

Abstract
A variety of cytokines and growth factors use the Janus kinase (Jak)–STAT signaling pathway to transmit extracellular signals to the nucleus. STATs (signal transducers and activators of transcription) are latent cytoplasmic transcription factors. There are seven mammalian STATs and they have critical, nonredundant roles in mediating cellular transcriptional responses to cytokines. The physiological roles of STATs have been elucidated by analysis of mice rendered deficient in STAT genes. STAT activation is regulated and can be modulated in a positive or negative fashion; it can be reprogrammed to drive different cellular responses. Several auto-regulatory and signaling crosstalk mechanisms for regulating Jak–STAT signaling have been described. Understanding and manipulation of the function of STATs will help in the development of therapeutic strategies for diseases that are regulated by cytokines.
Keywords: cytokine, inhibition, Jak, signal transduction, STAT
Basic Jak–STAT signaling
Cytokines are proteins that are secreted and mediate communication between cells. The biological functions of cytokines are achieved by binding with high affinity and specificity to cell-surface receptors, thereby triggering signal transduction cascades that regulate cellular activation, proliferation, differentiation, and survival. Many, if not most, cytokines use the Janus kinase (Jak)–signal transducer and activator of transcription (STAT) signaling pathway to mediate gene activation or repression. The discovery of the Jak–STAT pathway arose out of studies of mechanisms of gene induction by interferons (IFNs) [1]. Transcriptional activation of a number of IFN-inducible genes was rapid and independent of new protein synthesis, indicating that IFNs activated transcription by modifying the activity of pre-existing proteins and transcription factors. Subsequently, experiments from a large number of laboratories have defined the Jak–STAT signaling pathway.
Jaks are protein tyrosine kinases that are pre-associated with membrane-proximal regions (termed box 1 and box 2) of cytokine receptors. It seems that only four mammalian Jaks exist: Jak1, Jak2, Jak3, and Tyk2. Each contains a catalytically active kinase domain and a regulatory pseudokinase domain at the carboxy terminus of the protein. The domain responsible for the binding of Jaks to receptors is located at the amino terminus. The binding of cytokine ligands results in dimerization of receptor subunits, thus increasing the local concentration of Jaks and bringing these kinases into close proximity. The Jaks become activated as a result of tyrosine phosphorylation, and initiate the signal transduction cascade by phosphorylation of tyrosine motifs present in receptor cytoplasmic domains and in receptor-associated proteins. Phosphotyrosine-containing motifs in receptor cytoplasmic domains act as docking sites for many signaling proteins, including STATs (Fig. 1). Receptor-associated proteins activated by Jaks include kinases that then activate signaling pathways parallel to but distinct from the Jak–STAT pathway. However, Jaks are thought to lie at the apex of the signaling cascade and to be required for all cytokine signal transduction via type I and type II cytokine receptors. In some cases, Jak expression is required for appropriate cell surface expression of receptors [2], and thus Jaks can have a structural as well as a catalytic role in signal transduction.
Figure 1.
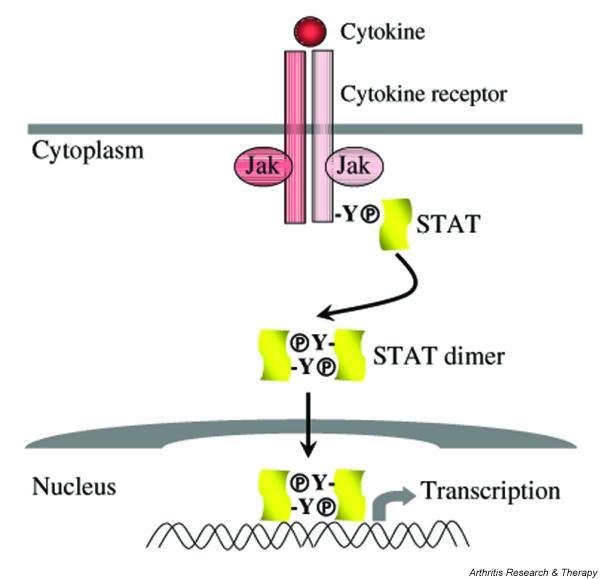
Cytokine activation of the Jak–STAT pathway. Cytokine ligation and dimerization of plasma membrane receptors results in activation of receptor-associated Jak kinases and phosphorylation of receptor cytoplasmic domain tyrosine residues. STATs dock on these phosphotyrosine motifs by means of their Src homology 2 (SH2) domains. STATs are then phosphorylated on a conserved carboxy-terminus tyrosine, dimerize, and translocate to the nucleus to activate gene transcription. Transcriptional responses can be detected within a few minutes of receptor ligation. The use of a small number of protein:protein or protein:DNA interactions between the extracellular ligand and gene promoters allows specificity in signal transduction. Jak, Janus kinase; STAT, signal transducer and activator of transcription.
STATs are present as latent monomeric proteins in the cytoplasm of most cells. After cytokine ligation of receptors, STATs are recruited to the receptor signaling complex by interactions between STAT Src homology 2 (SH2) domains and specific receptor phosphotyrosine sequences. One important determinant of the specificity of STAT activation by individual receptors is the affinity and specificity of the SH2–phosphotyrosine interaction [3]. Once recruited to the receptor, STATs themselves are phosphorylated on a unique conserved carboxy-terminal tyrosine. Tyrosine phosphorylation of STATs is associated with dissociation from the receptor and the formation of STAT:STAT dimers, which is mediated by reciprocal SH2–phosphotyrosine interactions between the dimer partners. The STATs can all form homodimers, and, in addition, STAT1:STAT3 and STAT5A:STAT5B heterodimers have been described; STAT1 can associate with STAT2 and interferon regulatory factor 9 (IRF9) in a heterotrimeric complex termed interferon-stimulated gene factor 3 (ISGF3).
STATs acquire DNA-binding activity through dimerization, and then translocate to the nucleus, where they bind to gene promoters and activate transcription. STAT transcriptional activity is potentiated by phosphorylation of a conserved carboxy-terminal serine residue [4], which increases interactions with additional proteins that are important in STAT-mediated transcription, such as mini-chromosome maintenance 5 (MCM5). Besides tyrosine and serine phosphorylation, arginine methylation has been described to be another post-translational modification mechanism that is required for optimal gene activation by STATs, at least in the IFN–STAT1 pathway [5]. STATs can interact directly with transcriptional coactivators such as CBP (CREB-binding protein)/p300, and may also need to interact with additional proteins such as MCM5 and N-Myc-interacting protein (Nmi) to achieve maximal transcriptional activation. STATs are often involved in the regulation of complex promoters or enhancers that bind multiple transcription factors, where they can bind as tetramers (a process mediated by the amino terminus), and interact with additional transcription factors such as Jun, specificity protein 1 (Sp1), and the glucocorticoid receptor.
Typically, cytokine stimulation involves the ligation of (at least) two different receptor subunits, and this results in the association of a pair of different Jaks. For example, IFN-γ activates Jak1 and Jak2, and IFN-α activates Jak1 and Tyk2. Although individual STAT proteins may be activated by multiple ligands, certain cytokines preferentially activate particular STATs. IFN-γ preferentially activates STAT1, interleukin (IL)-6 preferentially activates STAT3, and IL-4 preferentially activates STAT6. A series of biochemical and genetic experiments have revealed that the specificity of STAT activation is not determined by the Jaks that are activated, and the different kinases can substitute for each other. Instead, the specificity is likely to arise from specific interaction between STAT SH2 domain and receptor phosphotyrosine motifs [6]. This pattern of STAT activation does allow a certain level of specificity of cytokine action. However, in the circumstances in which multiple cytokines activate common STATs, one intriguing question is that of how the specificity of cytokine signaling is achieved. For example, both IFN-γ and IL-6 activate STAT1. However, IFN-γ and IL-6 trigger very distinct cellular responses. In such a case, what determines signaling specificity is the cross-regulation between STAT1 and other STATs such as STAT3 because in the cells lacking STAT3, IL-6 actually mediates an IFN-γ-like response [7]. Another level of specificity lies in the interaction of STATs with promoter sequences, because different STATs have different affinities for sequences that vary from the consensus sequence TTCCNGGAA either in individual base pairs or in the spacing between half-sites [3].
