

Abstract
Background and Aims
Some species of Genlisea possess ultrasmall nuclear genomes, the smallest known among angiosperms, and some have been found to have chromosomes of diminutive size, which may explain why chromosome numbers and karyotypes are not known for the majority of species of the genus. However, other members of the genus do not possess ultrasmall genomes, nor do most taxa studied in related genera of the family or order. This study therefore examined the evolution of genome sizes and chromosome numbers in Genlisea in a phylogenetic context. The correlations of genome size with chromosome number and size, with the phylogeny of the group and with growth forms and habitats were also examined.
Methods
Nuclear genome sizes were measured from cultivated plant material for a comprehensive sampling of taxa, including nearly half of all species of Genlisea and representing all major lineages. Flow cytometric measurements were conducted in parallel in two laboratories in order to compare the consistency of different methods and controls. Chromosome counts were performed for the majority of taxa, comparing different staining techniques for the ultrasmall chromosomes.
Key Results
Genome sizes of 15 taxa of Genlisea are presented and interpreted in a phylogenetic context. A high degree of congruence was found between genome size distribution and the major phylogenetic lineages. Ultrasmall genomes with 1C values of <100 Mbp were almost exclusively found in a derived lineage of South American species. The ancestral haploid chromosome number was inferred to be n = 8. Chromosome numbers in Genlisea ranged from 2n = 2x = 16 to 2n = 4x = 32. Ascendant dysploid series (2n = 36, 38) are documented for three derived taxa. The different ploidy levels corresponded to the two subgenera, but were not directly correlated to differences in genome size; the three different karyotype ranges mirrored the different sections of the genus. The smallest known plant genomes were not found in G. margaretae, as previously reported, but in G. tuberosa (1C ≈ 61 Mbp) and some strains of G. aurea (1C ≈ 64 Mbp).
Conclusions
Genlisea is an ideal candidate model organism for the understanding of genome reduction as the genus includes species with both relatively large (∼1700 Mbp) and ultrasmall (∼61 Mbp) genomes. This comparative, phylogeny-based analysis of genome sizes and karyotypes in Genlisea provides essential data for selection of suitable species for comparative whole-genome analyses, as well as for further studies on both the molecular and cytogenetic basis of genome reduction in plants.
Keywords: Bladderwort, carnivorous plant, chromosome number, flow cytometry, Genlisea, genome miniaturization, genome size, Lentibulariaceae, Lamiales
INTRODUCTION
The carnivorous corkscrew plants, Genlisea (Order Lamiales, Lentibulariaceae, bladderwort family) comprise 29 species (Fleischmann, 2012), which are distributed from South to Central America and in Africa. The plants are annual or perennial herbs, strictly heterophyllous, producing spathulate, green, photosynthetic epiterrestrial leaves and achlorophyllous subterranean tubular leaves (rhizophylls). The latter constitute the carnivorous traps of the plant, and consist of a basal stalk and a swollen vesicle (‘stomach’) followed by a long tubular ‘neck’, which is dichotomously forked at the apex into two helically twisted, hollow arms (Fig. 1A).
Fig. 1.
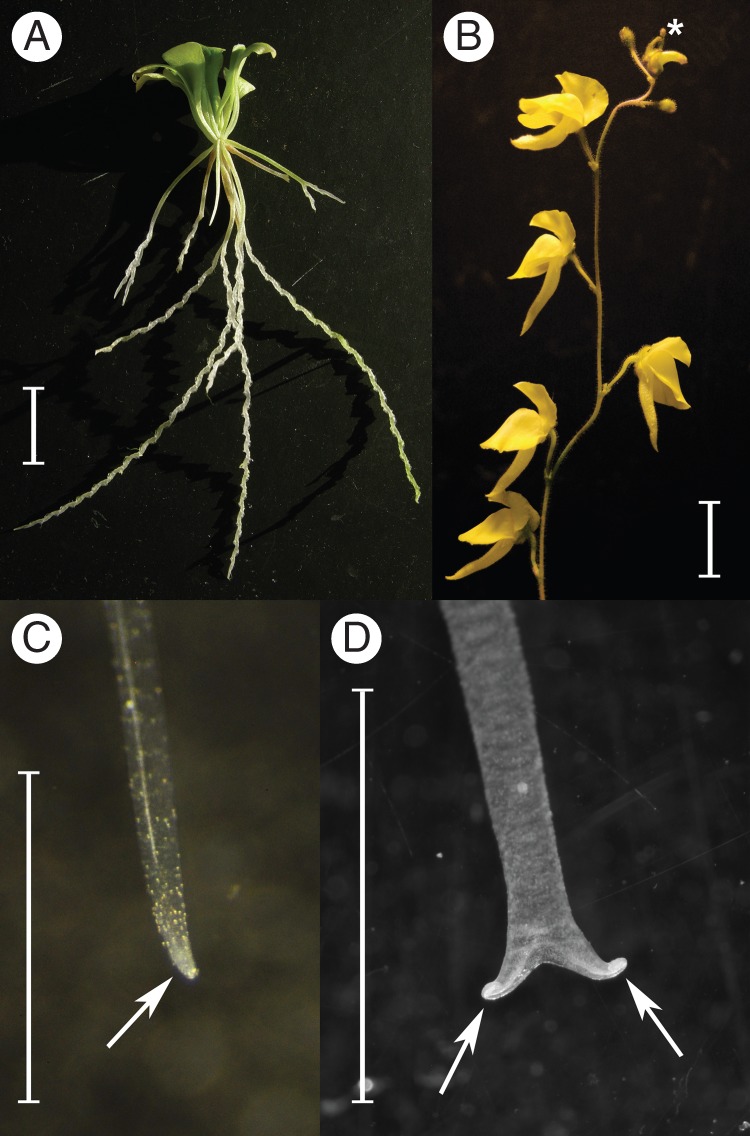
(A) Growth habit of Genlisea (an excavated plant of G. flexuosa is shown) illustrating the green photosynthetic leaves and the pale white, subterranean carnivorous trap leaves (=rhizophylls). (B) Inflorescence of Genlisea (G. aurea var. minor is shown). The most apical, juvenile flower buds (*, with the sepals still touching each other at their tips) bear anthers at the right stage of development, with pollen mother cells suitable for meiotic chromosome counts. (C, D) Two stages of young, developing rhizophylls used for mitotic chromosome counts. Only the apical tips of the rhizophylls (arrows) containing meristematic cells (visible as milky white tissue) were used for preparation. Scale bars = 1 cm.
Since its discovery >200 years ago, Genlisea has remained the most understudied and poorly known genus among otherwise well-studied carnivorous plant genera. This is mainly due to its distribution across remote areas and the difficulty of accessing living plant material. Although the carnivorous nature of Genlisea was postulated as early as 1875 by Charles Darwin (Darwin, 1875), with an alleged specialization for trapping protozoa and various other small soil organisms (Goebel, 1891; Barthlott et al., 1998; Płachno et al., 2008; Darnowski and Fritz, 2010; Fleischmann, 2012), the underlying mechanism of trap functioning remains unclear (Adamec, 2003; Fleischmann, 2012).
One of the biggest surprises, which brought the rather elusive genus to the interest of the scientific community, was the discovery that some species of Genlisea possess ultrasmall nuclear genomes (holoploid genome size ∼60–64 Mbp in some strains of G. aurea; Greilhuber et al., 2006; Albert et al., 2010; Leushkin et al., 2013), the smallest known among angiosperms (Greilhuber et al., 2006; Bennett and Leitch, 2011). Furthermore, some Genlisea species were found to have chromosomes of diminutive size (Greilhuber et al., 2006), which may explain why chromosome numbers and karyotypes are not known for the majority of species of the genus. However, other members of the same genus do not possess ultrasmall genomes, nor do most taxa studied in related genera of the family or order, except certain derived aquatic members of Utricularia section Utricularia, such as U. gibba, which has a genome of ∼80 Mbp (Greilhuber et al., 2006; Ibarra-Laclette et al., 2011a, b, 2013).
Among the three genera of Lentibulariaceae, both the smallest and the largest genomes known are found within Genlisea, with the ultrasmall genomes reported for G. aurea and G. margaretae representing one end of the scale and members of subgenus Tayloria and G. hispidula the other. This difference translates into an ∼24-fold variation in genome size (Greilhuber et al., 2006). Genome sizes have been recorded for several species of the sister genus Utricularia and the common sister Pinguicula, all falling within a range (of ∼1099 Mbp) that lies between the ultrasmall and the larger genomes of Genlisea (Greilhuber et al., 2006; Veleba et al., 2014). Within the genus Genlisea, genome sizes range from 63·4 to 1510 Mbp (Greilhuber et al., 2006; Veleba et al., 2014), and in different populations investigated for a few taxa (most notably G. aurea) genome size can vary up to 2-fold (Albert et al., 2010).
