

Abstract
The breakdown of the intestinal barrier is a common manifestation of many diseases. Recent evidence suggests that vitamin D and its receptor VDR may regulate intestinal barrier function. Claudin-2 is a tight junction protein that mediates paracellular water transport in intestinal epithelia, rendering them “leaky”. Using whole body VDR-/- mice, intestinal epithelial VDR conditional knockout (VDRΔIEC) mice, and cultured human intestinal epithelial cells, we demonstrate here that the CLDN2 gene is a direct target of the transcription factor VDR. The Caudal-Related Homeobox (Cdx) protein family is a group of the transcription factor proteins which bind to DNA to regulate the expression of genes. Our data showed that VDR-enhances Claudin-2 promoter activity in a Cdx1 binding site-dependent manner. We further identify a functional vitamin D response element (VDRE) 5΄-AGATAACAAAGGTCA-3΄ in the Cdx1 site of the Claudin-2 promoter. It is a VDRE required for the regulation of Claudin-2 by vitamin D. Absence of VDR decreased Claudin-2 expression by abolishing VDR/promoter binding. In vivo, VDR deletion in intestinal epithelial cells led to significant decreased Claudin-2 in VDR-/- and VDRΔIEC mice. The current study reveals an important and novel mechanism for VDR by regulation of epithelial barriers.
Tight junction (TJ) structural components determine epithelial polarization and intestinal barrier functions1,2,3,4. Claudins, with approximately 24 members, are integral membrane proteins and components of tight junctions5. The complex expression pattern of claudins creates diversity in the barrier/channel property of TJs, which varies depending on the type of epithelium6. Claudin-2 and -10 tend to make tight monolayers leakier5,7,8,9. Claudin-2, a “leak” protein uniquely restricted to the proliferative zone of the crypt base10,11 , forms a paracellular water channel that mediates paracellular water transport in epithelia and renders it more “leaky”11,12,13,14,15. Defective epithelial barrier function has been implicated in IBD16 and similarly, elevation of Claudin-2 is associated with active IBD12,17. In addition, Claudin-2 likely participates in cellular functions other than its known effects on TJ function. For example, we have demonstrated that Salmonella targets Claudin-2 to facilitate bacterial invasion18, and epithelial cells with Claudin-2 knockdown have significantly less internalized Salmonella than control cells with normal Claudin-2 expression. Claudin-2 has also been identified as a target of Wnt/β-catenin signaling19, which is essential for intestinal development, and a recent study reported that Claudin-2 and -12 contributed to vitamin D-dependent calcium homeostasis20. Because of this latter report, and because Vitamin D and its receptor (VDR) are implicated in the pathogenesis of various intestinal illness, including IBD 21,22,23,24,25,26,27,28,29,30,31, we sought to determine how Claudin-2 is regulated by VDR signaling.
In the current study, we hypothesize that Claudin-2 is a direct target of the VDR. Using whole body VDR-/- mice, intestinal epithelial VDR conditional knockout (VDRΔIEC) mice, and cultured human intestinal epithelial cells; we perform a series of molecular and biochemical experiments in vivo and in vitro to investigate VDR regulation of Claudin-2 expression in enterocytes.
Results
Intestinal VDR deficiency in epithelial cells leads to reduction of Claudin-2 at the mRNA and protein levels
In whole VDR-/- mice, we detected significantly decreased mRNA levels of Claudin-2 in intestine (Fig. 1A), whereas other Claudins, such as Claudin-1,-4,-7,-10, and - 15, were not altered by the absence of VDR (data not shown). By immuno-blots, we further found that VDR+/+ mice had the highest protein level of Claudin-2 in intestine. VDR+/- mice had intermediate levels of Claudin-2, whereas the VDR-/- had the lowest levels of Claudin-2 protein (Fig. 1B). Therefore, VDR expression correlates with protein levels of Claudin-2 in colonic epithelial cells in vivo (Fig. 1C).
Figure 1.

VDR status in intestinal epithelial cells leads to the change of Claudin-2 at both mRNA and protein levels in vivo. (A) Claudin-2 mRNA level and (B) Claudin-2 protein level in the intestinal epithelial cells of VDR+/+, VDR+/-, or VDR-/- mice. (C) Claudin-2/VDR protein relative in the intestinal epithelial cells of VDR+/+, VDR+/-, or VDR-/- mice. (D) Claudin-2 mRNA level and (E) Claudin-2 protein level in the intestinal epithelial cells of VDR KO (VDRΔIEC) mice. Data are expressed as mean ± SD. *P < 0.05. n = 3 mice/group. (F) Location and quantification of Claudin-2 protein in colons of mice in vivo. Images for each protein shown represent three separate experiments. n = 3 mice/group.
We further tested the specificity of intestinal VDR on expression levels of Claudin-2, in VDRΔIEC mice. We found that the mRNA levels of Claudin-2 were significantly lower in VDRΔIEC mice, compared to the VDR-lox mice (Fig. 1D). Claudin-2 protein was also significantly decreased in VDRΔIEC, where no VDR protein was detected in VDRΔIEC colon by Western blot (Fig. 1E). As expected, Claudin-3 and Claudin-7 were unchanged. These data indicate that intestinal VDR specifically regulates the expression levels of Claudin-2.
Colonic Claudin-2 expression is uniquely restricted to the proliferative zone10,11,18. In Fig.1F, Claudin-2 staining (Green) was observed at the crypt base in VDR+/+ mice. The density of Claudin-2 fluorescence staining was weak in the VDR+/- and VDR-/- intestinal epithelial cells. Claudin-7 was very stable in these mice and showed unchanged distribution and density (Fig. 1F).
Vitamin D3 treatment upregulates mRNA levels of Claudin-2
For molecular mechanism studies in vitro, we used the human colonic epithelial SKCO15 cell line, which is widely used in studying TJs32,33. Vitamin D3 is known to increase VDR expression and activate VDR signaling. Claudin-2 mRNA was significantly elevated in SKCO15 cells treated with 1, 25 vitamin D3 (20 nM) for 24 hours, whereas Claudin-7 mRNA was not altered by vitamin D3 treatment (Fig. 2A). Moreover, protein levels of Claudin-2 were increased by vitamin D3 treatment in a dose-dependent manner (Fig. 2B). In contrast, the expression of Claudin-3 and 7 was unchanged in cells receiving vitamin D3 treatment. These data suggest that the Claudin-2 gene could be a direct transcriptional target of the VDR.
Figure 2.
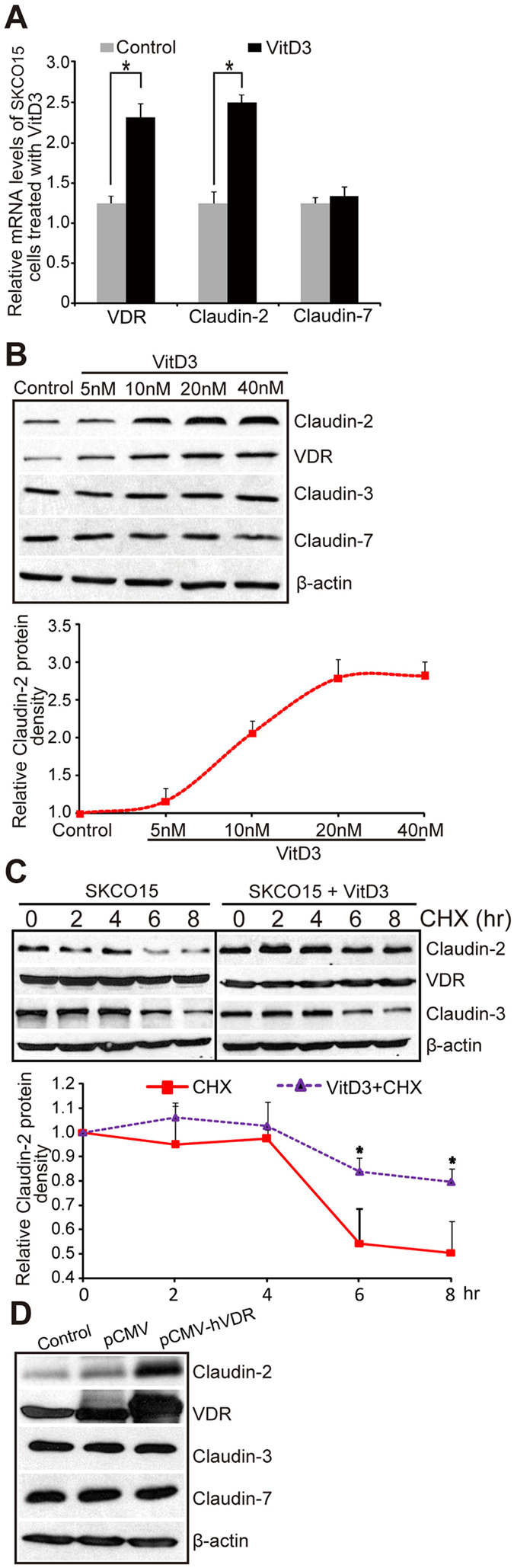
High levels VDR lead to increased Claudin-2 in human colonic epithelial SKCO15 cells in vitro. (A) Claudin-2 mRNA level increased post vitamin D3 treatment. SKCO15 cells were treated with vitamin D3 (20 nM) for 24 hours. Data are expressed as mean ± SD. *P < 0.05. n = 3 separate experiments. (B) Claudin-2 protein level and vitamin D3 dose-dependent curve. SKCO15 cells were treated with indicated vitamin D3 concentrations for 24 hours. Data are expressed as mean ± SD. *P < 0.05. n = 3 separate experiments. (C) Protein synthesis of Claudin-2 is high in vitamin D3-treated SKCO15 cells. Data are expressed as mean ± SD. *P < 0.05. n = 3 separate experiments. (D) Claudin-2 expression after SKCO15 cell transfection with human VDR in a pCMV-hVDR plasmid. Data are expressed as mean ± SD. *P < 0.05. n = 3 separate experiments.
VDR is generally present in the cytosol or bound to DNA in an inactive state and requires activation by binding ligand34. Upon binding to vitamin D, VDR translocates to the nucleus and binds to vitamin D response elements (VDREs) in target genes and induces gene expression. A previous study showed that ongoing protein synthesis is not required for this process to occur35. We treated human SKCO15 cells with vitamin D3 (20 nM) in the presence or absence of cyclohexamide (CHX) to block protein synthesis. We chose to treat cells with vitamin D3 at 20 nM because our dose-response data in Fig. 2B indicated that 20 nm is a suitable concentration to induce Claudin-2 expression. CHX is an inhibitor of eukaryotic protein biosynthesis and is commonly used to determine protein half-life. Therefore, in the cells treated with CHX only, the expression of Claudin-2 was significantly decreased (Fig. 2C SKCO15). CHX+vitamin D3 treatment was able to stabilize the expression of Claudin-2 (Fig. 2C SKCO15+Vit.D3). Whereas vitamin D3- induced Claudin-2 gene expression occurred in the absence of ongoing protein synthesis (presence of CHX), Vitamin D treatment did not induce Claudin-3 gene expression (Fig. 2C SKCO15+Vit.D3). These data further support the hypothesis that the Claudin-2 gene is a direct target of the VDR and not activated by secondary events, such as the synthesis of other transcription factors that are induced by VDR.
To study the effect of VDR overexpression on Claudin-2, we transiently transfected the human SKCO15 cells with a pCDNA-hVDR plasmid expressing human VDR. We found Claudin-2 expression increased after SKCO15 cells were transfected with pCMV-hVDR plasmids, whereas no change of Claudin-3 with VDR overexpression (Fig. 2D).
VDR binds the Claudin-2 promoter in vitro and in vivo
VDR is a nuclear receptor that acts as a transcription factor to regulate expression of its target genes21,36. We reasoned that VDR may bind to DNA promoters of Claudin-2, thus changing mRNA expression of the Claudin-2 genes. VDR’s effect on promoters of Claudin-2 was analyzed by CHIP assay. We designed primers to the nonrepetitive region near the transcriptional start site that specifically amplifies the Claudin-2 promoter. For negative controls, chromatin was immunoprecipitated with IgG or villin. The samples were amplified by conventional PCR. We found that VDR bound to the Claudin-2 promoter in vitro (SKCO15 cells, Fig. 3A) and in vivo (mouse colon, Fig. 3B). The expression of the other Claudin members, such as Claudin-1, was also tested. We found that VDR did not bind to the Claudin-1 promoter, either in vitro or in vivo (Figs. 3A,B). VDR is known to interact with nuclear receptor RXR in regulating gene expression. However there was no significant change in mRNA level of RXR in the VDR-/- intestine (Fig. 3C).
Figure 3.
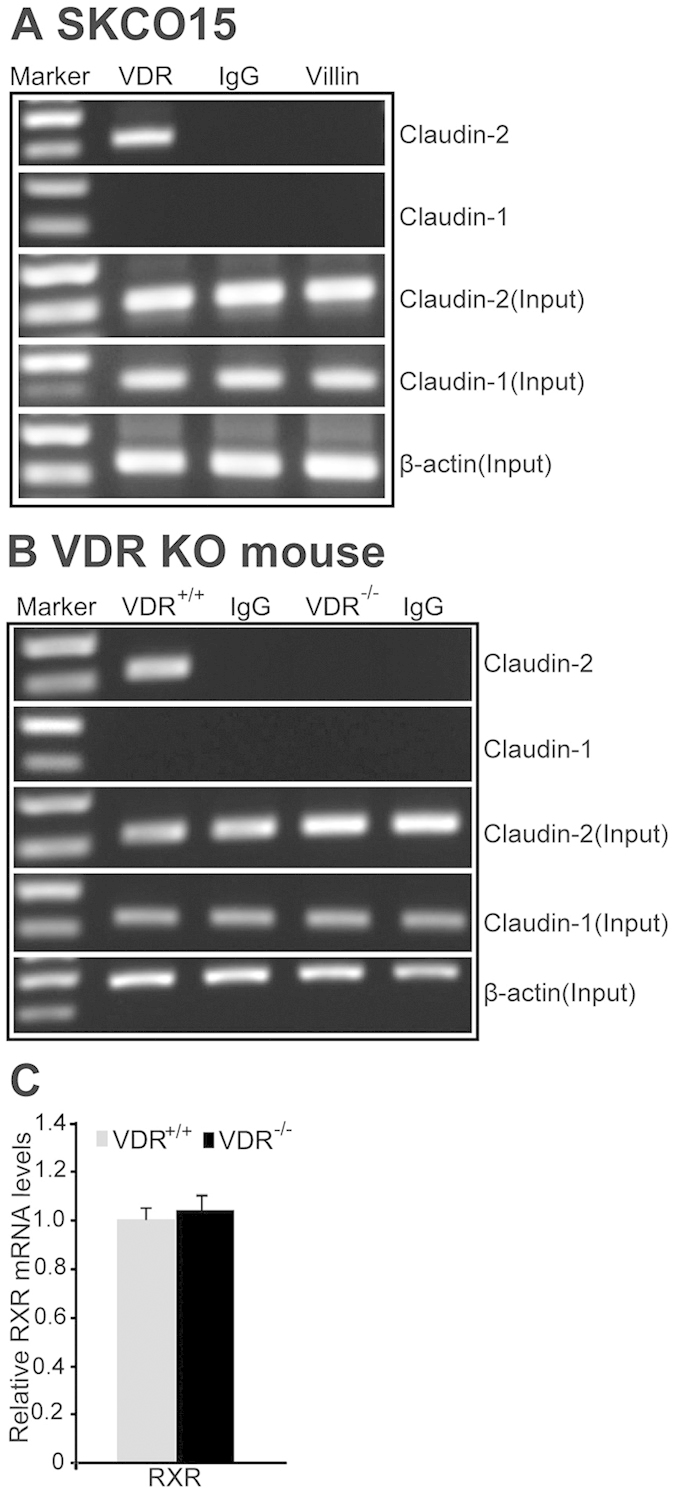
VDR binds to the Claudin-2 promoter. (A) CHIP-PCR amplification demonstrated binding of VDR to the promoter regions of human Claudin-2 in SKCO15 cells. PCR were performed including input-positive controls and IgG/villin-negative controls. n = 3 separate experiments. (B) ChIP-PCR assays of VDR binding to the Claudin-2 promoter in mouse colon. n = 3 separate experiments. (C) PCR analysis of RXR in VDR+/+ or VDR-/- mice. Data are expressed as mean ± SD. *P < 0.05. n = 3 separate experiments.
Lacking VDR decreases Claudin-2 by abolishing VDR/promoter binding
We reasoned that less Claudin-2 was generated in cells lacking VDR if VDR binds to the promoter of Claudin-2. We further tested the effects of VDR in regulating mRNA and protein levels of Claudin-2 in human colonic epithelial SKCO15 cells with VDR-siRNA. We found that Claudin-2 mRNA and protein expression were reduced when VDR was knocked down by siRNA (Fig. 4A,B). To examined the effect of one allele of VDR gene on the expression of Claudin-2, we chose VDR-/- and VDR+/- mouse embryonic fibroblast (MEF) cells37. We found that one allele of the VDR gene in the VDR+/- MEF cells was able to increase the expression of Claudin-2 protein (Fig. 4C). In contrast, Claudin-3 and -7 remained unchanged. At the transcriptional level, increased VDR mRNA was associated with elevated Claudin-2, but not Claudin-7 in VDR+/- MEFs. Claudin-2 mRNA was significantly decreased in VDR-/- MEF cells (Fig. 4D). This result suggests that VDR deletion affects Claudin-2 mRNA. Additionally, if we knocked down Claudin-2 by siRNA, there was no reduction of VDR at either the protein or mRNA level (Fig. 4E,F). These data suggest that Claudin-2 is downstream of VDR signaling.
Figure 4.

Lacking VDR decreases Claudin-2 protein and mRNA expression. (A) Claudin-2 protein and (B) mRNA were reduced by using siRNA against VDR. Data are expressed as mean ± SD. *P < 0.05. n = 3 separate experiments. (C) Claudin-2 protein and (D) mRNA were decreased in MEF VDR+/-/VDR-/- cells. Data are expressed as mean ± SD. *P < 0.05. n = 3 separate experiments. (E) VDR protein and (F) mRNA did not change after Claudin-2 was knocked down with Claudin-2 siRNA for 72 hours in SKCO15 cells. Data are expressed as mean ± SD. *P < 0.05. n = 3 separate experiments.
VDR-enhances Claudin-2 promoter activity in a Cdx1 binding site-dependent manner
Cdx is a member of the caudal-related homeobox gene family38,39. Suzuki et al. reports that IL-6-induced Claudin-2 promoter activity requires Cdx binding sites40. To assess whether vitamin D3 could enhance Claudin-2 promoter activity through Cdx binding sites, we used an in vitro reporter Luciferase assay. A schematic drawing of transcriptional binding sites in the wild-type (WT) Claudin-2 promoter and its mutants is shown in Fig. 5A. Plasmids with WT or deletions of NFκB, STAT, or Cdx1 in the Claudin-2 promoter binding site (of ΔNFκB, ΔSTAT, or ΔCdx) were transfected into cells, respectively, and then treated with vitamin D3. Vitamin D3 enhanced WT-Claudin-2 promoter activity in both HCT116 and CaCO2 cells (Fig. 5B,C). Deletions of NFκB and STAT binding sites did not affect the Claudin-2 promoter activity. In contrast, deletions of Cdx1 binding sites clearly suppressed the promoter activity (Fig. 5B,C). These results demonstrate that vitamin D3-induced Claudin-2 expression requires Cdx1 binding sites in the Claudin-2 promoter sequence.
Figure 5.
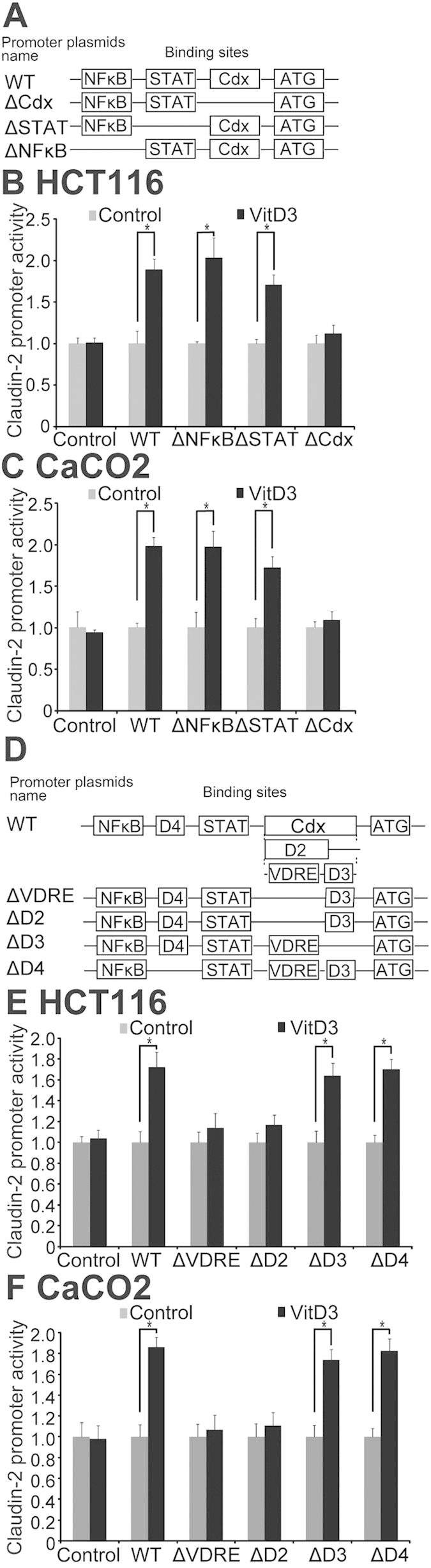
Identification of a functional VDRE sequence in the Claudin-2 promoter. (A) A schematic representation of transcriptional binding sites in the WT Claudin-2 promoter and deletion mutants. Plasmids include wild-type (WT), binding site deletions of NFκB (ΔNFκB), STAT (ΔSTAT), or Cdx1 (ΔCdx) in the Claudin-2 promoter. (B) WT or mutant Claudin-2 reporter plasmids were transfected in HCT116 and (C) CaCO2 cells. Luciferase activity was measured in the cell monolayers incubated in the absence or presence of vitamin D3 (20 nM) for 24 hours. Dual luciferase assays were performed and firefly luciferase activity was normalized to renilla luciferase activity. Data are expressed as mean ± SD. *P < 0.05. n = 3 separate experiments. (D) A schematic representation of VDRE deletion construct plasmids. Putative VDR-binding sites (containing AGATAACAAAGGTCA sequence) are designated as VDRE. Deletions of all VDRE binding sites (ΔVDRE), deletion of VDRE binding sites and adjacent bases (ΔD2), and non-VDRE deletion controls (ΔD3/ΔD4). (E) WT Claudin-2 reporter gene plasmids and the deletion mutant plasmids were transfected to HCT116 and (F) CaCO2 cells. Luciferase activity was measured in the cell monolayers incubated in the absence or presence of vitamin D3 (20 nM) for 24 hours. Data are expressed as mean ± SD. *P < 0.05. n = 3 separate experiments.
Identification of a functional VDRE sequence in the Claudin-2 promoter
As Claudin-2 promoter activity is strongly elevated by exposure to 1, 25 vitamin D3, we predicted the existence of a VDRE in the Claudin-2 promoter. Our results from the reporter assay suggested that Cdx binding sites are involved in vitamin D3-mediated increases in Claudin-2 expression. Therefore, studies were conducted to investigate the Cdx binding site region. VDRE sequence is AGATAACAAAGGTCA41. A search of the Cdx region revealed a DR3-type, which binds preferentially to directly arrangements of two hexameric binding sites with three spacing nucleotides. PCR was used to construct deletions of all VDRE binding sites (ΔVDRE), deletion of VDRE binding sites and adjacent bases (ΔD2), and non-VDRE deletion controls (ΔD3/ΔD4). These fragments were separately subcloned into the pGL3-basic firefly luciferase reporter plasmid. A schematic drawing of the VDRE deletion and control mutants is shown in Fig. 5D. The Claudin-2 promoter VDRE deletion constructs were transfected into cells and were subsequently treated with vitamin D3 (20 nM). Deletions of VDRE (ΔVDRE and ΔD2) clearly lower the promoter activity of vitamin D3 in HCT116 (Fig. 5E) and CaCO2 cells (Fig. 5F). In contrast, non-VDRE deletion controls (ΔD3 and ΔD4) did not affect the Claudin-2 promoter activity induced by vitamin D3 (Fig. 5E&5F). Our results demonstrate that deletion of the VDRE sequence 5΄-AGATAACAAAGGTCA-3΄ in the Claudin-2 promoter region causes loss of its responsiveness to vitamin D3, and thus confirm that Claudin-2 is a direct target of vitamin D receptor signaling in intestinal epithelial cells.
Discussion
The experimental focus of our current study was to investigate the molecular mechanisms whereby VDR may act as a transcriptional factor to regulate the expression of Claudin-2. First, we provide molecular biological evidence that the Claudin-2 gene is a direct target of the transcription factor VDR. A transcriptional reporter study demonstrated Claudin-2 up-regulation by over-expressed VDR. CHIP-PCR data demonstrated specific binding of VDR to the Claudin-2 promoter. VDR enhanced Claudin-2 promoter activity in a Cdx1 binding site-dependent manner. Next, we identified a functional VDRE sequence within in the Claudin-2 promoter. Knockout of VDR led to lower Claudin-2 at both mRNA and protein levels. Increased VDR by vitamin D3 pretreatment was associated with elevated Claudin-2 mRNA and protein levels. This study highlights an important and novel mechanism for VDR regulation of Claudin-2 critical to intestinal homeostasis.
Claudin-2 is a unique member of the Claudin family of transmembrane proteins as its expression is restricted to leaky epithelium in vivo and correlates with epithelial leakiness in vitro. VDR is a nuclear receptor that mediates most functions of vitamin D50,51,52. Our data showed that activation of the CLDN2 gene occurred via a consensus VDRE in the promoter that is bound by VDR. VDR is expressed in a wide range of tissues. Therefore, potentially, Claudin-2 can be induced in various tissues. We know that multiple factors contribute to the upregulation of Claudin-2 at the transcriptional level4,13,40. TNF-α and IL-1beta contribute to elevated Claudin-2 in vitro42,43. IL-6 enhances claudin-2 promoter activity in a Cdx binding site-dependent manner40. VDR has multiple critical functions in regulating innate and adaptive immunity, intestinal homeostasis, host response to invasive pathogens and commensal bacteria, and tight junction structure35,53,54,55,56,57,58,59,60. TJ structure plays a critical role in intestinal barrier and inflammation44,45,46,47,48,49. Claudin-2 is enhanced in the inflamed gut of patients with IBD12,17. The pathobiological importance of the VDR regulation of Claudin-2 could be complex. Hence, further insight into the mechanisms responsible for VDR and barrier dysfunction in mucosal inflammation is needed, especially in in vivo systems and disease models.
In summary, for the first time, we identify CLDN2 gene is a direct target of VDR. Our findings reveal a novel activity of VDR in regulation of TJs in primary cell structure and intestinal homeostasis. This study fills an existing gap by characterizing the precise molecular mechanism of VDR in regulating Claudin-2 and highlights the complex role of VDR in intestinal homeostasis61. It also brings up the possibility for restoration VDR-dependent functions and prevention of the intestinal barrier breakdown in patients with intestinal disorders.
Materials and Methods
Animals
VDR+/+, VDR+/- and VDR−/− mice on a C57BL6 background were obtained by breeding heterozygous VDR+/− mice62. VDR flox mice were originally reported by Dr. Geert Carmeliet 63. VDRΔIEC mice were obtained by crossing the VDR flox mice with villin-cre mice (Jackson Laboratory, 004586, Bar Harbor, Maine, USA), as we previously reported21. Experiments were performed on 2–3 months old mice. All animal work was approved by the Rush University Committee on Animal Resources. Euthanasia method was sodium pentobarbital (100 mg per kg body weight) I.P. followed by cervical dislocation.
Ethics statement
The methods in animal models were carried out accordance with the approved guidelines by the Rush University Committee on Animal Resources.
Mouse colonic epithelial cells
Mouse colonic epithelial cells were collected by scraping the tissue from the colon of the mouse, including the proximal and distal regions64,65. The cells were sonicated in lysis buffer (1% Triton X-100, 150 mM NaCl, 10 mM Tris, pH 7.4, 1 mM EDTA, 1 mM EGTA, pH 8.0, 0.2 mM sodium ortho-vanadate, and protease inhibitor cocktail). The protein concentration was measured using the BioRad Reagent (BioRad, Hercules, CA, USA).
Cell culture
Human epithelial CaCO2 and SKCO15 cells were maintained on transwell inserts (0.33 or 4.67 cm2, 0.4 mm pore. Costar, Cambridge, MA, USA) in DMEM supplemented with 10% fetal bovine serum, penicillin-streptomycin (Penicillin, 100 I.U./ml/Streptomycin, 100 μg/ml), and L-glutamine (4.5 g/L). Human colonic epithelial HCT116 cells, VDR+/− and VDR−/− MEF cells were cultured in DMEM medium supplemented with 10% (vol/vol) fetal bovine serum, as previously described62,65.
Immunofluorescence
Colonic tissues were freshly isolated and embedded in paraffin wax after fixation with 10% neutral buffered formalin. Immunofluorescence was performed on paraffin-embedded sections (4 μm), after preparation of the slides as described previously62 followed by incubation for 1 hour in blocking solution (2% bovine serum albumin, 1% goat serum in HBSS) to reduce nonspecific background. The tissue samples were incubated overnight with primary antibodies at 4 °C. The following antibodies were used: anti-Claudin-2, anti-Claudin-7 (Invitrogen, Grand Island, NY, USA). Samples were then incubated with secondary antibodies (goat anti-mouse Alexa Fluor 488 or goat anti-rabbit Alexa Fluor 488, Molecular Probes, CA; 1:200) for 1 hour at room temperature. Tissues were mounted with SlowFade Antifade Kit (Life technologies, s2828, Grand Island, NY, USA), followed by a coverslip, and the edges were sealed to prevent drying. Specimens were examined with a Zeiss laser scanning microscope (LSM) 710 (Carl Zeiss Inc., Oberkochen, Germany).
Analysis of claudins distribution
Fluorescence images were analyzed using image analysis software (LSM 710 META, version 4.2; Carl Zeiss Inc., Oberkochen, Germany). Each analysis was performed in triplicate from each tissue section on a total of 10 images per mouse sample (n = 5).
Transient transfections
Transient transfections were performed with Lipofectamine 2000 (Invitrogen, San Diego, CA, USA) in accordance with the manufacturer’s instructions. Cells were seeded on 60 mm dishes overnight before transfection with DNA and were mixed with liposome reagent at a ratio of 1:1 before addition to cells. After a 24-hour transfection period, the proteins were extracted for western-blot analysis.
Chromatin immunoprecipitation (ChIP) assays
The ChIP assays were performed essentially as described by the manufacturer (Upstate Inc., Chalottesville, VA, USA). Briefly, SKC015 cells or scraped VDR+/+/VDR-/- colonic epithelial cells were treated with 1% formaldehyde for 10 min at 37 0C. Cells were washed twice in ice-cold phosphate buffered saline containing protease inhibitor cocktail tablets (Roche, Nutley, NJ, USA). Cells were scraped into conical tubes, pelleted and lysed in SDS Lysis Buffer. The lysate was sonicated to shear DNA into fragments of 200–1000 bp (4 cycles of 10 s sonication, 10 s pausing, Branson Sonifier 250, Danbury, CT, USA). The chromatin samples were pre-cleared with salmon sperm DNA-bovine serum albumin-sepharose beads, then incubated overnight at 4 0C with VDR antibody (Santa Cruz Biotechnology Inc., Dallas, Texas, USA). Immune complexes were precipitated with salmon sperm DNA-bovine serum albumin-sepharose beads. DNA was prepared by treatment with proteinase K, extraction with phenol and chloroform, and ethanol precipitation, and was subjected to PCR (Primers see supplement table 1).
Western-blot analysis
Mouse epithelial cells were lysed in lysis buffer (1% Triton X-100, 150 mM NaCl, 10 mM Tris, pH 7.4, 1 mM EDTA, 1 mM EGTA, pH 8.0, 0.2 mM sodium ortho-vanadate, and protease inhibitor cocktail) and the protein concentration was measured. SKC015 and MEF cells were rinsed three times in ice-cold HBSS, lysed in protein loading buffer (50 mM Tris, pH 6.8, 100 mM dithiothreitol, 2% SDS, 0.1% bromophenol blue, and 10% glycerol) and sonicated (Branson Sonifier, 250). Equal amounts of protein were separated by SDS-polyacrylamide gel electrophoresis, transferred to nitrocellulose (162–0112, Bio-rad, Hercules, CA, USA), and immunoblotted with primary antibodies. The following antibodies were used: anti-Claudin-2, anti-Claudin-3, anti-Claudin-7 (Invitrogen, Grand Island, NY, USA), anti-VDR, anti-Villin (Santa Cruz Biotechnology Inc., Dallas, Texas, USA), or anti-β-actin (Sigma-Aldrich, St. Louis, MO, USA) and were visualized by ECL (Thermo Scientific, Rockford, IL, USA). Membranes that were probed with more than one antibody were stripped before reprobing.
Transcriptional activation
After a 24 hour transfection period, the cells were lysed and luciferase activity was determined using the Dual Luciferase Reporter Assay System (Promega, Madison, WI, USA) with a TD-20/20 luminometer (Turner Designs, Sunnyvale, CA, USA). Firefly luciferase activity was normalized to Renilla luminescence activity and the activity was expressed as relative units.
Identification of functional VDRE
PCR was used to construct deletion of entire VDRE binding sites (ΔVDRE), deletion of VDRE binding site with adjacent bases (ΔD2), and control (ΔD3/ΔD4). These fragments were separately subcloned into the firefly luciferase reporter plasmid pGL3-basic (Primers see supplement table 2). Deletions of different domains of the Claudin-2 promoter cloned into the in pGL3 vector, driving luciferase expression, were transfected into HCTC116/CaCO2 cells. Luciferase activity in cell lysates was assayed by the Dual Luciferase Reporter Assay System (Promega, Madison, WI, USA).
Real-time quantitative PCR analysis
Total RNA was extracted from mouse epithelial cells or cultured cells using TRIzol reagent (Invitrogen, Grand Island, NY, USA). The RNA integrity was verified by electrophoresis. RNA reverse transcription was performed using the iScript cDNA synthesis kit (Bio-Rad, Hercules, CA, USA) according to the manufacturer’s protocol. The RT cDNA reaction products were subjected to quantitative real-time PCR using CTFX 96 Real-time system (Bio-Rad, Hercules, CA, USA) and SYBR green supermix (Bio-Rad, Hercules, CA, USA) according to the manufacturer’s protocol. All expression levels were normalized to β-actin levels of the same sample. Percent expression was calculated as the ratio of the normalized value of each sample to that of the corresponding untreated control cells. All real-time PCR reactions were performed in triplicate. Optimal primer sequences were designed using Primer-BLAST or were obtained from Primer Bank primer pairs listed in Supplement Table 3.
Statistical Analysis
All of the data are expressed as means ± SD. All of the statistical tests were two-sided and P values of less than 0.05 were considered to be statistically significant. Differences between two samples were analyzed using Student’s t-test. The statistical analyses were performed using SAS version 9.2 (SAS Institute, Inc., Cary, NC).
Additional Information
How to cite this article: Zhang, Y.-g. et al. Tight junction CLDN2 gene is a direct target of the vitamin D receptor. Sci. Rep. 5, 10642; doi: 10.1038/srep10642 (2015).
Supplementary Material
Acknowledgments
We want to thank Dr. Takuya Suzuki from Hiroshima University, Japan for generously providing the Claudin-2 reporter plasmids. This work was supported by Brian Piccolo Cancer Award and Swim Across America Cancer Research Award to JS.
Footnotes
Author Contributions Y. Z., S. W. and R. L.: acquisition, analysis and interpretation of data; drafting of the manuscript; statistical analysis.D. Z., J. Z., G. C., E. P. and E. C. C.: technical or material support; drafting of the manuscript.J. S.: study concept and design; critical revision of the manuscript for important intellectual content; obtained funding; study supervision.
References
- Su L. et al. Targeted epithelial tight junction dysfunction causes immune activation and contributes to development of experimental colitis. Gastroenterology 136, 551–563, 10.1053/j.gastro.2008.10.081 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Sadi R. et al. Occludin regulates macromolecule flux across the intestinal epithelial tight junction barrier. American journal of physiology. Gastrointestinal and liver physiology 300, G1054–1064, 10.1152/ajpgi.00055.2011 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahner C., Mitic L. L. & Anderson J. M. Heterogeneity in expression and subcellular localization of claudins 2, 3, 4, and 5 in the rat liver, pancreas, and gut. Gastroenterology 120, 411–422, 10.1053/gast.2001.21736 (2001). [DOI] [PubMed] [Google Scholar]
- Sakaguchi T. et al. Cloning of the human claudin-2 5’-flanking region revealed a TATA-less promoter with conserved binding sites in mouse and human for caudal-related homeodomain proteins and hepatocyte nuclear factor-1alpha. J Biol Chem 277, 21361–21370, 10.1074/jbc.M110261200 (2002). [DOI] [PubMed] [Google Scholar]
- Furuse M. Molecular basis of the core structure of tight junctions. Cold Spring Harb Perspect Biol 2, a002907, 10.1101/cshperspect.a002907 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson J. M. & Van Itallie C. M. Physiology and function of the tight junction. Cold Spring Harb Perspect Biol 1, a002584, 10.1101/cshperspect.a002584 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amasheh S. et al. Claudin-2 expression induces cation-selective channels in tight junctions of epithelial cells. Journal of cell science 115, 4969–4976, 10.1242/jcs.00165 (2002). [DOI] [PubMed] [Google Scholar]
- Colegio O. R., Van Itallie C. M., McCrea H. J., Rahner C. & Anderson J. M. Claudins create charge-selective channels in the paracellular pathway between epithelial cells. Am J Physiol Cell Physiol 283, C142–147, 10.1152/ajpcell.00038.2002 (2002). [DOI] [PubMed] [Google Scholar]
- Van Itallie C. M. & Anderson J. M. Claudins and epithelial paracellular transport. Annual review of physiology 68, 403-429, 10.1146/annurev.physiol.68.040104.131404 (2006). [DOI] [PubMed] [Google Scholar]
- Escaffit F., Boudreau F. & Beaulieu J. F. Differential expression of claudin-2 along the human intestine: Implication of GATA-4 in the maintenance of claudin-2 in differentiating cells. Journal of cellular physiology 203, 15–26, 10.1002/jcp.20189 (2005). [DOI] [PubMed] [Google Scholar]
- Holmes J. L., Van Itallie C. M., Rasmussen J. E. & Anderson J. M. Claudin profiling in the mouse during postnatal intestinal development and along the gastrointestinal tract reveals complex expression patterns. Gene Expr Patterns 6, 581–588, 10.1016/j.modgep.2005.12.001 (2006). [DOI] [PubMed] [Google Scholar]
- Zeissig S. et al. Changes in expression and distribution of claudin 2, 5 and 8 lead to discontinuous tight junctions and barrier dysfunction in active Crohn’s disease. Gut 56, 61–72, 10.1136/gut.2006.094375 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenthal R. et al. Claudin-2, a component of the tight junction, forms a paracellular water channel. Journal of cell science 123, 1913–1921, 10.1242/jcs.060665 (2010). [DOI] [PubMed] [Google Scholar]
- Schulzke J. D. & Fromm M. Tight junctions: molecular structure meets function. Ann N Y Acad Sci 1165, 1–6, 10.1111/j.1749-6632.2009.04925.x (2009). [DOI] [PubMed] [Google Scholar]
- Van Itallie C. M., Holmes J., Bridges A. & Anderson J. M. Claudin-2-dependent changes in noncharged solute flux are mediated by the extracellular domains and require attachment to the PDZ-scaffold. Ann N Y Acad Sci 1165, 82–87, 10.1111/j.1749-6632.2009.04052.x (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulzke J. D. et al. Epithelial tight junctions in intestinal inflammation. Ann N Y Acad Sci 1165, 294–300, 10.1111/j.1749-6632.2009.04062.x (2009). [DOI] [PubMed] [Google Scholar]
- Weber C. R., Nalle S. C., Tretiakova M., Rubin D. T. & Turner J. R. Claudin-1 and claudin-2 expression is elevated in inflammatory bowel disease and may contribute to early neoplastic transformation. Lab Invest 88, 1110–1120, 10.1038/labinvest.2008.78 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y. G., Wu S., Xia Y. & Sun J. