the emission spectra are quite similar to the photoluminescence spectra obtained in acetonitrile solution, but showing much increased ΦPL. All the spectra have a maxima located between 570–640 nm, with the bluest emission corresponding to the 2a and 2b complexes at 575 nm and 580 nm, respectively, similar to those found in acetonitrile solution. These complexes present the highest PL intensities, exhibiting a ΦPL close to 10% and 12%, respectively. However, the rest of the complexes show a red-shifted broad band centered around 640 nm that is significantly blue-shifted compared to their corresponding solution emission maxima, with PL intensities decreased drastically from 4a and 4b (Fig. 7).
Theoretical calculations
In order to better understand the nature of the optoelectronic properties and the impact of methoxy substitution on the complexes, a combined density functional theory (DFT) and time-dependent DFT (TDDFT) study was undertaken59,60,61,62,63,64. All complexes were modeled using Gaussian 0965 using the following DFT protocol at the B3LYP66,67,68 level of theory with the SBKJC-DVZ69,70,71,72 basis set for iridium, 6–31 G* for heavy atoms directly coordinated to iridium and 3–21 G* for all other atoms69,73,74,75,76,77,78,79,80,81,82 in the presence of the solvent (ACN)83.
The geometry of the ground state structures was fully optimized without the imposition of symmetry restrictions. The geometry for each of the complexes is pseudo-octahedral. Selected structural parameters for 1a-4a are summarized in Table 2. Bond lengths from the bonded ligand atoms to iridium are significantly longer than those observed for [Ir(ppy)2(bpy)]PF6: Ir–Cppy = 2.013 Å, Ir–Nppy = 2.045 Å, and Ir–Nbpy = 2.133 Å84. Ligand bite angles for 1a-4a are similar to this archetype complex. Increasing the MeO-content on the C^N ligand results in a progressive contraction of the Ir-Nbpy bond. There is a modest elongation of the Ir-C^N bond in 4a, most likely due to accommodation of the methoxy group at C5 of the phenyl ring.
Table 2. Selected average structural parameters for 1a, 2a, 1b, and 2ba.
| Complexes | 1a |
2a |
3a |
4a |
||||
|---|---|---|---|---|---|---|---|---|
| S0 | T1 | S0 | T1 | S0 | T1 | S0 | T1 | |
| Ir-Nbpy | 2.187 | 2.179 | 2.186 | 2.175 | 2.184 | 2.175 | 2.178 | 2.169 |
| Ir-NC^N | 2.077 | 2.076 | 2.078 | 2.075 | 2.078 | 2.076 | 2.073 | 2.072 |
| Ir-CC^N | 2.030 | 2.002 | 2.028 | 2.008 | 2.028 | 2.003 | 2.039 | 2.011 |
| Nbpy-Ir-Nbpy | 75.5 | 76.0 | 75.6 | 76.1 | 75.7 | 76.1 | 75.9 | 76.4 |
| NC^N-Ir-NC^N | 80.2 | 80.9 | 80.3 | 81.2 | 80.2 | 81.0 | 79.9 | 80.3 |
aBond lengths in Å and bond angles in°.
Figure 8 provides a comparison of the relative energies of the five highest energy occupied and five lowest energy unoccupied molecular orbitals (MOs) for 1a-4b along with contour plots of HOMOs and LUMOs. As has been reported for analogous cationic systems9,37,49, the HOMO is located on both the aryl ring of the C^N ligands and the iridium atom. The 4-MeO group in 2a and 2b perturb this picture in that electron density is found also on the pyridyl moiety of the C^N ligand. The LUMO for each of the eight complexes is situated exclusively (electron density distribution >95%) on the diimine ligand.
Figure 8. Calculated energy level scheme for the Kohn-Sham orbitals between HOMO-4 to LUMO+4 of 1a-4b, and the associated DFT calculated HOMO-LUMO energy gap (in eV) and electron density contour plots for 1a-4a (0.002 e bohr−3).

The contour plots for 1b-4b mirror those of 1a-4a.
The energies of the HOMOs are sensitive to the methoxy substitution pattern on the C^N ligands. For instance, the HOMO for 1a resides at −5.37 eV whereas the HOMO for 2a is significantly stabilized at −5.70 eV. When two MeO-groups are present as in 3a there is only a slight destabilization to −5.64 eV compared to 2a. A further destabilization is predicted for 4a with the HOMO found at −5.53 eV. Replacement of bpy for dtBubpy results in a net destabilization of the HOMO by only about 0.03 eV. Generally, the picture provided by the DFT computations is somewhat consistent with the CV data presented in Table S1 in that 2a and 2b are the most anodically shifted in the series of complexes. However, from a survey of the first oxidation potentials the DFT calculations underestimate the magnitude of the destabilization of the HOMO in 3a, 3b, 4a and 4b. The LUMO energies for 1a-4a remain quite invariant at around −2.48 eV. In a similar fashion, the LUMO energies for 1b-4b are destabilized relative to 1a-4a owing to the electron-releasing tert-butyl substituents and are found around −2.29 eV. This trend is reproduced in the CVs though only a 0.08 V relative destabilization is observed experimentally. Similarly, the computations reproduce the principal features in the UV-Visible spectra for the complexes (cf. Figures S39-S42). The HOMO-LUMO energy gap is therefore smallest for 1a at 2.90 eV and largest for 2b at 3.40 eV.
The geometries of the triplet state were optimized using spin-unrestricted DFT calculations at the UB3LYP level. Notable deformations are modest contractions of the Ir-Nbpy and Ir-CC^N bonds. Otherwise there is little perturbation of the geometry upon migrating onto the triplet manifold (Table 2). The spin densities for the T1 state for 1a-4a are shown in Fig. 9; those for 1b-4b possess similar topologies. The T1 state is describe predominantly by a LUMO →HOMO transition implying an emission resulting from an admixture of 3MLCT/3LLCT. This assignment is consistent with the TD-DFT results (Table S3) and the observed broad and unstructured emission at 298 K in ACN.
Figure 9. Calculated spin density contours of the T1 state for 1a, 2a, 3a and 4a (isocontour value of 0.0004 au).

The emission energy was predicted using three different methodologies and these results are summarized in Table 3. The phosphorescence is estimated as the difference between the T1 and S0 states in their respective optimized geometries (E0,0), which is a good indicator of the E0,0 emission measured at 77 K. The emission predicted by TDDFT (ETDDFT) for the S0 → T1 monoexcitation are based on an optimized S0 geometry. The adiabatic electronic emission (EAE) is determined from the vertical energy difference between the T1 and S0 states at the optimized geometry of the T1 state. The EAE calculations reproduce accurately the solution state emission observed at 298 K, with relative errors ranging from 3.1–15.4% though the calculations for 3a (and 3b) underestimate more significantly the impact of the electron-releasing methoxy groups on red shifting the emission.
Table 3. Predicted Emission Energies.
| Theoreticala |
λem (298 K) /nm | Errord/% | |||
|---|---|---|---|---|---|
| ETDDFT/nm | E0,0/nm | EAE/nm | |||
| 1a | 550.7 | 593.0 | 688.4 | 710 | 3.1 |
| 2a | 483.8 | 507.9 | 566.0 | 618 | 8.4 |
| 3a | 499.6 | 535.5 | 618.0 | 730 | 15.4 |
| 4a | 524.1 | 574.0 | 676.6 | 720 | 6.0 |
aETDDFT = energy of S0 →T1 transition obtained by TDDFT at the S0 optimized geometry; E0,0 = E(T1)-E(S0) at their respective optimized geometries obtained by DFT; EAE = E(T1)-E(S0) at the T1 optimized geometries (adiabatic electronic emission) obtained by DFT. See experimental section for computational details. All values obtained are in the presence of ACN solvent; bHighest energy 77 K emission band reported and highest intensity 298 K emission band reported; cError = |λem(77 K)-E0,0/λem(77 K)| in eV; dError = |λem(298 K)-EAE/λem(298 K)| in eV.
