

Abstract
Background
Drug resistance hampers the efficient treatment of malignancies, including advanced stage ovarian cancer, which has a 5-year survival rate of only 30 %. The molecular processes underlying resistance have been extensively studied, however, not much is known about the involvement of microRNAs.
Methods
Differentially expressed microRNAs between cisplatin sensitive and resistant cancer cell line pairs were determined using microarrays. Mimics were used to study the role of microRNAs in drug sensitivity of ovarian cancer cell lines and patient derived tumor cells. Luciferase reporter constructs were used to establish regulation of target genes by microRNAs.
Results
MiR-634 downregulation was associated with cisplatin resistance. Overexpression of miR-634 affected cell cycle progression and enhanced apoptosis in ovarian cancer cells. miR-634 resensitized resistant ovarian cancer cell lines and patient derived drug resistant tumor cells to cisplatin. Similarly, miR-634 enhanced the response to carboplatin and doxorubicin, but not to paclitaxel. The cell cycle regulator CCND1, and Ras-MAPK pathway components GRB2, ERK2 and RSK2 were directly repressed by miR-634 overexpression. Repression of the Ras-MAPK pathway using a MEK inhibitor phenocopied the miR-634 effects on viability and chemosensitivity.
Conclusion
miR-634 levels determine chemosensitivity in ovarian cancer cells. We identify miR-634 as a therapeutic candidate to resensitize chemotherapy resistant ovarian tumors.
Electronic supplementary material
The online version of this article (doi:10.1186/s12943-015-0464-4) contains supplementary material, which is available to authorized users.
Keywords: microRNA, miRNA, Drug resistance, miR-634, Ras-MAPK pathway, Ovarian cancer, Chemotherapy, Cisplatin, RPS6KA3, RSK2
Background
Ovarian tumors are a group of molecularly and etiologically heterogeneous cancers [1], and are the fifth most common cause of cancer related mortality among women. The current standard of therapy is debulking surgery and combination chemotherapy, consisting of a taxane (e.g. paclitaxel) and a platinum-based compound (e.g cisplatin or carboplatin) [2, 3]. Despite an initial high chemo-responsiveness, with response rates over 80 % [4], most advanced epithelial ovarian cancer patients will relapse and ultimately die of drug-resistant disease [5].
In the cellular response to cytotoxic substances, cell signaling pathways, such as the MAPK pathways, play a pivotal role. The classical MAPK pathway is activated by receptor tyrosine kinases (RTK), which bind GRB2 and SOS, after which the kinases RAS, RAF, MEK and MAPK are sequentially activated. Three major MAPK routes have been identified: the p38 MAPK, Jun kinase and ERK pathway. The MAPK pathways regulate numerous targets, including p90 ribosomal S6 protein kinases (RSKs), which in turn can activate downstream proteins. Together, MAPK pathways regulate the activity of genes involved in cell proliferation, DNA damage repair, cell cycle progression and apoptosis [6–10].
Although the mechanisms behind chemotherapy resistance in ovarian cancer have been studied extensively, the involvement of microRNAs (miRNAs), small RNAs that regulate gene expression, is just beginning to be unraveled [11, 12]. We have recently shown that miR-141 targets KEAP1 in ovarian cancer cells, repression of which results in enhanced cisplatin resistance [13]. In the current study we aimed to identify additional miRNAs that play a role in cisplatin resistance. Here, we describe that miR-634 can sensitize both ovarian cancer cell lines and primary ovarian cancer cell cultures to chemotherapy. We show that miR-634 regulates cyclin D1 and several Ras-MAPK pathway components (GRB2, ERK2, RSK1 and RSK2), which may contribute to the effects of miR-634 on ovarian cancer cell survival and chemotherapy response.
Results
Comparison of miRNA expression profiles of cisplatin sensitive and resistant cell line pairs
In order to find miRNAs that play a role in cisplatin resistance, we compared miRNA expression profiles of cisplatin sensitive/resistant cell line pairs (IC50 values in Additional file 1: Table S1A). We hypothesized that in different cell types the same miRNAs play a role in cisplatin sensitivity, as has been reported for other factors involved in drug resistance [14]. Therefore, the miRNA expression pattern of an ovarian cancer cell line pair (A2780/A2780 DDP) was compared with expression patterns of a bladder cancer (T24/T24 DDP) and colon cancer (HCT8/HCT8 DDP) cell line pair. The only miRNA that showed a common pattern in all cell lines was miR-634 (Additional file 1: Figure S1, FDR = 0.000), which was downregulated ≥1.5 fold in all cisplatin resistant cell lines (Additional file 1: Table S2). We further investigated the role of miR-634 in ovarian cancer.
Effects of miR-634 overexpression on cell cycle and apoptosis
Before examining the effects of miR-634 on cisplatin sensitivity, we determined whether miR-634 overexpression affects the cell cycle and cell survival of A2780 DDP cells, which have a low basal miR-634 expression compared to the parental A2780 cells. Upon transfection of the miR-634 mimic, a slightly higher percentage of cells was observed in the G1 phase (p = 0.04) accompanied by a lower number of cells in the G2/M phase (p = 0.04) (Fig. 1a). These effects, which were observed at 48 h after transfection in multiple experiments, suggest that miR-634 overexpression may affect the G1-to-S phase transition. At 72 h after transfection, however, the cell cycle profile of miR-634 overexpressing cells was comparable to cells transfected with scrambled mimic (Fig. 1a).
Fig. 1.
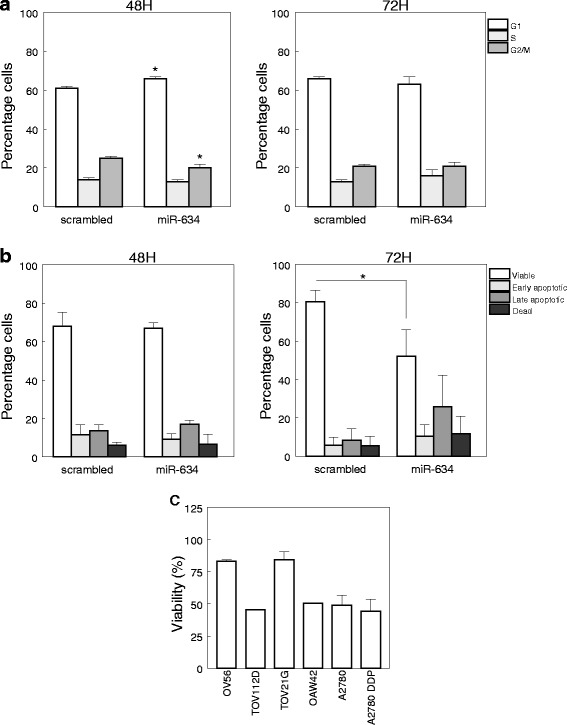
MiR-634 overexpression induces G1 arrest and causes cell death. a Percentage of A2780 DDP cells in G0/G1, S or G2/M phase 48 or 72 h after transfection with a miR-634 mimic or a scrambled control (n = 3), * = p < 0.05. b A2780 DDP cells were stained with PI and Annexin V 48 or 72 h after transfection with miR-634 mimic or a scrambled control. Depicted are viable (PI/Annexin V negative), early apoptotic (Annexin V positive/PI negative), late (Annexin V positive/PI positive) and dead (PI positive/Annexin V negative) cells (n = 3). c Viability of miR-634 mimic transfected ovarian cancer cells compared to cells transfected with a scrambled mimic (set at 100 %), as determined by an MTT assay 72 h after transfection. Depicted are average values ± SD (n = 3)
Next, we examined if miR-634 overexpression induces apoptosis. Whereas at 48 h after transfection the viability of control and miR-634 mimic transfected cells was comparable, at 72 h the percentage of viable cells was significantly lower (p = 0.03) in miR-634 transfectants, corresponding to increased numbers of apoptotic and dead cells (Fig. 1b). This effect of miR-634 on apoptosis was also detected by MTT assay in five other ovarian cancer cell lines, A2780 (parental line), OV56, OAW42, TOV21G and TOV112D. In these cells miR-634 gave rise to a 20–50 % reduction in viability, relative to control transfectants (Fig. 1c).
