

Abstract
Candida lusitaniae is usually susceptible to echinocandins. Beta-1,3-glucan synthase encoded by FKS genes is the target of echinocandins. A few missense mutations in the C. lusitaniae FKS1 hot spot 1 (HS1) have been reported. We report here the rapid emergence of antifungal resistance in C. lusitaniae isolated during therapy with amphotericin B (AMB), caspofungin (CAS), and azoles for treatment of persistent candidemia in an immunocompromised child with severe enterocolitis and visceral adenoviral disease. As documented from restriction fragment length polymorphism (RFLP) and random amplified polymorphic DNA (RAPD) analysis, the five C. lusitaniae isolates examined were related to each other. From antifungal susceptibility and molecular analyses, 5 different profiles (P) were obtained. These profiles included the following: profile 1 (P1) (CAS MIC [μg/ml], 0.5; fluconazole [FLC] MIC, 0.25), determined while the patient was being treated with liposomal AMB for 3 months; P2 (FLC MIC [μg/ml], 0.25; CAS MIC, 4), while the patient was being treated with CAS for 2 weeks; P3 (CAS MIC [μg/ml], 0.5; FLC MIC, 32), while the patient was being treated with azoles and CAS initially followed by azoles alone for a week; P4 (CAS MIC [μg/ml], 8; FLC MIC, 8), while the patient was being treated with both drugs for 3 weeks; and P5 (AMB MIC [μg/ml], 0.125; CAS MIC, 8), while the patient was being treated with AMB and FLC for 2 weeks. CAS resistance was associated with resistance not only to micafungin and anidulafungin but also to AMB. Analysis of CAS resistance revealed 3 novel FKS1 mutations in CAS-resistant isolates (S638Y in P2; S631Y in P4; S638P in P5). While S638Y and -P are within HS1, S631Y is in close proximity to this domain but was confirmed to confer candin resistance using a site-directed mutagenesis approach. FLC resistance could be linked with overexpression of major facilitator gene 7 (MFS7) in C. lusitaniae P2 and P4 and was associated with resistance to 5-flurocytosine. This clinical report describes resistance of C. lusitaniae to all common antifungals. While candins or azole resistance followed monotherapy, multidrug antifungal resistance emerged during combined therapy.
INTRODUCTION
Candida lusitaniae, an opportunistic haploid yeast, remains a rare cause of candidemia. While C. lusitaniae can develop amphotericin B (AMB) resistance (1, 2), it is considered generally susceptible to all systemic antifungal agents (3). Echinocandins are used as first-line therapy for candidemia due to C. lusitaniae. The target of echinocandins is β-1,3-glucan synthase and is encoded by FKS genes (4). Three echinocandins, anidulafungin (ANI), caspofungin (CAS), and micafungin (MICA), have been available and widely used for about a decade. As a result, emerging resistance to echinocandins has been reported in several species, including C. albicans, C. dubliniensis, C. kefyr, C. glabrata, C. krusei, C. tropicalis, and C. lusitaniae (5–12). Missense mutations in FKS genes (FKS1 and FKS2) that are situated in different regions (host spot 1 [HS1] and HS2) are responsible for the increase of drug MICs compared to the MICs seen with wild-type isolates. These MIC increases were shown to cause treatment failures in animal experiments similarly to those seen in clinical cases, thus suggesting the emergence of clinical resistance (13). In C. lusitaniae, a single missense mutation in C. lusitaniae FKS1 HS1 at position 645 (S645F) was reported in clinical isolates and resulted in increased MICs of several echinocandins. While recent data documented cross-resistance between echinocandins and azoles in C. glabrata (14), no cross-resistance has yet been reported in C. lusitaniae. The present paper reports the unusual emergence of clinical isolates of C. lusitaniae with documented cross-resistance to candins and azoles following exposure to various antifungal regimens for persistent candidemia.
MATERIALS AND METHODS
Strains and media.
C. lusitaniae strains were grown in complete yeast extract-peptone-dextrose (YEPD) medium (1% Bacto peptone [Difco Laboratories, Basel, Switzerland], 0.5% yeast extract [Difco]) with 2% (wt/vol) glucose (Fluka, Buchs, Switzerland). Saccharomyces cerevisiae was grown on YEPD medium for isolate precultures and on yeast nitrogen base (YNB) agar (Difco) with 2% (wt/vol) glucose. Species identification was performed using matrix-assisted laser desorption ionization–time of flight (MALDI-TOF) mass spectrometry (MS) Microflex LT systems (Bruker Daltonics GmbH, Leipzig, Germany) and with analysis of data using FlexControl (version 3.0) software (Bruker Daltonics) as described in reference 15.
Susceptibility assays.
Determinations of drug MICs for C. lusitaniae clinical isolates according to EUCAST guidelines were performed in RPMI 1640 medium (Sigma-Aldrich, Switzerland) with 2% glucose and in flat-well microtiter plates. RPMI 1640 buffered at pH 7.0 with MOPS (morpholinepropanesulfonic acid) was used for MIC tests of azoles, 5-fluorocytosine (5-FC), candins, and AMB. Cells were diluted to a density of 0.5 2 × 105 to 2 × 105 cells/ml. All compounds were dissolved to obtain final concentrations ranging from 128 μg/ml to 0.0162 μg/ml. Plates were incubated at 35°C for 24 h, and readings were carried out in a microplate reader at 540 nm. The MIC was defined as the drug concentration at which the optical density was ≤50% of that of the drug-free culture. Quality controls included C. albicans strain ATCC 928. Antifungal agents used in this study were provided as pure substances by pharmaceutical companies (CAS, Merck; micafungin [MICA], Astellas; anidulafungin [ANI] and FLC, Pfizer). AMB deoxycholate (Fungizone) was obtained from Bristol-Myers Squibb (Cham, Switzerland).
RLFP and RAPD analysis.
The recovered C. lusitaniae isolates were subjected to restriction fragment length polymorphism (RLFP) and random amplified polymorphic DNA (RAPD) analysis as described elsewhere (16). Genomic DNA was isolated by glass bead extraction from each isolate as previously described (17) and was subjected to EcoRI and MspI digestion. RAPD analysis was performed with primer OPE-18 (GGACTGCAGA) as previously recommended (16). Gel electrophoresis was carried out with 0.8% agarose followed by ethidium bromide staining. Additional software (ImageJ) (18) was used to corroborate our findings from the RFLP analysis (see Fig. S1 in the supplemental material).