Biology of STATs
There are seven mammalian STATs: STAT1, STAT2, STAT3, STAT4, STAT5a, STAT5b, and STAT6. STATs serve important functions by regulating the expression of effector genes and by regulating cell differentiation, survival, and apoptosis. Each STAT gene has been deleted in mice, and analysis of the phenotype of STAT-deficient mice has been helpful in unraveling the role of STATs in normal physiology and under certain pathological conditions. These gene targeting studies have shown that STATs have nonredundant functions in regulating various aspects of host immune responses as well as many non-immune processes. Overall, STATs are not required for the development or survival of mice; an exception is STAT3, whose targeted deletion results in embryonic lethality [8].
STAT1
Many of the important immune regulatory functions of STAT1 can be explained on the basis of its key role in mediating responses to IFNs. Gene targeting experiments [9,10] suggest that the primary role of STAT1 in vivo is to mediate IFN signaling, because responses to other cytokines that activate STAT1 seem preserved in STAT1-deficient mice. STAT1 deficiency results in enhanced susceptibility to viral infections and leads to an inability to handle infections with intracellular pathogens. STAT1 mediates the anti-viral and immune/inflammatory effects of IFNs through the induction of immune effector and inflammatory genes such as major histocompatibility complex (MHC), costimulatory molecule, chemokine, complement, IRF1, inducible nitric oxide synthase, and Fcγ RI genes. In contrast, STAT1 mediates anti-proliferative and pro-apoptotic effects of IFNs, suggesting that STAT1 also has the potential to restrain inflammation. Interestingly, infection of STAT1-deficient mice with an attenuated strain of influenza leads to a chronic pulmonary inflammatory condition culminating in death, despite normal clearance of the virus [11]. Moreover, STAT1-null mice exhibit increased susceptibility to experimental autoimmune encephalomyelitis and increased severity of arthritis [12,13], implying a role of STAT1 in vivo in suppressing certain autoimmune disorders.
These dual and opposing roles for STAT1 conform to a homeostatic paradigm in immune activation in which molecules that promote cell activation and effector function also have a role in limiting the response, such that excessive activation and autoimmunity are avoided. STAT1 is also important in host anti-tumor responses. Work with mice deficient in STAT1 demonstrates that endogenous STAT1 functions as part of the tumor suppressing system and shapes the immunogenic phenotype of developing tumors [14], the phenomenon termed 'cancer immunoediting'. Besides its unequivocal role in modulating immune responses, STAT1 also possesses non-immune functions, such as the regulation of bone formation and destruction under homeostatic and pathological conditions. STAT1's action on bone is complex because it attenuates both osteoclast formation and osteoblast differentiation [15-17]. Increased bone mass in STAT1-deficient mice suggests that during homeostasis the effect of STAT1 on osteoblasts is more significant than its effect on osteoclasts.
STAT2
So far, STAT2 has been shown to be activated only by IFN-α/β and IFN-λs [18,19], and to be necessary for IFN-α/β responses, such as the antiviral response. Accordingly, STAT2-null mice exhibit increased susceptibility to viral infections [20]. STAT2 together with STAT1 and IRF9 form the heterodimeric transcription complex ISGF3 and assembly of ISGF3 is necessary for IFN-α/β responses in a wide variety of cell types. STAT2 can also form complexes with STAT6 and IRF9 specifically in B cells; the significance of STAT2:STAT6 interactions in B cells is not yet clear.
STAT3
STAT3 is pleiotropic and can be activated by multiple cytokines including IL-6, IL-10, and IFN-α/β. Because STAT3 is essential for the early development of mouse embryos [8], conditional gene targeting has been employed to elucidate the biological function of STAT3 in various organs and cell types. In bone marrow progenitor cells, STAT3 negatively regulates granulopoiesis [21]. Deletion of STAT3 during hematopoiesis results in abnormalities in myeloid cells, leads to overactivation of innate immune responses, and causes inflammatory bowel disease-like pathogenesis [22]. In T cells, STAT3 is important in IL-6-mediated suppression of apoptosis, and potentiates proliferation through this mechanism [23].
STAT3 is also required for optimal IL-2-induced T cell proliferative responses by upregulating the expression of IL-2Rα [24], a component of the high-affinity IL-2 receptor. In contrast to its pro-immune action in T cells, deletion of STAT3 in myeloid cells results in hyperactivation of macrophages, marked increases in inflammatory cytokine production, and inflammatory bowel disease [25], demonstrating the immune-suppressive and anti-inflammatory function of STAT3 in the myeloid lineage. Because the response to IL-10 was completely abolished in STAT3-deficient macrophages and neutrophils, the hyperactive phenotype of these mice suggests that the in vivo action of STAT3 in macrophages and neutrophils is to mediate the anti-inflammatory effects of IL-10. The chronic enterocolitis in myeloid cell-specific STAT3-null animals might be a result of overproduction of IL-12 triggered by Toll-like receptor 4 (TLR4) signaling [26], suggesting a role for STAT3 in orchestrating innate and adaptive immunity. In addition, a recent report shows that STAT3 signaling in antigen-presenting cells is required for the induction of antigen-specific T cell anergy and thus immune tolerance [27], providing further insight into the immune regulatory action of STAT3.
Besides immune system cells, STAT3 gene has been conditionally ablated in several other cell types such as keratinocytes, epithelial cells, endothelial cells, mammary gland, and motor neurons. Deleting STAT3 in keratinocytes reveals that STAT3 is essential for skin remodeling and wound healing [28]. Interestingly, mammary-gland-specific ablation of STAT3 demonstrates that STAT3 is indispensable for the apoptosis of epithelial cells that is required for normal mammary gland involution [29], in contrast to the anti-apoptotic role of STAT3 in T cells and probably in fibroblasts. Thus, depending on cell type and cell context, STAT3 can contribute to either apoptosis or survival. A causal role for STAT3 in oncogenesis is supported by the finding that the introduction of a constitutively active form of STAT3 is sufficient for cellular transformation [30]. This result, together with the observation that constitutive activation of STAT3 has been detected in human cancer cells, focuses attention on the importance of STAT3 in malignancy. Collectively, the roles of STAT3 are complex, because STAT3 can have opposing effects on different cells, depending on cell type and activation status.
STAT4
STAT4 is activated by IL-12 and IL-23 in murine cells [31], and additionally is activated by IFN-α in human cells by being recruited to type I IFN receptor through interaction with STAT2 [32]. As might be expected for a STAT activated by canonical Th1 cytokines, STAT4-deficient mice exhibit impaired Th1 differentiation, IFN-γ production, and cell-mediated immune responses [33,34]. As a result, STAT4 knockout mice are resistant to Th1-dependent autoimmune diseases. However, the exact contribution of IL-12 versus IL-23 to STAT4 activation in vivo needs to be clarified in view of the recent discovery of IL-23 and especially its crucial role in certain autoimmune processes [35].
STAT5
Closely related STAT5 proteins, STAT5a and STAT5b, are encoded by different but chromosomally linked genes and seem to have overlapping yet distinct functions. STAT5a cannot entirely compensate for STAT5b deficiency, and vice versa. STAT5 is activated by a wide range of cytokines, including growth hormone, prolactin, epidermal growth factor, platelet-derived growth factor, the hematopoietic cytokines IL-3, granulocyte/macrophage colony-stimulating factor, IL-5, and erythropoietin, and also cytokines that act on lymphocytes and share a common gamma (γc) receptor chain (IL-2, IL-4, IL-7, IL-9, and IL-15). Results from mice singly deficient in either of the STAT5 genes demonstrate a specific and limited role for individual STAT5 proteins [36]. Mammary gland development is greatly impaired in STAT5a-deficient mice but not in STAT5b-null animals. T cells from STAT5a-deficient mice have decreased proliferation secondary to diminished expression of the IL-2Rα chain [37], similar to the phenotype of STAT3-null T cells. Thus, STAT3 and STAT5 have nonredundant roles in maintaining IL-2Rα expression because they cannot substitute for each other. STAT5b-deficient mice have a more significant proliferation defect that cannot be corrected by high-dose IL-2, and decreased numbers of NK cells with diminished cytolytic activity [38].