MiR-634 enhances cisplatin sensitivity of ovarian cancer cell lines
We next determined the effects of miR-634 overexpression on cisplatin sensitivity using a previously developed assay [13]. Briefly, cells were transfected with a miR-634 mimic or a scrambled control, and after 48 h exposed to various concentrations of cisplatin. After another 24 h cell viability was determined using an MTT assay. Because miRNA transfection was transient we used 24 h drug exposure intervals. Note that the difference in IC50 values observed between drug sensitive and resistant cell lines was similar to IC50 values determined in assays with longer drug exposure times [13] (Additional file 1: Table S1A, C). As is shown in Fig. 2a, transfection with miR-634 mimic gave rise to a marked increase in sensitivity after treatment with 80 μM (p = 0.006) and 125 μM cisplatin (p = 0.002) in the A2780 DDP cell line. A possible explanation for this sensitization may be a higher intracellular cisplatin accumulation and concomitantly increased cytotoxic activity. In fact, cisplatin resistance is frequently accompanied by reduced intracellular cisplatin levels [15] (Additional file 1: Table S1B). We measured the intracellular platinum content of miR-634 and scrambled control transfected cells after exposure to 80 and 125 μM cisplatin for 2 h, 6 h and 12 h. MiR-634 transfection did not affect the platinum uptake by A2780 DDP cells (Additional file 1: Figure S2) ruling out that miR-634 sensitizes ovarian cancer cells for cisplatin by increasing its uptake and/or reducing its efflux.
Fig. 2.
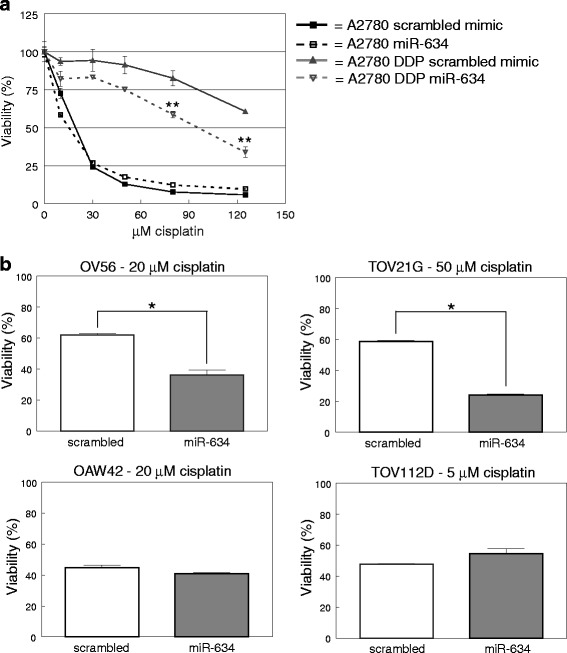
MiR-634 mimic enhances sensitivity for cisplatin in ovarian cancer cell lines. Ovarian cancer cell lines were transfected with miR-634 mimic or a scrambled control. 48 h after transfection, cisplatin was added and after 24H cell viability was determined with an MTT assay. a Overexpression of miR-634 miRNA in the sensitive A2780 and resistant A2780 DDP cell lines. Error bars represent the standard deviation within one experiment (n = 5). ** = p < 0.01. b Overexpression of miR-634 miRNA in the OV56, TOV21G, OAW42, TOV112D ovarian cancer cell lines. The cisplatin concentration is based on the IC25 value of mock transfected cells). Error bars represent the standard deviation within one experiment (n = 3). * = p < 0.05
In contrast to the resistant A2780 DDP cell line, enhanced expression of miR-634 in the sensitive A2780 cells did not alter the sensitivity for cisplatin. This might be due to the fact that miR-634 levels are already high in A2780 cells relative to A2780 DDP. We next examined the effect of miR-634 transfection on other ovarian cancer cell lines. Because the most significant effect was observed at the IC25 dose in the A2780 DDP cell line, the other ovarian cancer cell lines were treated with the IC25 dose of mock (transfection reagent only) transfected cells (Additional file 1: Table S1C). In two of the most resistant cell lines in this assay, OV56 and TOV21G, miR-634 overexpression significantly reduced viability (Fig. 2b). In contrast, in the more sensitive cell lines OAW42 and TOV112D, overexpression of miR-634 did not alter sensitivity. The effect of miR-634 on cisplatin response thus appears to be strongest in resistant cell lines.
MiR-634 sensitizes tumor cells from patients with drug resistant ovarian cancer to chemotherapy
Ovarian cancer is routinely treated with platinum (e.g. cisplatin, carboplatin) and taxane (e.g. paclitaxel) based combination chemotherapy. However, most tumors eventually become resistant. As miR-634 overexpression can increase cisplatin sensitivity of cell lines, we examined whether this treatment could also sensitize chemotherapy resistant primary ovarian tumor cells. Ascites was collected from 6 patients with serous and 1 patient with clear cell ovarian cancer (for patient characteristics, see Table 1). One patient was chemotherapy naïve, the others had received carboplatin/paclitaxel combination regimens and were or had become resistant. The ovarian cancer cells were isolated and cultured from ascites as described previously [16, 17], All cultures tested positive for the epithelial marker pan-keratin (Additional file 1: Figure S3). Another epithelial marker, EpCAM, was only positive in tumor cells from patient 2, however it is known that primary ovarian cancer cells may lose EpCAM expression in culture [16]. To verify that the cultures contain tumor cells, p53 staining was analyzed as p53 is often mutated in high and intermediate grade serous ovarian cancer [18]. A clear nuclear p53 staining was observed in all cultures, indicating the cultures do indeed consist of tumor cells.
Table 1.
Ovarian cancer patient characteristics
| Histological subtype | FIGO stage | Grade | Cytotoxic agents used | Response to last Pt containing treatment | |
|---|---|---|---|---|---|
| Patient 1 | Serous | IV | 2 | Carboplatin, Paclitaxel | PD within 1 month |
| Patient 2 | Serous | IIIc | UK | Carboplatin, Paclitaxel, Gemcitabine | PD within 4 months |
| Patient 3 | Serous | IIIc | 3 | Carboplatin, Paclitaxel, Gemcitabine | PD within 1 month |
| Patient 4 | Serous | IIIc | 2 | Carboplatin, Paclitaxel, Olaparib, Caelyx | PD during treatment |
| Patient 5 | Clear cell | IIIc | 3 | Carboplatin, Ifosfamide | PD within 1 month |
| Patient 6 | Serous | IIIc | 3 | Carboplatin, Paclitaxel, Caelyx | PD within 5 months |
| Patient 7 | Serous | IIIc | 2 | Chemotherapy naive |
UK: Unknown, Pt: Platinum, PD: Progressive disease, Caelyx: liposomal doxorubicin
We first tested whether miR-634 overexpression in primary tumor cells gave rise to a reduced viability, as was observed for the ovarian cancer cell lines. Remarkably, miR-634 overexpression in primary cell cultures only mildly diminished (5–25 %) the number of viable cells (Fig. 3a).
Fig. 3.
