

Summary
We present self-cloning CRISPR/Cas9 (scCRISPR), a technology that allows for CRISPR/Cas9-mediated genomic mutation and site-specific knockin transgene creation within several hours by circumventing the need to clone a site-specific single-guide RNA (sgRNA) or knockin homology construct for each target locus. We introduce a self-cleaving palindromic sgRNA plasmid and a short double-stranded DNA sequence encoding the desired locus-specific sgRNA into target cells, allowing them to produce a locus-specific sgRNA plasmid through homologous recombination. scCRISPR enables efficient generation of gene knockouts (∼88% mutation rate) at approximately one-sixth the cost of plasmid-based sgRNA construction with only 2 hr of preparation for each targeted site. Additionally, we demonstrate efficient site-specific knockin of GFP transgenes without any plasmid cloning or genome-integrated selection cassette in mouse and human embryonic stem cells (2%–4% knockin rate) through PCR-based addition of short homology arms. scCRISPR substantially lowers the bar on mouse and human transgenesis.
Graphical Abstract
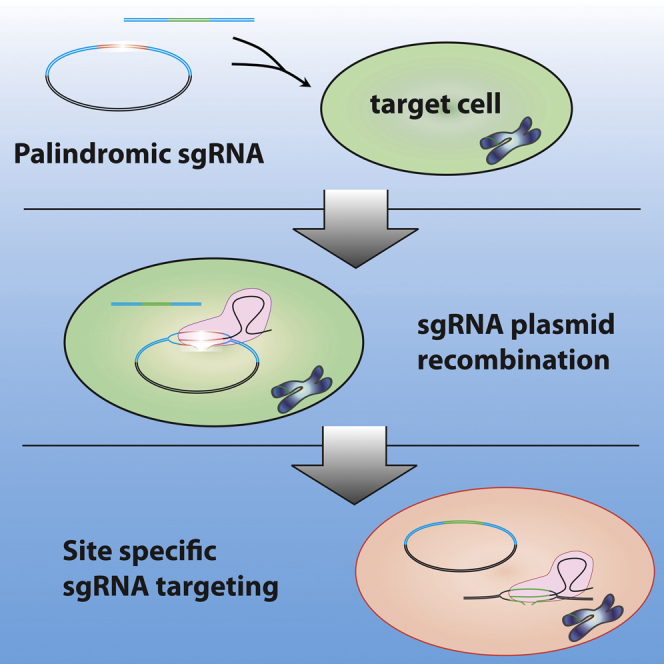
Highlights
-
•
scCRISPR circumvents the need to clone sgRNAs or knockin homology constructs
-
•
A self-cleaving palindromic plasmid recombines to produce a locus-specific sgRNA
-
•
scCRISPR targets via state-of-the-art efficiency for a fraction of the time and cost
-
•
scCRISPR benefits throughput of functional genomic screens and generation of reporter lines
In this article, Sherwood and colleagues present a novel method of CRISPR/Cas9 cell targeting that circumvents the need to generate site-specific sgRNA plasmids. Using a self-recognizing palindromic sgRNA plasmid, genome-targeting sgRNA sequences are recombined into the sgRNA hairpin within target cells, resulting in high-efficiency genome editing with significantly less preparation. This work provides a transformative tool for high-throughput functional genomic screening.
Introduction
The clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated protein 9 (Cas9) system has emerged as an efficient tool to mutate, delete, and insert genomic DNA sequences in a site-specific manner (Cong et al., 2013, Jinek et al., 2013, Mali et al., 2013). In CRISPR-mediated genome editing, Cas9 protein is directed to cleave DNA by an associated single-guide RNA (sgRNA) hairpin structure that can be designed to target almost any genomic site of interest (Jinek et al., 2012). Site-specific mutagenesis and targeted transgenesis are key applications for studying development and disease, and the ability to easily edit any genomic locus is revolutionizing genetics and stem cell research.
Currently, CRISPR/Cas9 targeting requires molecular cloning of a site-specific sgRNA plasmid for every new locus, which involves the time-consuming and costly steps of plasmid ligation, transformation, purification, and sequence verification over the course of about 1 week. This investment hinders large-scale sgRNA screening necessary for multiplexed and high-throughput genome editing applications. Additionally, knockin transgenesis of genes such as GFP using CRISPR/Cas9 still requires the time-consuming construction of homology constructs typically with 600 to 6,000 bp homology arms, a laborious process that impedes routine knockin line generation. These barriers are holding back the revolutionary potential of large-scale targeted genome manipulation. In this work, we provide alternative methods of sgRNA and homology construct generation that eliminate the need for plasmid cloning and, thus, substantially reduce the time, workload, and cost of CRISPR/Cas9-mediated genome editing, while maintaining high efficiency of site-specific mutation and transgene insertion (Table S1).
In the standard CRISPR/Cas9 method, once a site-specific sgRNA sequence is found, it is cloned into a plasmid containing a hairpin structure enabling Cas9 binding and a U6 promoter capable of transcribing the sgRNA hairpin in target cells (Ran et al., 2013, Yang et al., 2014). As each locus to be targeted requires a unique sgRNA sequence, this plasmid-cloning step must be performed for every new sgRNA to be used, providing a bottleneck to the throughput of CRISPR/Cas9-mediated genome editing and, thus, limiting mutation-based functional genomic screening applications. We have designed methods that circumvent any cloning steps in the gene editing process and demonstrate their efficacy at genome editing in both mouse and human embryonic stem cells (ESCs) as well as HEK293T cells. This method vastly simplifies the generation of targeted transgenic or knockout cell lines without compromising efficiency, creating an ideal platform for large-scale genome editing and screening applications.
Results
Self-cloning CRISPR/Cas9 (scCRISPR) relies on the target cells to clone the desired sgRNA sequence. Mammalian cells are known to repair introduced plasmid DNA through homologous recombination (HR) (Folger et al., 1982, Small and Scangos, 1983, Wake and Wilson, 1979). We asked whether we could take advantage of plasmid HR by introducing a template sgRNA plasmid into cells that could be recombined with a small DNA fragment containing the desired site-specific sgRNA sequence to form a functional site-specific sgRNA plasmid. The HR pathway is stimulated by double-stranded DNA breaks (Rouet et al., 1994), so we designed a self-cleaving palindromic template sgRNA plasmid that, upon transcription in cells, should induce a DNA break in its own sequence, which subsequently could be repaired into a functional site-specific sgRNA (Figure 1A).
Figure 1.

Simplified, Efficient Genome Editing Using scCRISPR
(A) Schematic shows the scCRISPR/Cas9 process that occurs inside target cells.
(B) Histograms show flow cytometric GFP fluorescence (x axis) in Hist1h3a mouse ESCs (left) after electroporation with Cas9 and plasmid sgRNA (second from left), sgPal1 plasmid alone (third from left), and sgPal1 plasmid and sgGFP homology fragment with standard-length arms (fourth from left).