Device Characterization
Despite the poor solution and solid state PL performance and taking into account the presence of several candidate red emitters we investigated the electroluminescence performance of the iridium complexes by preparing simple two-layer light emitting electrochemical cells (LEECs). The devices were prepared by spin-coating a thin layer (80 nm) of poly(3,4-ethylenedioxythiophene):poly(styrenesulfonate) (PEDOT:PSS) on top of an indium tin oxide (ITO)-coated glass substrate, followed by the active layer (100 nm) consisting of a mixture of each complex with the ionic liquid (IL) 1-butyl-3-methylimidazolium hexafluorophosphate [BMIM+:PF6–] at a molar ratio of 4:1. The ionic liquid was added to reduce the turn-on time of the device, as higher mobilities for the ionic species are expected. Finally a 70 nm aluminium layer was evaporated as the top electrode. More details concerning the device preparation can be found in the Experimental section.
The device lifetime was measured by applying a constant voltage (4 V) and monitoring the current density and luminance versus time. Most of the devices didn´t show good performances, presenting very short lifetimes and efficiencies. The best result, obtained for molecule 4b, is shown in Fig. 10.
Figure 10. Electroluminescence performance for ITO/PEDOT:PSS/4b:IL(4:1)/Al device driven at a constant voltage of 4 V.
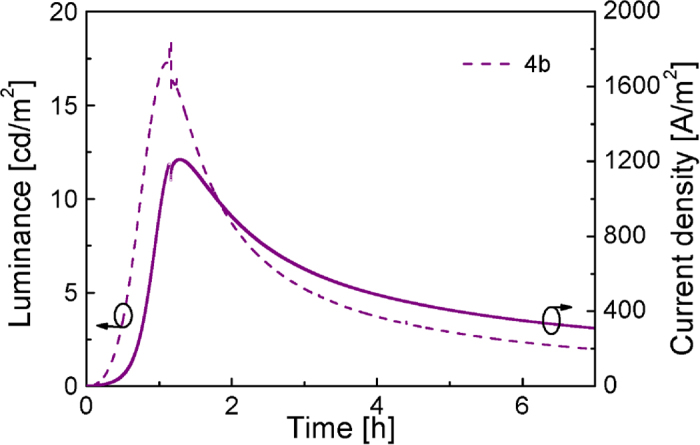
As can be seen in Fig. 10, the luminance reaches a maximum value of 18 cd m−2 after one hour running, and decays to a half-maximum (L1/2) in 2 hours. The current peaks at 1206 A m−2 after 1.28 hours, and decreases in a similar trend to the luminance. As a consequence, low efficiencies were reached in these devices, leading to values lower than 1 cd A−1, an external quantum efficiency (EQE %) of 0.05% and CIE(x,y) coordinates equal to (0.61, 0.38).
Figure 11 shows the typical electroluminescence spectra for the LEEC device prepared with the molecule 4b. Comparing with the previous data it resembles the PL spectra, which consist on a broad band centered at around 650 nm.
Figure 11. Electroluminescence spectra obtained for the LEEC device prepared with molecules 4b at 298 K.
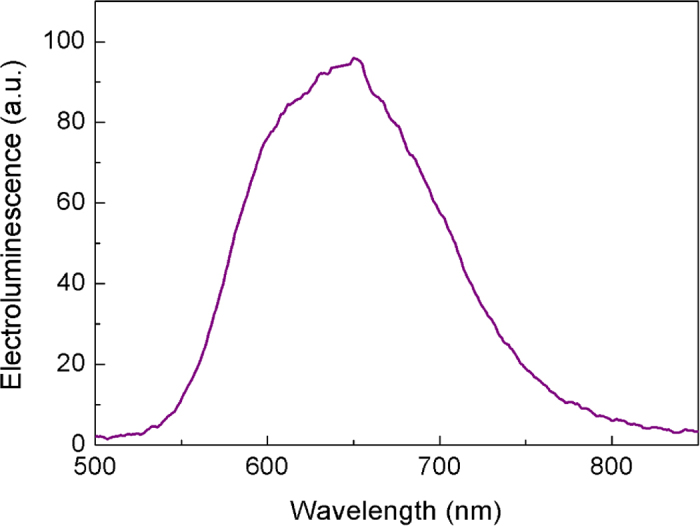
Device parameters for the other complexes in the series can be found in the supporting information (see Table S4). Compared to other recently reported LEECs, these performances are not very high85,86. In fact LEECs using complex 1a had such low performance that no electroluminescence data could be obtained. Nevertheless, the complexes that are presented in this work are interesting emitters as they show a very red-shifted emission spectra with maxima around 650 nm, leading to very deep-red CIE coordinates.
Conclusions
Herein, we reported eight new cationic iridium complexes bearing methoxy groups on the cyclometalating ligand. The goal of the study was to use mesomerically electron donating groups to promote a bathochromic shift in the emission and produce deep red emitters. Our measurements of solutions of these materials show that through incorporation of a methoxy group para to the Ir-C bond in the complex deep red to near infrared emission could be obtained in the complexes, however at dramatically reduced photoluminescence quantum yields and emission lifetimes. Experimental results are consistent with the DFT calculations. PL intensity increased somewhat in the thin film and LEEC devices were fabricated. Though these LEEC devices were also found to emit in the deep red they were poorly stable and not bright. From this study we can conclude that using electron donating groups on the cyclometallating ligands in cationic iridium complexes is not a good design strategy for producing bright and stable red emitters and rather highly conjugative C^N ligands should be employed.
Experimental Section
General Synthetic Procedures
Commercial chemicals were used as supplied. All reactions were performed using standard Schlenk techniques under inert (N2) atmosphere with freshly distilled anhydrous solvents obtained from a Pure SolvTM solvent purification system from Innovative Technologies except where specificallsy mentioned. Flash column chromatography was performed using silica gel (Silia-P from Silicycle, 60 Å, 40–63 μm). Analytical thin layer chromatography (TLC) was performed with silica plates with aluminum backings (250 μm with indicator F-254). Compounds were visualized under UV light. 1H and 13C NMR spectra were recorded on a Brucker Avance spectrometer at 400 MHz and 100 MHz, respectively. The following abbreviations have been used for multiplicity assignments: “s” for singlet, “d” for doublet, “t” for triplet, “m” for multiplet and “br” for broad. Deuterated chloroform (CDCl3) and deuterated actonitrile (CD3CN) were used as the solvent of record. Melting points (Mp’s) were recorded using open-ended capillaries on a Meltemp melting point apparatus and are uncorrected. High resolution mass spectra were recorded on a quadrupole time-of-flight (ESI-Q-TOF), model Maxis from Bruker in positive electrospray ionization mode and spectra were recorded using sodium formate solution as calibrantat at the Université de Sherbrooke. m-Methoxybenzeneboronic acid, p-Methoxybenzeneboronic acid, dimethoxybenzeneboronic acid and 3,4,5-Trimethoxybenzeneboronic acid were prepared in 75, 78, 36 and 65%, respectively from the corresponding bromomethoxybenzenes following the reported literature procedure87. The corresponding iridium(III) dimers, [(C^N)2Ir(Cl)]2 were prepared according to the procedure described by Nonoyama42 and isolated by filtration and used without further purification or characterization.
General Procedure for the Synthesis of cyclometallating (C^N) ligands
2-Bromo-5-methylp-yridine (1.0 equiv.), K2CO3 (3.0 equiv.) and arylboronic acid (1.25 equiv.) were dissolved in degassed dioxane/H2O (5:1 v/v) in order to obtain a 0.05 M concentration in 2-bromo-5-methylpyridine. Pd(PPh3)4 (0.008 g, 0.007 mmol, 5 mol%) was then added and the solution was repeatedly degassed. The reaction mixture was heated at reflux temperature for 19 h. Upon cooling, the solution was quenched with equal volume of H2O and diluted with equal volume of DCM. The organic layer was separated and dried over anhydrous MgSO4. Removal of solvent gave a dark slurry, which was purified by column chromatography with silica gel (EtOAc/hexane, 1:9 to 1:1)
2-(3’-Methoxyphenyl)-5-methylpyridine (3-MeOppy). 0.026 g was obtained as yellow oil-like liquid. Yield: 95%. Rf: 0.23 (10% EtOAc/hexanes on silica). 1H NMR (400 MHz, CDCl3) δ (ppm): 8.51 (dd, J = 0.70, 1.85 Hz, 1H), 7.61 (d, J = 8.06 Hz, 1H), 7.54 (m, 2H), 7.51 (ddd, J = 0.87, 1.46, 7.68, 1H), 6.94 (t, J = 8.17 Hz, 1H), 3.88 (s, 3H), 2.36 (s, 3H). The characterization matches that previously reported88.