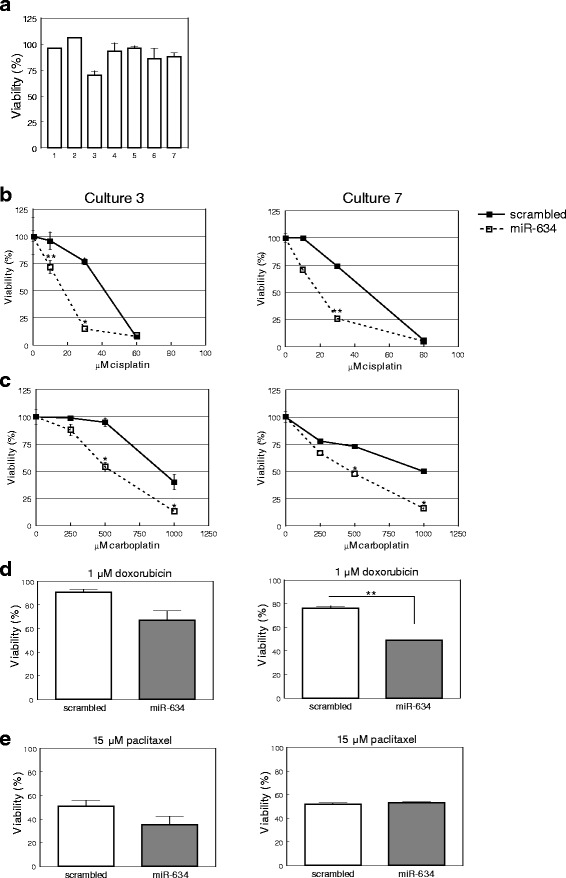
MiR-634 mimic enhances chemotherapy sensitivity in primary ovarian cancer cell cultures. a Overexpression of miR-634 in primary ovarian cancer cell cultures, derived from the ascites of ovarian cancer patients, has no or a moderate effect on cell viability 72 h after transfection. Indicated is the viability of miR-634 transfected cells relative to the viability of cells transfected with scrambled mimic (set at 100 %). The numbers on the X-axis refer to the number of the culture. Depicted are average values ± SD (n = 3). b MiR-634 enhances the sensitivity for cisplatin. Depicted are the results of a representative experiment for culture 3 (left, n = 4) and culture 7 (right, n = 3). An overview of the results obtained for all cultures is in Additional file 1 Figure S4. * = p < 0.05, ** = p < 0.01. c MiR-634 enhances the sensitivity for carboplatin. Depicted are the results of a representative experiment for culture 3 (left, n = 3) and culture 7 (right, n = 4). An overview of all cultures that were treated with carboplatin is in Additional file 1: Figure S5. * = p < 0.05, d MiR-634 enhances the sensitivity for doxorubicin. Depicted are the results of a representative experiment for culture 3 (left, n = 3) and culture 7 (right, n = 3). ** = p < 0.01. e MiR-634 does not significantly alter the sensitivity for paclitaxel. Depicted are the results of a representative experiment for culture 3 (left, n = 3) and culture 7 (right, n = 3)
miR-634 overexpression gave rise to a significant and reproducible decrease in cisplatin resistance in five cultures (Fig. 3b, Additional file 1: Figure S4). Note that in cultures 1 and 2 miR-634 overexpression gave rise to a similar phenotype, however, they could only be tested once due to low cell numbers. When the data of all primary cultures were combined, miR-634 overexpression was associated with a significant reduction in cellular viability of cells treated with 15 or 30 μM cisplatin (p = 0.002, p < 0.001, respectively).
Next, we tested if miR-634 could alter the response towards other anticancer compounds. miR-634 overexpression gave rise to significant increases in carboplatin sensitivity in cultures 3 and 7 (Fig. 3c, Additional file 1: Figure S5) as well as reduced resistance in two other cultures (which could only be tested once). We could not examine other cultures because of low cell numbers. In addition, miR-634 overexpression gave rise to a significant increase in doxorubicin sensitivity (p = 0.004), whereas there was no significant reduction in sensitivity for paclitaxel (p = 0.335) (Fig. 3d, e). These data indicate that miR-634 overexpression sensitizes resistant primary ovarian cancer cells to cisplatin, carboplatin and doxorubicin.
MiR-634 modulates the expression of key proliferation genes
MiRNAs mainly act by downregulating the expression of protein-coding genes. In order to identify target genes, we performed a pathway analysis on genes predicted to be a miR-634 target. Among the most enriched Gene Ontology (GO) terms were ‘positive regulation of cyclin-dependent protein kinase activity’ and ‘regulation of the G1-S transition of the mitotic cell cycle’ (Additional file 1: Figure S6, Table S3). Notably, Cyclin D isoforms CCND1 and CCND2 were associated with these GO terms, which activity is required for the G1 to S phase transition. Overexpression of miR-634 resulted in a decreased CCND1 protein level in A2780, A2780 DDP and primary tumor cells (derived from patient 3) (Fig. 4a) and this may explain the slight increase in G1 cells at 48 h after transfection (Fig. 1a).
Fig. 4.
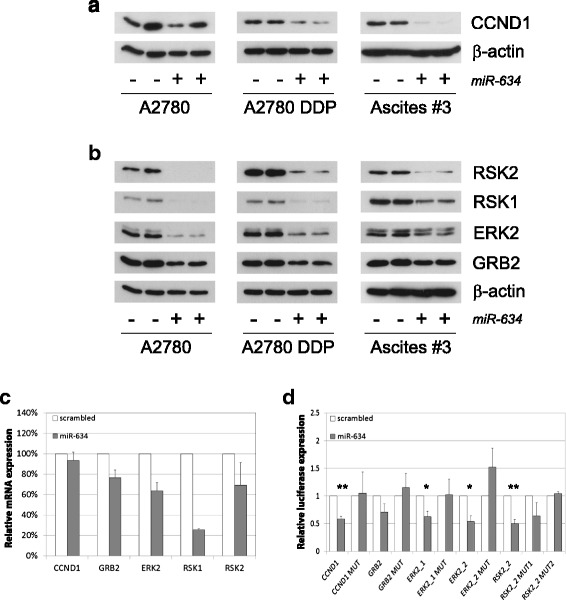
MiR-634 regulates genes involved in cell cycle regulation and the Ras-MAPK pathway. Ovarian cancer cell lines (A2780, A2780 DDP) and patient derived tumor cells (culture #3) were transiently transfected with scrambled control (-) or miR-634 mimic (+). 48 h after transfection protein lysates were analyzed for the expression of putative miR-634 target genes by Western blotting (A, B) or RT-PCR (C). a Transfection with miR-634 reduces Cyclin D1 protein levels in the A2780 cell lines and in primary ovarian cancer cell cultures. β-actin is used as a loading control. b MiR-634 overexpression reduces protein levels of GRB2, ERK2, RSK1 and RSK2, components of the Ras-MAPK pathway in the A2780 and A2780 DDP cell lines and primary ovarian cancer cell cultures. β-actin is used as a loading control. c MiR-634 overexpression lowers mRNA levels of CCND1, GRB2, ERK2, RSK1 and RSK2 in the A2780 DDP cell lines 48 h after transfection. Expression was normalized to HPRT and GAPDH expression. Depicted are average values ± SD (n = 2). The mRNA level in cells transfected with scrambled mimic is set at 100 %. d MiR-634 binds directly to elements of the 3’UTR of CCND1, GRB2, ERK2 and RSK2. A2780 DDP cells were transfected with a scrambled mimic or a miR-634 mimic. After 8 h, the same cells were transfected with a Renilla luciferase reporter construct (psiCHECK™-2) containing a region of 500 bp surrounding the predicted target sites (Additional file 1: Figure S7), or a luciferase construct in which the miR-634 binding sites were mutated. Since RSK2 contains both a canonical and a non-canonical miR-634 binding site, one construct was generated that contained a mutation in the canonical binding site (RSK2_2 mut 1) and another construct that contained mutations in both the canonical and non-canonical site (RSK2_2 mut 2). The Renilla luciferase activity was measured and normalized using the Firefly luciferase activity. For each construct, the relative luciferase activity of cells transfected with the scrambled mimic was set at 1. Depicted are the average values ± SD (n = 3). * p < 0.05, ** p < 0.01
Another GO term that was enriched was ‘positive regulation of proliferation’. Several proliferation pathways were predicted to be regulated by miR-634, most notably the Ras-MAPK pathway, of which GRB2, N-RAS, RAF, ERK2, RSK1 and RSK2 (Additional file 1: Table S3 and S4, Additional file 1: Figure S6) are potential targets. Concurringly, overexpression of miR-634 gave rise to reduced protein levels of GRB2, ERK2, RSK1 and RSK2 in the A2780 cell lines and primary cell culture 3 (Fig. 4b). Since miRNAs can regulate target expression by translational repression and/or mRNA degradation, we also examined target mRNA levels. Whereas there was only a slight decrease in CCND1 mRNA levels in miR-634 transfected cells, we observed a 25–75 % decrease in GRB2, ERK2, RSK1 and RSK2 mRNA levels (Fig. 4c).