(C) Fluorescence microscopy shows GFP fluorescence in Hist1h3a-GFP mouse ESCs (left) after targeting with Cas9 and plasmid sgRNA (second from left) and sgPal1 plasmid and sgGFP homology fragment (third from left).
(D) Histograms show flow cytometric GFP fluorescence (x axis) in Hist1h3a-GFP knockin mouse ESCs after electroporation with Cas9 and (from left to right) sgPal1, sgPal7, and sgPal8 plasmids together with a long sgGFP homology fragment.
(E) MiSeq plasmid copy numbers per cell of sgPal1, sgGFP, and the three most frequently mismatched sgGFP species 96 hr after co-electroporation of mouse ESCs are shown.
(F) Multiplexed mutation of GFP (x axis) and dsRed (y axis) in Hist1h3a-GFP Rosa26-dsRed mouse ESCs (left) after co-introduction of Cas9, sgPal1 plasmid, and sgGFP and sgDsRed long-armed homology fragments (right) is shown.
See also Figure S1.
To implement scCRISPR, we designed self-complementary palindromic sgRNA plasmids (sgPals) that should induce their own cleavage after complexing with Cas9 in cells. We used the improved “FE” sgRNA design that has been shown to increase Cas9 cleavage efficiency (Chen et al., 2013). To minimize off-target genomic DNA cleavage by sgPal, we designed an sgPal sequence with minimal predicted off-target cleavage potential (see the Supplemental Experimental Procedures). We also designed an oligonucleotide that, upon PCR amplification, contains an sgRNA sequence-targeting GFP flanked by arms of homology to the sgPal plasmid on either side (Figure 1A). We co-electroporated a Cas9 expression plasmid, our sgPal1 plasmid, and the GFP-targeting sgRNA homology fragment into Histone H3.1 (Hist1h3a)-GFP knockin mouse ESCs. The Cas9 plasmid encodes Blasticidin resistance and the sgPal1 plasmid encodes Hygromycin resistance, allowing transient antibiotic selection to enrich for cells that received both plasmids. All introduced components are transient and should not integrate into target cells such that the introduced mutation is the only lasting consequence of scCRISPR.
Electroporation of sgPal1, Cas9, and a GFP-targeting sgRNA homology fragment induced loss of GFP in 73% of cells 1 week after electroporation, while Cas9 and sgPal1 alone with no GFP-targeting sgRNA homology fragment produced minimal (0.3%) detectable GFP loss (Figures 1B and 1C). Comparatively, conventional CRISPR/Cas9 targeting with an sgRNA plasmid induced 99.9% loss of GFP. Sequence analysis confirmed loss of GFP was a result of genomic mutations at and around the target site of the sgGFP fragment (Figure S1A). scCRISPR-based treatment of Histone H2BJ (HIST1H2BJ)-GFP HEK293T cells also induced efficient (66%) GFP loss (Figure S1B). Thus, scCRISPR is an efficient method of inducing site-specific genomic mutation in mouse and human cell types, producing GFP loss in a majority of cells within the targeted population.
To determine whether scCRISPR indeed functions through plasmid HR, we varied the sgRNA plasmid and HR donor fragments. We found that substituting the sgPal plasmid with a non-self-cleaving sgRNA plasmid produced 9% GFP loss (Figure S1C), likely due to plasmid HR occurring in the absence of a double-strand break. When we varied the length of homology in the sgRNA homology fragment, we found that decreasing our standard homology arm length to short 60 bp arms of homology decreased the GFP loss after recombination with sgPal1 to 27% (Figure S1C), providing evidence that plasmid HR is required for scCRISPR. We designed nine additional sgPal plasmids and evaluated their efficiency in scCRISPR. All ten sgPals induced substantial GFP loss, although efficiencies ranged from 22% to 84% with sgPal7 yielding significantly more efficient GFP loss than sgPal1 (Figure S1D). The differences in efficiency between the distinct sgPals may be due to sequence characteristics affecting Cas9 cleavage, which are not yet well understood (Ren et al., 2014). Additional amplification of sgRNA fragments creating long homology arms further enhanced sgRNA targeting efficiency to 83% for sgPal1 and 88% for sgPal7 (Figure 1D). Thus, scCRISPR can achieve up to 88% mutation frequency with a self-cleaving sgRNA donor and an sgRNA acceptor amplified as a short double-stranded DNA homology fragment. Subsequent experiments reported here were carried out with sgPal1, using standard-length sgRNA homology fragments unless stated otherwise.
To assess sgPal plasmid recombination efficiency and accuracy inside target cells, we performed deep sequencing of sgRNA plasmid protospacer region in Hist1h3a-GFP mouse ESCs 4 days after electroporation of Cas9, sgPal1, and sgGFP homology fragment. The cells used for this experiment also contained a single-copy genomically integrated sgRNA cassette, which allowed us to calculate the average numbers of copies of each sgRNA species per cell. We found that by far the most abundant sgRNA plasmids inside cells were sgPal1 and sgGFP, with 15% of sgRNA plasmids having recombined from sgPal into sgGFP (Figure 1E). Copy-number analysis indicated that ∼100 copies of sgGFP plasmid are present at this time point as compared to 600 copies of sgPal. We estimated that plasmid copy number per cell was 5- to 10-fold higher than this during the peak CRISPR targeting period between 24 and 72 hr after electroporation and was diluted upon cell division. The dataset shows a low frequency of sequences similar to sgPal or sgGFP but with a single nucleotide mismatch (Figure 1E). These erroneous sequences occur at less than 0.5% of the frequency of the correct sequences, a rate that is indistinguishable from technical MiSeq sequencing error (Quail et al., 2012). While we cannot determine conclusively whether these mutant sgRNA reads are present inside cells or are artifacts of sequencing, this error rate represents at most fewer than 0.5% of correctly recombined sgRNA plasmids. Hence, scCRISPR induces efficient and faithful sgRNA recombination within target cells.
We next asked whether the HR of sgPal plasmids in cells occurs at a high enough frequency to target multiple sites in a single experiment. We designed sgRNA homology fragments targeting two additional locations within GFP and two within dsRed. All four additional sgRNAs produced >50% loss of GFP or dsRed (Figure S1E) in Hist1h3a-GFP or Rosa26-CAGGS-dsRed cells, respectively, suggesting that scCRISPR works with a variety of sgRNAs. We then introduced two sgRNAs simultaneously into mouse ESCs, finding high rates of GFP loss with two GFP-targeting sgRNAs in Hist1h3a-GFP cells (70%; Figure S1E), two dsRed-targeting oligos in Rosa26-CAGGS-dsRed cells (75%), or one GFP-targeting and one off-target dsRed-targeting sgRNA in single-positive Hist1h3a-GFP cells (51% loss of GFP; Figure S1E). Dual targeting with GFP-targeting sgRNAs led to deletion mutations as opposed to indels induced by single-targeted scCRISPR (Figure S1F), indicating that scCRISPR allows efficient site-specific deletion.