2-(4’-Methoxyphenyl)-5-methylpyridine (4-MeOppy): 0.675 g was obtained as a white solid. Yield: 95%. Rf: 0.25 (10% EtOAc/hexanes on silica). 1H NMR (400 MHz, CDCl3) δ (ppm): 8.48 (s, 1H), 7.92 (m, 2H), 7.54 (dt, J = 4.95, 8.17 Hz, 2H), 6.99 (m, 2H), 3.86 (s, 3H), 2.35 (s, 3H). The characterization matches that previously reported89,90.
2-(3’,4’-Dimethoxyphenyl)-5-methylpyridine (3,4-dMeO): 0.602 g was obtained as a brownish damp solid. Yield: 99%. Rf: 0.20 (10% EtOAc/hexanes on silica). 1H NMR (400 MHz, CDCl3) δ (ppm): 8.48 (s, 1H), 7.61 (m, 2H), 7.53 (dd, J = 2.05, 8.09 Hz, 1H), 7.47 (dd, J = 2.03, 8.35, 1H), 6.94 (d, J = 8.38, 1H), 3.99 (s, 3H), 3.93 (s, 3H), 2.26 (s, 3H). 13C NMR (100 MHz, CDCl3) δ (ppm): 154.7, 150.0, 149.9, 149.4, 137.6, 132.6, 131.3, 119.8, 119.3, 111.2, 109.9, 56.2, 18.4. HR-MS (EI, 70 eV): [M + Na]+ Calculated: (C14H15NO2Na) 252.0995; Found: 252.1003.
2-(3’,4’,5’-Trimethoxyphenyl)-5-methylpyridine (3,4,5-tMeO): 0.919 g was obtained as a yellow oil-like liquid. Yield: 99%. Rf: 0.15 (10% EtOAc/hexanes on silica). 1H NMR (400 MHz, CDCl3) δ (ppm): 8.49 (dd, J = 0.73, 1.30, 1H), 7.55 (m, 2H), 7.20 (s, 2H), 3.95 (s, 6H), 3.89 (s, 3H), 2.37 (s, 3H). 13C NMR (100 MHz, CDCl3) δ (ppm): 154.6, 153.7, 150.0, 138.9, 137.6, 135.3, 131.8, 120.1, 104.0, 93.2, 61.1, 56.4, 18.4. HR-MS (ES-Q-TOF): [M + Na]+ Calculated: (C15H17NO3Na) 282.1101; Found: 282.1110.
General procedure for the synthesis of [(C^N)2Ir(N^N)]PF6 complexes
Iridium dimer (0.07 mmol, equiv.) and N^N ligand (2,2’-bipyridine, bpy or 4,4’-di-tert-butyl-2,2’-bipyridine, dtbubpy) (0.16 mmol, 2.25 equiv.) were solubilized with 12 mL of DCM/MeOH (50:50 v/v). The mixture was degassed repeatedly and placed under N2 and heated to 55 °C for 19 h. Over the course of the reaction the mixture darkened in color. The solution was cooled to RT and the solvent removed under reduced pressure. The crude solid was re-dissolved in a minimum amount of MeOH and added slowly to an aqueous solution of NH4PF6 (10 mL, 6.13 mmol, 1 g/10 mL) under gentle stirring. The first drop caused the precipitation of a brownish yellow-to-red colored solid. The solid suspension was conserved at 0 °C for 2 h and then filtered through a Buckner funnel and the resulting solid was washed with water and Et2O. The residue was dried under vacuo to obtain the desired colored complex complex. The complex was then recrystallized in dichloromethane / diisopropylether (50:50 v/v) by slow evaporation.
Iridium (III) bis [2-(3’-methoxyphenyl)-5-methylpyridinato-N,C2’]-N,N’-(bipyridine) hexafluorophosphate: [(3-MeOppy)2Ir(bpy)](PF6) 1a: Red crystals (0.082 g). Yield: 94%. Mp: 310–311 °C. 1H NMR (400 MHz, CD3CN) δ (ppm): 8.53 (d, J = 8.17 Hz, 2H), 8.13 (t, J = 8.05 Hz, 2H), 8.02 (d, J = 5.43 Hz, 2H), 7.97 (d, J = 8.34 Hz, 2H), 7.69 (d, J = 8.32 Hz, 2H), 7.51 (dt, J = 1.17, 7.61 Hz, 2H), 7.38 (m, 4H), 6.62 (dd, J = 2.76, 8.29 Hz, 2H), 6.15 (d, J = 8.28, 2H), 3.79 (s, 6H), 2.12 (s, 6H). 13C NMR (100 MHz, CDCl3) δ (ppm): 165.2, 156.4, 156.0, 150.5, 148.2, 144.1, 139.9, 139.3, 139.2, 133.9, 132.0, 128.3, 125.4, 119.6, 117.3, 110.2, 55.6, 18.5. HR-MS (ES-Q-TOF): [M-PF6]+ Calculated: (C36H32IrN4O2) 745.2151; Found: 745.2160.
Iridium (III) bis [2-(3’-methoxyphenyl)-5-methylpyridinato-N,C2’]-N,N’-(di-tert-bipyridine) hexafluorophosphate: [(3-MeO-ppy)2Ir(dtbubpy)](PF6) 1b: Yellow powder (0.141 g). Yield: 88%. Mp: 209–210 °C. 1H NMR (400 MHz, CD3CN) δ (ppm): 8.49 (d, J = 1.83 Hz, 2H), 7.97 (d, J = 8.43 Hz, 2H), 7.89 (d, J = 5.87 Hz, 2H), 7.69 (dd, J = 1.10, 8.43 Hz, 2H), 7.51 (dd, J = 2.20, 5.87 Hz, 2H), 7.36 (m, 4H), 6.62 (dd, J = 2.57, 8.43 Hz, 2H), 6.13 (d, J = 8.07, 2H), 3.78 (s, 6H), 2.12 (s, 6H), 1.43 (s, 18H). 13C NMR (100 MHz, CDCl3) δ (ppm): 165.2, 164.0, 156.2, 156.1, 150.0, 148.5, 144.3, 139.8, 139.1, 133.8, 131.9, 125.5, 121.8, 119.4, 117.2, 110.1, 105.9, 55.6, 36.0, 30.9, 30.5, 18.5. HR-MS (ES-Q-TOF): [M-PF6]+ Calculated: (C44H48IrN4O2) 857.3404; Found: 857.3401.
Iridium (III) bis [2-(4’-methoxyphenyl)-5-methylpyridinato-N,C2’]-N,N’-(bipyridine) hexafluorophosphate: [(4-MeOppy)2Ir(bpy)](PF6) 2a: Yellow powder (0.134 g). Yield: 87%. Mp: 342 °C. 1H NMR (400 MHz, CD3CN) δ (ppm): 8.54 (d, J = 8.18 Hz, 2H), 8.14 (dt, J = 1.60, 8.00 Hz, 2H), 8.05 (d, J = 5.86 Hz, 2H), 7.84 (d, J = 8.39 Hz, 2H), 7.71 (d, J = 8.62 Hz, 2H), 7.64 (dd, J = 1.42, 8.36, 2H), 7.52 (dt, J = 1.16, 7.57, 2H), 7.33 (s, 2H), 6.64 (dd, J = 2.57, 8.60 Hz, 2H), 5.72 (d, J = 2.56 Hz, 2H), 3.59 (s, 6H), 2.09 (s, 6H). 13C NMR (100 MHz, CDCl3) δ (ppm): 165.1, 161.0, 155.9, 152.0, 150.4, 147.7, 140.0, 139.2, 137.0, 132.3, 128.2, 126.1, 125.6, 118.7, 117.6, 107.4, 55.0, 18.5. HR-MS (ES-Q-TOF): [M-PF6] + Calculated: (C36H32IrN4O2) 745.2151; Found: 745.2147. This photophysical characterization of this complex has been previously reported (λem = 593 nm, ΦPL = 2.19%, τe = 217 ns) but not the compound characterization41.