We next tested whether miR-634 could directly regulate these genes. For each of the potential target genes, luciferase constructs were generated that express part of the 3’UTR containing the miR-634 target site (Additional file 1: Figure S7). As ERK2 and RSK2 contain respectively three and two canonical miR-634 target sites, the 3’UTR surrounding these sites was cloned separately. As expected, miR-634 could inhibit the 3’UTR of CCND1 and GRB2, the ERK2 3’UTR (constructs ERK2_1 and ERK2_2) and the RSK2 3’UTR (construct RSK2_2) (Additional file 1: Figure S8). In contrast, we observed no repression of the miR-634 target site in the RSK1 3’UTR, suggesting that the effect of miR-634 on RSK1 protein and mRNA levels may be indirect.
To confirm specific binding, the miR-634 binding site was mutated in the relevant constructs (CCND1, GRB2, ERK2_1, ERK2_2, RSK2_2). Since construct RSK2_2 contained two potential binding sites (Additional file 1: Figure S7), we created one construct with a mutation in the canonical site only (RSK2_2 mut 1) and another construct with mutations in both sites (RSK2_2 mut 2). As indicated in Fig. 4d, miR-634 could no longer repress the mutated 3’UTRs of CCND1, GRB2, and ERK2. Interestingly, mutation of both sites in RSK2_2 was necessary to completely prevent miR-634 binding.
As an alternative approach to inhibit miR-634 function we assessed the effect of a specific antisense inhibitor on the luciferase activity of the wild-type 3’UTRs of miR-634 target genes. As shown in Additional file 1: Figure S9, inhibition of endogenous miR-634 led to increased luciferase activity with the RSK2_2 and, to a lesser extent, GRB2 constructs, indicating that endogenous miR-634 regulates the expression of these target genes. In this setup we did not find an increased luciferase activity for the ERK2 or CCND1 constructs, and therefore we cannot confirm that ERK2 and CCND1 are endogenous miR-634 targets in the A2780 DDP cell line. We next examined the relation between miR-634 expression and expression levels of its target genes in a large cohort of ovarian serous cystadenoma’s (TCGA dataset) (Additional file 1: Figure S10). Unfortunately, in the majority of tumors miR-634 expression was not detected above background. However, when we examined the 23 tumors with high miR-634 expression (expression > mean + stdev) we found a significant negative correlation between miR-634 levels and RSK2 (p = 0.002). We also found an inverse relation between miR-634 and ERK2 (p = 0.091 or p = 0.310 for different probes), RSK1 (p = 0.372) and GRB2 (p = 0.614), but in this set of 23 expressing tumors this relation was not significant. It is possible that in a larger dataset this relation becomes significant. We did not observe an inverse relation between miR-634 and CCND1 expression in this dataset.
Altogether, our findings show that overexpression of miR-634 leads to direct repression of RSK2, CCND1, GRB2 and ERK2 in ovarian cancer cell lines and ovarian cancer cells derived from patients. Repression of the Ras-MAPK pathway may contribute to the decrease in cellular proliferation observed upon miR-634 overexpression (Fig. 1b, c, d). In line with this, we observed that the MEK inhibitor PD0325901 reduced cellular viability at 48H after treatment (Additional file 1: Figure S11A). In addition, Ras-MAPK signaling may contribute to cisplatin resistance [19]. Our previous work has demonstrated that RSK2 depletion enhances cisplatin sensitivity [20]. Concurringly, PD0325901 augmented cisplatin toxicity at 24H after treatment (Additional file 1: Figure S11B).
Discussion
The aim of this study was to discover miRNAs that affect the response of ovarian cancer cells to cisplatin chemotherapy. We compared the expression of three cisplatin-sensitive and –resistant cell line pairs and identified miR-634 as a miRNA that can modulate the sensitivity to various drugs in ovarian cancer cells.
miR-634 is located on chromosome 17 within intron 15 of PRKCA (Protein Kinase C α), and is only conserved in primates. miR-634 has been first detected in colon cancer cells [21] via miRNA serial analysis of gene expression (miRAGE). Afterwards, miR-634 has been identified as a miRNA able to regulate the expression of the androgen receptor (AR) in prostate cancer cells [22]. Repression of AR resulted in a reduced viability, however, the effect of miR-634 overexpression was stronger than the effect of AR siRNA, suggesting that miR-634 may target other survival genes as well. We now report that miR-634 overexpression results in downregulation of multiple genes of the Ras-MAPK pathway, an important cell proliferation pathway that is activated in many types of cancer [8].
The observation that miR-634 is able to regulate multiple Ras-MAPK pathway genes instead of one key mediator is thought-provoking. Interestingly, many miRNAs appear to synergistically regulate a set of genes that participate in similar processes, such as the miR-17-92 cluster (regulates genes involved in growth control) [23], let-7 (represses Ras and its downstream target HMGA2) [24, 25] and the miR-200 family (represses the epithelial-to-mesenchymal transition (EMT) [26–29]. MiRNAs may thus allow cells to effectively switch off similar signaling pathways in response to changing circumstances, and this may be especially relevant for proliferative pathways, which activity may need to be tightly controlled. In addition, it may be advantageous for cells to switch off production of proteins for signaling pathways that are not active.
Repression of oncogene activity often leads to cell death in cancer cells, a phenomenon known as ‘oncogene addiction’ [30]. The reduction in cell viability observed in the ovarian cancer cell lines upon overexpression of miR-634 may be caused by repression of the Ras-MAPK pathway. Interestingly, the effects of miR-634 overexpression in primary cultures are much less pronounced, suggesting that these cells depend less on proliferation signaling. In support of this theory, the ascites derived tumor cells divided more slowly than the ovarian cancer cell lines, and the fastest growing cultures (cultures 3 and 7) showed the largest reduction in cell viability upon miR-634 overexpression.
We describe that miR-634 transfection results in enhanced cisplatin sensitivity. Intriguingly, this effect of miR-634 overexpression is most apparent in resistant ovarian cancer cell lines, and also occurs in tumor cells derived from ascites. The miR-634 mediated repression of the Ras-MAPK pathway might contribute to the sensitization [19], which is supported by our finding that a MEK inhibitor enhances cisplatin sensitivity. Both ERK2 and RSK2 can inhibit several pro-apoptotic genes [31–35], and as a consequence of repression by miR-634, downregulation of ERK2 and RSK2 might lower the threshold for apoptosis upon treatment with cytotoxic therapy (Fig. 5). Indeed, our previous study shows that depletion of RSK2 leads to increased cisplatin sensitivity [20].
Fig. 5.