Very few cytological studies have been carried out for Lentibulariaceae thus far, and these have predominantly focused on Pinguicula (summarized by Casper and Stimper, 2009) or aquatic species of Utricularia section Utricularia (summarized by Casper and Manitz, 1975 and Rahman et al., 2001). In the case of Genlisea, chromosome numbers have been reported for only five species so far (Greilhuber et al., 2006; Vu et al., 2012), but three of these represent approximations only. The general lack of karyotype data available for Utricularia and Genlisea is mainly due to the very small size of their metaphase chromosomes (Reese, 1951; Kondo, 1972a; Rahman et al., 2001; Greilhuber et al., 2006), because of difficulties in staining these chromosomes with standard dyes (Rahman et al., 2001; A. Fleischmann, A. Sousa and J. Greilhuber, pers. obs.), but also because of the difficulty of obtaining suitable living material of most taxa for chromosome counts. The present study was only possible because a large sampling of cultivated species of Genlisea was available to the authors. All species of Genlisea and Utricularia lack roots entirely, and therefore shoot apices have to be used to perform somatic chromosome counts in these two genera (Tanaka and Uchiyama, 1988; Rahman et al., 2001) when flowering material is not available for meiotic/mitotic chromosome counts (however, flower buds were used to obtain the majority of karyotypes published so far, by Subramanyam and Kamble, 1968; Kondo, 1972a, b; Casper and Manitz, 1975; Greilhuber et al., 2006). In the present work, rhizophyll shoot apices were used for cytological studies in Genlisea for the first time.
A major focus of the present work was to study the evolution of genome sizes and chromosome numbers in a phylogenetic context. The genome sizes, given as 1C values, were measured from cultivated plant material for 20 accessions (representing 12 taxa of the genus, or 40 % of the total species number), with representatives from both subgenera and all four sections (Fleischmann et al., 2010; Fleischmann, 2012), using flow cytometry. Chromosome counts were performed for the majority of taxa.
The aim of this study was to address the following questions in Genlisea. (1) How does genome size correlate with chromosome number/size in the genus? (2) What are the most likely ancestral states of ploidy level and what is the variation in genome size for major infrageneric lineages? (3) How does the observed pattern in ploidy level and variation in genome size correlate with growth forms, altitude and habitat preferences?
MATERIALS AND METHODS
Plant material
The Genlisea taxa used for this study were cultivated by the first author under greenhouse conditions and in a terrarium setup using artificial lights. Cultivation conditions and propagation of the plants were as described by Fleischmann (2012). Additional in vitro plant material from tissue culture was obtained from a commercial source (bestcarnivorousplants.com) for flow cytometric measurements.
Isolation and staining of nuclei and analysis of nuclear DNA content
Some of the species tested in this study were analysed using different, established methods at the Rutgers and Vienna laboratories, representing identical plant material. This seems reasonable because of the possibility, when measuring ultrasmall genome sizes, of laboratory-specific errors that may be associated with the equipment used, the stage of the tissue or secondary metabolites originating from the size standard plant material that interact with DNA or interfere with DNA staining. Additionally, different internal size standards were applied for the flow cytometric measurements in the two laboratories, to minimize the negative contribution of cytosolic compounds from the standard plant tissue to the variation in gene size measurements. At Rutgers, nuclei were isolated then mixed to reduce the effect of polysaccharides and polyphenols. Due to the abundance of polysaccharides and polyphenols in G. aurea tissue (Płachno et al., 2007; Fleischmann and Heubl, 2009), the protocol for measurement of nuclear DNA content by flow cytometry was modified (Peterson et al., 2000; Doležel et al., 2007). Briefly, 10 mg of fresh Genlisea plant tissue was placed in 1 ml of ice-cold 2-methyl-2,4-pentanediol (MPD)-based extraction buffer (Peterson et al., 2000) and immediately chopped into very fine slices, and the homogenate was filtered through a 30-μm nylon mesh. The nuclei were pelleted at 9000 rpm for 2 min, the supernatant was carefully removed and the nuclei were resuspended in 500 μl of Galbraith's buffer (Galbraith et al., 1983). The same steps were conducted for the internal controls Spirodela polyrhiza ‘7498’ (1C = 0·16 pg or 158 Mbp; Wang et al., 2011), Brachypodium distachyon ‘Bd21’ (1C = 0·31 pg or 300 Mbp; Bennett and Leitch, 2005), Selaginella apoda (1C = 0·10 pg or 88 Mbp; Little et al., 2007) and Arabidopsis thaliana ‘Columbia’ (1C = 0·16 pg or 157 Mbp; Bennett et al., 2003). The Genlisea sample and corresponding internal standard were combined (the ratio was determined empirically with respect to the concentration of nuclei in each plant, so that their G1 peaks were of similar height on the histograms of DNA content), 50 μl of stock solution (1 mg/ml) of propidium iodide (Sigma) and 5 μl of stock solution (5 mg/ml) of RNase A (Sigma) were added and the sample was incubated on ice before flow cytometry. Propidium iodide-stained nuclei were analysed for DNA content with a Coulter Cytomics FC500 Flow Cytometer (Beckman Coulter). In all experiments, the fluorescence of at least 3000 G1-phase nuclei was measured. DNA content of each target sample was calculated by comparing its mean nuclear fluorescence with an internal standard. At least two independent replicates for each sample were analysed on different days to obtain the mean DNA content, and the exact number of replicates is specified in Table 1.
Table 1.
Plant material used for the present study, including estimated average of genome sizes (Vienna, flow cytometry), size approximations (Rutgers, flow cytometry) and karyotypes of Genlisea. All genome size data represent at least two independent measurements (different preparations performed on different days; N = number of runs). The size standards used (abbreviation, value and reference) are explained in the text (Materials and methods). Cytological data for taxa with small chromosomes are based on at least ten different cells counted.