To further assess the capability to multiplex sgRNAs in scCRISPR, we targeted both GFP and dsRed simultaneously in Hist1h3a-GFP Rosa26-dsRed double-positive mouse ESCs by co-electroporation of sgPal1, Cas9, and two separate sgRNA homology fragments targeting GFP and dsRed. Simultaneous dual-site targeting with long-armed homology fragments induced 84% and 78% loss of GFP and dsRed, respectively, in the double-positive mouse ESCs with 75% double knockout (Figure 1F). Dual targeting with standard homology arms induced 69% and 64% loss of GFP and DsRed fluorescence, respectively, with both genes knocked out in 59% of cells (Figure S1G). These rates of mutation are similar to single-targeting rates, indicating that scCRISPR maintains equivalent efficiency in a multiplexed setting. Thus, scCRISPR is well suited to study the effects of compound mutations by simultaneous genome editing at multiple genomic loci in parallel.
One of the most transformative applications of CRISPR/Cas9 is the generation of gene knockins through site-specific HR to create fluorescent reporters of gene expression. Traditional knockin creation utilizing CRISPR/Cas9 requires the construction of a plasmid homology template with 600 to 6,000 bp homology arms flanking the insert sequence, a laborious undertaking requiring 1–2 weeks of molecular cloning for each targeted site and, thus, severely limiting the throughput of knockin generation. In the traditional approach, a gene-specific sgRNA plasmid (which also must be constructed), Cas9, and the plasmid homology template are co-electroporated into target cells, and screening is performed to purify the small percentage of clones that have undergone successful knockin. Having enabled cloning-free gene mutation, we asked whether we could perform plasmid-free GFP knockin.
To conduct plasmid-free GFP knockin, we designed an sgRNA targeting the C terminus of the Hist1h3a gene in wild-type mouse ESCs, and we performed PCR to generate a GFP homology template with a short Hist1h3a homology sequence on either side of GFP that should produce an in-frame C-terminal GFP fusion protein when recombined into the genome. We found that adding 80 bp of Hist1h3a homology sequence on either side of GFP allowed for quick and robust homology template generation in two PCR steps and under 2 hr total. To test PCR-based GFP knockin, we co-electroporated Cas9, Hist1h3a-targeting sgRNA plasmid, and Hist1h3a-GFP homology template fragment into mouse ESCs. One week after electroporation, 1.5% of cells expressed strong nuclear GFP and showed site-specific GFP integration by genomic DNA PCR (Figure 2A; Figure S2A). We achieved similar results constructing a Nanog-GFP knockin mouse ESC line (1.1%; Figure S2B). To demonstrate the reproducibility of mouse ESC knockin generation with PCR-based homology arms, we constructed nine additional site-specific GFP knockin lines, including C-terminal GFP fusion lines in the Esrrb, Fam25c, Gata6, Klf4, Nfya, Rpp25, and Sox2 loci and GFP replacements in the Tdgf1 and Zfp42 loci (Figure S2C). We successfully derived clonal GFP knockin lines in these 11 loci, demonstrating the dramatically increased throughput in mouse ESC knockin made possible by eliminating plasmid cloning from homology arm generation.
Figure 2.

Efficient, Cloning-Free Knockin Transgenesis Using PCR-Amplified Homology Arms
(A) Flow cytometric analysis shows efficient generation of Hist1h3a-GFP knockin mouse ESCs (y axis) using a PCR-amplified GFP fragment with 80 bp Hist1h3a homology arms and plasmid-based sgRNA (left) or scCRISPR sgRNA (right).
(B) Fluorescence microscopy shows Hist1h3a-GFP mouse ESCs generated through scCRISPR-based knockin.
(C) Flow cytometric analysis shows efficient generation of HIST1H2BJ-GFP knockin HUES2 human ESCs (y axis) with PCR-amplified homology arms and plasmid-based sgRNA (second from left) or scCRISPR-based sgRNA (right). Untargeted human ESC fluorescence is shown for comparison (left).
(D) Fluorescence microscopy shows HIST1H2BJ-GFP human ESCs generated through plasmid-based (left) or scCRISPR-based (right) knockin.
(E and F) Flow cytometric analysis shows that a cloning-free approach introducing a gBlock sgRNA and a PCR-amplified homology fragment leads to even more efficient generation of Hist1h3a-GFP knockin mouse ESCs (E) and HIST1H2BJ-GFP human ESCs (F).
See also Figure S2.
We carried out scCRISPR plasmid-free GFP knockin by co-electroporating Cas9, sgPal1, Hist1h3a-targeting sgRNA homology fragment, and Hist1h3a-GFP homology template fragment into mouse ESCs. One week after electroporation, 0.6% of cells expressed strong nuclear GFP and showed site-specific GFP integration by genomic DNA PCR (Figures 2A and 2B; Figure S2A). We achieved similar results constructing a Nanog-GFP knockin mouse ESC line (0.6%; Figure S2B). To ensure that the linear GFP homology fragment did not integrate promiscuously in the genome, we ascertained the number of GFP integrations in the genome in five scCRISPR-generated GFP knockin lines by Taqman qPCR copy-number assessment. We found that all five lines have one integration of GFP per cell (heterozygous) (Figure S2D), which is supported by PCR spanning the GFP integration site (Figure S2E). Thus, our plasmid-free GFP knockin method facilitates site-specific genomic integration of the transgene. Finally, we verified whether GFP expression faithfully reports on gene function in scCRISPR-generated Sox2-GFP mouse ESCs. After 96 hr in serum-deprived differentiation media, we saw a significant loss of Sox2-GFP fluorescence (Figure S2F). Additionally, scCRISPR-based mutation of an endogenous Sox2 promoter region resulted in loss of GFP expression (Figure S2G). Together, these results indicate that GFP expression accurately reflects endogenous gene expression.
We explored whether our approaches for fluorescent reporter generation perform just as efficiently in human ESCs, for which knockin line generation traditionally has been prohibitively difficult. We co-electroporated HUES2 human ESCs with Cas9 and sgPal1, this time in conjunction with an HIST1H2BJ-targeting homology fragment and HIST1H2BJ-GFP homology template fragment to target the C terminus of the human HIST1H2BJ locus in human ESCs. Fourteen days after electroporation, 1.1% of cells expressed GFP fluorescence, equivalent to targeting with conventional plasmid CRISPR/Cas9 (1.4%; Figures 2C and 2D). Thus, we present an approach that allows efficient construction of human ESC knockin lines with a total of 2 hr preparation time, a finding that will allow for a substantial increase in the throughput of human ESC knockin line generation.