FKS1 sequencing.
Primers were used to amplify FKS1 alleles encoding β-glucan synthase from C. lusitaniae isolates (19). These primers were designed to amplify conserved HS1 (hot spot region 1) and HS2 regions (for HS1, MDO002 [GCCTTTGGGTGGTTTGTTTA] and MDO003 [TCGGAATCTCTTGGGAAGAA]; for HS2, MDO004 [TGCTGGTATGGGTGAACAGA] and MDO005 [CGAACACTTCGAAGAATGGAG]). Sequencing procedures were performed with the same primers and are described elsewhere (20). Sequence alignments were performed with Geneious software (Biomatters Ltd., New Zealand).
qRT-PCR.
Quantitative reverse transcription-PCR (qRT-PCR) was performed as described elsewhere (21). Total RNA was extracted from log-phase cultures with an RNeasy Protect minikit (Qiagen) by a process involving mechanical disruption of the cells with glass beads and an RNase-free DNase treatment step as previously described (22). Gene expression levels were determined by real-time qRT-PCR in a StepOne real-time PCR system (Applied Biosystems) using a Mesa Blue quantitative PCR (qPCR) Mastermix Plus for Sybr assay kit (Eurogentec). Each reaction was run in triplicate on three separate occasions. Expression levels were normalized to ACT1 expression. Primers for C. lusitaniae ATP-binding cassette (ABC) and major facilitator superfamily (MFS) transporter genes were designed with Primer3Plus. The primers were chosen on the basis of the available genome sequences (Broad Institute). Primers are listed in Table S1 in the supplemental material. Gene names were given according to the work of Reboutier et al. (23). Primers ABC15-F and ABC15-R as well as primers ABC9-F and ABC9-R were selected from the C. lusitaniae genomes of the closest homologs of the C. albicans CDR1 gene. MFS7-R and MFS7-F as well as ABC12-R and ABC12-F were selected since the corresponding genes were previously shown to be differentially expressed in several C. lusitaniae isolates (23).
Construction of FKS1 mutants.
In order to introduce specific mutations in FKS1 for testing their effect on echinocandin susceptibility, the model yeast S. cerevisiae was used in combination with the clustered regularly interspaced short palindromic repeat (CRISPR)-Cas9 genome editing system. Briefly, a 20-nucleotide (nt) guide sequence adjacent to a protospacer-adjacent motif (PAM) sequence was selected within the region of FKS1 HS1. This region was selected using the online CHOPCHOP selection tool (24) (https://chopchop.rc.fas.harvard.edu) and is situated between positions 1892 and 1914 with respect to first ATG codon. The guide sequence was flanked by pMEL10 sequences (25) to allow homologous recombination in S. cerevisiae. The guide-pMEL10 sequence was produced by complementary assembly of primers FKS_crisp_R and FKS_crisp_F (see Table S2 in the supplemental material). pMEL10 was prepared by inverse PCR with primers p426 CRISPR rv and p426 CRISPR fw (see Table S2 in the supplemental material) followed by DpnI digestion as described by Mans et al. (25). Three different repair fragments were produced, with each containing the desired FKS1 mutation with overlapping primers for the FKS1 mutations S636Y, S643P, and S643Y. Primer pairs using left and right primers are indicated in Table S2 in the supplemental material and were used for PCR amplification to produce 120-bp repair fragments. Genome editing was performed by combining pMEL10, the guide-pMEL10 fragment, and each of the repair fragments and by transformation into S. cerevisiae IMX581 (25). Transformation of S. cerevisiae was performed as described previously (25), and selection was carried out in YNB agar lacking uracil. Verifications of introduced mutations were performed by PCR amplification with primers FKS1verif left and FKS1verif right (see Table S2 in the supplemental material) of the HS1 region and by sequence analysis as described above. Derivatives from IMX581 are described in Table S3 in the supplemental material.
β-Glucan measurements.
Patient blood samples were drawn, and sera were stored at −80°C and subjected to batch analysis with duplicate testing by Fungitell on an ELx808IU microplate reader (Associates of Cape Cod, East Falmouth, MA) per the manufacturer's package insert. Samples with β-glucan (BG) values above the upper limit of quantification (500 pg/ml) were diluted (58 of 921; 6%). The mean BG values of duplicates were used for data analysis.
Galactomannan assay.
The Bio-Rad Platelia Aspergillus antigen (Ag) assay was used to measure galactomannan levels. This immunoenzymatic sandwich microplate assay enabled the detection of Aspergillus galactomannan antigen in serum and bronchoalveolar lavage fluid samples through the use of rat EBA-2 monoclonal antibodies. Results are reported in standard international units (provided as index values with limits of 0.25 to 0.5), which refer to the absorbance (optical density) of specimens determined with a spectrophotometer set at 450 nm.
TDM.
Therapeutic drug monitoring (TDM) was performed according to published procedures with multiplex ultraperformance liquid chromatography-tandem mass spectrometry methods that enable simultaneous quantification in plasma of azoles and candins (26).
Nucleotide sequence accession numbers.
C. lusitaniae sequences were deposited in GenBank under accession no. JF304613 and JF304615. Sequences from isolates P1 to P5 were deposited in GenBank under accession numbers KM383792 to KM383795 and KP100692.
RESULTS
Case report.