Iridium (III) bis [2-(4’-methoxyphenyl)-5-methylpyridinato-N,C2’]-N,N’-(di-tert-bipyridine) hexafluorophosphate: [(4-MeOppy)2Ir(dtbubpy)](PF6) 2b: Yellow powder (0.134 g). Yield: 84%. Mp: 236–238 °C. 1H NMR (400 MHz, CD3CN) δ (ppm): 8.50 (d, J = 1.78 Hz, 2H), 7.92 (d, J = 5.86 Hz, 2H), 7.84 (d, J = 8.38 Hz, 2H), 7.70 (d, J = 8.61 Hz, 2H), 7.65 (m, 2H), 7.53 (dd, J = 1.88, 5.85 Hz, 2H), 7.28 (s, 2H), 6.63 (dd, J = 2.56, 8.59, 2H), 5.70 (d, J = 2.53 Hz, 2H), 3.59 (s, 6H), 2.10 (s, 6H), 1.44 (s, 18H). 13C NMR (100 MHz, CDCl3) δ (ppm): 165.1, 164.1, 160.9, 155.9, 152.6, 149.9, 148.0, 139.1, 137.2, 132.4, 126.0, 125.5, 122.0, 118.6, 117.3, 107.4, 105.9, 55.0, 36.0, 18.5. HR-MS (ES-Q-TOF): [M-PF6] + Calculated: (C44H48IrN4O2) 857.3404; Found: 857.3403. This photophysical characterization of this complex has been previously reported (λem = 576 nm, ΦPL = 5.87%, τe = 401 ns) but not the compound characterization41.
Iridium (III) bis [2-(3’,4’-dimethoxyphenyl)-5-methylpyridinato-N,C2’]-N,N’-(bipyridine) hexafluorophosphate: [(3,4-dMeOppy)2Ir(bpy)](PF6) 3a: Brownish yellow powder (0.132 g). Yield: 95%. Mp: 234–236 °C. 1H NMR (400 MHz, CD3CN) δ (ppm): 8.52 (d, J = 8.16 Hz, 2H), 8.13 (dt, J = 1.54, 8.02 Hz, 2H), 8.04 (d, J = 5.35 Hz, 2H), 7.84 (d, J = 8.39 Hz, 2H), 7.63 (dd, J = 1.32, 8.41 Hz, 2H), 7.53 (dt, J = 1.16, 7.57, 2H), 7.35 (s, 4H), 5.74 (s, 2H), 3.82 (s, 6H), 3.45 (s, 6H), 2.10 (s, 6H). 13C NMR (100 MHz, CD3CN) δ (ppm): 165.5, 156.3, 151.3, 151.1, 148.7, 145.8, 142.8, 139.2, 135.7, 132.4, 128.4, 124.8, 117.6, 113.6, 109.4, 100.6, 56.0, 54.9, 17.2. HR-MS (ES-Q-TOF): [M-PF6]+ Calculated: (C38H36IrN4O4) 805.2362; Found: 805.2361.
Iridium (III) bis [2-(3’,4’-dimethoxyphenyl)-5-methylpyridinato-N,C2’]-N,N’-(di-tert-bipyridine) hexafluorophosphate: [(3,4-dMeOppy)2Ir(dtbubpy)](PF6) 3b: Brownish yellow powder (0.158 g). Yield: 98%. Mp: 318 °C. 1H NMR (400 MHz, CD3CN) δ (ppm): 8.50 (d, J = 1.65 Hz, 2H), 7.92 (d, J = 5.84 Hz, 2H), 7.84 (d, J = 8.40 Hz, 2H), 7.63 (dd, J = 1.36, 8.43 Hz, 2H), 7.53 (dd, J = 1.88, 5.85, 2H), 7.34 (s, 2H), 7.32 (s, 2H), 5.73 (s, 2H), 3.82 (s, 6H), 3.44 (s, 6H), 2.11 (s, 6H), 1.44 (s, 18H). 13C NMR (100 MHz, CDCl3) δ (ppm): 165.8, 164.0, 156.0, 151.1, 150.0, 148.1, 145.5, 143.2, 139.0, 135.2, 132.0, 125.6, 121.7, 118.6, 113.1, 108.4, 105.9, 56.3, 55.5, 36.0, 30.8, 30.5, 18.4. HR-MS (ES-Q-TOF): [M-PF6]+ Calculated: (C46H52IrN4O4) 917.3615; Found: 917.3612.
Iridium (III) bis [2-(3’,4’,5’-trimethoxyphenyl)-5-methylpyridinato-N,C2’]-N,N’-(bipyridine) hexafluorophosphate: [(3,4,5-tMeOppy)2Ir(bpy)](PF6) 4a: Red powder (0.107 g). Yield: 98%. Mp: 203 °C. 1H NMR (400 MHz, CD3CN) δ (ppm): 8.49 (d, J = 8.14 Hz, 2H), 8.13 (t, J = 7.34 Hz, 2H), 8.05 (d, J = 5.16 Hz, 2H), 7.84 (d, J = 8.39 Hz, 2H), 7.52 (dd, J = 6.7712.22 Hz, 4H), 7.30 (s, 2H), 7.14 (s, 2H), 3.90 (s, 6H), 3.74 (s, 6H), 2.87 (s, 6H), 2.01 (s, 6H). 13C NMR (100 MHz, CDCl3) δ (ppm): 165.3, 159.1, 156.1, 150.4, 148.5, 144.2, 140.1, 140.0, 138.2, 131.7, 130.5, 128.4, 125.6, 118.5, 105.0, 61.0, 60.8, 56.5, 18.5. HR-MS (ES-Q-TOF): [M-PF6]+ Calculated: (C40H40IrN4O6) 865.2574; Found: 865.2572.
Iridium (III) bis [2-(3’,4’,5’-trimethoxyphenyl)-5-methylpyridinato-N,C2’]-N,N’-(di-tert-bipyri-dine) hexafluorophosphate: [(3,4,5-tMeOppy)2Ir(dtbubpy)](PF6) 4b: Brownish yellow powder (0.107 g). Yield: 75%. Mp: 186–188 °C. 1H NMR (400 MHz, CD3CN) δ (ppm): 8.46 (s, 2H), 7.92 (d, J = 5.79 Hz, 2H), 7.83 (d, J = 8.36 Hz, 2H), 7.53 (t, J = 6.16 Hz, 4H), 7.04 (s, 2H), 3.90 (s, 6H), 3.74 (s, 6H), 2.87 (s, 6H), 2.01 (s, 6H), 1.42 (s, 18H). 13C NMR (100 MHz, CDCl3) δ (ppm): 206.6, 165.4, 164.4, 159.1, 156.0, 150.2, 149.8, 148.6, 144.1, 140.3, 138.1, 131.6, 131.0, 125.8, 121.8, 118.6, 61.0, 60.8, 56.5, 36.0, 30.5 18.5. HR-MS (ES-Q-TOF): [M-PF6]+ Calculated: (C48H56IrN4O6) 977.3827; Found: 977.3829.
Photophysical measurements
All samples were prepared in HPLC grade acetonitrile (ACN) with varying concentrations on the order of μM. Absorption spectra were recorded at RT using a Shimadzu UV-1800 double beam spectrophotometer. Molar absorptivity determination was verified by linear least-squares fit of values obtained from at least three independent solutions at varying concentrations with absorbance ranging from 6.88 × 10−1 to 3.19 × 102 μM.