Mice doubly deficient in both STAT5a and STAT5b genes are infertile, and a significant proportion of these mice die within a few weeks of birth. Consistent with the phenotype of STAT5a and STAT5b singly deficient mice, the doubly deficient animals exhibit defective development of mammary glands and impaired T cell proliferation in response to IL-2. In addition, these mice lack NK cells, develop splenomegaly over time, and have peripheral T cells that exhibit an activated phenotype; these are not seen in animals lacking STAT5a or STAT5b alone. Compromised immune system function in mice doubly deficient in STAT5a and STAT5b genes demonstrates an important role for STAT5 in mediating the effects of cytokines that activate Jak–STAT signaling in lymphoid cells. STAT5 is also important in myeloid cell mobilization and survival, and thus in the formation of inflammatory exudates.
STAT6
STAT6 is activated by IL-4 and IL-13, and, in B cells, by IFN-α; STAT6 target genes include those encoding immunoglobulin heavy chain ε, CD23, MHC class II, GATA-3 and c-maf. STAT6 deficiency results in defective Th2 responses, with diminished Th2 differentiation, diminished IgE production, and diminished proliferative responses to IL-4 [39,40]. Interestingly, there is a STAT6-independent pathway of Th2 differentiation, which is revealed when BCL6 is absent.
Negative regulation of STAT signaling
Ligation of cytokine receptors typically results in a transient activation of the Jak–STAT signaling pathway. STAT activity often peaks about 5–30 min after cytokine stimulation, followed by a decay back to baseline over the subsequent 1–4 hours, although there are also several examples of sustained STAT activation. In addition, it has become clear that signaling by the Jak–STAT pathway can be blocked after preincubation with antagonistic agents. These observations suggest that there must exist mechanisms for downregulating cytokine Jak–STAT signaling, and multiple inhibitory mechanisms have been described. Downregulation of cytokine signaling is likely to be important for homeostasis and the prevention of chronic inflammation or autoimmunity. Both constitutive inhibitory pathways and inducible mechanisms have been described. These mechanisms can act at several levels in cytokine signaling, targeting the receptors, Jaks, or the STATs themselves. Constitutive inhibitory mechanisms include proteolysis, dephosphorylation, and interaction with inhibitory molecules termed protein inhibitors of activated STATs (PIAS). Three major regulated or inducible inhibitory mechanisms have been identified, mediated by downregulation of receptor expression, through the induction of inhibitory molecules termed suppressors of cytokine signaling (SOCS) proteins, and by rapid mitogen-activated protein kinase (MAPK) or protein kinase C (PKC)-dependent modification of pre-existing signaling components.
Inhibition of receptor expression
One effective way of turning off cytokine signaling is by turning off expression of the cytokine receptor. This mechanism is important in the regulation of T cell differentiation. Expression of the IL-12 receptor β 2 subunit is extinguished during Th2 development, with a concomitant loss of IL-12 signaling [41]. IL-2 signaling is also modulated during Th2 development by IL-4-dependent suppression of IL-2 receptor β chain expression, with the resulting suppression of STAT5 and its target gene activation, but preservation of proliferative responses [42]. During Th1 development or during T cell expansion in the presence of IFN-γ, expression of the IFN-γ receptor beta subunit is suppressed at the level of transcription, possibly to allow escape from the anti-proliferative effects of IFN-γ and allow cell expansion [43,44]. Inhibition of IL-2 receptor and Jak3 expression by glucocorticoids is one mechanism by which glucocorticoids suppress T cell expansion [45].
Proteolysis
One proposed mechanism for degrading cytokine signaling components is the coupling of signaling proteins to proteasomes by SOCS molecules (see also the section on SOCS proteins below). SOCS can bind to ubiquitin ligase complexes and target receptors and Jaks for degradation [46,47]. For example, the stability of Jak2 seems to be regulated through a SOCS1-mediated proteasome pathway [48]. Proteasome inhibitors have also been found to stabilize STAT5 activity, but ubiquitinated forms of STAT5 were not detected, and the possibility of an indirect mechanism was raised [49]. Interestingly, several viruses are capable of destabilizing STAT molecules and thus antagonizing host immune responses. The phenomenon of virus-induced STAT degradation has been reported for STAT1, STAT2, and STAT3 [50-53].
Dephosphorylation
The SH2 domain-containing tyrosine phosphatases SHP-1 and SHP-2 can be recruited to receptor complexes and downregulate signaling. SHP-1 inhibits signaling by erythropoietin and IFN-γ. Mice with defective SHP-1 (motheaten) exhibit an autoimmune/inflammatory phenotype, and hyperactive Jak–STAT signaling might contribute to this phenotype. Genetic studies indicate that SHP-2 is involved in the negative regulation of IFN-stimulated Jak1 activation [54]. CD45, a protein tyrosine phosphatase (PTP) previously characterized in antigen receptor signaling, has been recently described to be a Jak phosphatase, and gene targeting experiments support an important role of CD45 in downregulating erythropoietin and IFN signaling [55]. PTP1B is another phosphatase that has been reported to dephosphorylate Jaks, especially Jak2 and Tyk2 [56]. STATs can also serve as targets for phosphatases, both in the cytoplasm and in the nucleus. STAT1 is dephosphorylated in the nucleus and returns to the cytoplasm [57], and TC45 and SHP-2 are proposed STAT1 phosphatases in the nucleus [58,59]. In contrast, STAT5 can be dephosphorylated in the cytoplasm, possibly by SHP-2 [60]. It seems that there is substrate specificity in the dephosphorylation of STATs by PTPs, and it is not clear how this specificity is achieved.
PIAS proteins
PIAS proteins constitute a family of related proteins expressed both in the cytoplasm and in the nucleus. So far, there are four mammalian members in this family: PIAS1, PIAS3, PIASX and PIASY. PIAS1, PIAS3, and PIASX have been shown to interact with STAT1, STAT3, and STAT4, respectively [61-63]. In addition, PIASY can also associate with STAT1 [64]. PIAS proteins interfere with STAT-dependent gene activation by several distinct mechanisms. The best-characterized activity of PIAS is its ability to block STAT DNA binding. However, PIAS can inhibit STAT transcriptional activity without affecting STAT DNA binding. The physiological role of PIAS in regulating STAT signaling has been demonstrated in Drosophila [65]. The function of mammalian PIAS proteins in vivo remains to be determined.
SOCS proteins
SOCS, also termed JAB, SSI, or CIS molecules, are proteins that inhibit signaling by a large number of cytokines that use the Jak–STAT signaling pathway [66-68]. So far, CIS (the original family member to be cloned), SOCS1, SOCS2, and SOCS3 have been shown to inhibit cytokine signaling, and the functions of SOCS4–SOCS7 are largely unknown. SOCS1 and SOCS3 interact with Jaks by means of an SH2–phosphotyrosine interaction, and SOCS1 is an effective inhibitor of Jak catalytic activity; SOCS3 is much less effective at inhibiting Jaks directly. SOCS3 has been shown to interact with gp130 receptor chain, and this interaction is necessary for the effective inhibition of signaling to occur. CIS interacts with the IL-3 receptor and inhibits signaling, possibly by competing for and blocking phosphotyrosine docking sites that are important for signal transduction.
SOCS proteins are short-lived and ubiquitinated, and it is possible that they also work by targeting receptors and other signaling components for degradation. Except for a few tissues such as thymus and fetal liver [69,70], SOCS are not expressed at baseline and are rapidly induced as immediate early genes in response to cytokines that themselves activate the Jak–STAT pathway, or inflammatory cytokines such as IL-1 and tumor necrosis factor (TNF). Thus, SOCS might be important in feedback inhibition, and in cytokine antagonism. Gene targeting experiments demonstrate that SOCS1 and SOCS3 are essential in the regulation of immune responses. SOCS1-deficient mice exhibit hyper-responsiveness to IFN-γ and IL-4 stimulation. The pathology in SOCS1-null animals resembles that of IFN-γ transgenic mice and can be prevented by crossing with IFN-γ-deficient mice or by using IFN-γ neutralizing antibodies [71]. Thus, an important role for SOCS1 in vivo is the regulation of IFN-γ signaling, although SOCS1 also functions as a regulator of innate immunity by modulating TLR signaling [72,73]. SOCS3 is induced by multiple cytokines and has been suggested to be inhibitory for both IL-6 and IL-10 in vitro. However, gene targeting studies reveal unexpected specificity in regulating cytokine signaling by SOCS3. In SOCS3-deficient macrophages, only IL-6 signaling was augmented and altered, whereas IL-10 activation of STAT3 remained untouched [74-76], indicating that SOCS3 preferentially regulates IL-6 responsiveness in vivo.