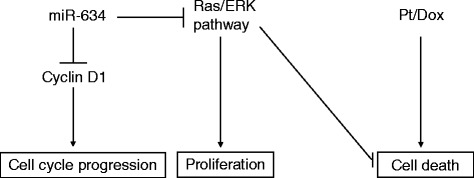
MiR-634 enhances the sensitivity for chemotherapy. By regulation of Cyclin D1 and the Ras-MAPK pathway, miR-634 overexpression may affect the cell cycle profile and apoptosis. Furthermore, repression of the Ras-MAPK pathway, which inhibits several pro-apoptotic factors (e.g. Bad [55], BimEL [56, 57]), can indirectly sensitize ovarian tumors to chemotherapeutics, such as cisplatin/carboplatin (Pt) and doxorubicin (dox)
Because of the pivotal role of the Ras-MAPK pathway in cell proliferation and cancer, inhibitors have been developed that target this pathway [36]. However, the objective response rates are modest [37], perhaps because tumors may rewire their signaling pathways [38]. Since miR-634 can inhibit several key proliferation factors simultaneously, acquired resistance is less likely to be a problem for a miR-634 based therapy than it is for targeted agents. A next step would be to study the effect of miR-634 overexpression on tumor growth and therapy response in a genetic mouse model for ovarian cancer. However, such a study is complicated by the fact that there is no high grade serous mouse model for ovarian cancer [39, 40], and miR-634 has no murine orthologue.
Furthermore, miR-634 levels in tumors may correlate with response to chemotherapy. Although some people have detected miR-634 expression using miRAGE and microarrays [22, 41], these studies do not report quantitative PCR validation. In addition, we and a few others were unable to detect endogenous miR-634 by RT-PCR [21, 42], despite evidence that the assay is functional (e.g. housekeepers RNU43, RNU48 were readily detected in our samples and we could find a miR-634 signal in mimic-transfected cells; Additional file 1: Figure S12). However, some publications describe successful miR-634 detection in RT-PCR assays [43–45]. An explanation for the discrepancy between studies could be that miR-634 is modified under certain cell-type specific conditions. Of note, modifications at the 3’ end are not uncommon for microRNAs [46, 47] and may prevent miR-634 detection by RT-PCR. Therefore, in order to monitor miR-634 levels in tumors, a specific high throughput miR-634 detection tool needs to be developed.
Conclusions
In summary our data indicate that miR-634 is an important player in cisplatin-resistance. First of all, miR-634 was the only miRNA that was commonly downregulated in three cisplatin/sensitive cell line pairs. Overexpression of miR-634 transiently inhibited G1-S cycle progression and enhanced apoptosis of ovarian cancer cells. Furthermore, miR-634 enhanced the chemotherapy response of cisplatin-resistant ovarian cancer cell lines and drug resistant patient-derived primary tumor cells. In addition, we observed that miR-634 overexpression in ovarian cancer cell lines and patient samples negatively regulates important cell-cycle genes (CCND1) and Ras-MAPK pathway components (GRB2, ERK2, RSK1 and RSK2). Inhibition of the Ras-MAPK pathway resulted in increased sensitivity to cisplatin, suggesting that the miR-634-mediated repression of this pathway is responsible for the effect of miR-634 on cisplatin resistance. In the future, therapeutic delivery of this miRNA to drug resistant ovarian cancer cells may help to resensitize patients to treatment.
Methods
Cell culture and reagents
The ovarian carcinoma cell line A2780, colon carcinoma cell line HCT8, bladder carcinoma cell line T24 and their cisplatin-resistant derivatives A2870 DDP, HCT8 DDP, and T24 DDP10 have been described before [14, 15, 48, 49]. Resistant cell lines were routinely challenged with cisplatin. Ovarian cancer cell lines OV56 and OAW42 were purchased from the ECACC (Salisbury, UK) and TOV112D, TOV21G were obtained from the ATCC (Manassas, VA, USA). Characteristics of the ovarian cancer cells were published in [50] and the authenticity of cell lines was verified by STR analysis. All cell lines were cultured in RPMI 1640 Glutamax (Invitrogen, Bleiswijk, The Netherlands) supplemented with 10 % FBS (Greiner, Alphen a/d Rijn, The Netherlands) and Penicillin/Streptomycin (final concentration 100 IE each, Sigma, Zwijndrecht, The Netherlands). Cisplatin, Carboplatin, Paclitaxel and Doxorubicin were obtained from Pharmachemie, Haarlem, the Netherlands. PD0325901 (cat # PZ0162) was obtained from Sigma-Aldrich.
Isolation and culture of primary ovarian tumor cell cultures occurred as described before [16, 17]. Briefly, 25 mL ascites fluid was mixed 1:1 with MCDB105/M199 medium supplemented with 0.5 μg/mL Fungizone and 50 μg/mL Gentamycine (all obtained from Sigma-Aldrich), and incubated at 37 °C, 5 % CO2, in T75 flasks for 4 days. Afterwards, the medium was replaced with fresh MCDB105/M199 medium and replaced twice weekly until cells were confluent. Cells were split 1:2-1:3 for up to 6 passages. To verify that the cultures contain tumor cells, cytospins were analyzed for epithelial markers (pan-keratin and EpCAM) and p53.
The study was approved by the Medical Ethical Committee of the Erasmus University MC (MEC-2008-183).
Microarray and data analysis
Total RNA was isolated with RNA Bee (BioConnect, Huissen, the Netherlands), and 1 μg RNA was labeled with the Cy3-TM ULS labeling kit (Kreatech Biotechnology, Amsterdam, The Netherlands), according to the manufacturer’s instructions. The RNA was hybridized with the LNA-based capture probe set (Exiqon, Vedbaek, Denmark) version 10 (annotation version 13). This probe set consists of 1344 probes including 725 human miRNAs, which are spotted in duplicate. Spots were quantified with the Imagene software (BioDiscovery), obvious outliers were removed and quantile normalization was performed.
miRNA profiling experiments were performed with RNA isolated at two (HCT8 and T24) or three (A2780) different passages. The average miRNA expression in the sensitive cell line was compared with the average expression in the resistant cell line and the fold-change was calculated. The microarray expression data has been deposited to the Gene Expression Omnibus (GEO) data repository (accession number GSE54665).
RT-PCR
0.5-1 μg RNA was reverse transcribed using random primers (Applied Biosystems, Bleiswijk, The Netherlands). 45 ng cDNA was used in a real-time PCR reaction using Taqman® assays-on-demand (CCND1, ERK2, GRB2, RSK1, RSK2). Expression was normalized to HPRT and GAPDH expression using the comparative CT-method [51]. MiRNA expression was determined using Taqman® miRNA assays (Applied Biosystems) according to the manufacturer’s protocol. In brief, 50 ng of total RNA was reverse transcribed using specific miRNA primers. The cDNA was used as input in a quantitative real-time PCR. RNU43 and RNU48 expression were used for normalization using the comparative CT- method [51].
Transfection
A miRIDIAN mimic for miR-634 (C-300961-01), a scrambled mimic (Mimic negative control #1; CN-001000-01), a miRIDIAN hairpin inhibitor for miR-634 (IH-300961-03), a scrambled miRIDIAN hairpin inhibitor (negative control; IN-001005-01) and transfection controls (miRIDIAN mimic with Dy547; CP-004500-01/miRIDIAN hairpin inhibitor with Dy547; IP-004500-01) were obtained from Dharmacon (Epsom, UK). The seeding concentrations (cells/well) in 24 well plates were for A2780 and A2780 DDP 4.5*104, for OV56 and TOV21G 4*104, for OAW42 3*104, for TOV112D 6*104 and for primary ovarian cell cultures 4*104 in a final volume of 450 μL medium without antibiotics. On the day after seeding, 50 μL of a mixture of Dharmafect 1 (final concentration 0.3 % (v/v)) and mimic (final concentration 50 nM) or inhibitor (final concentration 25 nM) in serum-free medium was added dropwise to each well. Under these conditions, the transfection efficiency was over 90 % as determined using fluorescently labeled mimics. 48 h after transfection, drugs were added in varying concentrations. After 24 h of continuous drug exposure, an MTT assay was performed [52]. psiCHECK™-2 constructs were transfected using Fugene HD (Promega) according to recommendations by the manufacturer.