The sample solutions for the emission spectra were prepared in N2-degassed dry ACN. Emission spectra were recorded at room temperature using a Cary Eclipse 300 fluorimeter. The samples were excited at the absorption maxima of the dominant low-energy 1MLLCT band as indicated in Table 1. Excited state lifetimes were measured with an Edinburgh Instruments Mini Tau lifetime fluorimeter with an EPL 405 laser (exciting at 405 nm). Melting points were measured with a BI Barnsted Electrothermal 9100 apparatus and were quoted referencing the decomposition temperature. Emission quantum yields were determined using the optically dilute method56,91. A stock solution with absorbance of ca. 0.5 was prepared and then four dilutions were prepared with dilution factors of 40, 20, 13.3 and 10 to obtain solutions with absorbances of ca. 0.013 0.025, 0.038 and 0.05, respectively. The Beer-Lambert law was found to be linear at the concentrations of the solutions. The emission spectra were then measured after the solutions were rigorously degassed with solvent-saturated nitrogen gas (N2) for 20 minutes prior to spectrum acquisition using septa-sealed quartz cells from Starna. For each sample, linearity between absorption and emission intensity was verified through linear regression analysis and additional measurements were acquired until the Pearson regression factor (R2) for the linear fit of the data set surpassed 0.9. Individual relative quantum yield values were calculated for each solution and the values reported represent the slope value. The equation Φs = Φr(Ar/As)(Is/Ir)(ns/nr)2 was used to calculate the relative quantum yield of each of the sample, where Φr is the absolute quantum yield of the reference, n is the refractive index of the solvent, A is the absorbance at the excitation wavelength, and I is the integrated area under the corrected emission curve. The subscripts s and r refer to the sample and reference, respectively. A solution of [Ru(bpy)3](PF6)2 in ACN (Φr = 0.095) was used as the external reference55.
Electrochemistry measurements
Cyclic voltammetry (CV) measurements were performed on an Electrochemical Analyzer potentiostat model 600D from CH Instruments. Solutions for cyclic voltammetry were prepared in ACN and degassed with ACN-saturated nitrogen bubbling for about 10 min prior to scanning. Tetra(n-butyl)ammoniumhexafluorophosphate (TBAPF6; ca. 0.1 M in ACN) was used as the supporting electrolyte. A non-aqueous Ag/Ag+ electrode (silver wire in a solution of 0.1 M AgNO3 in ACN) was used as the pseudoreference electrode; a glassy-carbon electrode was used for the working electrode and a Pt electrode was used as the counter electrode. The redox potentials are reported relative to a normal hydrogen electrode (NHE) electrode with a ferrocenium/ferrocene (Fc+ /Fc) redox couple as an internal reference (0.38 V vs SCE)43.
Density Functional Theory (DFT) Calculations
All calculations were performed with the Gaussian 0992 suite. The level of theory for all DFT61,93,94,95 and TD-DFT62,63,64 calculations was B3LYP; excited-state triplet geometries were calculated using the unrestricted B3LYP method (UB3LYP)67,68,96. The 6-31G* basis set97 was used for C, H and N directly linked to Iridium while the other C, H, N and F atoms where undertaken with 3-21G* basis set69,73,74,75,76,77, and the VDZ (valence double ζ) with SBKJC effective core potential basis set69,70,71,72 was used for Iridium. The predicted phosphorescence wavelengths were obtained by energy difference between the Triplet and Singlet states at their respective optimized geometries41,49. The energy, oscillator strength and related MO contributions for the 100 lowest singlet-singlet and 5 lowest singlet-triplet excitations were obtained from the TD-DFT/Singlets and the TD-DFT/Triplets output files, respectively. The calculated absorption spectra were visualized with GaussSum 2.1 (fwhm: 1000 cm−1)98.
Device Fabrication
An aqueous dispersion of Poly(3,4-ethylenedioxythiophene): poly-styrenesulfonate (PEDOT:PSS) was purchased from Hereaus and used as received. The ionicliquid 1-butyl-3-methyl-imidazolium hexafluorophosphate [BMIM+][PF−6] and the solvents used were obtained from Aldrich. Photolithographically patterned ITO covered glass substrates were purchased from Naranjo-Substrates (www.naranjosubstrates.com) and extensively cleaned before using by ultrasonic treatment in water-soap, water, and 2-propanol baths. After drying, the substrates were placed in a UV-ozone cleaner (Jelight 42-220) for 20 min.
Devices were prepared on the cleaned ITO substrates by depositing an 80 nm layer of PEDOT:PSS and annealing it at 150 °C during 15 minutes. On top of this layer the active film was deposited from an acetonitrile solution of the complex mixed with the ionic liquid in a molar ratio 4 to 1. All the layers were prepared by spin-coating the organic solutions. A concentration of 20 mg mL−1 of iTMC at 1000 rpm for 30 seconds leads to 100 nm thicknes. The thickness of the films was determined using an Ambios XP1 profilometer. Then, the samples were transferred to an inert atmosphere glovebox (<0.1 ppm O2 and H2O, MBraun). Finally, aluminum metal electrodes (70 nm) were thermally evaporated using a shadow mask under a vacuum (<1 × 10−6 mbar) using an Edwards Auto500 evaporator integrated into an inert atmosphere glovebox. Lifetime data were obtained by applying pulsed currents and monitoring the voltage and simultaneously the luminance by a True Colour Sensor MAZeT (MTCSICT Sensor) using a Lifetime Test System designed by BoTEST (Botest OLT OLED Lifetime-Test System). Electroluminescence spectra were recorded using an Avantis fiberoptics photospectrometer. The devices were not encapsulated and were characterized inside the glovebox.
Additional Information
How to cite this article: Hasan, K. et al. Tuning the Emission of Cationic Iridium (III) Complexes Towards the Red Through Methoxy Substitution of the Cyclometalating Ligand. Sci. Rep. 5, 12325; doi: 10.1038/srep12325 (2015).
Supplementary Material
Acknowledgments
EZ-C acknowledges CFI (Canadian Foundation for Innovation), NSERC (the Natural Sciences and Engineering Research Council of Canada), FQRNT (Le Fonds québécois de la recherche sur la nature et les technologies) and the University of St Andrews for financial support. We acknowledge Prof. Garry Hanan for providing access to facilities at the Université de Montréal. C. R-C. would like to thank the MINECO for the financial support of this research in the framework of project CTQ2010-17481, the Junta de Andalucía (CICyE) for special financial support (P08-FQM-4011 and P10-FQM-6703) and the MECD (Spanish Ministry of Education, Culture, and Sport) for an FPU grant. This work has been supported by the Spanish Ministry of Economy and Competitiveness (MINECO) (MAT2014-55200), the Generalitat Valenciana (Prometeo/2012/053). IDWS and AKB acknowledge financial support from the EPSRC of the UK (grant: EP/J01771).
Footnotes
Author Contributions E.Z.-C. conceived of the design of the study and contributed to the analysis of the data and writing of the manuscript. K.H. synthesized all the materials and contributed to the optoelectronic characterization and analysis of the data and writing of the manuscript. A.K.B. contributed to the optoelectronic characterization and analysis of the data and writing of the manuscript. I.D.W. Samuel contributed to the analysis of the data and writing of the manuscript. C.R.-C. prepared the devices and analysis of the data and writing of the manuscript. H.J.B. contributed to the analysis of the data and writing of the manuscript. All authors have reviewed the manuscript.