It remains, however, that plasmid sgRNAs enable slightly more efficient GFP knockin than scCRISPR. Therefore, we devised a strategy to achieve high-efficiency gene insertion with a wholly plasmid-free technique. We reasoned that introducing only the sgRNA expression cassette without the entirety of the sgRNA plasmid should allow efficient sgRNA production from a minimally sized DNA sequence. We thus PCR amplified 500 bp gBlock fragments composed of a U6 promoter, GFP-targeting sgRNA sequence, and sgRNA hairpin sequence, which can be commercially synthesized cost-effectively (Table S1). We co-electroporated this GFP-targeting gBlock sgRNA into Hist1h3a-GFP knockin mouse ESCs along with a Cas9 expression plasmid. The GFP-targeting gBlock sgRNA knocked out GFP fluorescence in 93.5% of targeted cells (Figures S2H and S2I), equivalent to the standard plasmid sgRNA CRISPR/Cas9 method.
We then performed a wholly plasmid-free GFP knockin using a gBlock sgRNA and a PCR-based GFP homology fragment. We achieved 3.6% GFP knockin at the Hist1h3a locus and 2.5% Nanog-GFP knockin (Figure 2E; Figures S2A, S2B, S2I, and S2J) in mouse ESCs. gBlock sgRNA also yielded efficient (2.9%) HIST1H2BJ-GFP fusion in human ESCs (Figure 2F; Figure S2J), over double the efficiency of conventional plasmid-targeted cells. GFP insertion by gBlock sgRNA also yielded highly efficient (12%) HIST1H2BJ-GFP gene insertion in HEK293T cells (Figure S2K). Thus, we show that genomic knockin can be performed without any molecular cloning at enhanced efficiency to the traditional plasmid-based method. Our approaches of sgRNA generation and construction of short homology sequences for gene integration dramatically decrease the time, cost, and labor involved in transgenesis.
As a proof of principle of the transformative capacity of scCRISPR in enabling functional genomic screens that are otherwise costly and time consuming, we asked whether mutation of genes involved in non-homologous end joining (NHEJ) could improve mouse ESC HR efficiency. Transient inhibition of NHEJ is known to improve HR (Chu et al., 2015, Maruyama et al., 2015), but a comprehensive screen to determine which genes are most important in the NHEJ/HR decision in mouse ESCs has not been carried out. We designed scCRISPR sgRNAs targeting 13 genes reported to regulate NHEJ in mouse ESCs and generated bulk mutant lines for each gene. We then tested Hist1h3a-GFP knockin efficiency in all 13 lines as compared to controls (Figure 3). Mutants in Prkdc, Lig4, and Xrcc4 led to the most significant increases in GFP knockin efficiency, a finding that meshes with the enhanced HR efficiency after small molecule and shRNA-based Lig4 knockdown (Böttcher et al., 2014, Chu et al., 2015, Maruyama et al., 2015). We then generated dual knockouts for all combinations of Prkdc, Lig4, and Xrcc4 using scCRISPR targeting, finding that dual mutation of Prkdc and Lig4 elevated the level of GFP integration by more than 3-fold as compared to wild-type cells to over 10% of cells (Figure 3). This NHEJ-impaired mouse ESC line capable of 3-fold more efficient HR should facilitate high-throughput knockin screens. More importantly, this experiment proves that the ease of scCRISPR combined with its high-efficiency mutation rate enables quick and cheap functional genomic screening. Considering cost and effort of sgRNA cloning compound for every targeted gene in an arrayed screen such as this, scCRISPR represents a transformative tool for functional genomic screening of large gene sets.
Figure 3.

An scCRISPR-Based NHEJ Gene Knockout Screen Improves HR Efficiency 3-Fold
Hist1h3a-GFP knockin efficiency (y axis) is shown for 13 scCRISPR-generated bulk knockout lines of genes reported to play a role in NHEJ (x axis). Values and SDs are averaged from three independent biological experiments. A clonal double knockout line for Prkdc and Lig4 (red) exhibits 3-fold more efficient HR than wild-type mouse ESCs (dotted line).
Discussion
We present scCRISPR as a unique tool for rapid (3 hr from oligonucleotide arrival versus 6 days for conventional CRISPR/Cas9), cost-effective (approximately one-sixth the cost), and efficient (up to 88% mutation rate) application of CRISPR/Cas9, optimally suited for high-throughput comparison and multiplexing of sgRNA sequences. We demonstrate that scCRISPR works efficiently in mouse and human ESCs as well as in HEK293 cells, and we expect it will show efficacy in any cell line or in vivo cell type capable of efficient homologous recombination. We show that 80 bp of homology is sufficient for efficient insertion of DNA up to 1 kb with the help of CRISPR/Cas9-mediated genome editing without the risk of off-target genome integration. These methodologies advance CRISPR/Cas9 technology by substantially reducing the effort and increasing the throughput of CRISPR/Cas9-mediated genomic mutation and gene knockin in mouse and human cell lines. By eliminating molecular cloning, these methods lower the bar for targeted genome editing, opening up opportunities for novel high-throughput genome editing and knockin screening applications.
Experimental Procedures
Cell Culture
Mouse embryonic stem cell culture was performed according to previously published protocols (Sherwood et al., 2014). All experiments were performed with 129P2/OlaHsd mouse ESCs except for the DsRed targeting, which was performed using the IB10 mouse ESC line. Mouse ESCs were maintained on gelatin-coated plates feeder-free in mouse ESC media composed of Knockout DMEM (Life Technologies) supplemented with 15% defined fetal bovine serum (FBS, HyClone), 0.1 mM nonessential amino acids (NEAA, Life Technologies), Glutamax (GM, Life Technologies), 0.55 mM 2-mercaptoethanol (b-ME, Sigma), 1X ESGRO LIF (Millipore), 5 nM GSK-3 inhibitor XV, and 500 nM UO126. Cells were regularly tested for mycoplasma. Mouse ESC differentiation was performed by switching to serum-deprived differentiation media consisting of Advanced DMEM (Life Technologies) supplemented with 2% FBS and GM for 96 hr.
Hist1h3a-GFP fusion mouse ESCs were created using the gBlock-CRISPR method described in this work and cloned such that >99.5% of cells expressed strong nuclear GFP. Rosa26-CAGGS-DsRed IB10 mouse ESCs were created using plasmid-based knockin and also cloned to enrich for DsRed-expressing cells.
HEK293FT cells were cultured using DMEM (Life Technologies) supplemented with 10% FBS (HyClone).
Human ESC culture was performed according to previously published protocols. All experiments were performed with HUES2 human ESCs. Human ESCs were maintained on gelatin-coated plates on a feeder layer of irradiated murine embryonic fibroblasts (MEFs) in complete human ESC media composed of 1:1 DMEM:F12 (Life Technologies) supplemented with 15% KOSR, 0.1 mM NEAA (Life Technologies), GM (Life Technologies), 3.2 mM b-ME (Sigma), 20 ng/ml bFGF (R&D Systems), 5 nM GSK-3 inhibitor XV, and 500 nM UO126. Cells were regularly tested for mycoplasma.