Rapidly inducible mechanisms (dependent on MAPK or PKC and independent of de novo protein synthesis)
SOCS-dependent inhibition of cytokine signaling is delayed by the necessity of producing SOCS mRNA and protein in response to an extracellular stimulus. Cytokine signaling can also be inhibited by rapidly inducible mechanisms that act within a few minutes [77] and are not dependent on de novo protein synthesis or on SOCS proteins. IL-6 signaling (and, in some cases, IL-10 signaling in myeloid cells) is rapidly inhibited by preincubation with IL-1, TNF, phorbol 12-myristate 13-acetate (PMA), or ionomycin [77-79], and IL-2 signaling is inhibited in preactivated, cycling T cells after religation of the T cell receptor [80]. One mechanism of rapid inhibition of cytokine signaling is mediated by MAPKs. For example, extracellular regulated kinases are important in the inhibition of IL-6 signaling by PMA [78], and of IL-2 signaling by T cell receptor religation [80]. In contrast, the p38 stress kinase (a member of a subfamily of MAPKs) is necessary for the inhibition of IL-6 signaling by IL-1 [79]. Rapid inhibition of Jak–STAT signaling can also occur through PKC-dependent pathways. Ligation of Fc receptors by immune complexes inhibits IL-10 STAT activation. The mechanism of inhibition involves decreased expression of cell-surface IL-10 receptor and is dependent on PKCδ [81]. It is likely that rapidly inducible, MAPK-dependent or PKC-dependent inhibitory mechanisms work together with SOCS-dependent pathways to achieve effective inhibition of signaling over a broad time frame.
Positive regulation of STAT signaling
Although much attention has focused on the negative regulatory pathways of Jak–STAT signaling, it is increasingly becoming apparent that cytokine signal transduction can also be positively modulated or sensitized. One report shows that IFN-γ signaling is positively modulated by previous exposure to low subthreshold concentrations of type I IFNs (IFN-α/β). The mechanism of sensitization of IFN-γ signaling by type I IFNs depends on low-level IFN-α/β signaling that leads to an association between the two nonligand-binding receptor subunits, IFN-αR1 and IFN-γR2, and increased dimerization of tyrosine-phosphorylated STAT1 [82]. In addition to signaling crosstalk with IFN-α/β, IFN-γ signaling can also be sensitized in a positive feedforward loop by low doses of IFN-γ that do not themselves activate detectable signaling events [83]. Sensitized IFN-γ signaling is mediated by sustained elevation of STAT1 expression induced by low concentrations of IFN-γ that do not effectively engage feedback inhibition by SOCS proteins. IFN-γ is a major activator of macrophages, and sensitization of IFN-γ signaling might be particularly important to achieve full macrophage activation early in immune responses when IFN-γ levels are low. Another example of such a positive self-regulatory loop is the induction of IFN-stimulated gene 15 (ISG15) by IFNs and the positive regulation of IFN signaling by IFN-activated ISG15. ISG15, a small protein homologous to ubiquitin, can be conjugated to cellular proteins in a process called ISGylation, and ISGylation in turn greatly enhances IFN-activated Jak–STAT signaling [84]. Positive and negative regulatory mechanisms of IFN signaling are illustrated schematically in Fig. 2.
Figure 2.
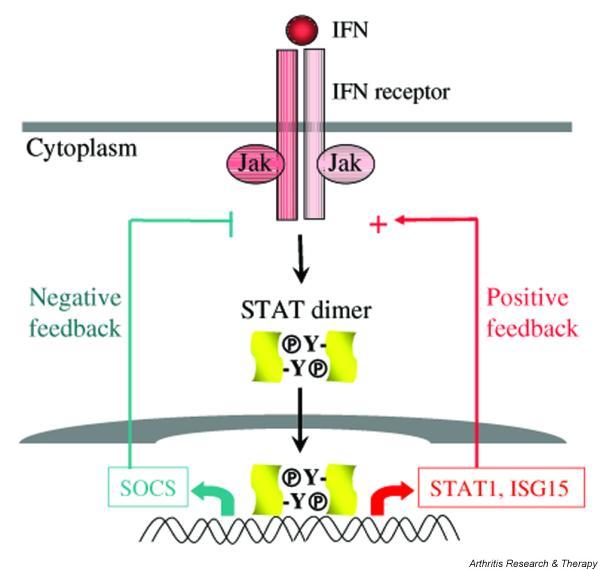
Activation of positive and negative feedback loops by interferons (IFNs). Low priming doses of IFNs will preferentially activate positive feedback loops, such as association with other receptors (not shown) and the induction of expression of IFN-stimulated genes that function as positive regulators of signaling, such as STAT1 and interferon-stimulated gene 15 (ISG15) (right). Higher doses of IFNs will also activate feedback inhibition, such as the induction of expression of suppressors of cytokine signaling (SOCS) (left). Jak, Janus kinase; STAT, signal transducer and activator of transcription.
Reprogramming of cytokine signaling
One emerging concept in Jak–STAT signaling is that not only can the quantity of signal transduction be positively or negatively regulated, but also the nature of cytokine responses can be altered or reprogrammed. Thus, the exact cellular response to a cytokine is determined by the presence of a particular stimulus as well as the microenvironment to which cells are exposed. One such example is the reprogramming of IL-10 signaling by IFN-α and IFN-γ [85,86]. IL-10 is a weak activator of STAT1 in myeloid cells and does not normally induce STAT1 target genes, which IFN-γ does. However, pre-exposure to type I IFNs reprograms STAT activation by IL-10 such that STAT1 is preferentially activated by IL-10 in these cells, resulting in the induction of a group of STAT1-dependent genes and thereby a gain of inflammatory function [85]. IFN-γ is also able to switch the balance of IL-10 STAT activation from STAT3 to STAT1, with concomitant downregulation of STAT3-dependent gene expression, and partial attenuation of IL-10 anti-inflammatory function [86]. The mechanisms of reprogramming of IL-10 signaling mediated by IFN-α and IFN-γ might be distinct. It seems that IFNs operate a switch that rapidly regulates STAT activation by IL-10 and alters macrophage responses to IL-10. Dynamic regulation of the activation of different STATs by the same cytokine provides a mechanism by which cells can integrate and balance signals delivered by opposing cytokines, and extends our understanding of cross-regulation by opposing cytokines to include reprogramming of signaling and alteration of function.
Conclusions
The activation of Jak–STAT signaling involves only two groups of proteins, Jaks and STATs, and does not require the synthesis of any new molecules. Ligation of the cytokine receptor on the cell surface is directly translated into transcriptional responses in the nucleus. However, such a seemingly simple pathway is subject to sophisticated modulation by multiple factors at multiple levels of signal transduction. The net outcome of a cytokine stimulation is orchestrated by several cellular events that involve positive regulation, negative regulation, and reprogramming of STAT activity. The physiological functions of STATs have been unraveled using mouse genetics. The involvement of STATs in rheumatic disease pathogenesis is an emerging area of research and is reviewed elsewhere [87,88]. Understanding the pathological roles of STATs would certainly help in the development of novel therapeutic strategies. For example, STAT3's oncogenic property might be a valuable target for novel tumor therapeutics. At the same time, potentiating STAT3's anti-inflammatory action in myeloid cells might be beneficial in treating inflammatory disorders. The expression of STAT1 along with several putative STAT1 target genes is elevated during synovitis in rheumatoid arthritis [83,89], but it is not known whether this overexpression of STAT1 is pathogenic or protective given the pleiotropic nature of STAT1. Indeed, STAT1 is protective in one animal model of rheumatoid arthritis [13]. Thus, STATs might have divergent effects on the pathogenesis of arthritis, depending on cell type and possibly on the stage of disease.
Competing interests
None declared.
Abbreviations
IFN = interferon; IL = interleukin; IRF = interferon regulatory factor; ISG15 = interferon-stimulated gene 15; ISGF3 = interferon-stimulated gene factor 3; Jak = Janus kinase; MAPK = mitogen-activated protein kinase; MCM5 = mini-chromosome maintenance 5; MHC = major histocompatibility complex; NK = natural killer; PIAS = protein inhibitor of activated STAT; PKC = protein kinase C; PMA = phorbol 12-myristate 13-acetate; PTP = protein tyrosine phosphatase; SH2 = Src homology 2; SHP = SH2 domain-containing tyrosine phosphatase; SOCS = suppressor of cytokine signaling; STAT = signal transducer and activator of transcription; TLR = Toll-like receptor; TNF = tumor necrosis factor.
Acknowledgments
Acknowledgements
This work was supported by NIH grants (LBI), a Cancer Research Institute Predoctoral Fellowship Training Grant (XH), and a Hospital for Special Surgery CSO Pilot and Feasibility Grant (XH).
Contributor Information
Lionel B Ivashkiv, Email: ivashkivl@hss.edu.
Xiaoyu Hu, Email: xih2001@med.cornell.edu.
References
- Darnell JE, Jr, Kerr IM, Stark GR. Jak–STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science. 1994;264:1415–1421. doi: 10.1126/science.8197455. [DOI] [PubMed] [Google Scholar]
- Gauzzi MC, Barbieri G, Richter MF, Uze G, Ling L, Fellous M, Pellegrini S. The amino-terminal region of Tyk2 sustains the level of interferon alpha receptor 1, a component of the interferon alpha/beta receptor. Proc Natl Acad Sci USA. 1997;94:11839–11844. doi: 10.1073/pnas.94.22.11839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivashkiv LB. Cytokines and STATs: how can signals achieve specificity? Immunity. 1995;3:1–4. doi: 10.1016/1074-7613(95)90152-3. [DOI] [PubMed] [Google Scholar]
- Decker T, Kovarik P. Serine phosphorylation of STATs. Oncogene. 2000;19:2628–2637. doi: 10.1038/sj.onc.1203481. [DOI] [PubMed] [Google Scholar]
- Mowen KA, Tang J, Zhu W, Schurter BT, Shuai K, Herschman HR, David M. Arginine methylation of STAT1 modulates IFNalpha/beta-induced transcription. Cell. 2001;104:731–741. doi: 10.1016/s0092-8674(01)00269-0. [DOI] [PubMed] [Google Scholar]
- Strobl B, Arulampalam V, Is'harc H, Newman SJ, Schlaak JF, Watling D, Costa-Pereira AP, Schaper F, Behrmann I, Sheehan KC, Schreiber RD, Horn F, Heinrich PC, Kerr IM. A completely foreign receptor can mediate an interferon-gamma-like response. EMBO J. 2001;20:5431–5442. doi: 10.1093/emboj/20.19.5431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa-Pereira AP, Tininini S, Strobl B, Alonzi T, Schlaak JF, Is'harc H, Gesualdo I, Newman SJ, Kerr IM, Poli V. Mutational switch of an IL-6 response to an interferon-gamma-like response. Proc Natl Acad Sci USA. 2002;99:8043–8047. doi: 10.1073/pnas.122236099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda K, Noguchi K, Shi W, Tanaka T, Matsumoto M, Yoshida N, Kishimoto T, Akira S. Targeted disruption of the mouse Stat3 gene leads to early embryonic lethality. Proc Natl Acad Sci USA. 1997;94:3801–3804. doi: 10.1073/pnas.94.8.3801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meraz MA, White JM, Sheehan KC, Bach EA, Rodig SJ, Dighe AS, Kaplan DH, Riley JK, Greenlund AC, Campbell D, Carver-Moore K, DuBois RN, Clark R, Aguet M, Schreiber RD. Targeted disruption of the Stat1 gene in mice reveals unexpected physiologic specificity in the JAK–STAT signaling pathway. Cell. 1996;84:431–442. doi: 10.1016/S0092-8674(00)81288-X. [DOI] [PubMed] [Google Scholar]
- Durbin JE, Hackenmiller R, Simon MC, Levy DE. Targeted disruption of the mouse Stat1 gene results in compromised innate immunity to viral disease. Cell. 1996;84:443–450. doi: 10.1016/S0092-8674(00)81289-1. [DOI] [PubMed] [Google Scholar]
- Durbin JE, Fernandez-Sesma A, Lee CK, Rao TD, Frey AB, Moran TM, Vukmanovic S, Garcia-Sastre A, Levy DE. Type I IFN modulates innate and specific antiviral immunity. J Immunol. 2000;164:4220–4228. doi: 10.4049/jimmunol.164.8.4220. [DOI] [PubMed] [Google Scholar]
- Nishibori T, Tanabe Y, Su L, David M. Impaired development of CD4+ CD25+ regulatory T cells in the absence of STAT1: increased susceptibility to autoimmune disease. J Exp Med. 2004;199:25–34. doi: 10.1084/jem.20020509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Hooge A, van de Loo F, Kolbe T, Muller M, Levy D, Bennink M, Arntz O, van den Berg WB. STAT1 and STAT3 activation during the course of experimental arthritis. Arthritis Rheum. 2002;46:S250. doi: 10.1002/1529-0131(200201)46:1<250::AID-ART10064>3.0.CO;2-T. [DOI] [Google Scholar]
- Shankaran V, Ikeda H, Bruce AT, White JM, Swanson PE, Old LJ, Schreiber RD. IFNγ and lymphocytes prevent primary tumour development and shape tumour immunogenicity. Nature. 2001;410:1107–1111. doi: 10.1038/35074122. [DOI] [PubMed] [Google Scholar]
- Kim S, Koga T, Isobe M, Kern BE, Yokochi T, Chin YE, Karsenty G, Taniguchi T, Takayanagi H. Stat1 functions as a cytoplasmic attenuator of Runx2 in the transcriptional program of osteoblast differentiation. Genes Dev. 2003;17:1979–1991. doi: 10.1101/gad.1119303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takayanagi H, Ogasawara K, Hida S, Chiba T, Murata S, Sato K, Takaoka A, Yokochi T, Oda H, Tanaka K, Nakamura K, Taniguchi T. T-cell-mediated regulation of osteoclastogenesis by signalling cross-talk between RANKL and IFN-γ. Nature. 2000;408:600–605. doi: 10.1038/35046102. [DOI] [PubMed] [Google Scholar]
- Takayanagi H, Kim S, Matsuo K, Suzuki H, Suzuki T, Sato K, Yokochi T, Oda H, Nakamura K, Ida N, Wagner EF, Taniguchi T. RANKL maintains bone homeostasis through c-Fos-dependent induction of interferon-β. Nature. 2002;416:744–749. doi: 10.1038/416744a. [DOI] [PubMed] [Google Scholar]
- Sheppard P, Kindsvogel W, Xu W, Henderson K, Schlutsmeyer S, Whitmore TE, Kuestner R, Garrigues U, Birks C, Roraback J, Ostrander C, Dong D, Shin J, Presnell S, Fox B, Haldeman B, Cooper E, Taft D, Gilbert T, Grant FJ, Tackett M, Krivan W, McKnight G, Clegg C, Foster D, Klucher KM. IL-28, IL-29 and their class II cytokine receptor IL-28R. Nat Immunol. 2003;4:63–68. doi: 10.1038/ni873. [DOI] [PubMed] [Google Scholar]
- Kotenko SV, Gallagher G, Baurin VV, Lewis-Antes A, Shen M, Shah NK, Langer JA, Sheikh F, Dickensheets H, Donnelly RP. IFN-λs mediate antiviral protection through a distinct class II cytokine receptor complex. Nat Immunol. 2003;4:69–77. doi: 10.1038/ni875. [DOI] [PubMed] [Google Scholar]
- Park C, Li S, Cha E, Schindler C. Immune response in Stat2 knockout mice. Immunity. 2000;13:795–804. doi: 10.1016/S1074-7613(00)00077-7. [DOI] [PubMed] [Google Scholar]