References
- For recent reviews on LEECs see: Costa R. D. et al. Angew. Chem. Int. Ed. 51 8178–8211, 10.1002/anie.201201471 (2012). [DOI] [PubMed] [Google Scholar]
- Slinker J. D. et al. Electroluminescent Devices from Ionic Transition Metal Complexes. J. Mater. Chem. 17, 2976–2988, 10.1039/B704017B (2007). [DOI] [Google Scholar]
- Bernards D. A., Slinker J. D., Malliaras G. G., Flores-Torres S. & Abruña H. D. Cascaded light emitting devices based on ruthenium complexes. Appl. Phys. Lett. 84, 4980 (2004). [Google Scholar]
- Slinker J. D. et al. Direct 120 volt, 60 hertz operation of an organic light emitting device. J. Appl. Phys. 99, 074502 (2006). [Google Scholar]
- Slinker J. D. et al. Solid-state electroluminescent devices based on transition metal complexes. Chem. Commun. 19, 2392–2399 (2003). [DOI] [PubMed] [Google Scholar]
- Neve F., Crispini A., Campagna S. & Serroni S. Synthesis, Structure, Photophysical Properties, and Redox Behavior of Cyclometalated Complexes of Iridium(III) with Functionalized 2,2’-Bipyridines. Inorg. Chem. 38, 2250–2258 (1999). [Google Scholar]
- Cunningham G. B., Li Y., Liu S. & Schanze K. S. Photoluminescence and Electroluminescence of d6 Metal-Organic Conjugated Oligomers: Correlation of Photophysics and Device Performance. J. Phys. Chem. B 107, 12569–12572, 10.1021/jp0358228 (2003). [DOI] [Google Scholar]
- Neve F. et al. Cationic Cyclometalated Iridium Luminophores: Photophysical, Redox, and Structural Characterization. Organometallics 23, 5856–5863 (2004). [Google Scholar]
- Tamayo A. B. et al. Cationic Bis-cyclometalated Iridium(III) Diimine Complexes and Their Use in Efficient Blue, Green, and Red Electroluminescent Devices. Inorg. Chem. 44, 8723–8732, 10.1021/ic050970t (2005). [DOI] [PubMed] [Google Scholar]
- Zhao Q. et al. Series of New Cationic Iridium(III) Complexes with Tunable Emission Wavelength and Excited State Properties: Structures, Theoretical Calculations, and Photophysical and Electrochemical Properties. Inorg. Chem. 45, 6152–6160, 10.1021/ic052034j (2006). [DOI] [PubMed] [Google Scholar]
- Kim K.-Y., Farley R. T. & Schanze K. S. An Iridium(III) Complex that Exhibits Dual Mechanism Nonlinear Absorption. J. Phys. Chem. B 110, 17302–17304, 10.1021/jp063916m (2006). [DOI] [PubMed] [Google Scholar]
- Shao F., Elias B., Lu W. & Barton J. K. Synthesis and Characterization of Iridium(III) Cyclometalated Complexes with Oligonucleotides: Insights into Redox Reactions with DNA. Inorg. Chem. 46, 10187–10199, 10.1021/ic7014012 (2007). [DOI] [PubMed] [Google Scholar]
- Yu M. et al. Cationic iridium(iii) complexes for phosphorescence staining in the cytoplasm of living cells. Chem. Commun. 2115–2117, 10.1039/B800939B (2008). [DOI] [PubMed] [Google Scholar]
- He L. et al. Toward Highly Efficient Solid-State White Light-Emitting Electrochemical Cells: Blue-Green to Red Emitting Cationic Iridium Complexes with Imidazole-Type Ancillary Ligands. Adv. Funct. Mater. 19, 2950–2960, 10.1002/adfm.200900723 (2009). [DOI] [Google Scholar]
- Rachford A. A., Ziessel R., Bura T., Retailleau P. & Castellano F. N. Boron Dipyrromethene (Bodipy) Phosphorescence Revealed in [Ir(ppy)2(bpy-C‚â°C-Bodipy)] + . Inorg. Chem. 49, 3730–3736, 10.1021/ic901996u (2010). [DOI] [PubMed] [Google Scholar]
- Zhao Q. et al. Cationic Iridium(III) Complexes with Tunable Emission Color as Phosphorescent Dyes for Live Cell Imaging. Organometallics 29, 1085–1091, 10.1021/om900691r (2010). [DOI] [Google Scholar]
- Lincker F. d. r. et al. Rodlike Fluorescent π-Conjugated 3,3′-Bipyridazine Ligand: Optical, Electronic, and Complexation Properties. Inorg. Chem. 49, 3991–4001, 10.1021/ic901925w (2010). [DOI] [PubMed] [Google Scholar]
- Hwang A.-R. et al. Photodynamic Behavior of Heteroleptic Ir(III) Complexes with Carbazole-Functionalized Dendrons Associated with Efficient Electron Transfer Processes. J. Phys. Chem. C 116, 1973–1986, 10.1021/jp2093077 (2011). [DOI] [Google Scholar]
- Soliman A. M., Fortin D., Harvey P. D. & Zysman-Colman E. Hybrid charged heterometallic Pt-Ir complexes: tailoring excited states by taking the best of both worlds. Chem. Commun. 48, 1120–1122 (2012). [DOI] [PubMed] [Google Scholar]
- Smith R. A. et al. Cyclometalated cinchophen ligands on iridium(iii): towards water-soluble complexes with visible luminescence. Dalton Trans. 42, 10347–10357, 10.1039/C3DT51098K (2013). [DOI] [PubMed] [Google Scholar]
- Hu T. et al. Efficient doped red light-emitting electrochemical cells based on cationic iridium complexes. Synth. Met. 163, 33–37, 10.1016/j.synthmet.2012.12.017 (2013). [DOI] [Google Scholar]
- Bunzli A. M. et al. Thienylpyridine-based cyclometallated iridium(iii) complexes and their use in solid state light-emitting electrochemical cells. Dalton Trans. 43, 738–750, 10.1039/C3DT52622D (2014). [DOI] [PubMed] [Google Scholar]
- Zhang K. Y. & Lo K. K.-W. Synthesis, Properties, and Live-Cell Imaging Studies of Luminescent Cyclometalated Iridium(III) Polypyridine Complexes Containing Two or Three Biotin Pendants. Inorg. Chem. 48, 6011–6025, 10.1021/ic900412n (2009). [DOI] [PubMed] [Google Scholar]
- Jiang W. et al. Zwitterionic Iridium Complexes: Synthesis, Luminescent Properties, and Their Application in Cell Imaging. Inorg. Chem. 49, 3252–3260, 10.1021/ic9021239 (2010). [DOI] [PubMed] [Google Scholar]
- Byun Y. et al. Highly efficient red electrophosphorescence from a solution-processed zwitterionic cyclometalated iridium(III) complex. Appl. Phys. Lett. 91, 211106, 10.1063/1.2809375 (2007). [DOI] [Google Scholar]
- Kessler F. et al. Near-UV to red-emitting charged bis-cyclometallated iridium(iii) complexes for light-emitting electrochemical cells. Dalton Trans. 41, 180–191 (2012). [DOI] [PubMed] [Google Scholar]
- Langdon-Jones E. E. et al. Using Substituted Cyclometalated Quinoxaline Ligands To Finely Tune the Luminescence Properties of Iridium(III) Complexes. Inorg. Chem. 52, 448–456, 10.1021/ic301853t (2013). [DOI] [PubMed] [Google Scholar]
- Zhang G. et al. Near-Infrared-Emitting Iridium(III) Complexes as Phosphorescent Dyes for Live Cell Imaging. Organometallics, 10.1021/om400676h (2013). [DOI] [Google Scholar]
- Costa R. D. et al. Efficient deep-red light-emitting electrochemical cells based on a perylenediimide-iridium-complex dyad. Chem. Commun. 3886–3888, 10.1039/b905367k (2009). [DOI] [PubMed] [Google Scholar]
- Shavaleev N. M., Scopelliti R., Grätzel M. & Nazeeruddin M. K. Phosphorescence of iridium(III) complexes with 2-(2-pyridyl)-1,3,4-oxadiazoles. Inorg. Chim. Acta 394, 295–299, 10.1016/j.ica.2012.07.026 (2013). [DOI] [Google Scholar]
- Donato L., McCusker C. E., Castellano F. N. & Zysman-Colman E. Mono- and Dinuclear Cationic Iridium(III) Complexes Bearing a 2,5-Dipyridylpyrazine (2,5-dpp) Ligand. Inorg. Chem. 52, 8495–8504, 10.1021/ic400478d (2013). [DOI] [PubMed] [Google Scholar]
- Hu T., He L., Duan L. & Qiu Y. Solid-state light-emitting electrochemical cells based on ionic iridium(iii) complexes. J. Mater. Chem. 22, 4206–4215, 10.1039/C2JM16185K (2012). [DOI] [Google Scholar]