FACS analysis
Forty-eight and seventy-two hours after miRNA transfection, cells were harvested and stained with FITC-Annexin V and Propidium iodide (PI) (FITC Annexin V Apoptosis Detection Kit I (BD Pharmingen, Breda, The Netherlands)) or, for cell cycle analysis, fixed in 70 % ethanol on ice, washed with PBS, then stained with PI (20 μg/mL in PBS-0.5 mL).
Platinum measurements
Platinum measurements in cell lysates were carried out essentially as described before using Atomic Absorption Spectrophotometry (AAS) [53].
Cytospins and immunohistochemistry
Primary ovarian cancer cell cultures were harvested using a cell scraper and collected in tubes, washed and resuspended in PBS containing 1 % BSA. 50-100 μL (depending on cell number) was used for cytospin preparation. Slides were air-dried for 30 min, fixed O/N in 10 % formalin, then stored in 70 % ethanol at 4 °C, until IHC staining. IHC staining was performed using Ventana Benchmark ULTRA automated slide stainers, and antibodies against pan keratin (Neomarkers, Fremont,CA, USA, MS-343-P), EpCAM (DAKO, Heverlee, Belgium, M0804) and p53 (DAKO, M7001).
Pathway analysis
The entire list of predicted miR-634 targets in Targetscan v6.0 (http://www.targetscan.org) was used for pathway analysis using the analysis wizard of DAVID Bioinformatics Resources (http://david.abcc.ncifcrf.gov/tools.jsp). Analyzed were enriched GO-terms (BP_fat; Additional file 1: Table S3) and enriched pathways (BIOCARTA, KEGG; Additional file 1: Table S4).
Western blot analysis
Fifteen to twenty μg of total protein of each sample was subjected to SDS-PAGE/Western blotting. Specific proteins were detected with antibodies against mouse GRB2 (BD Pharmingen, 610111), mouse RSK2 (Santa Cruz, Heerhugowaard, The Netherlands, SC-9986), rabbit ERK1/2 (Cell signaling, Leiden, The Netherlands, #9102), rabbit RSK1 (Santa Cruz, SC-231), rabbit CCND1 (Thermoscientific, Etten-Leur, The Netherlands, RM-9104-S) and mouse anti-β-actin (Sigma, A5441). Secondary HRP-conjugated antibodies used were Goat-anti-mouse (Santa Cruz, SC-2005) and Goat-anti-rabbit (Jackson ImmunoResearch, Westgrove, PA, USA, 111-035-144).
Cloning
Parts of the 3’UTR of CCND1, GRB2, ERK2, RSK1 and RSK2 (see Additional file 1: Figure S7) were PCR amplified from human genomic DNA (Promega) introducing a XhoI (5’-end) and a NotI site (3’-end). The PCR products were cloned in pCR®-Blunt, followed by XhoI and NotI restriction digests and ligation in psiCHECK™-2 (Promega). The constructs were verified by sequencing. The putative miR-634 target sites were mutated, forming a SmaI site to efficiently screen for mutants, in the CCND1, GRB2, ERK2 and RSK2 3’UTR constructs, using the Quickchange site-directed mutagenesis kit (Stratagene; Additional file 1: Figure S7).
Luciferase assay
Forty-eight hours after transfection with the miRNA mimics/inhibitors and the psiCHECK2 constructs, a luciferase assay (Promega; Dual-Luciferase Reporter assay) was carried out according to the manufacturer’s instructions. The Renilla luciferase expression was normalized on the Firefly luciferase signal.
Statistical analysis
A paired SAM analysis [54] was performed to compare miRNA expression profiles of cisplatin sensitive and resistant ovarian (A2780/A2780 DDP), colon (HCT8/HCT8 DDP) and bladder cancer cell lines (T24/T24 DDP). Two-tailed paired sample T-tests were used to assess whether differences were consistent between the scrambled mimic and miR-634 (Figs. 1, 2, 4d). Two way ANOVA’s were used to examine whether miR-634 overexpression had a significant effect on drug sensitivity, independent of the ascites batch. Furthermore, the effect of miR-634 overexpression on drug sensitivity in individual ascites cultures was assessed using paired sample T-tests (Fig. 3, Additional file 1: Figure S3 and S4). miR-634 and CCND1, ERK2, RSK1 and RSK2 expression was examined in 535 ovarian serous cystadenoma’s (TCGA dataset, (http://cancergenome.nih.gov/). Spearman rank tests were used to correlate target gene expression in tumors with high miR-634 expression (expression > mean + stdev) (Additional file 1: Figure S10).
Acknowledgements
The authors are grateful to Dr. Seiji Nato (Kyushu University, Fukuoka, Japan) and to Dr. Kevin Scanlon (Keck Graduate Institute of Applied Life Sciences, Claremont, CA, USA) for the kind gifts of the cisplatin sensitive/resistant cell line pairs. Thanks are due to Mariël Brok for technical assistance in early experiments, Peter de Bruijn for platinum measurements, the department of Pathology for performing immunohistochemistry on primary ovarian cancer cells, Patricia Ewing for pathological analysis of the ovarian cancer tumors, and Wendy Onstenk and Bianca Mostert for providing us with the ascites samples. The results shown here are in part based upon data generated by the TCGA Research Network: http://cancergenome.nih.gov/. This project was financially supported by a grant from the Dutch Cancer Society EMCR 2007-3794.
Additional file
Cisplatin sensitivity of cell lines. Table S2. miRNAs that show a ≥1.5 fold change of expression in cisplatin sensitive and resistant cell lines. Table S3. GO-terms that are enriched for miR-634 targets. Table S4. An overview of KEGG and Biocarta pathways that are enriched for miR-634 targets. Figure S1. miR-634 is the only miRNA showing consistent changes in cisplatin sensitive and resistant cell lines. Figure S2. Platinum levels in A2780 DDP cells transfected with miR-634 or scrambled controls. Figure S3. Immunohistochemical staining of primary ovarian cancer cell cultures. Figure S4. miR-634 enhances cisplatin sensitivity. Figure S5. miR-634 enhances carboplatin sensitivity. Figure S6. Predicted miR-634 targets. Figure S7. Overview of the location of the miR-634 target sites in the 3’UTR of its potential target genes. Figure S8. Effect of miR-634 overexpression on luciferase activity of psiCHECK™-2 reporter constructs containing part of the 3’UTR of miR-634 predicted targets. Figure S9. Effect of miR-634 inhibitors on luciferase activity of psiCHECK™-2 reporter constructs containing part of the 3’UTR of miR-634 predicted targets. Figure S10. Correlation between miR-634 expression and its target gene expression in 23 miR-634 expressing primary ovarian cancer samples (TCGA dataset). Figure S11. Effect of PD0325901 on cell survival and cisplatin sensitivity. Figure S12. Detection of miR-634, RNU43 and RNU48 by RT-PCR. (PDF 1609 kb)
Footnotes
Competing interests
The authors declare that they have no competing interests.
Author’s contributions
MvJ designed and executed experiments, analyzed and interpreted the data and drafted the manuscript. PvK and AB performed experiments. WvIJ provided the micro-array facility. JH, RM, JP, EB and JV contributed to the design of the experiments and interpretation of the data. EW conceived, designed and coordinated experiments, helped interpreting the data and drafting the manuscript. All authors have read and approved the manuscript.