| Sample |
Rutgers, flow cytometry |
Vienna, flow cytometry |
Cytology |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Species | Voucher* | 1C value (Mbp) | s.d. (N) | Standard | 1C value, size approximation (Mbp) | s.d. (N) | Standard | Chromosome number (this study) | Chromosome size range (based on mean of 10 cells) | Previous report |
| G. aurea var. aurea CdJ | Brazil, São Paulo, Campos do Jordão (LE366) | 83 | 8·5 (3) | Sa | 64·4 | 2·2 (5) | At | – | 0·2–0·4 μm | – |
| G. aurea var. aurea Cap | Brazil, Minas Gerais, Serra do Caparaó | 73 | 9·9 (10) | Bd, Sp, Sa | 67·2 | 1·2 (3) | At | – | – | – |
| G. aurea var. aurea ChdV | Brazil, Goiás, Chapada dos Veadeiros (LE368) | – | – | – | 63·6 | 2·4 (5) | At | – | – | – |
| G. aurea var. minor Ita | Brazil, Minas Gerais, Itacambira | 119 (estimated size) | – (2) | Bd | – | – | – | 2n ∼46 | 0·4–0·8 μm | G. aurea (without location data) 2n ∼50 (Greilhuber et al., 2006); 2n = 46 (Vu et al., 2012) |
| G. aurea var. minor ChdG | Brazil, Mato Grosso, Chapada dos Guimarães (LE360) | 131 | 6·1 (3) | Bd | 117·1 | 3·9 (3) | Sc | – | – | – |
| G. flexuosa | Brazil, Minas Gerais, Grão Mogol (LE364, as G. aff. violacea ‘giant’) | – | – | – | 1140·3 | 18·4 (4) | Zm | 2n = 16 | 1–2·3 μm | – |
| G. glandulosissima | Zambia, Northern, Kasama (LE263) | – | – | – | 189·3 | 1·2 (3) | Rs | 2n ∼38 | ∼0·5 μm | – |
| G. guianensis | Venezuela | – | – | – | 298·1 | 2·4 (3) | Sc | 2n ∼40 | 0·3–0·7 μm | |
| G. hispidula | South Africa, Pretoria, cult. BG Munich (LE294) | – | – | – | – | – | – | 2n = 32 | 0·8–1 μm | 2n = 40 (Vu et al., 2012), but pictured are 32 chromosomes |
| G. lobata | Brazil, Minas Gerais, Serra da Araponga (LE296) | – | – | – | 1722·4 | 2·7 (3) | Ps | 2n = 16 | – | 2n = 16 (Greilhuber et al., 2006) |
| G. margaretae | Madagascar (LE309) | 143·0 | 2·6 (3) | Bd, Sa | 195·2 | 2·3 (3) | Rs | 2n = 36 | – | 2n ∼ 40 (Greilhuber et al., 2006); voucher doubtful); 2n = 36 (Vu et al., 2012) |
| G. margaretae | Zambia, Luapula, Mansa (LE260) | 113 | 11·3 (2) | Bd, Sa | 180·8 | 0·9 (3) | Rs | 2n = 36, 38 (meiotic counts; n = 18, 19) | 0·5–0·8 μm | – |
| G. metallica | Brazil, Minas Gerais, Itacambira (LE365, as ‘G. sp. Itacambira’) | – | – | – | 1056·8 | 7·6 (3) | Sc | 2n = 16 | 1·2–2·3 μm | – |
| G. nigrocaulis | – | – | – | – | – | – | – | – | – | 2n ∼30 (as G. pygmaea; Vu et al., 2012) |
| G. oxycentron | Brazil, Pará, Vigia (LE321) | – | – | – | 74·6 | 0·5 (3) | At | – | – | – |
| G. repens | Venezuela, Amazonas, Aracamuni; Rivadavia et al., 1903 (SPF) | 86·1 | 3·6 (3) | At, Bd | – | – | – | – | – | – |
| G. repens | Chapada dos Veadeiros | – | – | – | 141·9 | 2·2 (4) | Sc | – | – | – |
| G. repens | Roraima | – | – | – | 80·9 | 1·0 (5) | At | – | – | – |
| G. repens | Cerro Avispa | – | – | – | 78·0 | 1·0 (3) | At | – | – | – |
| G. repens | Brazil, Minas Gerais, Uberaba (LE371) | – | – | – | 149·7 | 1·7 (3) | Sc | – | – | – |
| G. subglabra | Zambia, Kasama (LE266) | – | – | – | – | – | – | 2n = 32 | ∼1 μm | – |
| G. tuberosa | Brazil, Goiás, Chapada dos Veadeiros; Rivadavia & Batista 2611 (SPF) | 61 | 5·7 (2) | Sa | 65·0 | – (1) | At | – | – | – |
| G. uncinata | Brazil, Bahia, Mucugê (LE367) | – | – | – | – | – | – | 2n = 16 | – | 995 Mbp (Greilhuber et al., 2006; from Feulgen) |
| G. violacea | Brazil, Minas Gerais, Couto de Magalhães de Minas (LE303) | – | – | – | – | – | – | 2n = 16 | ∼1 μm | – |
| G. violacea | Brazil, Minas Gerais, Serra do Cipó (LE312) | – | – | – | 1609·7 | 3·3 (3) | Ps | 2n = 16 | 1·5–4 μm | – |
At, Arabidopsis thaliana; Bp, Brachypodium distachyon; Ps, Pisum sativum; Rs, Raphanus sativus; Sa, Selaginella apoda; Sc, Solanum pseudocapsicum; Sp, Spirodela polyrhiza; Zm, Zea mays.
*Laboratory code number. LE indicates identical material used by Fleischmann et al. (2010) for phylogenetic reconstruction.
The absolute DNA content of a sample was calculated on the basis of the G1 peak means: 1C nuclear DNA content of the sample = (sample G1 peak mean)/(standard G1 peak mean) × 1C DNA content of standard (Mbp).
At Vienna, ∼25 mg of fresh leaves was co-chopped as described by Galbraith et al. (1983) with the internal standard material A. thaliana (1C = 0·16 pg or 157 Mbp; Bennett et al., 2003), Solanum pseudocapsicum (1C = 1·29 pg or 1266 Mbp; Temsch et al., 2010), Pisum sativum ‘Kleine Rheinländerin’ (1C = 4·42 pg or 4322·8 Mbp; Greilhuber and Ebert, 1994), Raphanus sativus ‘Saxa’ (1C = 0·61 pg or 596·6 Mbp; Doležel et al., 1998) and Zea mays ‘CE-777’ (1C = 2·73 pg or 2670 Mbp; Doležel et al., 1998) in Otto's buffer I (Otto et al., 1981), and the preparation was filtered and incubated at 37 °C for 30 min with RNase (RNase A; Sigma). Subsequently, the isolates were stained in a propidium iodide (50 mg/L) solution containing Otto's buffer II (Otto et al., 1981). The measurement was performed with a Cyflow ML flow cytometer (Partec, Münster, Germany) equipped with a green laser (100 mW, 532 nm; Cobolt Samba, Cobolt, Stockholm, Sweden). For each preparation, three runs were done with a total of 15 000 particles, recording the fluorescence intensity and side scatter for each particle. The coefficients of variation of the G1 nuclei peaks were usually <3 %; if higher, up to five runs were measured. The 1C values of the Genlisea taxa were calculated for each run according to the formula described above. The run results were averaged to obtain the preparation's C value.
A few taxa (G. tuberosa, G. aurea var. minor from Itacambira; Table 1) that yielded interesting preliminary results with flow cytometry were included for ‘size estimations, based on one or two runs.
Chromosome preparation from rhizophylls
The apical tips of developing rhizophylls [before the apical end forked (Fig. 1C) or at a stage just after the two arms branched dichotomously, but before they began twisting helically (Fig. 1D)] were taken from cultivated plants grown under artificial light, in the late afternoon, and pre-treated with 0·002 m 8-hydroxyquinoline for 15 h at 4 °C to achieve mitotic arrest. The tips were then fixed in ethanol:glacial acetic acid (3:1 v/v) and stored at 4 °C. Fixed rhizophyll tips were hydrolysed in 2 N hydrochloric acid at 60 °C for 30 min, then rinsed with distilled water and squashed in 45 % acetic acid on glass slides. The prepared rhizophyll tip meristems were air-dried after removing the cover slip by the dry ice technique, and stained with aceto-orcein (Orcein; Roth) or DAPI (4′-6-diamidino-2-phenylindole; Merck, 2 μg/ml in McIlvaine's buffer, pH 7·0). Slides for DAPI staining were pre-incubated in McIlvaine's buffer for 30 min at room temperature before staining. DAPI-stained slides were incubated in the dark at room temperature for at least 15 min before use.
Chromosome preparation from Genlisea flower buds for species with very small chromosomes
Floral buds at an early stage of development (at a size when the sepal tips are still apically touching; Fig. 1B) were taken from young inflorescences of cultivated plants and fixed in freshly prepared ethanol:glacial acetic acid (3:1 v/v) at room temperature overnight and kept at –20 °C. Fixed anthers were prepared from the dissected flower buds and washed in distilled water, and the thecae were dissected in a drop of 45 % acetic acid and squashed. Coverslips were removed after freezing and preparations were air-dried at room temperature.
To access the DNA for staining, we used the first steps of the fluorescence in situ hybridization protocol modified by Sousa et al. (2013) before staining slides with DAPI. Firstly, slides were pre-fixed in a solution of 3:1 (v/v) ethanol:glacial acetic acid for 15 min, dehydrated for 5 min in a 70 % and 100 % ethanol series at room temperature, and incubated for 30 min at 60 °C. After cooling the slides at room temperature for ∼10 min, they were pre-treated with 100 μg/ml RNase A (Sigma) in 2× saline–sodium citrate (SSC) buffer for 1 h at 37 °C in a wet chamber and washed three times for 5 min in 2× SSC. Then, they were treated with 10 μg/ml pepsin (Sigma) in 0·01 N HCl for 20 min at 37 °C in a wet chamber, washed twice for 5 min in 2× SSC, post-fixed in 4 % formaldehyde solution (Roth) for 5 min at room temperature, washed again three times for 5 min in 2× SSC, dehydrated for 5 min in a 70 % and 100 % ethanol series and air-dried for at least 1 h at room temperature before being stained. The chromosomes were stained with DAPI (2 μg/ml) and mounted in Vectashield (Vector).
Chromosome counting
Chromosome counts were made using a fluorescence light microscope (DMR RXE; Leica) under 1000× magnification, and slides were documented photographically using a CCD camera (Kappa) and by camera lucida drawings, which were drawn combining different focus layers. The digital zoom of the Kappa camera was used for the very small chromosomes of most species, and digital images were optimized by manually improving colour contrast, brightness and noise rendering using Photoshop CS5 version 10·0 (Adobe).