- De Angelis F. et al. Controlling Phosphorescence Color and Quantum Yields in Cationic Iridium Complexes: A Combined Experimental and Theoretical Study. Inorg. Chem. 46, 5989–6001, 10.1021/ic700435c (2007). [DOI] [PubMed] [Google Scholar]
- He L. et al. Blue-Emitting Cationic Iridium Complexes with 2-(1H-Pyrazol-1-yl)pyridine as the Ancillary Ligand for Efficient Light-Emitting Electrochemical Cells. Adv. Funct. Mater. 18, 2123–2131, 10.1002/adfm.200701505 (2008). [DOI] [Google Scholar]
- Costa R. D., Viruela P. M., Bolink H. J. & OrtÌ E. Lowest triplet excited states of a novel heteroleptic iridium(III) complex and their role in the emission colour. J. Mol. Struct. THEOCHEM 912, 21–26 (2009). [Google Scholar]
- Yang C.-H. et al. Iridium Metal Complexes Containing N-Heterocyclic Carbene Ligands for Blue-Light-Emitting Electrochemical Cells. Inorg. Chem. 49, 9891–9901, 10.1021/ic1009253 (2010). [DOI] [PubMed] [Google Scholar]
- Baranoff E. et al. Tuning the photophysical properties of cationic iridium(iii) complexes containing cyclometallated 1-(2,4-difluorophenyl)-1H-pyrazole through functionalized 2,2[prime or minute]-bipyridine ligands: blue but not blue enough. Dalton Trans. 42, 1073–1087 (2013). [DOI] [PubMed] [Google Scholar]
- Meier S. B. et al. A deep-blue emitting charged bis-cyclometallated iridium(iii) complex for light-emitting electrochemical cells. J. Mater. Chem. C 1, 58, 10.1039/c2tc00251e (2013). [DOI] [PubMed] [Google Scholar]
- Fernandez-Hernandez J. M. et al. Blue light emitting electrochemical cells incorporating triazole-based luminophores. J. Mater. Chem. C 1, 7440–7452, 10.1039/C3TC31307G (2013). [DOI] [Google Scholar]
- For an example of methoxy substitution on the C^N ligand of cationic iridium complexes see: Davies D. L., Lowe M. P., Ryder K. S., Singh K. & Singh S. Dalton Trans. 40, 1028–1030 (2011). [DOI] [PubMed] [Google Scholar]
- Lowry M. S., Hudson W. R., Pascal R. A. Jr. & Bernhard S. Accelerated Luminophore Discovery through Combinatorial Synthesis. J. Am. Chem. Soc. 126, 14129–14135 (2004). [DOI] [PubMed] [Google Scholar]
- Nonoyama M. Benzo[h]quinolin-10-yl-N Iridium(III) Complexes. Bull. Chem. Soc. Jpn. 47, 767–768, 10.1246/bcsj.47.767 (1974). [DOI] [Google Scholar]
- Pavlishchuk V. V. & Addison A. W. Conversion constants for redox potentials measured versus different reference electrodes in acetonitrile solutions at 25°C. Inorg. Chim. Acta 298, 97–102, 10.1016/s0020-1693(99)00407-7 (2000). [DOI] [Google Scholar]
- Ladouceur S., Fortin D. & Zysman-Colman E. Enhanced Luminescent Iridium(III) Complexes Bearing Aryltriazole Cyclometallated Ligands. Inorg. Chem. 50, 11514–11526, 10.1021/ic2014013 (2011). [DOI] [PubMed] [Google Scholar]
- Dixon I. M. et al. A family of luminescent coordination compounds: iridium(III) polyimine complexes. Chem. Soc. Rev. 29, 385–391 (2000). [Google Scholar]
- Colombo M. G. & Güdel H. U. Synthesis and high-resolution optical spectroscopy of bis[2-(2-thienyl)pyridinato-C3,N’](2,2’-bipyridine)iridium(III). Inorg. Chem. 32, 3081–3087 (1993). [Google Scholar]
- Colombo M. G., Hauser A. & Güdel H. U. Evidence for strong mixing between the LC and MLCT excited states in bis(2-phenylpyridinato-C2,N’)(2,2’-bipyridine)iridium(III). Inorg. Chem. 32, 3088–3092 (1993). [Google Scholar]
- Ho C.-L. et al. Solution-Processible Multi-component Cyclometalated Iridium Phosphors for High-Efficiency Orange-Emitting OLEDs and Their Potential Use as White Light Sources. Adv. Funct. Mater. 17, 2925–2936 (2007). [Google Scholar]
- Ladouceur S., Fortin D. & Zysman-Colman E. The role of substitution on the photophysical propertiesof 5,5’-diaryl-2,2’-bipyridine (bpy*) in [Ir(ppy)2(bpy*)]PF6 complexes: A combined experimental and theoretical study. Inorg. Chem. 49, 5625–5641, 10.1021/ic100521t/ (2010). [DOI] [PubMed] [Google Scholar]
- Flamigni L., Barbieri A., Sabatini C., Ventura B. & Barigelletti F. Photochemistry and Photophysics of Coordination Compounds: Iridium. Top. Curr. Chem. 281, 143–203 (2007). [Google Scholar]
- Ladouceur S. & Zysman-Colman E. A Comprehensive Survey of Cationic Iridium(III) Complexes Bearing Nontraditional Ligand Chelation Motifs. Eur. J. Inorg. Chem. 2013, 2985–3007, 10.1002/ejic.201300171 (2013). [DOI] [Google Scholar]
- Ohsawa Y. et al. Electrochemistry and spectroscopy of ortho-metalated complexes of iridium(III) and rhodium(III). J. Phys. Chem. 91, 1047–1054, 10.1021/j100289a009 (1987). [DOI] [Google Scholar]
- Lo K. K.-W., Chan J. S.-W., Lui L.-H. & Chung C.-K. Novel Luminescent Cyclometalated Iridium(III) Diimine Complexes That Contain a Biotin Moiety. Organometallics 23, 3108–3116, 10.1021/om0499355 (2004). [DOI] [Google Scholar]
- Plummer E. A., Hofstraat J. W. & De Cola L. Mono- and di-nuclear iridium(iii) complexes. Synthesis and photophysics. Dalton Trans. 2080–2084, 10.1039/B300704A (2003). [DOI] [Google Scholar]
- Ishida H., Tobita S., Hasegawa Y., Katoh R. & Nozaki K. Recent advances in instrumentation for absolute emission quantum yield measurements. Coord. Chem. Rev. 254, 2449–2458, 10.1016/j.ccr.2010.04.006 (2010). [DOI] [Google Scholar]
- Crosby G. A. & Demas J. N. Measurement of photoluminescence quantum yields. Review. J. Phys. Chem. 75, 991–1024, 10.1021/j100678a001 (1971). [DOI] [Google Scholar]
- Slinker J. D. et al. Efficient Yellow Electroluminescence From a Single Layer of a Cyclometalated Iridium Complex. J. Am. Chem. Soc. 126, 2763 (2004). [DOI] [PubMed] [Google Scholar]
- Aoki S. et al. Regioselective Aromatic Substitution Reactions of Cyclometalated Ir(III) Complexes: Synthesis and Photochemical Properties of Substituted Ir(III) Complexes That Exhibit Blue, Green, and Red Color Luminescence Emission. Inorg. Chem. 50, 806–818, 10.1021/ic101164g (2011). [DOI] [PubMed] [Google Scholar]
- Hohenberg P. & Kohn W. Inhomogeneous Electron Gas. Phys. Rev. 136, B864 (1964). [Google Scholar]
- Kohn W. & Sham L. J. Self-Consistent Equations Including Exchange and Correlation Effects. Phys. Rev. 140, A1133 (1965). [Google Scholar]
- in The Challenge of d and f Electrons, (eds Salahub D.R. & Zerner M. C.) (ACS, 1989). [Google Scholar]
- Stratmann R. E., Scuseria G. E. & Frisch M. J. An efficient implementation of time-dependent density-functional theory for the calculation of excitation energies of large molecules. J. Chem. Phys. 109, 8218 (1998). [Google Scholar]
- Bauernschmitt R. & Ahlrichs R. Treatment of electronic excitations within the adiabatic approximation of time dependent density functional theory. Chem. Phys. Lett. 256, 454 (1996). [Google Scholar]
- Casida M. E., Jamorski C., Casida K. C. & Salahub D. R. Molecular excitation energies to high lying bound states from time dependent density-functional response theory: Characterization and correction of the time-dependent local density approximation ionization threshold. J. Chem. Phys. 108, 4439 (1998). [Google Scholar]
- Gaussian 98 (Revision A.6), (Gaussian Inc., Pittsburgh, PA, 1998).
- Becke A. D. Densityfunctional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 98, 5648 (1993). [Google Scholar]
- Lee C., Yang W. & Parr R. G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 37, 785–789 (1988). [DOI] [PubMed] [Google Scholar]
- Miehlich B., Savin A., Stoll H. & Preuss H. Results obtained with the correlation energy density functionals of becke and Lee, Yang and Parr. Chem. Phys. Lett. 157, 200–206 (1989). [Google Scholar]
- Binkley J. S., Pople J. A. & Hehre W. J. Self-consistent molecular orbital methods. 21. Small split-valence basis sets for first-row elements. J. Am. Chem. Soc. 102, 939 (1980). [Google Scholar]
- Stevens W. J., Basch W. J. & Krauss M. Compact effective potentials and efficient shared‐exponent basis sets for the first‐ and second‐row atoms. J. Chem. Phys. 81, 6026 (1984). [Google Scholar]
- Stevens W. J., Krauss M., Basch H. & Jasien P. G. Relativistic compact effective potentials and efficient, shared-exponent basis sets for the third-, fourth-, and fifth-row atoms. Can. J. Chem. 70, 612, 10.1139/v92-085 (1992). [DOI] [Google Scholar]
- Cundari T. R. & Stevens W. J. Effective core potential methods for the lanthanides. J. Chem. Phys. 98, 5555–5565 (1993). [Google Scholar]
- Gordon M. S., Binkley J. S., Pople J. A., Pietro W. J. & Hehre W. J. Self-consistent molecular-orbital methods. 22. Small split-valence basis sets for second-row elements. J. Am. Chem. Soc. 104, 2797 (1982). [Google Scholar]
- Pietro W. J. et al. Self-consistent Molecular Orbital Methods. 24. Supplemented Small Split-Valence Basis Sets for Second-Row Elements. J. Am. Chem. Soc. 104, 5039 (1982). [Google Scholar]
- Dobbs K. D. & Hehre W. J. Molecular orbital theory of the properties of inorganic and organometallic compounds 4. Extended basis sets for third-and fourth-row, main-group elements. J. Comput. Chem. 7, 359 (1986). [Google Scholar]
- Dobbs K. D. & Hehre W. J. You have full text access to this content Molecular orbital theory of the properties of inorganic and organometallic compounds 5. Extended basis sets for first-row transition metals. J. Comput. Chem. 8, 861–879 (1987). [Google Scholar]
- Dobbs K. D. & Hehre W. J. Molecular orbital theory of the properties of inorganic and organometallic compounds. 6. Extended basis sets for second-row transition metals. J. Comput. Chem. 8, 880 (1987). [Google Scholar]
- Ditchfield R., Hehre W. J. & Pople J. A. An Extended Gaussian-Type Basis for Molecular-Orbital Studies of Organic Molecules. J. Chem. Phys. 54, 724 (1971). [Google Scholar]
- Hehre W. J., Ditchfield R. & Pople J. A. Type Basis Sets for Use in Molecular Orbital Studies of Organic Molecules. J. Chem. Phys. 56, 2257 (1972). [Google Scholar]
- Hariharan P. C. & Pople J. A. Influence of polarization functions on MO hydrogenation energies. Theor. Chim. Acta 28, 213 (1973). [Google Scholar]
- Hariharan P. C. & Pople J. A. Accuracy of AH, Equilibrium Geometries by Single Determinant Molecular Orbital Theory. Mol. Phys. 27, 209 (1974). [Google Scholar]
- Gordon M. S. The isomers of silacyclopropane. Chem. Phys. Lett. 76, 163 (1980). [Google Scholar]
- Tomasi J., Mennucci B. & Cammi R. Quantum Mechanical Continuum Solvation Models. Chem. Rev. 105, 2999–3094, 10.1021/cr9904009 (2005). [DOI] [PubMed] [Google Scholar]
- Costa R. D. et al. Archetype Cationic Iridium Complexes and Their Use in Solid-State Light-Emitting Electrochemical Cells. Adv. Funct. Mater. 19, 3456–3463, 10.1002/adfm.200900911 (2009). [DOI] [Google Scholar]
- Zhang J. et al. Efficient Light-Emitting Electrochemical Cells (LECs) Based on Ionic Iridium(III) Complexes with 1,3,4-Oxadiazole Ligands. Adv. Funct. Mater. 23, 4667–4677, 10.1002/adfm.201300344 (2013). [DOI] [Google Scholar]
- Zeng X. et al. Cationic Bis-cyclometallated Iridium(III) Phenanthroline Complexes with Pendant Fluorenyl Substituents: Synthesis, Redox, Photophysical Properties and Light-Emitting Cells. Chem. Eur. J. 14, 933–943 (2008). [DOI] [PubMed] [Google Scholar]
- Li W. et al. An Improved Protocol for the Preparation of 3-Pyridyl- and Some Arylboronic Acids. J. Org. Chem. 67, 5394–5397, 10.1021/jo025792p (2002). [DOI] [PubMed] [Google Scholar]
- Sasaki I., Vendier L., Sournia-Saquet A. & Lacroix P. G. Facile Synthesis of Cyclometalated Ruthenium Complexes with Substituted Phenylpyridines. Eur. J. Inorg. Chem. 2006, 3294–3302, 10.1002/ejic.200600359 (2006). [DOI] [Google Scholar]
- Deng J. Z. et al. Copper-Facilitated Suzuki Reactions: Application to 2-Heterocyclic Boronates. Org. Lett. 11, 345–347, 10.1021/ol802556f (2008). [DOI] [PubMed] [Google Scholar]
- Sindhuja E., Ramesh R. & Liu Y. Palladium(ii) thiocarboxamide complexes: synthesis, characterisation and application to catalytic Suzuki coupling reactions. Dalton Trans. 41, 5351–5361, 10.1039/C2DT12243J (2012). [DOI] [PubMed] [Google Scholar]
- Fery-Forgues S. & Lavabre D. Are Fluorescence Quantum Yields So Tricky to Measure ? A Demonstration Using Familiar Stationery Products. J. Chem. Educ. 76, 1260, 10.1021/ed076p1260 (1999). [DOI] [Google Scholar]
- Gaussian 09, Revision A.02 v. 7.0 (Gaussian Inc., Wallingford, CT, 2009).
- Hohenberg P. & Kohn W. Inhomogeneous Electron Gas. Phys. Rev. B136, 864 (1964). [Google Scholar]
- Kohn W. & Sham L. J. Self-Consistent Equations Including Exchange and Correlation Effects. Phys. Rev. A140, 1133 (1965). [Google Scholar]
- Parr R. G. & Yang W. Density-functional theory of atoms and molecules. (Oxford Univ. Press, 1989). [Google Scholar]
- Becke A. D. Density-functional Thermochemistry. III. The role of exact exchange. J. Chem. Phys. 98, 5648–5652 (1993). [Google Scholar]
- Rassolov V. A., Pople J. A., Ratner M. A. & Windus T. L. 6-31G[sup *] basis set for atoms K through Zn. J. Chem. Phys. 109, 1223–1229 (1998). [Google Scholar]
- O’Boyle N. M., Tenderholt A. L. & Langner K. M. cclib: A library for package-independent computational chemistry algorithms. J. Comput. Chem. 29, 839–845, 10.1002/jcc.20823 (2008). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.