References
- 1.Berns EM, Bowtell DD. The changing view of high-grade serous ovarian cancer. Cancer Res. 2012;72(11):2701–4. doi: 10.1158/0008-5472.CAN-11-3911. [DOI] [PubMed] [Google Scholar]
- 2.Ledermann JA, Marth C, Carey MS, Birrer M, Bowtell DD, Kaye S, et al. Role of molecular agents and targeted therapy in clinical trials for women with ovarian cancer. Int J Gynecol Cancer. 2011;21(4):763–70. doi: 10.1097/IGC.0b013e31821b2669. [DOI] [PubMed] [Google Scholar]
- 3.Vergote I, Trope CG, Amant F, Kristensen GB, Ehlen T, Johnson N, et al. Neoadjuvant chemotherapy or primary surgery in stage IIIC or IV ovarian cancer. N Engl J Med. 2010;363(10):943–53. doi: 10.1056/NEJMoa0908806. [DOI] [PubMed] [Google Scholar]
- 4.Jemal A, Siegel R, Xu J, Ward E. Cancer statistics, 2010. CA Cancer J Clin. 2010;60(5):277–300. doi: 10.3322/caac.20073. [DOI] [PubMed] [Google Scholar]
- 5.Ozols RF. Epithelial Ovarian Cancer. In: Hoskins WJYR, Markman M, Perez CA, Barakat R, Randall M, editors. Principles and Practice of Gynaecologic Oncology. Philadelphia, PA: Lippincott Williams & Wilkins; 2005. pp. 895–985. [Google Scholar]
- 6.Zhang W, Liu HT. MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res. 2002;12(1):9–18. doi: 10.1038/sj.cr.7290105. [DOI] [PubMed] [Google Scholar]
- 7.Li W, Melton DW. Cisplatin regulates the MAPK kinase pathway to induce increased expression of DNA repair gene ERCC1 and increase melanoma chemoresistance. Oncogene. 2012;31(19):2412–22. doi: 10.1038/onc.2011.426. [DOI] [PubMed] [Google Scholar]
- 8.Dhillon AS, Hagan S, Rath O, Kolch W. MAP kinase signalling pathways in cancer. Oncogene. 2007;26(22):3279–90. doi: 10.1038/sj.onc.1210421. [DOI] [PubMed] [Google Scholar]
- 9.MacCorkle RA, Tan TH. Mitogen-activated protein kinases in cell-cycle control. Cell Biochem Biophys. 2005;43(3):451–61. doi: 10.1385/CBB:43:3:451. [DOI] [PubMed] [Google Scholar]
- 10.Cagnol S, Chambard JC. ERK and cell death: mechanisms of ERK-induced cell death--apoptosis, autophagy and senescence. Febs J. 2010;277(1):2–21. doi: 10.1111/j.1742-4658.2009.07366.x. [DOI] [PubMed] [Google Scholar]
- 11.Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136(2):215–33. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.van Jaarsveld MT, Helleman J, Berns EM, Wiemer EA. MicroRNAs in ovarian cancer biology and therapy resistance. Int J Biochem Cell Biol. 2010;42(8):1282–90. doi: 10.1016/j.biocel.2010.01.014. [DOI] [PubMed] [Google Scholar]
- 13.van Jaarsveld MT, Helleman J, Boersma AW, van Kuijk PF, van Ijcken WF, Despierre E, et al. miR-141 regulates KEAP1 and modulates cisplatin sensitivity in ovarian cancer cells. Oncogene. 2013;32(36):4284–93. doi: 10.1038/onc.2012.433. [DOI] [PubMed] [Google Scholar]
- 14.Kool M, de Haas M, Scheffer GL, Scheper RJ, van Eijk MJ, Juijn JA, et al. Analysis of expression of cMOAT (MRP2), MRP3, MRP4, and MRP5, homologues of the multidrug resistance-associated protein gene (MRP1), in human cancer cell lines. Cancer Res. 1997;57(16):3537–47. [PubMed] [Google Scholar]
- 15.Schmidt W, Chaney SG. Role of carrier ligand in platinum resistance of human carcinoma cell lines. Cancer Res. 1993;53(4):799–805. [PubMed] [Google Scholar]
- 16.Dunfield LD, Shepherd TG, Nachtigal MW. Primary culture and mRNA analysis of human ovarian cells. Biol Proced Online. 2002;4:55–61. doi: 10.1251/bpo34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shepherd TG, Theriault BL, Campbell EJ, Nachtigal MW. Primary culture of ovarian surface epithelial cells and ascites-derived ovarian cancer cells from patients. Nat Protoc. 2006;1(6):2643–9. doi: 10.1038/nprot.2006.328. [DOI] [PubMed] [Google Scholar]
- 18.Ayhan A, Kurman RJ, Yemelyanova A, Vang R, Logani S, Seidman JD, et al. Defining the cut point between low-grade and high-grade ovarian serous carcinomas: a clinicopathologic and molecular genetic analysis. Am J Surg Pathol. 2009;33(8):1220–4. doi: 10.1097/PAS.0b013e3181a24354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brozovic A, Osmak M. Activation of mitogen-activated protein kinases by cisplatin and their role in cisplatin-resistance. Cancer Lett. 2007;251(1):1–16. doi: 10.1016/j.canlet.2006.10.007. [DOI] [PubMed] [Google Scholar]
- 20.van Jaarsveld MT, Blijdorp IC, Boersma AW, Pothof J, Mathijssen RH, Verweij J, et al. The kinase RSK2 modulates the sensitivity of ovarian cancer cells to cisplatin. Eur J Cancer. 2013;49(2):345–51. doi: 10.1016/j.ejca.2012.08.024. [DOI] [PubMed] [Google Scholar]
- 21.Cummins JM, He Y, Leary RJ, Pagliarini R, Diaz LA, Jr, Sjoblom T, et al. The colorectal microRNAome. Proc Natl Acad Sci U S A. 2006;103(10):3687–92. doi: 10.1073/pnas.0511155103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ostling P, Leivonen SK, Aakula A, Kohonen P, Makela R, Hagman Z, et al. Systematic analysis of microRNAs targeting the androgen receptor in prostate cancer cells. Cancer Res. 2011;71(5):1956–67. doi: 10.1158/0008-5472.CAN-10-2421. [DOI] [PubMed] [Google Scholar]
- 23.Li Y, Choi PS, Casey SC, Dill DL, Felsher DW. MYC through miR-17-92 suppresses specific target genes to maintain survival, autonomous proliferation, and a neoplastic state. Cancer Cell. 2014;26(2):262–72. doi: 10.1016/j.ccr.2014.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Johnson SM, Grosshans H, Shingara J, Byrom M, Jarvis R, Cheng A, et al. RAS is regulated by the let-7 microRNA family. Cell. 2005;120(5):635–47. doi: 10.1016/j.cell.2005.01.014. [DOI] [PubMed] [Google Scholar]
- 25.Lee YS, Dutta A. The tumor suppressor microRNA let-7 represses the HMGA2 oncogene. Genes Dev. 2007;21(9):1025–30. doi: 10.1101/gad.1540407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gregory PA, Bert AG, Paterson EL, Barry SC, Tsykin A, Farshid G, et al. The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat Cell Biol. 2008;10(5):593–601. doi: 10.1038/ncb1722. [DOI] [PubMed] [Google Scholar]
- 27.Park SM, Gaur AB, Lengyel E, Peter ME. The miR-200 family determines the epithelial phenotype of cancer cells by targeting the E-cadherin repressors ZEB1 and ZEB2. Genes Dev. 2008;22(7):894–907. doi: 10.1101/gad.1640608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Burk U, Schubert J, Wellner U, Schmalhofer O, Vincan E, Spaderna S, et al. A reciprocal repression between ZEB1 and members of the miR-200 family promotes EMT and invasion in cancer cells. EMBO Rep. 2008;9(6):582–9. doi: 10.1038/embor.2008.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu YN, Yin JJ, Abou-Kheir W, Hynes PG, Casey OM, Fang L, et al. MiR-1 and miR-200 inhibit EMT via Slug-dependent and tumorigenesis via Slug-independent mechanisms. Oncogene. 2013;32(3):296–306. doi: 10.1038/onc.2012.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Weinstein IB, Joe A. Oncogene addiction. Cancer Res. 2008;68(9):3077–80. doi: 10.1158/0008-5472.CAN-07-3293. [DOI] [PubMed] [Google Scholar]