Prior to electroporation, human ESCs were enzymatically passaged using 0.05% trypsin and quenched with complete human ESC media supplemented with 1% FBS (HyClone) and 10 μM Y-27632 (Tocris). For depletion of the cell suspension of feeders, the cells were plated onto a 15-cm dish in 7 ml quenching media and incubated at 37°C for 30 min. The media were then carefully transferred to a 15-ml tube and pelleted to remove excess serum.
scCRISPR Off-Target Effect Analysis
For the CRISPR in genome editing, a site-specific sgRNA sequence must be designed by a set of rules that determines both the efficiency and specificity of CRISPR targeting. sgRNAs are typically 20 bp long although 17- to 21 bp sgRNAs have been reported to be functional (Cong et al., 2013, Mali et al., 2013, Fu et al., 2014, Ran et al., 2013). Cas9 will recognize and cleave DNA only when there is a PAM sequence (-NGG) in the genome that is directly 3′ of the sgRNA sequence (Cho et al., 2014, Gilbert et al., 2013, Fu et al., 2013). Lastly, Cas9 can generate off-target DNA cleavage at sites bearing close similarity to the sgRNA sequence, especially in the 10 bp PAM-adjacent sequence (Fu et al., 2013, Wu et al., 2014, Kuscu et al., 2014), so sgRNAs with high similarity to other genomic sequences should be avoided.
To avoid unwanted off-target effects of sgPal in human and mouse applications, we searched for 10 bp sequences largely unique to the mouse and human genomes. CRISPR is highly specific but can tolerate up to five nucleotide mismatches between the sgRNA and template DNA (Cho et al., 2014). Cas9 will cleave at non-specific sites with a low efficiency so long as no more than two nucleotide differences occur within the final 11 nt, and crucially a PAM sequence must be present at the 3 bp directly downstream of the complementary region (Kuscu et al., 2014, Lin et al., 2014). sgPal sequence similarity to off-target genomic loci was determined by BLAST comparison of the 10 bp mirrored sequences to the mouse and human genomes. BLAST hits in coding regions for all palindromic sgRNAs used in this work are listed in the Supplemental Experimental Procedures.
scCRISPR
We ordered sets of oligonucleotides to clone palindromic sgRNA sequences for use in scCRISPR (all sequences are listed in the Supplemental Experimental Procedures). scCRISPR palindromic sgRNAs have an initial G nucleotide followed by an 18 or 20 bp palindromic sequence. We used a published cloning protocol (Ran et al., 2013) to clone these sequences into a BbsI-digested plasmid subcloned from the pX330 sgRNA expression cassette into a plasmid with a pT2AL200R175 backbone (Urasaki et al., 2006), Hygromycin resistance, and a modified hairpin structure to incorporate the FE alterations shown to improve sgRNA hairpin stability (Chen et al., 2013). Because the 2 nt at the end of the U6 promoter immediately upstream of the sgRNA sequence are CC, the cloned palindromic sgRNA is of the form CCG(18 to 20 bp palindromic sgRNA sequence). The reverse complement of this sequence is (18 to 20 bp palindromic sgRNA sequence)CGG, so palindromic sgRNAs of this form are capable of self-cleaving once they are transcribed in target cells and complex with Cas9.
We also subcloned the CBh Cas9 expression cassette from pX330 (Ran et al., 2013) into a plasmid with a pT2AL200R175 backbone (Urasaki et al., 2006) and Blasticidin resistance.
To prepare site-specific sgRNA homology fragments, we designed a two-step PCR amplification protocol. First, we ordered an oligonucleotide from Integrated DNA Technologies (IDT) that contains the sgRNA sequence and ∼20 bp of homology to the upstream and downstream regions of the sgRNA expression cassette. Homology arm lengths used in this paper varied from short (60 bp of homology on either side), to standard (150 and 122 bp of homology on the left and right, respectively), to long (210 and 148 bp of homology on the left and right, respectively). All specific oligonucleotides are listed in the Supplemental Experimental Procedures and are of the following form: for 20 bp sgRNA, TGGAAAGGACGAAACACCGN19GTTTAAGAGCTATGCTGGAAAC; for 21 bp sgRNA, GGAAAGGACGAAACACCGN20GTTTAAGAGCTATGCTGGAAAC; and for 19 bp sgRNA, TGGAAAGGACGAAACACCGN18GTTTAAGAGCTATGCTGGAAACA.
To create standard-length homology fragments, we performed 35 cycles of Onetaq PCR using a three-step protocol (94°C for 15 s followed by 60°C for 30 s followed by 68°C for 30 s) using the following reaction mix that contains two primer sets that combine to add standard homology arms to the sgRNA oligonucleotide: 2X Onetaq master mix with standard buffer (New England Biolabs), 50% of reaction volume; 20 μM sg[LocusX], 2.5% of reaction volume; 20 μM scCRISPR_homology_fw, 2.5% of reaction volume; 20 μM scCRISPR_homology_rv, 2.5% of reaction volume; 20 μM scCRISPR_homology_extension_fw, 2.5% of reaction volume; 20 μM scCRISPR_homology_extension_rv, 2.5% of reaction volume; and dH2O, 37.5% of reaction volume.
We used a reaction volume of 100 μl per electroporation to be performed. A 2 μl aliquot of this second PCR product was run on 2% agarose to test for the expected ∼292 bp product.
To create long homology fragments, we performed the first PCR but for only ten cycles, using at least 15 μl reaction volume for this first PCR.
We then performed a second PCR using the first PCR reaction as the template without purification. For this PCR, we performed 35 cycles of Onetaq PCR using a three-step protocol (94°C for 15 s followed by 60°C for 30 s followed by 68°C for 30 s) using the following reaction mix: 2X Onetaq master mix with standard buffer, 50% of reaction volume; unpurified first PCR product, 5% of reaction volume; 20 μM scCRISPR_homology_double extension_fw, 2.5% of reaction volume; 20 μM scCRISPR_homology_double extension_rv, 2.5% of reaction volume; and dH2O, 40% of reaction volume.
We used a reaction volume of 100 μl per electroporation to be performed. A 2 μl aliquot of this second PCR product was run on 2% agarose to test for the expected ∼378 bp product. The products of standard and long homology fragment PCRs with different formatting to denote the initial oligonucleotide (bold), standard homology primers (underline), and long homology primers (italic) are as follows: CGATACAAGGCTGTTAGAGAGATAATTGGAATTAATTTGACTGTAAACACAAAGATATTAGTACAAAATACGTGACGTAGAAAGTAATAATTTCTTGGGTAGTTTGCAGTTTTAAAATTATGTTTTAAAATGGACTATCATATGCTTACCGTAACTTGAAAGTATTTCGATTTCTTGGCTTTATATATCTTGTGGAAAGGACGAAACACCG[N18-20] GTTTAAGAGCTATGCTGGAAACAGCATAGCAAGTTTAAATAAGGCTAGTCCGTTATCAACTTGAAAAAGTGGCACCGAGTCGGTGCTTTTTTGTTTTAGAGCTAGAAATAGCAAGTTAAAATCGAGCAGACATGATAAGATACATTGA.