- Lee CK, Raz R, Gimeno R, Gertner R, Wistinghausen B, Takeshita K, DePinho RA, Levy DE. STAT3 is a negative regulator of granulopoiesis but is not required for G-CSF-dependent differentiation. Immunity. 2002;17:63–72. doi: 10.1016/S1074-7613(02)00336-9. [DOI] [PubMed] [Google Scholar]
- Welte T, Zhang SS, Wang T, Zhang Z, Hesslein DG, Yin Z, Kano A, Iwamoto Y, Li E, Craft JE, Bothwell AL, Fikrig E, Koni PA, Flavell RA, Fu XY. STAT3 deletion during hematopoiesis causes Crohn's disease-like pathogenesis and lethality: a critical role of STAT3 in innate immunity. Proc Natl Acad Sci USA. 2003;100:1879–1884. doi: 10.1073/pnas.0237137100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda K, Kaisho T, Yoshida N, Takeda J, Kishimoto T, Akira S. Stat3 activation is responsible for IL-6-dependent T cell proliferation through preventing apoptosis: generation and characterization of T cell-specific Stat3-deficient mice. J Immunol. 1998;161:4652–4660. [PubMed] [Google Scholar]
- Akaishi H, Takeda K, Kaisho T, Shineha R, Satomi S, Takeda J, Akira S. Defective IL-2-mediated IL-2 receptor alpha chain expression in Stat3-deficient T lymphocytes. Int Immunol. 1998;10:1747–1751. doi: 10.1093/intimm/10.11.1747. [DOI] [PubMed] [Google Scholar]
- Takeda K, Clausen BE, Kaisho T, Tsujimura T, Terada N, Forster I, Akira S. Enhanced Th1 activity and development of chronic enterocolitis in mice devoid of Stat3 in macrophages and neutrophils. Immunity. 1999;10:39–49. doi: 10.1016/S1074-7613(00)80005-9. [DOI] [PubMed] [Google Scholar]
- Kobayashi M, Kweon MN, Kuwata H, Schreiber RD, Kiyono H, Takeda K, Akira S. Toll-like receptor-dependent production of IL-12p40 causes chronic enterocolitis in myeloid cell-specific Stat3-deficient mice. J Clin Invest. 2003;111:1297–1308. doi: 10.1172/JCI200317085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng F, Wang HW, Cuenca A, Huang M, Ghansah T, Brayer J, Kerr WG, Takeda K, Akira S, Schoenberger SP, Yu H, Jove R, Sotomayor EM. A critical role for Stat3 signaling in immune tolerance. Immunity. 2003;19:425–436. doi: 10.1016/S1074-7613(03)00232-2. [DOI] [PubMed] [Google Scholar]
- Sano S, Itami S, Takeda K, Tarutani M, Yamaguchi Y, Miura H, Yoshikawa K, Akira S, Takeda J. Keratinocyte-specific ablation of Stat3 exhibits impaired skin remodeling, but does not affect skin morphogenesis. EMBO J. 1999;18:4657–4668. doi: 10.1093/emboj/18.17.4657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman RS, Lourenco PC, Tonner E, Flint DJ, Selbert S, Takeda K, Akira S, Clarke AR, Watson CJ. Suppression of epithelial apoptosis and delayed mammary gland involution in mice with a conditional knockout of Stat3. Genes Dev. 1999;13:2604–2616. doi: 10.1101/gad.13.19.2604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bromberg JF, Wrzeszczynska MH, Devgan G, Zhao Y, Pestell RG, Albanese C, Darnell JE., Jr Stat3 as an oncogene. Cell. 1999;98:295–303. doi: 10.1016/S0092-8674(00)81959-5. [DOI] [PubMed] [Google Scholar]
- Oppmann B, Lesley R, Blom B, Timans JC, Xu Y, Hunte B, Vega F, Yu N, Wang J, Singh K, Zonin F, Vaisberg E, Churakova T, Liu M, Gorman D, Wagner J, Zurawski S, Liu Y, Abrams JS, Moore KW, Rennick D, de Waal-Malefyt R, Hannum C, Bazan JF, Kastelein RA. Novel p19 protein engages IL-12p40 to form a cytokine, IL-23, with biological activities similar as well as distinct from IL-12. Immunity. 2000;13:715–725. doi: 10.1016/S1074-7613(00)00070-4. [DOI] [PubMed] [Google Scholar]
- Farrar JD, Smith JD, Murphy TL, Leung S, Stark GR, Murphy KM. Selective loss of type I interferon-induced STAT4 activation caused by a minisatellite insertion in mouse Stat2. Nat Immunol. 2000;1:65–69. doi: 10.1038/76932. [DOI] [PubMed] [Google Scholar]
- Kaplan MH, Sun YL, Hoey T, Grusby MJ. Impaired IL-12 responses and enhanced development of Th2 cells in Stat4-deficient mice. Nature. 1996;382:174–177. doi: 10.1038/382174a0. [DOI] [PubMed] [Google Scholar]
- Thierfelder WE, van Deursen JM, Yamamoto K, Tripp RA, Sarawar SR, Carson RT, Sangster MY, Vignali DA, Doherty PC, Grosveld GC, Ihle JN. Requirement for Stat4 in interleukin-12-mediated responses of natural killer and T cells. Nature. 1996;382:171–174. doi: 10.1038/382171a0. [DOI] [PubMed] [Google Scholar]
- Cua DJ, Sherlock J, Chen Y, Murphy CA, Joyce B, Seymour B, Lucian L, To W, Kwan S, Churakova T, Zurawski S, Wiekowski M, Lira SA, Gorman D, Kastelein RA, Sedgwick JD. Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature. 2003;421:744–748. doi: 10.1038/nature01355. [DOI] [PubMed] [Google Scholar]
- Teglund S, McKay C, Schuetz E, van Deursen JM, Stravopodis D, Wang D, Brown M, Bodner S, Grosveld G, Ihle JN. Stat5a and Stat5b proteins have essential and nonessential, or redundant, roles in cytokine responses. Cell. 1998;93:841–850. doi: 10.1016/S0092-8674(00)81444-0. [DOI] [PubMed] [Google Scholar]
- Nakajima H, Liu XW, Wynshaw-Boris A, Rosenthal LA, Imada K, Finbloom DS, Hennighausen L, Leonard WJ. An indirect effect of Stat5a in IL-2-induced proliferation: a critical role for Stat5a in IL-2-mediated IL-2 receptor alpha chain induction. Immunity. 1997;7:691–701. doi: 10.1016/S1074-7613(00)80389-1. [DOI] [PubMed] [Google Scholar]
- Imada K, Bloom ET, Nakajima H, Horvath-Arcidiacono JA, Udy GB, Davey HW, Leonard WJ. Stat5b is essential for natural killer cell-mediated proliferation and cytolytic activity. J Exp Med. 1998;188:2067–2074. doi: 10.1084/jem.188.11.2067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimoda K, van Deursen J, Sangster MY, Sarawar SR, Carson RT, Tripp RA, Chu C, Quelle FW, Nosaka T, Vignali DA, Doherty PC, Grosveld G, Paul WE, Ihle JN. Lack of IL-4-induced Th2 response and IgE class switching in mice with disrupted Stat6 gene. Nature. 1996;380:630–633. doi: 10.1038/380630a0. [DOI] [PubMed] [Google Scholar]
- Takeda K, Tanaka T, Shi W, Matsumoto M, Minami M, Kashiwamura S, Nakanishi K, Yoshida N, Kishimoto T, Akira S. Essential role of Stat6 in IL-4 signalling. Nature. 1996;380:627–630. doi: 10.1038/380627a0. [DOI] [PubMed] [Google Scholar]
- Szabo SJ, Dighe AS, Gubler U, Murphy KM. Regulation of the interleukin (IL)-12R beta 2 subunit expression in developing T helper 1 (Th1) and Th2 cells. J Exp Med. 1997;185:817–824. doi: 10.1084/jem.185.5.817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro A, Sengupta TK, Ruiz DC, Yang E, Ivashkiv LB. IL-4 selectively inhibits IL-2-triggered Stat5 activation, but not proliferation, in human T cells. J Immunol. 1999;162:1261–1269. [PubMed] [Google Scholar]