A 3-year-old female with hematologic and central nervous system (CNS) relapse of acute myeloid leukemia (AML) was started on high-dose cytarabine and clofarabine as second-line induction. She remained profoundly neutropenic over the following 4 months until her death. Three weeks after induction chemotherapy, she presented with fever and diarrhea. She had been on prolonged prophylactic treatment with intravenous (i.v.) liposomal AMB (3 mg/kg of body weight/day) and broad-spectrum antibiotics for over a month. Clinical and radiological examination showed severe enterocolitis. As such invasive C. lusitaniae candidiasis was suspected based on the documentation of C. lusitaniae in her stools and a positive mannan assay result (immunoenzyme assay, 500 pg/ml) (27, 28). Blood culture results were, however, negative. Given the lack of validated clinical breakpoint definitions for C. lusitaniae, we used those available for C. albicans (29, 30). Given the profile (P1; see Fig. 1 and Table 2 for susceptibility profiles) of the susceptibility of the recovered C. lusitaniae isolate to echinocandins (CAS, micafungin [MICA], and anidulafungin [ANI] MICs, 0.5 μg/ml, 0.03 μg/ml, and 0.06 μg/ml, respectively), to azoles (FLC MIC, 0.25 μg/ml) and to AMB (MIC, 0.06 μg/ml), she was started on CAS at 100 mg/m2/day, which resulted in clinical improvement. While she was on CAS for 2 weeks with measured plasma levels of 4.3 mg/liter, she presented with C. lusitaniae candidemia (isolate P2; see Fig. 1), which was resistant to all echinocandins (CAS, MICA, and ANI MICs, 4 μg/ml, 16 μg/ml, and 2 μg/ml, respectively) and AMB (MIC, 2 μg/ml) but not to azoles (FLC MIC, 0.25 μg/ml) or 5-fluorocytosine (5-FC) (MIC, 0.5 μg/ml). At the same time, she presented with visceral adenovirus disease (subtype 41 F) with high viral loads in her blood (2 × 105 copies [cp]/ml) and stool (4 × 109 cp/ml) (31). She was therefore started on intravenous cidofovir. No endovascular source was documented, and repeat computed tomography (CT) showed severe enterocolitis but no hepatosplenic nor pulmonary lesions. Intravenous FLC (12 mg/kg/day) was added to CAS, with documented plasma levels of 12.1 mg/liter and 4.4 mg/liter, respectively (26). While she was on combined therapy for a week, she presented with a new onset of fever and diarrhea with simultaneous positive blood cultures for C. lusitaniae (isolate P2). Combined therapy with CAS was maintained pending synergistic testing results as we suspected the presence of different strains, among which some could still have been CAS sensitive. In addition, we preferred maintaining a fungicidal drug in a profoundly neutropenic host. Synergistic testing showed no benefit of combined CAS/FLC therapy (data not shown). Therefore, CAS was stopped after an overall duration of 6 weeks. Her blood culture results remained persistently positive (P2) for a week despite clinical improvement, adequate drug levels, and no documented endovascular source. All intravenous lines were changed. While being on combined therapy (CAS/FLC) for 3 weeks, followed by FLC monotherapy for 1 week with adequate drug levels, she presented with a new onset of fever, a maculopapular rash, profuse diarrhea, and candidemia. Surprisingly, her new C. lusitaniae isolate (P3; see Fig. 1) was susceptible to echinocandins (CAS and MICA MICs, 0.5 μg/ml and 0.03 μg/ml, respectively) and AMB (MIC, 0.25 μg/ml) but resistant to azoles (FLC MIC, 32 μg/ml) and 5-FC (MIC, 64 μg/ml). CAS (100 mg/m2/day) was added again to FLC to avoid the emergence of C. lusitaniae strains exhibiting either echinocandin resistance or azole resistance. While being on combined therapy for 3 weeks (CAS plasma level, 2.5 mg/liter; FLC plasma level, 9.3 mg/liter), she presented with a new onset of candidemia, with a C. lusitaniae isolate (P4; see Fig. 1) resistant to echinocandins (CAS, MICA, and ANI MICs, 8 μg/ml, 16 μg/ml, and 4 μg/ml, respectively), FLC (MIC, 8 μg/ml), and 5-FC (MIC, 32 μg/ml) but susceptible to AMB (MIC, 0.06 μg/ml). CAS was therefore replaced by AMB (5 mg/kg/day). FLC was replaced by voriconazole (VORI) (9 mg/kg twice a day [b.i.d.]; plasma level, 1.5 mg/liter), as probable invasive pulmonary aspergillosis (32) was suspected based on new pulmonary infiltrates on a repeat CT and a positive blood galactomannan assay result (enzyme-linked immunoassay [EIA], 6.54 pg/ml) (32). Although AMB monotherapy would have covered both fungal infections, combined therapy was preferred because of the severity of both infections. Further disease evolution was marked by persistent fever and diarrhea associated with persistent adenovirus viremia (106 cp/ml). A week later, she underwent allogeneic hematopoietic stem cell transplantation (HSCT) after receiving conditioning chemotherapy with busulfan, anti-thymocyte globulins (ATG), and fludarabine. While she was on AMB and VORI at subtherapeutic (0.7 mg/liter) levels for almost 2 weeks, her blood cultures were again found to be positive for C. lusitaniae. At that point, she was continued on the same antifungal regimen. An isolate (P5) with a susceptibility profile similar to that of P2 (CAS, AMB, and FLC MICs, 8, 2.0, and 0.125 μg/ml, respectively) was recovered during this period. All her lines were changed, an endovascular source was ruled out, and a repeat CT scan still evidenced severe enterocolitis. The further evolution of her disease state was marked with progressive fulminant hepatitis, renal dysfunction, and death, mainly attributed to drug toxicities and disseminated adenoviral infection. The latter was corroborated by persistently high-level viremia (>5 × 108 cp/ml) despite her having received 11 doses of intravenous cidofovir (5 mg/kg) but no administration of adenovirus-specific cytotoxic T lymphocytes. Invasive candidiasis and pulmonary aspergillosis probably also contributed to her death. Autopsy was refused by the family.
FIG 1.

Summary of the susceptibility profiles of C. lusitaniae isolates. MIC values were obtained with the EUCAST method as described in Materials and Methods. Dates of isolate collection are given at the top of the figure as follows: 9.06, 9 June 2013; 1-8-15.07, 1, 8, and 15 July 2013; 1.08, 1 August 2013; 20.08, 20 August 2013; 20.09, 20 September 2013. Resistance and susceptibility are highlighted by red and yellow sectors, respectively. Details of the types of treatments and their durations and therapeutic drug monitoring (TDM) are given at the bottom of the figure. BC, blood culture; NA, not available.
TABLE 2.