- 31.Ewings KE, Wiggins CM, Cook SJ. Bim and the pro-survival Bcl-2 proteins: opposites attract. ERK repels Cell Cycle. 2007;6(18):2236–40. doi: 10.4161/cc.6.18.4728. [DOI] [PubMed] [Google Scholar]
- 32.Romeo Y, Zhang X, Roux PP. Regulation and function of the RSK family of protein kinases. Biochem J. 2012;441(2):553–69. doi: 10.1042/BJ20110289. [DOI] [PubMed] [Google Scholar]
- 33.Ray-David H, Romeo Y, Lavoie G, Deleris P, Tcherkezian J, Galan JA, et al. RSK promotes G2 DNA damage checkpoint silencing and participates in melanoma chemoresistance. Oncogene. 2013;32(38):4480–9. doi: 10.1038/onc.2012.472. [DOI] [PubMed] [Google Scholar]
- 34.Sulzmaier FJ, Ramos JW. RSK isoforms in cancer cell invasion and metastasis. Cancer Res. 2013;73(20):6099–105. doi: 10.1158/0008-5472.CAN-13-1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen C, Zhang L, Huang NJ, Huang B, Kornbluth S. Suppression of DNA-damage checkpoint signaling by Rsk-mediated phosphorylation of Mre11. Proc Natl Acad Sci U S A. 2013;110(51):20605–10. doi: 10.1073/pnas.1306328110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Santarpia L, Lippman SM, El-Naggar AK. Targeting the MAPK-RAS-RAF signaling pathway in cancer therapy. Expert Opin Ther Targets. 2012;16(1):103–19. doi: 10.1517/14728222.2011.645805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pritchard AL, Hayward NK. Molecular pathways: mitogen-activated protein kinase pathway mutations and drug resistance. Clin Cancer Res. 2013;19(9):2301–9. doi: 10.1158/1078-0432.CCR-12-0383. [DOI] [PubMed] [Google Scholar]
- 38.Seton-Rogers S. Therapeutics: Delving deeper into resistance. Nat Rev Cancer. 2013;14(1):7. doi: 10.1038/nrc3653. [DOI] [PubMed] [Google Scholar]
- 39.Shan W, Liu J. Epithelial ovarian cancer: focus on genetics and animal models. Cell Cycle. 2009;8(5):731–5. doi: 10.4161/cc.8.5.7848. [DOI] [PubMed] [Google Scholar]
- 40.Karst AM, Drapkin R. The new face of ovarian cancer modeling: better prospects for detection and treatment. F1000 Med Rep. 2011;3:22. doi: 10.3410/M3-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Leivonen SK, Sahlberg KK, Makela R, Due EU, Kallioniemi O, Borresen-Dale AL, et al. High-throughput screens identify microRNAs essential for HER2 positive breast cancer cell growth. Mol Oncol. 2014;8(1):93–104. doi: 10.1016/j.molonc.2013.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rainer J, Ploner C, Jesacher S, Ploner A, Eduardoff M, Mansha M, et al. Glucocorticoid-regulated microRNAs and mirtrons in acute lymphoblastic leukemia. Leukemia. 2009;23(4):746–52. doi: 10.1038/leu.2008.370. [DOI] [PubMed] [Google Scholar]
- 43.Bach D, Fuereder J, Karbiener M, Scheideler M, Ress AL, Neureiter D, et al. Comprehensive analysis of alterations in the miRNome in response to photodynamic treatment. J Photochem Photobiol B. 2013;120:74–81. doi: 10.1016/j.jphotobiol.2013.01.012. [DOI] [PubMed] [Google Scholar]
- 44.Peng X, Cao P, He D, Han S, Zhou J, Tan G, et al. MiR-634 sensitizes nasopharyngeal carcinoma cells to paclitaxel and inhibits cell growth both in vitro and in vivo. Int J Clin Exp Pathol. 2014;7(10):6784–91. [PMC free article] [PubMed] [Google Scholar]
- 45.Yamamoto S, Inoue J, Kawano T, Kozaki K, Omura K, Inazawa J. The impact of miRNA-based molecular diagnostics and treatment of NRF2-stabilized tumors. Mol Cancer Res. 2014;12(1):58–68. doi: 10.1158/1541-7786.MCR-13-0246-T. [DOI] [PubMed] [Google Scholar]
- 46.Newman MA, Mani V, Hammond SM. Deep sequencing of microRNA precursors reveals extensive 3’ end modification. RNA. 2011;17(10):1795–803. doi: 10.1261/rna.2713611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wyman SK, Knouf EC, Parkin RK, Fritz BR, Lin DW, Dennis LM, et al. Post-transcriptional generation of miRNA variants by multiple nucleotidyl transferases contributes to miRNA transcriptome complexity. Genome Res. 2011;21(9):1450–61. doi: 10.1101/gr.118059.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kotoh S, Naito S, Yokomizo A, Kumazawa J, Asakuno K, Kohno K, et al. Increased expression of DNA topoisomerase I gene and collateral sensitivity to camptothecin in human cisplatin-resistant bladder cancer cells. Cancer Res. 1994;54(12):3248–52. [PubMed] [Google Scholar]
- 49.Goto S, Yoshida K, Morikawa T, Urata Y, Suzuki K, Kondo T. Augmentation of transport for cisplatin-glutathione adduct in cisplatin-resistant cancer cells. Cancer Res. 1995;55(19):4297–301. [PubMed] [Google Scholar]
- 50.Beaufort CM, Helmijr JC, Piskorz AM, Hoogstraat M, Ruigrok-Ritstier K, Besselink N, et al. Ovarian cancer cell line panel (OCCP): clinical importance of in vitro morphological subtypes. PLoS One. 2014;9(9):e103988. doi: 10.1371/journal.pone.0103988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc. 2008;3(6):1101–8. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
- 52.Carmichael J, DeGraff WG, Gazdar AF, Minna JD, Mitchell JB. Evaluation of a tetrazolium-based semiautomated colorimetric assay: assessment of chemosensitivity testing. Cancer Res. 1987;47(4):936–42. [PubMed] [Google Scholar]
- 53.Burger H, Zoumaro-Djayoon A, Boersma AW, Helleman J, Berns EM, Mathijssen RH, et al. Differential transport of platinum compounds by the human organic cation transporter hOCT2 (hSLC22A2) Br J Pharmacol. 2010;159(4):898–908. doi: 10.1111/j.1476-5381.2009.00569.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tusher VG, Tibshirani R, Chu G. Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci U S A. 2001;98(9):5116–21. doi: 10.1073/pnas.091062498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bonni A, Brunet A, West AE, Datta SR, Takasu MA, Greenberg ME. Cell survival promoted by the Ras-MAPK signaling pathway by transcription-dependent and -independent mechanisms. Science. 1999;286(5443):1358–62. doi: 10.1126/science.286.5443.1358. [DOI] [PubMed] [Google Scholar]
- 56.Ewings KE, Hadfield-Moorhouse K, Wiggins CM, Wickenden JA, Balmanno K, Gilley R, et al. ERK1/2-dependent phosphorylation of BimEL promotes its rapid dissociation from Mcl-1 and Bcl-xL. Embo J. 2007;26(12):2856–67. doi: 10.1038/sj.emboj.7601723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dehan E, Bassermann F, Guardavaccaro D, Vasiliver-Shamis G, Cohen M, Lowes KN, et al. betaTrCP- and Rsk1/2-mediated degradation of BimEL inhibits apoptosis. Mol Cell. 2009;33(1):109–16. doi: 10.1016/j.molcel.2008.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]