Once verified, we performed minElute PCR purification (QIAGEN) on the product, loading a maximum of 200 μl of PCR product into a single minElute column.
For targeting of mouse ESCs, we then electroporated a mixture of 5 μg CBh Cas9-BlastR plasmid, 5 μg sgPal plasmid, and minElute purified product of 100 μl sg(LocusX) homology fragment into ∼106 mouse ESCs. For control experiments using sgRNA plasmid, a mixture of 5 μg CBh Cas9-BlastR plasmid and 5 μg sgLocusX plasmid was used. We vacuum centrifuged the DNA mixture to a final volume of <20 μl and added 120 μl EmbryoMax Electroporation Buffer (ES-003-D, Millipore) to the mouse ESCs. DNA mixture and mouse ESC suspension were mixed and electroporated in a 0.4-cm electroporation cuvette using a BioRad electroporator at 230 V, 0.500 μF, and maximum resistance.
Electroporated cells were plated onto a single well of a 12-well tissue culture plate (BD Falcon) in >2 ml mouse ESC media supplemented with 7.5 μM Y-27632 (Tocris). From 24 to 72 hr after electroporation, media were refreshed daily with mouse ESC media supplemented with 10 μg/ml Blasticidin (Life Technologies) and 66 μg/ml (1:666) Hygromycin (Cellgro). After selection, media were refreshed every day and cells were trypsinized and replated when confluent. Testing of CRISPR mutation or homologous recombination efficiency was performed 7 days after electroporation.
We found that transfection using Lipofectamine 3000 (Life Technologies) using the standard protocol was slightly less effective (∼80%–90% as efficient) than electroporation at scCRISPR and gBlock-CRISPR in mouse ESCs. For 293FT experiments, we used Lipofectamine transfection, as this cell line is known to be particularly amenable to transfection.
For targeting of human ESCs, we electroporated a mixture of 5 μg CBh Cas9-BlastR plasmid, 5 μg sgPal plasmid, and minElute purified product of 100 μl sg(LocusX) homology fragment into ∼106 human ESCs depleted of feeder cells. For control experiments using sgRNA plasmid, a mixture of 5 μg CBh Cas9-BlastR plasmid and 5 μg sgLocusX plasmid was used. We vacuum centrifuged the DNA mixture to a final volume of <20 μl and added 100 μl electroporation buffer from the Amaxa Human Stem Cell Nucleofector kit 1 to the human ESCs. DNA mixture and human ESC suspension were mixed and electroporated in an Amaxa Nucleofector II with program B-16.
Electroporated cells were plated onto a single well of a six-well tissue culture plate (BD Falcon) previously coated with gelatin and irradiated MEFs in >2 ml complete human ESC media supplemented with 10 μM Y-27632 (Tocris). From 24 to 72 hr after electroporation, media were refreshed daily with complete human ESC media supplemented with 2 μg/ml Blasticidin (Life Technologies) and 66 μg/ml (1:666) Hygromycin (Cellgro). After selection, media were refreshed every day and cells were trypsinized and replated when confluent. Testing of CRISPR mutation or homologous recombination efficiency was performed at the first and second passages, circa 10 and 14 days after electroporation.
gBlock-Mediated CRISPR
The gBlock sequences containing the full U6 promoter, locus-specific sgRNA, and FE-modified sgRNA hairpin were ordered from IDT as gBlocks using the following template: AGTATTACGGCATGTGAGGGCCTATTTCCCATGATTCCTTCATATTTGCATATACGATACAAGGCTGTTAGAGAGATAATTGGAATTAATTTGACTGTAAACACAAAGATATTAGTACAAAATACGTGACGTAGAAAGTAATAATTTCTTGGGTAGTTTGCAGTTTTAAAATTATGTTTTAAAATGGACTATCATATGCTTACCGTAACTTGAAAGTATTTCGATTTCTTGGCTTTATATATCTTGTGGAAAGGACGAAACACC G[N18–20] GTTTAAGAGCTATGCTGGAAACAGCATAGCAAGTTTAAATAAGGCTAGTCCGTTATCAACTTGAAAAAGTGGCACCGAGTCGGTGCTTTTTTGTTTTAGAGCTAGAAATAGCAAGTTAAAATAAGGCTAGTCCGTTTTTAGCGCGTGCGCCAATTCTGCAGACAAATGGCTCTAGAGGTACGGCCGCTTCGAGCAGACATGATAAGATACATTGA.
For 21 bp sgRNAs, the final A was omitted, and for 19 bp sgRNAs, a T was added at the beginning. We then performed 35 cycles of Onetaq PCR amplification on the gBlock using a three-step protocol (94°C for 15 s followed by 60°C for 30 s followed by 68°C for 30 s) using the following reaction mix: 2X Onetaq master mix with standard buffer, 50% of reaction volume; gBlock resuspended at 1 ng/μl, 0.25% of reaction volume; 20 μM gBlock-CRISPR_fw, 2.5% of reaction volume; 20 μM gBlock-CRISPR_rv, 2.5% of reaction volume; and dH2O, 44.75% of reaction volume.
We used a reaction volume of 100 μl per electroporation to be performed. A 2 μl aliquot of this PCR product was run on 2% agarose to test for the expected 500 bp product. Once verified, we performed minElute PCR purification (QIAGEN) on the product, loading a maximum of 200 μl PCR product into a single minElute column. Alternatively, we achieved equivalent results when we PCR amplified existing sgRNA plasmids with the same gBlock-CRISPR fw and rv primers, which also occur in our sgRNA plasmid.
For targeting of mouse ESCs, we electroporated a mixture of 5 μg CBh Cas9-BlastR plasmid and minElute purified product of 100 μl sg(LocusX) gBlock fragment into ∼106 mouse ESCs using the same protocol as above. Electroporated cells were plated onto a single well of a 12-well tissue culture plate (BD Falcon) in >2 ml mouse ESC media supplemented with 7.5 μM Y-27632 (Tocris). From 24 to 72 hr after electroporation, media were refreshed daily with mouse ESC media supplemented with 10 μg/ml Blasticidin (Life Technologies) only since no Hygromycin plasmid was added. After selection, media were refreshed every day and cells were trypsinized and replated when confluent. Testing of CRISPR mutation or homologous recombination efficiency was performed 7 days after electroporation.