Candin MICs of S. cerevisiae FKS1 mutants
| Isolate | MIC (μg/ml)a |
||
|---|---|---|---|
| CAS | MICA | ANI | |
| S. cerevisiae wild-type IMX581 | 0.03 | 0.015 | 0.03 |
| DSY4762 (FKS1S636Y) | 1 (32) | 0.5 (32) | 0.5 (16) |
| DSY4763 (FKS1S643Y) | 2 (64) | 4 (256) | 1 (32) |
| DSY4764 (FKS1S643P) | 8 (256) | 4 (256) | 1 (32) |
MIC assays were performed according to the EUCAST protocol but at 30°C and with YEPD medium. Numbers in parentheses represent relative fold increases in MICs compared to the MIC value of the wild type.
Molecular analysis of C. lusitaniae strains.
The RFLP and RAPD profiles of recovered isolates P1 to P5 were identical (Fig. 2; see also Fig. S1 in the supplemental material) and thus suggest that the strains originated from the same parent. Molecular analyses of candin resistance revealed two novel FKS1 mutations in resistant isolates (S638Y in P2 and S631Y in P4). These mutations correspond to positions S638 and S645 in FKS1 of C. albicans; the latter position is known to be involved in CAS resistance (S645F, -P, and -Y) (Fig. 3) (8, 33). Interestingly, the last recovered isolate, P5, with a drug susceptibility profile similar to that of P2, exhibited the FKS1 S638P substitution (corresponding to S645P in C. albicans), which was different from that exhibited by P2 (S638Y). Thus, P2 and P5 are of distinct genotypes. No mutations were observed in HS2 of FKS1 (data not shown). As summarized in Table 1, CAS resistance was associated with cross-resistance to other candins (MICA MICs, 8 to 16 μg/ml; ANI MICs, 2 to 4 μg/ml) and, surprisingly, with resistance to AMB (MIC, 2 μg/ml) in P2.
FIG 2.
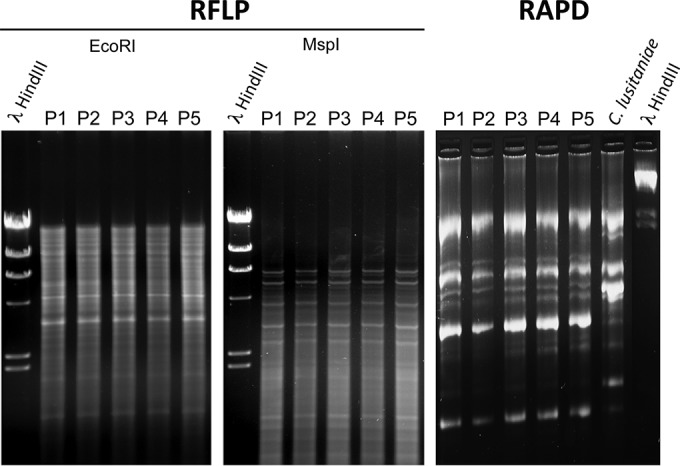
RFLP and RAPD analysis of C. lusitaniae isolates P1 to P5. RFLP analysis was carried out with EcoRI and MspI. RFLP profiles are shown in Fig. S1 in the supplemental material. RAPD analysis was performed as described in Materials and Methods. Identical patterns of ethidium bromide-stained profiles suggest a high-level relationship between the strains. Lambda phage DNAs digested by HindIII were loaded as standard sizes. A separate C. lusitaniae isolate (Sanglard laboratory collection) was used as a control for RAPD analysis.
FIG 3.

Alignments of FKS1 HS1 regions from Candida spp. The C. lusitaniae and C. albicans SC5314 data were aligned with chromatograms of FKS1 HS1 regions from C. lusitaniae isolates P1 to P5 as indicated. Sequences from isolates P1 to P5 were deposited in GenBank.
TABLE 1.
Antifungal susceptibility profiles of C. lusitaniae isolatesa
| Date of isolation | Sample origin | Profile | MIC (μg/ml)b |
FKS1 mutation | MFS7 expression | AF | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| FLC | CAS | MICA | ANI | AMB | 5-FC | ||||||
| 9 June 2013 | Stool | 1 | 0.25 | 0.5 | 0.03 | 0.06 | 0.06 | 0.5 | WT | No | AMB |
| 1 July 2013 | Blood | 2 | 0.25 | 4 (8) | 16 (512) | 2 (32) | 2 | 0.5 | S638Y | No | CAS |
| 1 August 2013 | Blood | 3 | 32 | 0.5 (1) | 0.03 (1) | 0.06 (1) | 0.25 | 64 | WT | Yes | FLC |
| 23 August 2013 | Blood | 4 | 8 | 8 (16) | 16 (512) | 4 (64) | 0.06 | 32 | S631Y | Yes | VORI/CAS |
| 2 September 2013 | Stool | 4 | 32 | 4 (8) | 8 (256) | 4 (64) | 0.125 | 32 | S631Y | Yes | VORI/AMB |
| 20 September 2013 | NA | 5 | 0.125 | 8 (16) | 16 (512) | 4 (64) | 2 | 2 | S638P | No | VORI/AMB |
AF, antifungal treatment; NA, not available; WT, wild-type HS1 FKS1 sequence.
Numbers in parentheses represent relative fold increases in MICs compared to the MIC value of the isolate with profile 1.
Since mutation S631Y in C. lusitaniae and the equivalent mutation, Ser638, in C. albicans have not yet been reported to be involved in candin resistance, we performed site-directed mutagenesis analysis in FKS1 from S. cerevisiae at the equivalent position (Ser636) to produce a S636Y variant. FKS1 variants at position Ser643 (S643Y and S643P), which is equivalent to position Ser638 in C. lusitaniae, were also produced as comparisons. A recent genome editing system (CRISPR-Cas) was used for this purpose (25) and thus introduced the desired mutations at the S. cerevisiae genomic FKS1 locus. The resulting isolates exhibited resistance to all three candins compared to the wild type (Table 2). The FKS1 S636Y mutation showed CAS, MICA, and ANI MICs that had increased by 32-, 32-, and 16-fold, respectively, compared to the wild type (Table 2). The mutations S643Y and S643P increased candin MICs from 32- to 256-fold compared to the wild type (Table 2); thus, the data suggest that they have a greater impact on candin resistance than S636Y. In any case, position Ser636 (Ser631 in C. lusitaniae) can be added as another novel site relevant for candin resistance.