Correlation of genome size and chromosome numbers
We used PhyloCom version 4.2 (Webb et al., 2008) to test for a correlation of genome size and chromosome numbers. Only taxa for which both chromosome counts and genome size estimates were available were included in the analysis. Since Phylocom turned out to be unable to parse Newick strings with non-numeric characters in branch lengths that used scientific notation, branches were uniformly scaled using the ‘calculate node/branch data’ function in TreeGraph 2 (Stöver and Müller, 2010). The significance of the correlation coefficient for independent contrasts was tested using tables of critical values for the Pearson correlation coefficient, using N – 1 degrees of freedom (d.f.), where N is the number of internal nodes providing contrasts.
RESULTS
Genome size in phylogenetic context
A tendency to reduce the genome size was not only generally evident in the genus Genlisea as a whole, but genome miniaturization was also repeatedly observed in all clades, except for G. subgenus Tayloria (Fig. 4). Within the subgenus Genlisea, the first-branching taxa of each clade had distinctly larger genomes than taxa in a more derived phylogenetic position in the same clade (e.g. G. margaretae from Madagascar in Genlisea section Recurvatae, G. guianensis in Genlisea section Genlisea and G. repens from Venezuela in the clade comprising G. repens, G. nigrocaulis, G. oxycentron and G. tuberosa).
Fig. 4.

Genome size variation and karyotype evolution in Genlisea. DNA 1C values of studied species and a phylogenetic tree based on molecular sequence data of three chloroplast loci (Fleischmann et al., 2010); the phylogenetic position of G. tuberosa is based on unpublished rps16 and trnQ-rps16 sequence data (deposited at GenBank, accession numbers KF952604 and KF952605); only tree branches that had maximal statistic support (bootstrap value >90, Bayesian posterior probability = 1) are shown as resolved; chromosome base numbers and suggested ploidy levels are shown on the respective branches of monophyletic groups. The range of 1C DNA content and chromosome numbers for the Lentibulariaceae outgroup taxa Pinguicula and Utricularia are taken from Greilhuber et al. (2006) and from Casper and Manitz (1975), Rahman et al. (2001) and Casper and Stimper (2009), respectively, 1C values for Genlisea uncinata are from Greilhuber et al. (2006) and those for G. nigrocaulis are from Vu et al. (2012).
Chromosome staining
Aceto-orcein stained chromosomes of Genlisea very well (Fig. 2), equivalent to Feulgen staining, which was used by Greilhuber et al. (2006). However the chromosomes of most species (all except members of subgenus Tayloria) were too small for accurate counting under a light microscope at 1000× magnification, as their size range of 0·2–0·8 μm was close to the maximum size resolution of light microscopes (the Abbé limit). Furthermore, the metaphase chromosomes were usually found clustered in a bundle in the nucleus, so that adjacent chromosomes could not be reliably differentiated. When chromosome preparations of species possessing very small chromosomes were stained with classical dyes (e.g. Giemsa, Feulgen and orcein), the cytoplasm was also stained by these dyes and a complete rounded cell was seen, and differentiation of the very small chromosomes was not possible.
Fig. 2.

Light microscope photographs and camera lucida drawings of orcein-stained mitotic prophase plates from rhizophyll tip meristems of Genlisea spp. (A, B) G. uncinata, 2n = 16. (C, D) G. metallica, 2n = 16. (E, F) G. violacea from Couto de Magalhães, 2n = 16. (G, H) G. flexuosa, 2n = 16. (J, K) G. hispidula, 2n = 32. (L, M) G. subglabra, 2n = 32. (N, O) G. guianensis, 2n ∼40. Scale bars = 3 μm.
The fluorochrome DAPI, which preferentially stains DNA regions rich in AT bases, did not stain the chromosomes of Genlisea species well without additional pre-treatment to remove DNA background noise as described above; we believe that Genlisea species have a high density of cytoplasmic components that are stained by these dyes and thus are only accessible after treatment with RNase A (which degrades RNA into smaller components) and pepsin. The GC content in Genlisea is not exceptionally high, but differs strongly between species (Veleba et al., 2014). The species with ultrasmall genomes, as studied by Greilhuber et al. (2006) and Ibbarra-Laclette et al. (2013), had DNA of a relatively high GC content, which could make them generally less receptive to staining. Nucleoli of G. aurea could be easily identified as DAPI-negative regions surrounded by more DAPI-dense DNA (Fig. 3).
Fig. 3.

Metaphase chromosomes from pollen mother cells of Genlisea spp., stained with DAPI. (A–C) G. margaretae from Zambia, n = 18, 19. (C) Two meiotic metaphase cells. (D–F) G. aurea var. minor from Itacambira, pre-meiotic mitosis cells, 2n = 46. Note the smaller chromosome size compared with G. margaretae. (F) DAPI-stained interphase nuclei. Nucleoli (*) can be identified as DAPI-negative regions surrounded by more DAPI-positive DNA staining. Scale bars = 10 μm.
Genome sizes and karyotypes
The known Genlisea genome sizes, chromosome numbers and chromosome size ranges (previously published or newly presented here) are summarized in Table 1.
Chromosome counts made for selected species of Genlisea, representing different phylogenetic clades, displayed variation between 2n = 16 and 2n∼46. Species with 2n = 16 were restricted to Genlisea subgenus Tayloria (Figs 2 and 4) and displayed larger chromosomes (1–4 μm) than species with higher chromosome numbers (all <1 μm) (Table 1). In Genlisea section Africanae, the two studied species showed 2n = 32, likely resulting from polyploidization, while the consecutive sister clade, section Recurvatae, exhibited dysploid series of 2n = 36 and 38 (Figs 2–4). The single member of section Genlisea with ultrasmall chromosomes for which chromosome counts could be performed was G. aurea var. minor with 2n∼46 (Table 1, Fig. 3).
Correlation of genome size and chromosome numbers
The correlation of independent contrasts was 0·131, which is statistically insignificant (number of internal nodes providing contrasts = 8, d.f. = 7, P > 0·05).
DISCUSSION
Comparison of 1C values from flow cytometry and Feulgen densitometry
The flow cytometric measurements of identical material showed a few laboratory-specific differences between Vienna and Rutgers (Table 2, Figs 4 and 5). The values obtained at Vienna were notably higher than the Rutgers size estimates, with the exception of G. aurea, which had a slightly lower 1C value for all three accessions run in parallel. This is most likely due to the different genome size standard species used in the different laboratories, and the Rutgers values are here considered as size estimates, compared with the Vienna flow cytometric data.
Table 2.
Comparison of 1C DNA contents of the same taxa (identical plant material used) measured independently by flow cytometry at the Vienna and at Rutgers laboratories
| Voucher | Species | Vienna 1C value (Mbp) | Rutgers 1C value (Mbp) | Deviation from Vienna value (100 %) |
|---|---|---|---|---|
| – | G. aurea var. aurea Serra do Caparaó | 67·2 | 73 | 108·6 |
| LE366 | G. aurea var. aurea Campos do Jordão | 64·4 | 83 | 128·9 |
| LE360 | G. aurea var. minor Chapada dos Guimarães | 117·1 | 131 | 111·9 |
| – | G. tuberosa | 65·0 | 61 | 93·8 |
| LE309 | G. margaretae Madagascar | 195·2 | 143·0 | 73·3 |
| LE260 | G. margaretae Zambia | 180·8 | 113 | 62·5 |
Fig. 5.

Evolutionary decrease in genome size in Genlisea. The species are arranged in phylogenetic order on the x-axis, from basal branching (left) to derived taxa (right; following Fleischmann et al. 2010); their average 1C values are shown on the y-axis. The 1C values obtained as size estimates from flow cytometry measurements at Rutgers are marked ‘(R)’. The respective 2n number is indicated where known. Genome sizes of taxa marked with an asterisk are taken from the literature (Greilhuber et al., 2006; Vu et al., 2012). For abbreviations of location data, see vouchers in Table 1.
As becomes evident from comparison of the 1C values obtained by Feulgen densitometry (published in Greilhuber et al., 2006) and flow cytometry, the measured sizes for some taxa (considering the identical material used) differ slightly to significantly. The cases in which significant discrepancies were observed are discussed below. The genome size values we obtained are generally supported by those published by Veleba et al. (2014), with the notable exceptions of G. lobata and G. violacea, for which they obtained significantly lower 1C values using flow cytometry (see below).