- Bach EA, Szabo SJ, Dighe AS, Ashkenazi A, Aguet M, Murphy KM, Schreiber RD. Ligand-induced autoregulation of IFN-gamma receptor beta chain expression in T helper cell subsets. Science. 1995;270:1215–1218. doi: 10.1126/science.270.5239.1215. [DOI] [PubMed] [Google Scholar]
- Pernis A, Gupta S, Gollob KJ, Garfein E, Coffman RL, Schindler C, Rothman P. Lack of interferon gamma receptor beta chain and the prevention of interferon gamma signaling in TH1 cells. Science. 1995;269:245–247. doi: 10.1126/science.7618088. [DOI] [PubMed] [Google Scholar]
- Bianchi M, Meng C, Ivashkiv LB. Inhibition of IL-2-induced Jak–STAT signaling by glucocorticoids. Proc Natl Acad Sci USA. 2000;97:9573–9578. doi: 10.1073/pnas.160099797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamura T, Sato S, Haque D, Liu L, Kaelin WG, Jr, Conaway RC, Conaway JW. The Elongin BC complex interacts with the conserved SOCS-box motif present in members of the SOCS, ras, WD-40 repeat, and ankyrin repeat families. Genes Dev. 1998;12:3872–3881. doi: 10.1101/gad.12.24.3872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang JG, Farley A, Nicholson SE, Willson TA, Zugaro LM, Simpson RJ, Moritz RL, Cary D, Richardson R, Hausmann G, Kile BJ, Kent SB, Alexander WS, Metcalf D, Hilton DJ, Nicola NA, Baca M. The conserved SOCS box motif in suppressors of cytokine signaling binds to elongins B and C and may couple bound proteins to proteasomal degradation. Proc Natl Acad Sci USA. 1999;96:2071–2076. doi: 10.1073/pnas.96.5.2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ungureanu D, Saharinen P, Junttila I, Hilton DJ, Silvennoinen O. Regulation of Jak2 through the ubiquitin-proteasome pathway involves phosphorylation of Jak2 on Y1007 and interaction with SOCS-1. Mol Cell Biol. 2002;22:3316–3326. doi: 10.1128/MCB.22.10.3316-3326.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D, Moriggl R, Stravopodis D, Carpino N, Marine JC, Teglund S, Feng J, Ihle JN. A small amphipathic alpha-helical region is required for transcriptional activities and proteasome-dependent turnover of the tyrosine-phosphorylated Stat5. EMBO J. 2000;19:392–399. doi: 10.1093/emboj/19.3.392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parisien JP, Lau JF, Rodriguez JJ, Sullivan BM, Moscona A, Parks GD, Lamb RA, Horvath CM. The V protein of human parainfluenza virus 2 antagonizes type I interferon responses by destabilizing signal transducer and activator of transcription 2. Virology. 2001;283:230–239. doi: 10.1006/viro.2001.0856. [DOI] [PubMed] [Google Scholar]
- Parisien JP, Lau JF, Rodriguez JJ, Ulane CM, Horvath CM. Selective STAT protein degradation induced by paramyxoviruses requires both STAT1 and STAT2 but is independent of alpha/beta interferon signal transduction. J Virol. 2002;76:4190–4198. doi: 10.1128/JVI.76.9.4190-4198.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulane CM, Horvath CM. Paramyxoviruses SV5 and HPIV2 assemble STAT protein ubiquitin ligase complexes from cellular components. Virology. 2002;304:160–166. doi: 10.1006/viro.2002.1773. [DOI] [PubMed] [Google Scholar]
- Ulane CM, Rodriguez JJ, Parisien JP, Horvath CM. STAT3 ubiquitylation and degradation by mumps virus suppress cytokine and oncogene signaling. J Virol. 2003;77:6385–6393. doi: 10.1128/JVI.77.11.6385-6393.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You M, Yu DH, Feng GS. Shp-2 tyrosine phosphatase functions as a negative regulator of the interferon-stimulated Jak/STAT pathway. Mol Cell Biol. 1999;19:2416–2424. doi: 10.1128/mcb.19.3.2416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irie-Sasaki J, Sasaki T, Matsumoto W, Opavsky A, Cheng M, Welstead G, Griffiths E, Krawczyk C, Richardson CD, Aitken K, Iscove N, Koretzky G, Johnson P, Liu P, Rothstein DM, Penninger JM. CD45 is a JAK phosphatase and negatively regulates cytokine receptor signalling. Nature. 2001;409:349–354. doi: 10.1038/35053086. [DOI] [PubMed] [Google Scholar]
- Myers MP, Andersen JN, Cheng A, Tremblay ML, Horvath CM, Parisien JP, Salmeen A, Barford D, Tonks NK. TYK2 and JAK2 are substrates of protein-tyrosine phosphatase 1B. J Biol Chem. 2001;276:47771–47774. doi: 10.1074/jbc.M009472200. [DOI] [PubMed] [Google Scholar]
- Haspel RL, Darnell JE., Jr A nuclear protein tyrosine phosphatase is required for the inactivation of Stat1. Proc Natl Acad Sci USA. 1999;96:10188–10193. doi: 10.1073/pnas.96.18.10188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu TR, Hong YK, Wang XD, Ling MY, Dragoi AM, Chung AS, Campbell AG, Han ZY, Feng GS, Chin YE. SHP-2 is a dual-specificity phosphatase involved in Stat1 dephosphorylation at both tyrosine and serine residues in nuclei. J Biol Chem. 2002;277:47572–47580. doi: 10.1074/jbc.M207536200. [DOI] [PubMed] [Google Scholar]
- ten Hoeve J, de Jesus Ibarra-Sanchez M, Fu Y, Zhu W, Tremblay M, David M, Shuai K. Identification of a nuclear Stat1 protein tyrosine phosphatase. Mol Cell Biol. 2002;22:5662–5668. doi: 10.1128/MCB.22.16.5662-5668.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu CL, Jin YJ, Burakoff SJ. Cytosolic tyrosine dephosphorylation of STAT5. Potential role of SHP-2 in STAT5 regulation. J Biol Chem. 2000;275:599–604. doi: 10.1074/jbc.275.1.599. [DOI] [PubMed] [Google Scholar]
- Liu B, Liao J, Rao X, Kushner SA, Chung CD, Chang DD, Shuai K. Inhibition of Stat1-mediated gene activation by PIAS1. Proc Natl Acad Sci USA. 1998;95:10626–10631. doi: 10.1073/pnas.95.18.10626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung CD, Liao J, Liu B, Rao X, Jay P, Berta P, Shuai K. Specific inhibition of Stat3 signal transduction by PIAS3. Science. 1997;278:1803–1805. doi: 10.1126/science.278.5344.1803. [DOI] [PubMed] [Google Scholar]
- Arora T, Liu B, He H, Kim J, Murphy TL, Murphy KM, Modlin RL, Shuai K. PIASx is a transcriptional co-repressor of signal transducer and activator of transcription 4. J Biol Chem. 2003;278:21327–21330. doi: 10.1074/jbc.C300119200. [DOI] [PubMed] [Google Scholar]
- Liu B, Gross M, ten Hoeve J, Shuai K. A transcriptional co-repressor of Stat1 with an essential LXXLL signature motif. Proc Natl Acad Sci USA. 2001;98:3203–3207. doi: 10.1073/pnas.051489598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betz A, Lampen N, Martinek S, Young MW, Darnell JE., Jr A Drosophila PIAS homologue negatively regulates stat92E. Proc Natl Acad Sci USA. 2001;98:9563–9568. doi: 10.1073/pnas.171302098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endo TA, Masuhara M, Yokouchi M, Suzuki R, Sakamoto H, Mitsui K, Matsumoto A, Tanimura S, Ohtsubo M, Misawa H, Miyazaki T, Leonor N, Taniguchi T, Fujita T, Kanakura Y, Komiya S, Yoshimura A. A new protein containing an SH2 domain that inhibits JAK kinases. Nature. 1997;387:921–924. doi: 10.1038/43213. [DOI] [PubMed] [Google Scholar]