Azole resistance in C. albicans is mediated by several mechanisms, among which transport-related mechanisms involving either major facilitator superfamily (MFS) genes or ATP-binding cassette (ABC) transporter genes are the most frequently reported (34). Few studies have explored azole resistance and the involvement of major drug transporters. Among these, a recent report by Reboutier et al. (23) suggested upregulation of MFS7 in documented FLC-resistant isolates. As illustrated in Fig. 4, we corroborated the overexpression of MFS7 in FLC-resistant C. lusitaniae isolates P3 and P4 (50- and 32-fold compared to P1, respectively). No expression variations in azole resistance genes belonging to the ABC transporter family or to ERG11 (target of azoles) were identified among our C. lusitaniae isolates (Fig. 4). Interestingly, FLC resistance in P3 and P4 correlated with an elevated VORI MIC (0.25 μg/ml, compared with 0.008 μg/ml for P1, P2, and P5) but also with resistance to 5-FC (MIC, 32 μg/ml).
FIG 4.
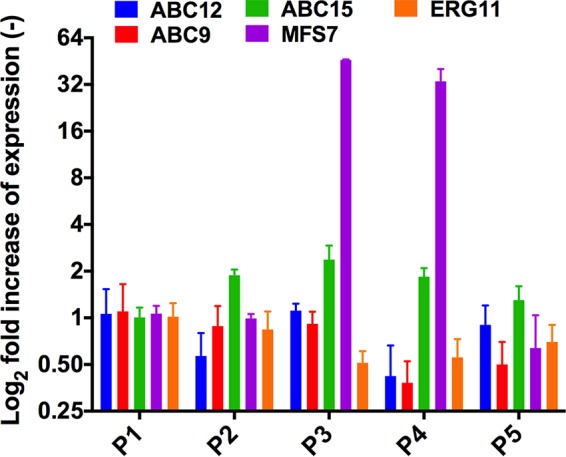
qRT-PCR of C. lusitaniae genes potentially involved in azole resistance. Results are expressed as means of the results from triplicate biological experiments relative to isolate P1 data.
In contrast to our findings, other studies reported 5-FC/azole cross-resistance correlating with mutations of FCY1 and FCY2 genes encoding cytosine deaminase and purine-cytosine permease involved in 5-FC transport and metabolism (35). These mutations were documented following simultaneous use of 5-FC and azoles in susceptibility assays, thus suggesting a different mechanism for 5-FC-azole cross-resistance. Indeed, no mutations in 5-FC resistance genes (FCY1 and FCY2) were detected in the P3 and P4 isolates (data not shown). Analysis of FUR1, encoding uracil phosphoribosyl transferase, was not conducted as isolates P1 to P5 were not resistant to 5-fluorouracil, which is commonly reported among FUR1-deficient isolates (data not shown) (36).
DISCUSSION
This clinical report describes acquired resistance of C. lusitaniae to all common antifungals in a profoundly neutropenic host with severe enterocolitis. When simultaneous combinations of resistance to 2 or more different drug classes occur, which was the case in the present study, the phenotype is referred to as multidrug resistance (MDR). While MDR is not a common phenotype among fungal pathogens, it was reported earlier in C. glabrata, with simultaneous acquisition of resistance to echinocandins and to azoles and separate acquisition of resistance to 5-FC (37). In the United States, a significant proportion (30% to 40%) of echinocandin-resistant isolates are also resistant to azoles (38, 39). While a recent study (40) suggested an association between MDR and the use of echinocandins and azoles, another investigation (41) described MDR to echinocandins, azoles, and amphotericin B in C. glabrata isolates recovered from a neutropenic patient with prolonged fever.
Antifungal resistance is a growing concern worldwide (42–44); however, less is known about the mechanism of resistance to echinocandins in C. lusitaniae. In the present paper, resistance to CAS was correlated to the identification of 3 novel FKS1 mutations (S638Y, S638P, and S631Y). Among these, FKS1 mutations S638Y and -P corresponded to C. albicans and S. cerevisiae positions Ser645 and Ser643, respectively, which are commonly attributed to echinocandin resistance (45), whereas the remaining FKS1 S631Y mutation corresponded to position Ser638 in C. albicans and position Ser636 in S. cerevisiae. Here we confirmed that a mutation at this position can alter candin susceptibility in S. cerevisiae. The FKS1 S636Y mutation yields lower candin MICs than S643Y and S643P; thus, one may conclude that Ser636 is less effective for candin resistance development. In evaluating the relative increases of candin MICs in C. lusitaniae and S. cerevisiae compared to their respective wild types (Table 1 and 2), the trends seen with the two yeast species are globally similar, with the exception of the relative MICA MIC increases (32-fold versus 256-fold and 256-fold for C. lusitaniae and S. cerevisiae, respectively). Such differences might be due to differences in the intrinsic FKS1 structures of the two species. Similarly to our case report, a recent study (19) documented isolates of C. lusitaniae exhibiting missense mutation S645F in FKS1, which resulted in increased MICs of several echinocandins (CAS, MICA, and ANI) following CAS exposure. In contrast to our report, those isolates were not cross-resistant to other classes of antifungal drugs (5-FC, FLC, and AMB).
Overexpression of a major facilitator gene (MFS7) was documented among our FLC-resistant isolates. MFS transporter upregulation in Candida spp. is associated with mutations in the MRR1 transcriptional activator in C. albicans (46). Similar mutations could be suspected in C. lusitaniae strains. In the present report, FLC resistance was coupled to 5-FC resistance, despite the lack of 5-FC exposure, thus suggesting that the 5-FC resistance resulted from FLC resistance. Given that MFS7 upregulation results in FLC efflux in FLC-resistant strains, a similar mechanism might be involved in 5-FC-resistant strains. While this hypothesis remains speculative, mutations responsible for FLC/5-FC cross-resistance differ from the usual nonsense and missense mutations in FCY2 and FCY1 reported in C. lusitaniae strains and should thus be further explored (35).
Cross-resistance to AMB resistance and candins occurred without ongoing exposure to AMB (see Fig. 1). The molecular basis of AMB resistance in C. lusitaniae has not yet been well documented. While some studies attributed AMB resistance to a rapidly switching phenotype occurring at a frequency of 10−2 to 10−4 (47), other studies attributed it to cell wall reorganization (48). FKS mutations documented in C. lusitaniae P2 could have induced cell wall stress, which then could result in AMB resistance. Even if CAS resistance was not associated with AMB resistance in P4 (FKS1 mutation S631Y), this hypothesis should be further investigated. Such issues could now be addressed using the genetic tools that have become accessible for use in C. lusitaniae studies.