For targeting of human ESCs, we electroporated a mixture of 5 μg CBh Cas9-BlastR plasmid and minElute purified product of 100 μl sg(LocusX) gBlock fragment into ∼106 human ESCs depleted of feeder cells using the same protocol as above. Electroporated cells were plated onto a single well of a six-well tissue culture plate (BD Falcon) previously coated with gelatin and irradiated MEFs in >2 ml complete human ESC media supplemented with 10 μM Y-27632 (Tocris). From 24 to 72 hr after electroporation, media were refreshed daily with complete human ESC media supplemented with 2 μg/ml Blasticidin (Life Technologies). After selection, media were refreshed every day and cells were trypsinized and replated when confluent. Testing of CRISPR mutation or homologous recombination efficiency was performed at the first and second passages, circa 10 and 14 days after electroporation.
Homologous Recombination
GFP was amplified using two successive PCR reactions to add ∼70 to 80 bp homology arms to each side. Homology arms were designed to encode GFP in frame immediately upstream of the stop codon of the Hist1h3a and Nanog genes and to include a stop codon after the GFP open reading frame (ORF). The sgRNA sequences were designed to cleave DNA as close as possible to the endogenous stop codon of the gene to be targeted. Homology arms were designed so as not to overlap with the sgRNA sequence by more than the 10 bp on the side opposite the PAM sequence, and no overlap was ever allowed on the PAM side to avoid CRISPR cleavage of the GFP homology template. The first homology primer pair is of the following format: LocusX_GFPhomologyarm_fw (LocusX pre-stop40bp)GTGAGCAAGGGCGAGGAGCT, and LocusX_GFPhomologyarm_rv (LocusX post-stop reverse complement40bp)TGAGGAGTGAATTGCGGCCG.
The common 20 bp sequences allow amplification of the entire GFP ORF and include the stop codon. These primers produce an 819 bp product. We PCR amplified GFP using 25 cycles of Phusion (NEB) PCR amplification using a two-step protocol (98°C for 10 s followed by 72°C for 45 s) using the following reaction mix: 2X Phusion master mix with standard buffer, 50% of reaction volume; GFP plasmid at 100 ng/μl, 0.5% of reaction volume; 20 μM LocusX_GFPhomologyarm_fw, 2.5% of reaction volume; 20 μM LocusX_GFPhomologyarm_rv, 2.5% of reaction volume; DMSO, 3% of reaction volume; and dH2O, 41.5% of reaction volume. For each electroporation to be performed, we used at least 10 μl reaction volume for this first PCR.
We then performed a second PCR using the first PCR reaction as the template without purification. For this PCR, we ordered 60 bp primers that extend the locus-specific homology by 30–40 bp on each end. To do so, we designed a set of PCR primers that overlapped with the first homology arm by 20–30 bp. We chose the minimal overlap such that the overlapping region was estimated to have a Tm >65°C using the NEB Tm calculator (http://tmcalculator.neb.com/#!/). We then PCR amplified the unpurified product of the previous reaction using 35 cycles of Phusion PCR amplification using a two-step protocol (98° for 10 s followed by 72° for 45 s) using the following reaction mix: 2X Phusion master mix with standard buffer, 50% of reaction volume; unpurified product of PCR1, 5% of reaction volume; 20 μM LocusX_homologyarmextension_fw, 2.5% of reaction volume; 20 μM LocusX_homologyarmextension_rv, 2.5% of reaction volume; DMSO, 3% of reaction volume; and dH2O, 37% of reaction volume.
For each electroporation to be performed, we used at least 100 μl reaction volume for this second PCR. A 2 μl aliquot of this PCR product was run on 2% agarose to test for the expected ∼900 bp product. Once verified, we performed minElute PCR purification (QIAGEN) on the product, loading a maximum of 200 μl PCR product into a single minElute column.
For targeting mouse ESCs, we then electroporated a mixture of 5 μg CBh Cas9-BlastR plasmid, minElute purified product of 100 μl GFP LocusX homology arm fragment, and either gBlock or sgPal and homology fragment at the same amounts as mentioned above into ∼106 mouse ESCs using the same protocol as above. Electroporated cells were plated onto a single well of a 12-well tissue culture plate (BD Falcon) in >2 ml mouse ESC media supplemented with 7.5 μM Y-27632 (Tocris). From 24 to 72 hr after electroporation, media were refreshed daily with mouse ESC media supplemented with 10 μg/ml Blasticidin and 66 μg/ml (1:666) Hygromycin (only with sgPal, not with gBlock). After selection, media were refreshed every day and cells were trypsinized and replated when confluent. Testing of homologous recombination efficiency was performed 7 days after electroporation.
For human ESCs, we electroporated a mixture of 5 μg CBh Cas9-BlastR plasmid, minElute purified product of 100 μl GFP LocusX homology arm fragment, and either gBlock or sgPal and homology fragment at the same amounts as mentioned above into ∼106 human ESCs depleted of feeder cells using the same protocol as above. Electroporated cells were plated onto a single well of a six-well tissue culture plate (BD Falcon) previously coated with gelatin and irradiated MEFs in >2 ml complete human ESC media supplemented with 10 μM Y-27632 (Tocris). From 24 to 72 hr after electroporation, media were refreshed daily with complete human ESC media supplemented with 2 μg/ml Blasticidin and 66 μg/ml (1:666) Hygromycin (only with sgPal, not with gBlock). After selection, media were refreshed every day and cells were trypsinized and replated when confluent. Testing of CRISPR mutation or homologous recombination efficiency was performed at the first and second passages, circa 10 and 14 days after electroporation.
Flow Cytometry
Cells to be analyzed by flow cytometry were trypsinized, quenched, and fluorescence of 2 × 104 cells was measured using a BD Accuri C6 flow cytometer and accompanying software (BD Biosciences).
Fluorescence Imaging
Live-cell imaging was performed using a DMI 6000b inverted fluorescence microscope (Leica), and image analysis was performed with the Leica AF6000 software package.
Author Contributions
Experiments were designed by M.A. and R.I.S. Experiments were carried out and analyzed by M.A., S.S., and R.I.S. Computational analysis was performed by T.H. The manuscript was prepared by M.A., N.G., and R.I.S.
Acknowledgments
The authors thank Koichi Kawakami and Feng Zhang for reagents, Lendy Chu and Nisha Rajagopal for technical assistance, and Yiling Qiu for flow cytometric assistance. The authors acknowledge funding from NIH 5UL1DE019581, RL1DE019021, 1K01DK101684-01, 1U01HG007037, and 5P01NS055923; the Harvard Stem Cell Institute’s Sternlicht Director’s Fund award; and Human Frontier Science Program grant to R.I.S.
Published: October 29, 2015
Footnotes
This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/4.0/).
Supplemental Information includes Supplemental Experimental Procedures, two figures, and one table and can be found with this article online at http://dx.doi.org/10.1016/j.stemcr.2015.09.022.