For G. lobata, flow cytometry yielded a 1C DNA content of 1722 Mbp, while 1277 Mbp (74 %) was previously obtained with Feulgen densitometry (Greilhuber et al., 2006). This points to some stoichiometric error in the published Feulgen data, according to Greilhuber; the new, higher 1C value obtained by flow cytometry was used for the species in the present analyses, although the data of Veleba et al. (2014) would indicate a flow cytometric 1C value of 1200 Mbp for this species.
In G. violacea, Greilhuber et al. (2006) reported 1C = 1005 Mbp, based on Feulgen measurements, while our flow cytometric measurements published here show a 1C value of ∼1609 Mbp. This species, however, is widespread and morphologically quite variable, and was shown to be paraphyletic (Fleischmann et al., 2010), recently being split into several distinct species (Fleischmann et al., 2011). Unfortunately, we cannot trace back whether G. violacea s.str. or another closely related species (e.g. G. flexuosa) was used for the Feulgen measurements published by Greilhuber et al. (2006), as no voucher was made from the plant material used. The new flow cytometric value for the species was used in this study, accompanied by measurements for two closely related species, G. flexuosa (1C ∼1140 Mbp) and G. metallica (1C ∼1057 Mbp). The notably lower 1C value of 460 Mbp for G. violacea published by Veleba et al. (2014) seems very implausible in the phylogenetic and cytological context, and might be based on a measurement error or misidentification. Plant material from an identical source was found in the present study to be of the same karyotype as all other accessions of G. violacea (Table 1). Moreover, all members of Genlisea subgenus Tayloria studied thus far (including G. violacea) are commonly characterized by large genome sizes of ∼1000 Mbp or higher (Table 1). Thus the different, much lower value is unlikely to have resulted from ‘unrecognized taxonomic diversity’ or ‘unrecognized karyological variability’, as used in explanation by Veleba et al. (2014).
A general conclusion from the genome size data available now is the necessity to revise the published Feulgen densitometric data in Lentibulariaceae (Greilhuber et al., 2006), using flow cytometry together with a unified selection of internal standards, and to combine these data with reliable chromosome counts in defined individuals. On this basis, deviating genome sizes and ploidy levels can then be identified. While Feulgen densitometry has proved to be a reliable method in many investigations (reviewed by Greilhuber, 1998, 2008), the method is considerably more sensitive than flow cytometry in regard to stoichiometric errors, due to secondary metabolites and also mechanical barriers against the reagents. Secondary metabolites, which are found in abundance in plant tissues of certain species of Genlisea, such as G. aurea, with its numerous foliar mucilage glands (Płachno et al., 2007; Fleischmann and Heubl, 2009; Fleischmann, 2012), were shown to negatively affect the Feulgen reaction, and therefore hamper densitometry measurements of the DNA content (Greilhuber, 1998, 2008). However, other cytosolic compounds were also recently shown to interfere with flow cytometric measurements (Greilhuber et al., 2007; Temsch et al., 2008). Generally, however, flow cytometry is not only much faster but also less prone to interference, since isolated nuclei are stained.
Karyotype evolution
All members of Genlisea subgenus Tayloria that were analysed were diploids, with 2n = 16, and possessed comparatively large chromosomes 1–4 μm in length. In contrast, the sister group Genlisea subgenus Genlisea seemed to be derived from a tetraploid lineage with 32 small chromosomes. We suggest that the haploid number n = 8 represents the ancestral chromosome number for the genus Genlisea. Due to the lack of cytological data for the sister genus Utricularia, however, inference of the ancestral chromosome number of Genlisea is difficult.
Reported base numbers for Utricularia are x = 6, 7, 9, 10 and 11 (Subramanyam and Kamble, 1968; Casper and Manitz, 1975); however, only ∼10 % of the species of the large genus Utricularia have been studied cytologically thus far, while those in the common sister genus Pinguicula have x = 6, 8, 9, 11 and 14 (Casper and Stimper, 2009). Common evidence of n = 8 was found in many species of Genlisea; however, cytogenetic support for a common ancestral base number of x = 8 is still lacking for the sister genus Utricularia. Some support for the hypothesis of x = 8 in the clade is given by the fact that the supposed primary basic number in the common sister genus Pinguicula is also x = 8 according to Casper and Stimper (2009). As the cytology of Genlisea and its sister genus Utricularia is poorly studied and few chromosome counts are known at present, the estimated haploid number n = 8 and its evolutionary meaning obviously may change as more cytogenetic studies become available and are interpreted in the phylogenetic context (Cusimano et al., 2012).
Tetraploids with 2n = 4x = 32 can be found in the basal branching Genlisea section Africanae, here represented by G. hispidula and G. subglabra (Fig. 4). In the derived sections Recurvatae and Genlisea, likely dysploidy led to chromosome numbers of 2n = 36–38. Greilhuber et al. (2006) assumed that differences in chromosome number were a result of fission–fusion rearrangements, but not polyploidy; however, this hypothesis could not be tested in the focus of the present study. At least in the basalmost branching lineages of Genlisea subgenus Genlisea, we found what could be the true tetraploid group, with 2n = 32.
In Genlisea, the chromosome number was found to be negatively correlated with chromosome size. We observed that species with fewer chromosomes (diploids of Genlisea subgenus Tayloria with 2n = 16 and the tetraploids G. hispidula and G. subglabra with 2n = 32) have comparatively large chromosomes (especially evident in members of Genlisea subgenus Tayloria if compared with the rest of the genus; Fig. 2) than the remainder of Genlisea species of subgenus Genlisea, which possess very small chromosomes (dysploids with 2n = 36, 38 and ∼46). The euploid species with larger chromosomes also displayed large genome sizes, while ultrasmall genomes could only be found in the dysploid species with very small chromosomes (Table 1, Fig. 4).
In G. aurea, flow cytometry measurements performed in Vienna for four accessions showed two DNA levels: one shared by three accessions yielded a 1C DNA content of ∼65 Mbp and the other accession 117 Mbp (1C). In the Rutgers laboratory the values were similar but somewhat higher, at 73–83 and 119–131 Mbp, respectively. The value of 131 Mbp has been confirmed independently for G. aurea by Veleba et al. (2014) – the accession they used was G. aurea from Itacambira (only variety minor occurs in that area), but their value does not correspond to our results for the same taxon from that region. Feulgen densitometry (Greilhuber et al., 2006) had revealed 63·6 Mbp for G. aurea. A later Feulgen test in material from Gatersleben yielded 150 Mbp (1C) (I. Schubert, IPK Gatersleben, Germany, pers. comm.), and thus was similar to the flow cytometry data in Gatersleben and about twice the value measured previously by Feulgen staining, and even higher than that measured by flow cytometry in the present study. As genome size is not generally correlated to ploidy levels (Leitch and Bennett, 2007; Leong-Škorničková et al., 2007), this does not necessarily indicate the occurrence of two ploidy levels (e.g. as put forward by Veleba et al., 2014), but can rather be explained by the inclusion of two infraspecific taxa of G. aurea in the study (see below).
Genome size evolution
Interestingly, the ploidy shift from diploids (2n = 16 in Genlisea subgenus Tayloria) to likely tetraploids (2n = 32 at least in the basal-branching G. hispidula of subgenus Genlisea) does not directly correspond to a notable decrease in genome size (995–1722 Mbp in subgenus Tayloria compared with ∼1500 Mbp in G. hispidula), as repeatedly observed in several angiosperm groups, where polyploidy often goes along with genome miniaturization (e.g. Leitch and Bennett 2004; Weiss-Schneeweiss et al., 2006). However, in the case of the two Genlisea subgenera we are dealing with two immediate sister clades but not a phylogenetic grade, which makes the reconstruction of the ancestral karyotype difficult. Thus, it is best to examine both subgenera independently, as previously done with contrasting morphological traits (Fleischmann et al., 2010).
In Genlisea subgenus Tayloria an apparent evolutionary increase in genome size is evident (Figs 4 and 5), ranging from 995 Mbp in the basalmost branching G. uncinata to ∼1600–1700 Mbp in the more derived species G. violacea and G. lobata. This increase in 1C DNA content is consistent with the shift from perennial to annual life history (Fleischmann et al., 2010). This correlation of genome size with generation time is remarkable, as this tendency is opposite to what occurs in the majority of angiosperms, in which annual taxa generally show smaller genome sizes than perennial taxa (Bennett, 1972; Bennett and Leitch, 2011).
In the sister group Genlisea subgenus Genlisea, a similar situation was not observed, as annual and ephemeral taxa (such as G. oxycentron, 1C ∼75 Mbp) do not show any significant difference in genome size compared with their perennial sisters (e.g. G. nigrocaulis, 1C = ∼ 86 Mbp; Vu et al., 2012). However, the taxon sampling for 1C DNA content of annual species of Genlisea (e.g. G. stapfii, G. barthlottii and G. filiformis) was rather deficient in this study due to difficulties in obtaining enough plant material of these delicate short-lived species.
Within G. aurea, two distinct groups of genome size range were evident (one with ultrasmall genomes of 63·5–83 Mbp and another group displaying an almost 2-fold size range, of 117–131 Mbp). These two groups correlate with the two morphologically distinct, geographically more or less separated varieties, G. aurea var. aurea (ultrasmall genomes) and G. aurea var. minor (small genomes; Fleischmann, 2012). The wide span of genome sizes observed in G. aurea populations was assumed by Albert et al. (2010) to result from DNA double-strand breaks caused by reactive oxygen species (ROS) as by-products of increased oxidative phosphorylation due to a unique cytochrome c oxidase modification found in Lentibulariaceae (Jobson et al., 2004; Laakkonen et al., 2006). This ROS hypothesis was also used by Albert et al. (2010) to explain the small genome sizes and the very high nucleotide substitution rates observed in Genlisea and Utricularia (Jobson and Albert, 2002; Müller et al., 2004), as a result of error-prone DNA repair mechanisms and chromatin breaks. The same mutagenic action of self-produced ROS was postulated by Ibarra-Laclette et al. (2011a, b, 2013) for Utricularia gibba, likewise a species with a minimal genome. This would be consistent with the shorter non-coding sequences and introns and less repetitive sequences that characterize the ultrasmall genomes of G. aurea (Leushkin et al., 2013) and U. gibba (Ibarra-Laclette et al., 2013).
It is generally recognized that in plants, but also other organisms, genome sizes increase due to the proliferation of long terminal repeat (LTR) retrotransposons (Kellogg and Bennetzen, 2004). In contrast, the mechanisms of genome size reduction are less well characterized, but evidence is emerging from whole-genome analyses in Arabidopsis, rice (Oryza) and recently the miniature genomes of G. aurea and U. gibba. In a comparison of two closely related Arabidopsis species, A. thaliana and A. lyrata, the smaller genome of the former could be explained by many small deletions (Hu et al., 2011). In contrast, genome size differences in rice are explained by both LTR retrotransposon expansion and DNA loss as a result of double-strand break repair through non-homologous end joining (Ma and Bennetzen, 2004; Chen et al., 2013). Utricularia gibba has few, if any, full-length and presumably active LTRs, consistent with its reduction in genome size (Ibarra-Laclette et al., 2013). Interestingly, U. gibba has undergone three rounds of whole-genome duplication since ancestry with tomato (Solanum), and its genome has decreased to one-ninth the size while maintaining the standard number of genes for a plant (Ibarra-Laclette et al., 2013). It is compelling to speculate that there is a force actively removing LTR retrotransposons or excess DNA from whole-genome duplication, resulting in miniature genome sizes. The mechanism driving a genome to lose DNA is still unknown, and more in-depth studies of Genlisea genomes could therefore provide clues about the underlying mechanisms.
Further correlations between geography and genome size are evident in Genlisea. Accessions of G. repens from Brazil south of the Amazon (from Uberaba, Minas Gerais state, and from the Chapada dos Veadeiros in Goiás state) have relatively large genomes (Table 1, Fig. 5) compared with accessions from lower latitudes and high-altitude mountain summits of Venezuelan tepuis north of the Amazon basin (from Roraima tepui in Bolívar state and Cerro Aracamuni and Cerro Avispa in Amazonas state). This geographical difference in genome size is also mirrored in phylogenetic reconstructions (Fleischmann et al., 2010). Interestingly, G. repens with the larger genomes are phylogenetically close to G. oxycentron and G. nigrocaulis, species that exhibit ultrasmall genomes.
A geographical correlation of horizontal (latitudinal) and vertical (altitude) distribution with infraspecific DNA content was previously noticed in some plant species (Ohri and Khoshoo, 1986; Rayburn and Auger, 1990; Reeves et al., 1998; Bottini et al., 2000; Leong-Škorničková et al., 2007; Díez et al., 2013), while other studies found no such correlation (summarized in Wang et al., 2011). It is assumed that variation in genome size might have importance for the adaptation of plants to different ecological and environmental conditions (Bottini et al., 2000). All three accessions of G. repens from high-altitude tepui summits in Venezuela (from ∼1350 m on Cerro Avispa and Aracamuni to 2700 m on Roraima tepui) had smaller genomes than the two accessions from upland savannah of central Brazil (altitude ∼1000 m); this trend, however, is opposite to an observed increase in DNA content in Zea mays growing at increasing altitudes (Rayburn and Auger, 1990). Regarding the correlation of genome size with chromosome number, more data, particularly on chromosome counts, are needed to clearly establish a putative relationship between chromosome number and genome size.
Genlisea aurea and G. tuberosa have the smallest genomes, not G. margaretae
The smallest genome sizes currently known in angiosperms were obtained in the Feulgen densitometry measurements of the two sister species G. aurea var. aurea (1C values ranged from 63·4 to 67·2 Mbp; flow cytometry results were slightly higher, 1C ∼73–83 Mbp) and G. tuberosa (1C ∼65 Mbp, size estimation was based on a single run, but the low range was confirmed by flow cytometric results of 1C ∼61 Mbp). The ultrasmall genome size of 63·4 Mbp reported from Feulgen measurements by Greilhuber et al. (2006) for G. margaretae from Madagascar (from a cultivated plant in Bonn Botanical Gardens) actually belongs to a specimen of G. aurea, probably even the same accession that yielded an identical value in this study. The ultrasmall 1C value of G. margaretae could not be reproduced with Feulgen densitometry or flow cytometric measurements, either for material of G. margaretae from Bonn or for any of the other accessions of the same species used in the present study (vouchers LE309 and LE260). All accessions of G. margaretae independently had an ∼3-fold genome size of ∼180–195 Mbp with both flow cytometry and Feulgen measurements. This was confirmed by flow cytometric measurements of material of identical strains of G. margaretae sent to the IPK Gatersleben (Vu et al., 2012; I. Schubert, pers. comm.). A later test made by J. Greilhuber with Feulgen densitometry on G. margaretae (LE309) revealed the same larger value, combined with a microscopic appearance of relatively dense euchromatin in the interphase nuclei even in differentiated tissue. A young trap fixed in ethanol-acetic acid (3:1) sent to Vienna yielded 185 and 189 Mbp (1C) in meristem and differentiated tissues, respectively.
Genlisea aurea and G. margaretae have a very similar vegetative morphology, with dense rosettes consisting of narrowly spathulate leaves, and are difficult to tell apart when not in flower. Although no herbarium vouchers are available, it seems likely that plants were mixed up in cultivation in Bonn, and material of G. aurea was erroneously used as ‘G. margaretae’ by Greilhuber et al. (2006). Potted plants in Bonn Botanic Garden labelled as the respective voucher ‘11400’ from Madagascar were studied personally by the first author in spring 2011, and these did in fact represent G. aurea. A photo-voucher made in Vienna by Eva Temsch from the material sent from Bonn for Feulgen measurements, for the published value from 2006, could clearly also be identified as G. aurea by the first author and Fernando Rivadavia.
Conclusions
The present study represents the first comprehensive analysis of genome size variation in Genlisea, evaluated in a phylogenetic context and including members of all sections, representing half of the known species diversity of the genus. For several species, cytological data are reported for the first time, while for some species we were able to confirm or correct previously published chromosome numbers. Currently, the genus Genlisea contains species with the smallest known angiosperm genome sizes, and a new record holder was found during this study with a DNA 1C value of 61 Mbp in G. tuberosa, which is slightly smaller than the previously published smallest angiosperm genome of G. aurea (1C = 63·4 Mbp, previously assigned erroneously to G. margaretae). Genlisea is an interesting study case, because the genus contains species with both the largest and the smallest known genome sizes in Lentibulariaceae, spanning ∼1440 Mbp between the largest and smallest known 1C values. However, the underlying mechanism of genome size miniaturization in Genlisea is still unknown. As more high-throughput whole-genome sequencing data for Genlisea species are being compiled, it may become possible to infer mechanisms of genome reduction by comparative genome analysis, and Genlisea may become a suitable model genus for the study of genome reduction. Likewise, as soon as more karyotypes become available for Genlisea, the cytogenetic basis of genome reduction, chromosome miniaturization and increase in chromosome number could be examined. Finally, with more cytological data from the sister genus Utricularia, the common ancestral chromosome base number of the Genlisea–Utricularia lineage could be verified. With our comparative analysis of genome sizes and karyotypes and their phylogenetic distribution in the genus Genlisea, we provide an essential basis for the selection of suitable species for comparative genome analyses and for further studies on the molecular and cytogenetic base of genome reduction in plants.
ACKNOWLEDGEMENTS
Kamil Pasek, Thomas Carow, Matt Hochberg, Markus Welge and Matthias Teichert are thanked for providing additional plant material and Ingo Schubert, Jörg Fuchs, Lubomír Adamec and Adam Veleba for helpful correspondence. Two anonymous reviewers and the handling editor are thanked for valuable comments on the manuscript. This study was in part supported by the Deutsche Forschungsgemeinschaft, DFG Grant MU2875/2 to K.F.M. and by the DOE Plant Feedstock Genomics for Bioenergy Program (DE-FG02-08ER6430) to T.P.M.
LITERATURE CITED
- Adamec L. Zero water flows in the carnivorous genus Genlisea. Carnivorous Plant Newsletter. 2003;32:46–48. [Google Scholar]
- Albert VA, Jobson RW, Michael TP, Taylor DJ. The carnivorous bladderwort (Utricularia, Lentibulariaceae): a system inflates. Journal of Experimental Botany. 2010;61:5–9. doi: 10.1093/jxb/erp349. [DOI] [PubMed] [Google Scholar]
- Barthlott W, Porembski S, Fischer E, Gemmel B. First protozoa-trapping plant found. Nature. 1998;392:447. [Google Scholar]
- Bennett MD. Nuclear DNA content and minimum generation time in herbaceous plants. Proceedings of the Royal Society of London. Series B, Biological Sciences. 1972;181:109–135. doi: 10.1098/rspb.1972.0042. [DOI] [PubMed] [Google Scholar]
- Bennett MD, Leitch IJ. Nuclear DNA amounts in angiosperms: progress, problems and prospects. Annals of Botany. 2005;95:45–90. doi: 10.1093/aob/mci003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett MD, Leitch IJ. Nuclear DNA amounts in angiosperms: targets, trends and tomorrow. Annals of Botany. 2011;107:467–590. doi: 10.1093/aob/mcq258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett MD, Leitch IJ, Price HJ, Johnston JS. Comparisons with Caenorhabditis (∼100 Mb) and Drosophila (∼175 Mb) using flow cytometry show genome size in Arabidopsis to be ∼157 Mb and thus ∼25 % larger than the Arabidopsis Genome Initiative estimate of ∼125 Mb. Annals of Botany. 2003;91:547–557. doi: 10.1093/aob/mcg057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bottini MCJ, Greizerstein EJ, Aulicino MB, Poggio L. Relationships among genome size, environmental conditions and geographical distribution in natural populations of NW Patagonian species of Berberis L. (Berberidaceae) Annals of Botany. 2000;86:565–573. [Google Scholar]
- Casper SJ, Manitz H. Beiträge zur Taxonomie und Chorologie der mitteleuropäischen Utricularia-Arten. Feddes Repertorium. 1975;86:211–232. [Google Scholar]
- Casper SJ, Stimper R. Chromosome numbers in Pinguicula (Lentibulariaceae): survey, atlas, and taxonomic conclusions. Plant Systematics and Evolution. 2009;277:21–60. [Google Scholar]
- Chen J, Huang Q, Gao D, et al. Whole-genome sequencing of Oryza brachyantha reveals mechanisms underlying Oryza genome evolution. Nature Communications. 2013;4:1595. doi: 10.1038/ncomms2596. doi:10.1038/ncomms2596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cusimano N, Sousa A, Renner SS. Maximum likelihood inference implies a high, not a low, ancestral haploid chromosome number in Araceae, with a critique of the bias introduced by ‘x. Annals of Botany. 2012;109:681–692. doi: 10.1093/aob/mcr302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darnowski DW, Fritz S. Prey preference in Genlisea. Small crustaceans, not protozoa. Carnivorous Plant Newsletter. 2010;39:114–116. [Google Scholar]
- Darwin C. Insectivorous plants. London: Murray; 1875. [Google Scholar]
- Díez CM, Gaut BS, Meca E, et al. Genome size variation in wild and cultivated maize along altitudinal gradients. New Phytologist. 2013;199:264–276. doi: 10.1111/nph.12247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doležel J, Greilhuber J, Lucretti S, et al. Plant genome size estimation by flow cytometry; inter-laboratory comparison. Annals of Botany. 1998;82(Suppl A):17–26. [Google Scholar]
- Doležel J, Greilhuber J, Suda J. Estimation of nuclear DNA content in plants using flow cytometry. Nature Protocols. 2007;2:2233–2244. doi: 10.1038/nprot.2007.310. [DOI] [PubMed] [Google Scholar]
- Fleischmann A. Monograph of the genus Genlisea. Dorset: Redfern National History Publications; 2012. [Google Scholar]
- Fleischmann A, Heubl G. Overcoming DNA extraction problems from carnivorous plants. Anales del Jardín Botánico de Madrid. 2009;66:209–215. [Google Scholar]
- Fleischmann A, Schäferhoff B, Heubl G, Rivadavia F, Barthlott W, Müller KF. Phylogenetics and character evolution in the carnivorous plant genus Genlisea A. St.-Hil. (Lentibulariaceae) Molecular Phylogenetics and Evolution. 2010;56:768–783. doi: 10.1016/j.ympev.2010.03.009. [DOI] [PubMed] [Google Scholar]
- Fleischmann A, Rivadavia F, Gonella PM, Heubl G. A revision of Genlisea subgenus Tayloria (Lentibulariaceae) Phytotaxa. 2011;33:1–40. [Google Scholar]
- Galbraith DW, Harkins KR, Maddox JM, Ayres NM, Sharma DP, Firoozabady E. Rapid flow cytometric analysis of the cell cycle in intact plant tissues. Science. 1983;220:1049–1051. doi: 10.1126/science.220.4601.1049. [DOI] [PubMed] [Google Scholar]
- Goebel K. Pflanzenbiologische Schilderungen 2. Marburg: NG Elwert; 1891. [Google Scholar]
- Greilhuber J. Intraspecific variation in genome size: a critical reassessment. Annals of Botany. 1998;82(Suppl A):27–35. [Google Scholar]
- Greilhuber J. Cytochemistry and C values: the less-well-known world of nuclear DNA amounts. Annals of Botany. 2008;101:791–804. doi: 10.1093/aob/mcm250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greilhuber J, Ebert I. Genome size variation in Pisum sativum. Genome. 1994;37:646–655. doi: 10.1139/g94-092. [DOI] [PubMed] [Google Scholar]
- Greilhuber J, Borsch T, Müller KF, Worberg A, Porembski S, Barthlott W. Smallest angiosperm genomes found in Lentibulariaceae, with chromosomes of bacterial size. Plant Biology. 2006;8:770–777. doi: 10.1055/s-2006-924101. [DOI] [PubMed] [Google Scholar]
- Greilhuber J, Temsch EM, Loureiro J. Nuclear DNA content measurement. In: Doležel J, Greilhuber J, Suda J, editors. Flow cytometry with plant cells. Analysis of genes, chromosomes and genomes. Weinheim: Wiley-VCH; 2007. pp. 67–101. [Google Scholar]
- Hu TT, Pattyn P, Bakker EG, et al. The Arabidopsis lyrata genome sequence and the basis of rapid genome size change. Nature Genetics. 2011;43:476–481. doi: 10.1038/ng.807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibarra-Laclette E, Albert VA, Pérez-Torres CA, et al. Transcriptomics and molecular evolutionary rate analysis of the bladderwort (Utricularia), a carnivorous plant with a minimal genome. BMC Plant Biology. 2011a;11:101. doi: 10.1186/1471-2229-11-101. doi:10.1186/1471-2229-11-101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibarra-Laclette E, Albert VA, Herrera-Estrella A, Herrera-Estrella L. Is GC bias in the nuclear genome of the carnivorous plant Utricularia driven by ROS-based mutation and biased gene conversion? Plant Signaling & Behavior. 2011b;6:1631–1634. doi: 10.4161/psb.6.11.17657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibarra-Laclette E, Lyons E, Hernandez-Guzman G, et al. Architecture and evolution of a minute plant genome. Nature. 2013;498:94–98. doi: 10.1038/nature12132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jobson RW, Albert VA. Molecular rates parallel diversification contrasts between carnivorous plant sister lineages. Cladistics. 2002;18:127–136. doi: 10.1111/j.1096-0031.2002.tb00145.x. [DOI] [PubMed] [Google Scholar]
- Jobson RW, Nielsen R, Laakkonen L, Wikström M, Albert VA. Adaptive evolution of cytochrome c oxidase: infrastructure for a carnivorous plant radiation. Proceedings of the National Academy of Sciences of the USA. 2004;101:18064–18068. doi: 10.1073/pnas.0408092101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellogg EA, Bennetzen JL. The evolution and phylogenetic relevance of nuclear genome structure in plants. American Journal of Botany. 2004;91:1709–1725. doi: 10.3732/ajb.91.10.1709. [DOI] [PubMed] [Google Scholar]
- Kondo K. A comparison of variability in Utricularia cornuta and Utricularia juncea. American Journal of Botany. 1972a;59:23–37. [Google Scholar]
- Kondo K. Chromosome numbers of some angiosperms in the United States II. Phyton. 1972b;30:47–51. [Google Scholar]
- Laakkonen L, Jobson RW, Albert VA. A new model for the evolution of carnivory in the bladderwort plant (Utricularia): adaptive changes in cytochrome c oxidase (COX) provide respiratory power. Plant Biology. 2006;8:758–764. doi: 10.1055/s-2006-924459. 2006. [DOI] [PubMed] [Google Scholar]
- Leitch IJ, Bennett MD. Genome downsizing in polyploid plants. Biological Journal of the Linnean Society. 2004;82:651–663. [Google Scholar]
- Leitch IJ, Bennett MD. Genome size and its uses: the impact of flow cytometry. In: Doležel J, Greilhuber J, Suda J, editors. Flow cytometry with plant cells. Analysis of genes, chromosomes and genomes. Weinheim: Wiley-VCH; 2007. pp. 153–176. [Google Scholar]
- Leong-Škorničková J, Šída O, Jarolímová V, et al. Chromosome numbers and genome size variation in Indian species of Curcuma (Zingiberaceae) Annals of Botany. 2007;100:505–526. doi: 10.1093/aob/mcm144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leushkin EV, Sutormin RA, Nabieva ER, Penin AA, Kondrashov AS, Logacheva MD. The miniature genome of a carnivorous plant Genlisea aurea contains a low number of genes and short non-coding sequences. BMC Genomics. 2013;14:476. doi: 10.1186/1471-2164-14-476. doi:10.1186/1471-2164-14-476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Little DP, Moran RC, Brenner ED, Stevenson DW. Nuclear genome size in Selaginella. Genome. 2007;50:351–356. doi: 10.1139/g06-138. [DOI] [PubMed] [Google Scholar]
- Ma J, Bennetzen JL. Rapid recent growth and divergence of rice nuclear genomes. Proceedings of the National Academy of Sciences of the USA. 2004;101:12404–12410. doi: 10.1073/pnas.0403715101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller KF, Borsch T, Legendre L, Porembski S, Theisen I, Barthlott W. Evolution of carnivory in Lentibulariaceae and the Lamiales. Plant Biology. 2004;6:477–490. doi: 10.1055/s-2004-817909. [DOI] [PubMed] [Google Scholar]
- Ohri D, Khoshoo TN. Genome size in gymnosperms. Plant Systematics and Evolution. 1986;153:119–132. [Google Scholar]
- Otto F, Oldiges H, Goehde W, Jain VK. Flow cytometric measurement of nuclear DNA content variations as a potential in vivo mutagenicity test. Cytometry. 1981;2:189–191. doi: 10.1002/cyto.990020311. [DOI] [PubMed] [Google Scholar]
- Peterson DG, Tomkins JP, Frisch DA, Wing RA, Paterson AH. Construction of plant bacterial artificial chromosome (BAC) libraries: an illustrated guide. Journal of Agricultural Genomics. 2000;5:1–100. [Google Scholar]
- Płachno BJ, Kozieradzka-Kiszkurno M, Swiatek P. Functional ultrastructure of Genlisea (Lentibulariaceae) digestive hairs. Annals of Botany. 2007;100:195–203. doi: 10.1093/aob/mcm109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Płachno BJ, Kozieradzka-Kiszkurno M, Swiatek P, Darnowski DW. Prey attraction in carnivorous Genlisea (Lentibulariaceae) Acta Biologica Cracoviensia, Series Botanica. 2008;50:87–94. [Google Scholar]
- Rahman MO, Adamec L, Kondo K. Chromosome numbers of Utricularia bremii and Utricularia dimorphantha (Lentibulariaceae) Chromosome Science. 2001;5:105–108. [Google Scholar]
- Rayburn AL, Auger JA. Genome size variation in Zea mays ssp. mays adapted to different altitudes. Theoretical and Applied Genetics. 1990;79:470–474. doi: 10.1007/BF00226155. [DOI] [PubMed] [Google Scholar]
- Reese G. Ergänzende Mitteilungen über die Chromosomenzahlen mitteleuropäischer Gefäβpflanzen I. Berichte der Deutschen Botanischen Gesellschaft. 1951;64:240–255. [Google Scholar]
- Reeves G, Francis D, Davies MS, Rogers HJ, Hodkinson TR. Genome size is negatively correlated with altitude in natural populations of Dactylis glomerata. Annals of Botany. 1998;82(Suppl A):99–105. [Google Scholar]
- Sousa A, Fuchs J, Renner SS. Molecular cytogenetics (FISH, GISH) of Coccinia grandis: a ca. 3 myr-old species of Cucurbitaceae with the largest Y/autosome divergence in flowering plants. Cytogenetic and Genome Research. 2013;139:107–118. doi: 10.1159/000345370. [DOI] [PubMed] [Google Scholar]
- Stöver BC, Müller KF. TreeGraph 2: Combining and visualizing evidence from different phylogenetic analyses. BMC Bioinformatics. 2010;11:7. doi: 10.1186/1471-2105-11-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanyam K, Kamble NP. Chromosome numbers in certain Indian species of Utricularia L. (Lentibulariaceae) Proceedings of the Indian Academy of Sciences, Section B. 1968;68:221–224. [Google Scholar]
- Tanaka R, Uchiyama H. Chromosomes of four species of Utricularia in Japan. Journal of Japanese Botany. 1988;63:219–223. [Google Scholar]
- Temsch EM, Greilhuber J, Hammett KRW, Murray BG. Genome size in Dahlia Cav. (Asteraceae–Coreopsideae) Plant Systematics and Evolution. 2008;276:157–166. [Google Scholar]
- Temsch EM, Greilhuber J, Krisai R. Genome size in liverworts. Preslia. 2010;82:63–80. [Google Scholar]
- Veleba A, Bureš P, Adamec L, Šmarda P, Lipnerová I, Horová L. Genome size and genomic GC content evolution in the miniature genome-sized family Lentibulariaceae. New Phytologist. 2014;203:22–28. doi: 10.1111/nph.12790. [DOI] [PubMed] [Google Scholar]
- Vu GTH, Cao HX, Bull F, et al. San Diego: Poster at Plant & Animal Genome Conference XX; 2012. Genome size evolution in the genus Genlisea – from half of the A. thaliana genome to the twenty-fold. 2012. www.opgen.com/wp-content/uploads/36_-Vu-et-al-PAG2012.pdf . [Google Scholar]
- Wang W, Kerstetter RA, Michael TP. Estimation of nuclear DNA content of duckweeds (Lemnaceae) Journal of Botany. 2011;2011:570319. doi:10.1155/2011/570319. [Google Scholar]
- Webb CO, Ackerly DD, Kembel SW. Phylocom: software for the analysis of phylogenetic community structure and trait evolution. Bioinformatics. 2008;24:2098–2100. doi: 10.1093/bioinformatics/btn358. [DOI] [PubMed] [Google Scholar]
- Weiss-Schneeweiss H, Greilhuber J, Schneeweiss GM. Genome size evolution in holoparasitic Orobanche (Orobanchaceae) and related genera. American Journal of Botany. 2006;93:148–156. doi: 10.3732/ajb.91.3.439. [DOI] [PubMed] [Google Scholar]