The present report illustrates rapid selection of resistant mutants under conditions of drug pressure. The sequential administration of specific agents resulted in the emergence of isolates resistant to a specific molecule as illustrated in Fig. 1. Treatments using CAS and azole and their combination were followed within days by the selection of CAS- and/or azole-resistant isolates. This rapid emergence can be facilitated by the haploid nature of C. lusitaniae. It is also possible that several resistant populations with P1 to P5 profiles may have coexisted in the patient and that dominant resistance profiles were selected and emerged under conditions of exposure to a specific antifungal agent. As such, the colon may have been a colonizing reservoir which then seeded infection and different resistance phenotypes. Long-lasting neutropenia and enterocolitis certainly also contributed to this mechanism. Rapid emergence of multidrug-resistant mutants under conditions of combined therapy reinforces the idea of a need for limiting dual therapy to exceptional situations.
Supplementary Material
ACKNOWLEDGMENTS
We are thankful for technical assistance from F. Ischer and for isolate collection and maintenance by C. Durussel. We are indebted to R. Zbinden of the University of Zurich, Institut für Medizinische Mikrobiologie, for helping in isolate collection.
This study was partially financed by a Swiss Research National Foundation grant (31003A_146936/1) to D.S. We thank Astellas, Pfizer, and Merck for providing pure antifungal substances.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.02204-15.
REFERENCES
- 1.Favel A, Michel-Nguyen A, Peyron F, Martin C, Thomachot L, Datry A, Bouchara JP, Challier S, Noel T, Chastin C, Regli P. 2003. Colony morphology switching of Candida lusitaniae and acquisition of multidrug resistance during treatment of a renal infection in a newborn: case report and review of the literature. Diagn Microbiol Infect Dis 47:331–339. doi: 10.1016/S0732-8893(03)00094-4. [DOI] [PubMed] [Google Scholar]
- 2.Atkinson BJ, Lewis RE, Kontoyiannis DP. 2008. Candida lusitaniae fungemia in cancer patients: risk factors for amphotericin B failure and outcome. Med Mycol 46:541–546. doi: 10.1080/13693780801968571. [DOI] [PubMed] [Google Scholar]
- 3.Lockhart SR, Iqbal N, Cleveland AA, Farley MM, Harrison LH, Bolden CB, Baughman W, Stein B, Hollick R, Park BJ, Chiller T. 2012. Species identification and antifungal susceptibility testing of Candida bloodstream isolates from population-based surveillance studies in two U.S. cities from 2008 to 2011. J Clin Microbiol 50:3435–3442. doi: 10.1128/JCM.01283-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Abruzzo GK, Flattery AM, Gill CJ, Kong L, Smith JG, Pikounis VB, Balkovec JM, Bouffard AF, Dropinski JF, Rosen H, Kropp H, Bartizal K. 1997. Evaluation of the echinocandin antifungal MK-0991 (L-743,872): efficacies in mouse models of disseminated aspergillosis, candidiasis, and cryptococcosis. Antimicrob Agents Chemother 41:2333–2338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Perlin DS. 2011. Echinocandin-resistant Candida: molecular methods and phenotypes. Curr Fungal Infect Rep 5:113–119. doi: 10.1007/s12281-011-0054-x. [DOI] [Google Scholar]
- 6.Baixench MT, Aoun N, Desnos-Ollivier M, Garcia-Hermoso D, Bretagne S, Ramires S, Piketty C, Dannaoui E. 2007. Acquired resistance to echinocandins in Candida albicans: case report and review. J Antimicrob Chemother 59:1076–1083. doi: 10.1093/jac/dkm095. [DOI] [PubMed] [Google Scholar]
- 7.Balashov SV, Park S, Perlin DS. 2006. Assessing resistance to the echinocandin antifungal drug caspofungin in Candida albicans by profiling mutations in FKS1. Antimicrob Agents Chemother 50:2058–2063. doi: 10.1128/AAC.01653-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Desnos-Ollivier M, Bretagne S, Raoux D, Hoinard D, Dromer F, Dannaoui E; European Committee on Antibiotic Susceptibility Testing. 2008. Mutations in the fks1 gene in Candida albicans, C. tropicalis, and C. krusei correlate with elevated caspofungin MICs uncovered in AM3 medium using the method of the European Committee on Antibiotic Susceptibility Testing. Antimicrob Agents Chemother 52:3092–3098. doi: 10.1128/AAC.00088-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Garcia-Effron G, Chua DJ, Tomada JR, DiPersio J, Perlin DS, Ghannoum M, Bonilla H. 2010. Novel FKS mutations associated with echinocandin resistance in Candida species. Antimicrob Agents Chemother 54:2225–2227. doi: 10.1128/AAC.00998-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kahn JN, Garcia-Effron G, Hsu MJ, Park S, Marr KA, Perlin DS. 2007. Acquired echinocandin resistance in a Candida krusei isolate due to modification of glucan synthase. Antimicrob Agents Chemother 51:1876–1878. doi: 10.1128/AAC.00067-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Park S, Kelly R, Kahn JN, Robles J, Hsu MJ, Register E, Li W, Vyas V, Fan H, Abruzzo G, Flattery A, Gill C, Chrebet G, Parent SA, Kurtz M, Teppler H, Douglas CM, Perlin DS. 2005. Specific substitutions in the echinocandin target Fks1p account for reduced susceptibility of rare laboratory and clinical Candida sp. isolates. Antimicrob Agents Chemother 49:3264–3273. doi: 10.1128/AAC.49.8.3264-3273.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Arendrup MC, Garcia-Effron G, Lass-Florl C, Lopez AG, Rodriguez-Tudela JL, Cuenca-Estrella M, Perlin DS. 2010. Echinocandin susceptibility testing of Candida species: comparison of EUCAST EDef 7.1, CLSI M27-A3, Etest, disk diffusion, and agar dilution methods with RPMI and isosensitest media. Antimicrob Agents Chemother 54:426–439. doi: 10.1128/AAC.01256-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Arendrup MC, Perlin DS. 2014. Echinocandin resistance: an emerging clinical problem? Curr Opin Infect Dis 27:484–492. doi: 10.1097/QCO.0000000000000111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pfaller MA, Moet GJ, Messer SA, Jones RN, Castanheira M. 2011. Candida bloodstream infections: comparison of species distributions and antifungal resistance patterns in community-onset and nosocomial isolates in the SENTRY Antimicrobial Surveillance Program, 2008–2009. Antimicrob Agents Chemother 55:561–566. doi: 10.1128/AAC.01079-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Eddouzi J, Hofstetter V, Groenewald M, Manai M, Sanglard D. 2013. Characterization of a new clinical yeast species, Candida tunisiensis sp. nov., isolated from a strain collection from Tunisian hospitals. J Clin Microbiol 51:31–39. doi: 10.1128/JCM.01627-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sanchez V, Vazquez JA, Barth-Jones D, Dembry L, Sobel JD, Zervos MJ. 1992. Epidemiology of nosocomial acquisition of Candida lusitaniae. J Clin Microbiol 30:3005–3008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sanglard D, Ischer F, Monod M, Bille J. 1996. Susceptibilities of Candida albicans multidrug transporter mutants to various antifungal agents and other metabolic inhibitors. Antimicrob Agents Chemother 40:2300–2305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schneider CA, Rasband WS, Eliceiri KW. 2012. NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Desnos-Ollivier M, Moquet O, Chouaki T, Guerin AM, Dromer F. 2011. Development of echinocandin resistance in Clavispora lusitaniae during caspofungin treatment. J Clin Microbiol 49:2304–2306. doi: 10.1128/JCM.00325-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Eddouzi J, Parker JE, Vale-Silva LA, Coste A, Ischer F, Kelly S, Manai M, Sanglard D. 2013. Molecular mechanisms of drug resistance in clinical Candida species isolated from Tunisian hospitals. Antimicrob Agents Chemother 57:3182–3193. doi: 10.1128/AAC.00555-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ferrari S, Sanguinetti M, Torelli R, Posteraro B, Sanglard D. 2011. Contribution of CgPDR1-regulated genes in enhanced virulence of azole-resistant Candida glabrata. PLoS One 6:e17589. doi: 10.1371/journal.pone.0017589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sanguinetti M, Posteraro B, Fiori B, Ranno S, Torelli R, Fadda G. 2005. Mechanisms of azole resistance in clinical isolates of Candida glabrata collected during a hospital survey of antifungal resistance. Antimicrob Agents Chemother 49:668–679. doi: 10.1128/AAC.49.2.668-679.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Reboutier D, Piednoel M, Boisnard S, Conti A, Chevalier V, Florent M, Gibot-Leclerc S, Da Silva B, Chastin C, Fallague K, Favel A, Noel T, Ruprich-Robert G, Chapeland-Leclerc F, Papon N. 2009. Combination of different molecular mechanisms leading to fluconazole resistance in a Candida lusitaniae clinical isolate. Diagn Microbiol Infect Dis 63:188–193. doi: 10.1016/j.diagmicrobio.2008.10.019. [DOI] [PubMed] [Google Scholar]
- 24.Montague TG, Cruz JM, Gagnon JA, Church GM, Valen E. 2014. CHOPCHOP: a CRISPR/Cas9 and TALEN web tool for genome editing. Nucleic Acids Res 42:W401–W407. doi: 10.1093/nar/gku410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mans R, van Rossum HM, Wijsman M, Backx A, Kuijpers NG, van den Broek M, Daran-Lapujade P, Pronk JT, van Maris AJ, Daran JM. 2015. CRISPR/Cas9: a molecular Swiss army knife for simultaneous introduction of multiple genetic modifications in Saccharomyces cerevisiae. FEMS Yeast Res 15:fov004. doi: 10.1093/femsyr/fov004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Decosterd LA, Rochat B, Pesse B, Mercier T, Tissot F, Widmer N, Bille J, Calandra T, Zanolari B, Marchetti O. 2010. Multiplex ultra-performance liquid chromatography-tandem mass spectrometry method for simultaneous quantification in human plasma of fluconazole, itraconazole, hydroxyitraconazole, posaconazole, voriconazole, voriconazole-N-oxide, anidulafungin, and caspofungin. Antimicrob Agents Chemother 54:5303–5315. doi: 10.1128/AAC.00404-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Marchetti O, Lamoth F, Mikulska M, Viscoli C, Verweij P, Bretagne S; European Conference on Infections in Leukemia (ECIL) Laboratory Working Groups. 2012. ECIL recommendations for the use of biological markers for the diagnosis of invasive fungal diseases in leukemic patients and hematopoietic SCT recipients. Bone Marrow Transplant 47:846–854. doi: 10.1038/bmt.2011.178. [DOI] [PubMed] [Google Scholar]
- 28.Mikulska M, Calandra T, Sanguinetti M, Poulain D, Viscoli C; Third European Conference on Infections in Leukemia Group. 2010. The use of mannan antigen and anti-mannan antibodies in the diagnosis of invasive candidiasis: recommendations from the Third European Conference on Infections in Leukemia. Crit Care 14:R222. doi: 10.1186/cc9365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pfaller MA, Andes D, Diekema DJ, Espinel-Ingroff A, Sheehan D; CLSI Subcommittee for Antifungal Susceptibility Testing. 2010. Wild-type MIC distributions, epidemiological cutoff values and species-specific clinical breakpoints for fluconazole and Candida: time for harmonization of CLSI and EUCAST broth microdilution methods. Drug Resist Updat 13:180–195. doi: 10.1016/j.drup.2010.09.002. [DOI] [PubMed] [Google Scholar]
- 30.Fothergill AW, Sutton DA, McCarthy DI, Wiederhold NP. 2014. Impact of new antifungal breakpoints on antifungal resistance in Candida species. J Clin Microbiol 52:994–997. doi: 10.1128/JCM.03044-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lion T. 2014. Adenovirus infections in immunocompetent and immunocompromised patients. Clin Microbiol Rev 27:441–462. doi: 10.1128/CMR.00116-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.De Pauw B, Walsh TJ, Donnelly JP, Stevens DA, Edwards JE, Calandra T, Pappas PG, Maertens J, Lortholary O, Kauffman CA, Denning DW, Patterson TF, Maschmeyer G, Bille J, Dismukes WE, Herbrecht R, Hope WW, Kibbler CC, Kullberg BJ, Marr KA, Muñoz P, Odds FC, Perfect JR, Restrepo A, Ruhnke M, Segal BH, Sobel JD, Sorrell TC, Viscoli C, Wingard JR, Zaoutis T, Bennett JE; European Organization for Research and Treatment of Cancer/Invasive Fungal Infections Cooperative Group; National Institute of Allergy and Infectious Diseases Mycoses Study Group (EORTC/MSG) Consensus Group. 2008. Revised definitions of invasive fungal disease from the European Organization for Research and Treatment of Cancer/Invasive Fungal Infections Cooperative Group and the National Institute of Allergy and Infectious Diseases Mycoses Study Group (EORTC/MSG) Consensus Group. Clin Infect Dis 46:1813–1821. doi: 10.1086/588660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Desnos-Ollivier M, Dromer F, Dannaoui E. 2008. Detection of caspofungin resistance in Candida spp. by Etest. J Clin Microbiol 46:2389–2392. doi: 10.1128/JCM.00053-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sanglard D, Odds FC. 2002. Resistance of Candida species to antifungal agents: molecular mechanisms and clinical consequences. Lancet Infect Dis 2:73–85. doi: 10.1016/S1473-3099(02)00181-0. [DOI] [PubMed] [Google Scholar]
- 35.Florent M, Noel T, Ruprich-Robert G, Da Silva B, Fitton-Ouhabi V, Chastin C, Papon N, Chapeland-Leclerc F. 2009. Nonsense and missense mutations in FCY2 and FCY1 genes are responsible for flucytosine resistance and flucytosine-fluconazole cross-resistance in clinical isolates of Candida lusitaniae. Antimicrob Agents Chemother 53:2982–2990. doi: 10.1128/AAC.00880-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Papon N, Noel T, Florent M, Gibot-Leclerc S, Jean D, Chastin C, Villard J, Chapeland-Leclerc F. 2007. Molecular mechanism of flucytosine resistance in Candida lusitaniae: contribution of the FCY2, FCY1, and FUR1 genes to 5-fluorouracil and fluconazole cross-resistance. Antimicrob Agents Chemother 51:369–371. doi: 10.1128/AAC.00824-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chapeland-Leclerc F, Hennequin C, Papon N, Noel T, Girard A, Socie G, Ribaud P, Lacroix C. 2010. Acquisition of flucytosine, azole, and caspofungin resistance in Candida glabrata bloodstream isolates serially obtained from a hematopoietic stem cell transplant recipient. Antimicrob Agents Chemother 54:1360–1362. doi: 10.1128/AAC.01138-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Grossman NT, Chiller TM, Lockhart SR. 2014. Epidemiology of echinocandin resistance in Candida. Curr Fungal Infect Rep 8:243–248. doi: 10.1007/s12281-014-0209-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pham CD, Iqbal N, Bolden CB, Kuykendall RJ, Harrison LH, Farley MM, Schaffner W, Beldavs ZG, Chiller TM, Park BJ, Cleveland AA, Lockhart SR. 2014. Role of FKS mutations in Candida glabrata: MIC values, echinocandin resistance, and multidrug resistance. Antimicrob Agents Chemother 58:4690–4696. doi: 10.1128/AAC.03255-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Farmakiotis D, Tarrand JJ, Kontoyiannis DP. 2014. Drug-resistant Candida glabrata infection in cancer patients. Emerg Infect Dis 20:1833–1840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cho EJ, Shin JH, Kim SH, Kim HK, Park JS, Sung H, Kim MN, Im HJ. 2015. Emergence of multiple resistance profiles involving azoles, echinocandins and amphotericin B in Candida glabrata isolates from a neutropenia patient with prolonged fungaemia. J Antimicrob Chemother 70:1268–1270. doi: 10.1093/jac/dku518. [DOI] [PubMed] [Google Scholar]
- 42.Laverdière M, Lalonde RG, Baril JG, Sheppard DC, Park S, Perlin DS. 2006. Progressive loss of echinocandin activity following prolonged use for treatment of Candida albicans oesophagitis. J Antimicrob Chemother 57:705–708. doi: 10.1093/jac/dkl022. [DOI] [PubMed] [Google Scholar]
- 43.Perlin DS. 2007. Resistance to echinocandin-class antifungal drugs. Drug Resist Updat 10:121–130. doi: 10.1016/j.drup.2007.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Snelders E, van der Lee HA, Kuijpers J, Rijs AJ, Varga J, Samson RA, Mellado E, Donders AR, Melchers WJ, Verweij PE. 2008. Emergence of azole resistance in Aspergillus fumigatus and spread of a single resistance mechanism. PLoS Med 5:e219. doi: 10.1371/journal.pmed.0050219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pfaller MA, Diekema DJ, Andes D, Arendrup MC, Brown SD, Lockhart SR, Motyl M, Perlin DS, CLSI Subcommittee for Antifungal Testing. 2011. Clinical breakpoints for the echinocandins and Candida revisited: integration of molecular, clinical, and microbiological data to arrive at species-specific interpretive criteria. Drug Resist Updat 14:164–176. doi: 10.1016/j.drup.2011.01.004. [DOI] [PubMed] [Google Scholar]
- 46.Dunkel N, Blass J, Rogers PD, Morschhauser J. 2008. Mutations in the multi-drug resistance regulator MRR1, followed by loss of heterozygosity, are the main cause of MDR1 overexpression in fluconazole-resistant Candida albicans strains. Mol Microbiol 69:827–840. doi: 10.1111/j.1365-2958.2008.06309.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Miller NS, Dick JD, Merz WG. 2006. Phenotypic switching in Candida lusitaniae on copper sulfate indicator agar: association with amphotericin B resistance and filamentation. J Clin Microbiol 44:1536–1539. doi: 10.1128/JCM.44.4.1536-1539.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ghannoum MA, Rice LB. 1999. Antifungal agents: mode of action, mechanisms of resistance, and correlation of these mechanisms with bacterial resistance. Clin Microbiol Rev 12:501–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.