- Naka T, Narazaki M, Hirata M, Matsumoto T, Minamoto S, Aono A, Nishimoto N, Kajita T, Taga T, Yoshizaki K, Akira S, Kishimoto T. Structure and function of a new STAT-induced STAT inhibitor. Nature. 1997;387:924–929. doi: 10.1038/43219. [DOI] [PubMed] [Google Scholar]
- Starr R, Willson TA, Viney EM, Murray LJ, Rayner JR, Jenkins BJ, Gonda TJ, Alexander WS, Metcalf D, Nicola NA, Hilton DJ. A family of cytokine-inducible inhibitors of signalling. Nature. 1997;387:917–921. doi: 10.1038/43206. [DOI] [PubMed] [Google Scholar]
- Marine JC, McKay C, Wang D, Topham DJ, Parganas E, Nakajima H, Pendeville H, Yasukawa H, Sasaki A, Yoshimura A, Ihle J. SOCS3 is essential in the regulation of fetal liver erythropoiesis. Cell. 1999;98:617–627. doi: 10.1016/S0092-8674(00)80049-5. [DOI] [PubMed] [Google Scholar]
- Marine JC, Topham DJ, McKay C, Wang D, Parganas E, Stravopodis D, Yoshimura A, Ihle JN. SOCS1 deficiency causes a lymphocyte-dependent perinatal lethality. Cell. 1999;98:609–616. doi: 10.1016/S0092-8674(00)80048-3. [DOI] [PubMed] [Google Scholar]
- Alexander WS, Starr R, Fenner JE, Scott CL, Handman E, Sprigg NS, Corbin JE, Cornish AL, Darwiche R, Owczarek CM, Kay TW, Nicola NA, Hertzog PJ, Metcalf D, Hilton DJ. SOCS1 is a critical inhibitor of interferon gamma signaling and prevents the potentially fatal neonatal actions of this cytokine. Cell. 1999;98:597–608. doi: 10.1016/S0092-8674(00)80047-1. [DOI] [PubMed] [Google Scholar]
- Nakagawa R, Naka T, Tsutsui H, Fujimoto M, Kimura A, Abe T, Seki E, Sato S, Takeuchi O, Takeda K, Akira S, Yamanishi K, Kawase I, Nakanishi K, Kishimoto T. SOCS-1 participates in negative regulation of LPS responses. Immunity. 2002;17:677–687. doi: 10.1016/S1074-7613(02)00449-1. [DOI] [PubMed] [Google Scholar]
- Kinjyo I, Hanada T, Inagaki-Ohara K, Mori H, Aki D, Ohishi M, Yoshida H, Kubo M, Yoshimura A. SOCS1/JAB is a negative regulator of LPS-induced macrophage activation. Immunity. 2002;17:583–591. doi: 10.1016/S1074-7613(02)00446-6. [DOI] [PubMed] [Google Scholar]
- Yasukawa H, Ohishi M, Mori H, Murakami M, Chinen T, Aki D, Hanada T, Takeda K, Akira S, Hoshijima M, Hirano T, Chien KR, Yoshimura A. IL-6 induces an anti-inflammatory response in the absence of SOCS3 in macrophages. Nat Immunol. 2003;4:551–556. doi: 10.1038/ni938. [DOI] [PubMed] [Google Scholar]
- Lang R, Pauleau AL, Parganas E, Takahashi Y, Mages J, Ihle JN, Rutschman R, Murray PJ. SOCS3 regulates the plasticity of gp130 signaling. Nat Immunol. 2003;4:546–550. doi: 10.1038/ni932. [DOI] [PubMed] [Google Scholar]
- Croker BA, Krebs DL, Zhang JG, Wormald S, Willson TA, Stanley EG, Robb L, Greenhalgh CJ, Forster I, Clausen BE, Nicola NA, Metcalf D, Hilton DJ, Roberts AW, Alexander WS. SOCS3 negatively regulates IL-6 signaling in vivo. Nat Immunol. 2003;4:540–545. doi: 10.1038/ni931. [DOI] [PubMed] [Google Scholar]
- Sengupta TK, Schmitt EM, Ivashkiv LB. Inhibition of cytokines and JAK–STAT activation by distinct signaling pathways. Proc Natl Acad Sci USA. 1996;93:9499–9504. doi: 10.1073/pnas.93.18.9499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sengupta TK, Talbot ES, Scherle PA, Ivashkiv LB. Rapid inhibition of interleukin-6 signaling and Stat3 activation mediated by mitogen-activated protein kinases. Proc Natl Acad Sci USA. 1998;95:11107–11112. doi: 10.1073/pnas.95.19.11107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed ST, Ivashkiv LB. Inhibition of IL-6 and IL-10 signaling and Stat activation by inflammatory and stress pathways. J Immunol. 2000;165:5227–5237. doi: 10.4049/jimmunol.165.9.5227. [DOI] [PubMed] [Google Scholar]
- Lee IH, Li WP, Hisert KB, Ivashkiv LB. Inhibition of interleukin 2 signaling and signal transducer and activator of transcription (STAT)5 activation during T cell receptor-mediated feedback inhibition of T cell expansion. J Exp Med. 1999;190:1263–1274. doi: 10.1084/jem.190.9.1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji JD, Tassiulas I, Park-Min KH, Aydin A, Mecklenbrauker I, Tarakhovsky A, Pricop L, Salmon JE, Ivashkiv LB. Inhibition of interleukin 10 signaling after Fc receptor ligation and during rheumatoid arthritis. J Exp Med. 2003;197:1573–1583. doi: 10.1084/jem.20021820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takaoka A, Mitani Y, Suemori H, Sato M, Yokochi T, Noguchi S, Tanaka N, Taniguchi T. Cross talk between interferon-γ and -α/β signaling components in caveolar membrane domains. Science. 2000;288:2357–2360. doi: 10.1126/science.288.5475.2357. [DOI] [PubMed] [Google Scholar]
- Hu X, Herrero C, Li WP, Antoniv TT, Falck-Pedersen E, Koch AE, Woods JM, Haines GK, Ivashkiv LB. Sensitization of IFN-γ Jak–STAT signaling during macrophage activation. Nat Immunol. 2002;3:859–866. doi: 10.1038/ni828. [DOI] [PubMed] [Google Scholar]
- Malakhova OA, Yan M, Malakhov MP, Yuan Y, Ritchie KJ, Kim KI, Peterson LF, Shuai K, Zhang DE. Protein ISGylation modulates the JAK–STAT signaling pathway. Genes Dev. 2003;17:455–460. doi: 10.1101/gad.1056303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharif MN, Tassiulas I, Hu Y, Mecklenbrauker I, Tarakhovsky A, Ivashkiv LB. IFN-alpha priming results in a gain of proinflammatory function by IL-10: implications for systemic lupus erythematosus pathogenesis. J Immunol. 2004;172:6476–6481. doi: 10.4049/jimmunol.172.10.6476. [DOI] [PubMed] [Google Scholar]
- Herrero C, Hu X, Li WP, Samuels S, Sharif MN, Kotenko S, Ivashkiv LB. Reprogramming of IL-10 activity and signaling by IFN-gamma. J Immunol. 2003;171:5034–5041. doi: 10.4049/jimmunol.171.10.5034. [DOI] [PubMed] [Google Scholar]
- Ivashkiv LB. Type I interferon modulation of cellular responses to cytokines and infectious pathogens: potential role in SLE pathogenesis. Autoimmunity. 2003;36:473–479. doi: 10.1080/08916930310001605882. [DOI] [PubMed] [Google Scholar]
- Ivashkiv LB, Hu X. The JAK/STAT pathway in rheumatoid arthritis: pathogenic or protective? Arthritis Rheum. 2003;48:2092–2096. doi: 10.1002/art.11095. [DOI] [PubMed] [Google Scholar]
- van der Pouw Kraan TC, van Gaalen FA, Kasperkovitz PV, Verbeet NL, Smeets TJ, Kraan MC, Fero M, Tak PP, Huizinga TW, Pieterman E, Breedveld FC, Alizadeh AA, Verweij CL. Rheumatoid arthritis is a heterogeneous disease: evidence for differences in the activation of the STAT-1 pathway between rheumatoid tissues. Arthritis Rheum. 2003;48:2132–2145. doi: 10.1002/art.11096. [DOI] [PubMed] [Google Scholar]