Supplemental Information
References
- Böttcher R., Hollmann M., Merk K., Nitschko V., Obermaier C., Philippou-Massier J., Wieland I., Gaul U., Förstemann K. Efficient chromosomal gene modification with CRISPR/cas9 and PCR-based homologous recombination donors in cultured Drosophila cells. Nucleic Acids Res. 2014;42:e89. doi: 10.1093/nar/gku289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B., Gilbert L.A., Cimini B.A., Schnitzbauer J., Zhang W., Li G.W., Park J., Blackburn E.H., Weissman J.S., Qi L.S., Huang B. Dynamic imaging of genomic loci in living human cells by an optimized CRISPR/Cas system. Cell. 2013;155:1479–1491. doi: 10.1016/j.cell.2013.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho S.W., Kim S., Kim Y., Kweon J., Kim H.S., Bae S., Kim J.S. Analysis of off-target effects of CRISPR/Cas-derived RNA-guided endonucleases and nickases. Genome Res. 2014;24:132–141. doi: 10.1101/gr.162339.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu V.T., Weber T., Wefers B., Wurst W., Sander S., Rajewsky K., Kühn R. Increasing the efficiency of homology-directed repair for CRISPR-Cas9-induced precise gene editing in mammalian cells. Nat. Biotechnol. 2015;33:543–548. doi: 10.1038/nbt.3198. [DOI] [PubMed] [Google Scholar]
- Cong L., Ran F.A., Cox D., Lin S., Barretto R., Habib N., Hsu P.D., Wu X., Jiang W., Marraffini L.A., Zhang F. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folger K.R., Wong E.A., Wahl G., Capecchi M.R. Patterns of integration of DNA microinjected into cultured mammalian cells: evidence for homologous recombination between injected plasmid DNA molecules. Mol. Cell. Biol. 1982;2:1372–1387. doi: 10.1128/mcb.2.11.1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y., Foden J.A., Khayter C., Maeder M.L., Reyon D., Joung J.K., Sander J.D. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat. Biotechnol. 2013;31:822–826. doi: 10.1038/nbt.2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y., Sander J.D., Reyon D., Cascio V.M., Joung J.K. Improving CRISPR-Cas nuclease specificity using truncated guide RNAs. Nat. Biotechnol. 2014;32:279–284. doi: 10.1038/nbt.2808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert L.A., Larson M.H., Morsut L., Liu Z., Brar G.A., Torres S.E., Stern-Ginossar N., Brandman O., Whitehead E.H., Doudna J.A. CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell. 2013;154:442–451. doi: 10.1016/j.cell.2013.06.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinek M., Chylinski K., Fonfara I., Hauer M., Doudna J.A., Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816–821. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinek M., East A., Cheng A., Lin S., Ma E., Doudna J. RNA-programmed genome editing in human cells. eLife. 2013;2:e00471. doi: 10.7554/eLife.00471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuscu C., Arslan S., Singh R., Thorpe J., Adli M. Genome-wide analysis reveals characteristics of off-target sites bound by the Cas9 endonuclease. Nat. Biotechnol. 2014;32:677–683. doi: 10.1038/nbt.2916. [DOI] [PubMed] [Google Scholar]
- Lin Y., Cradick T.J., Brown M.T., Deshmukh H., Ranjan P., Sarode N., Wile B.M., Vertino P.M., Stewart F.J., Bao G. CRISPR/Cas9 systems have off-target activity with insertions or deletions between target DNA and guide RNA sequences. Nucleic Acids Res. 2014;42:7473–7485. doi: 10.1093/nar/gku402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mali P., Yang L., Esvelt K.M., Aach J., Guell M., DiCarlo J.E., Norville J.E., Church G.M. RNA-guided human genome engineering via Cas9. Science. 2013;339:823–826. doi: 10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maruyama T., Dougan S.K., Truttmann M.C., Bilate A.M., Ingram J.R., Ploegh H.L. Increasing the efficiency of precise genome editing with CRISPR-Cas9 by inhibition of nonhomologous end joining. Nat. Biotechnol. 2015;33:538–542. doi: 10.1038/nbt.3190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quail M.A., Smith M., Coupland P., Otto T.D., Harris S.R., Connor T.R., Bertoni A., Swerdlow H.P., Gu Y. A tale of three next generation sequencing platforms: comparison of Ion Torrent, Pacific Biosciences and Illumina MiSeq sequencers. BMC Genomics. 2012;13:341. doi: 10.1186/1471-2164-13-341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran F.A., Hsu P.D., Wright J., Agarwala V., Scott D.A., Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013;8:2281–2308. doi: 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren X., Yang Z., Xu J., Sun J., Mao D., Hu Y., Yang S.J., Qiao H.H., Wang X., Hu Q. Enhanced specificity and efficiency of the CRISPR/Cas9 system with optimized sgRNA parameters in Drosophila. Cell Rep. 2014;9:1151–1162. doi: 10.1016/j.celrep.2014.09.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouet P., Smih F., Jasin M. Expression of a site-specific endonuclease stimulates homologous recombination in mammalian cells. Proc. Natl. Acad. Sci. USA. 1994;91:6064–6068. doi: 10.1073/pnas.91.13.6064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherwood R.I., Hashimoto T., O’Donnell C.W., Lewis S., Barkal A.A., van Hoff J.P., Karun V., Jaakkola T., Gifford D.K. Discovery of directional and nondirectional pioneer transcription factors by modeling DNase profile magnitude and shape. Nat. Biotechnol. 2014;32:171–178. doi: 10.1038/nbt.2798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Small J., Scangos G. Recombination during gene transfer into mouse cells can restore the function of deleted genes. Science. 1983;219:174–176. doi: 10.1126/science.6294829. [DOI] [PubMed] [Google Scholar]
- Urasaki A., Morvan G., Kawakami K. Functional dissection of the Tol2 transposable element identified the minimal cis-sequence and a highly repetitive sequence in the subterminal region essential for transposition. Genetics. 2006;174:639–649. doi: 10.1534/genetics.106.060244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wake C.T., Wilson J.H. Simian virus 40 recombinants are produced at high frequency during infection with genetically mixed oligomeric DNA. Proc. Natl. Acad. Sci. USA. 1979;76:2876–2880. doi: 10.1073/pnas.76.6.2876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X., Scott D.A., Kriz A.J., Chiu A.C., Hsu P.D., Dadon D.B., Cheng A.W., Trevino A.E., Konermann S., Chen S. Genome-wide binding of the CRISPR endonuclease Cas9 in mammalian cells. Nat. Biotechnol. 2014;32:670–676. doi: 10.1038/nbt.2889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L., Yang J.L., Byrne S., Pan J., Church G.M. CRISPR/Cas9-directed genome editing of cultured cells. Curr. Protoc. Mol. Biol. 2014;107 doi: 10.1002/0471142727.mb3101s107. 31.1.1–31.1.17. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.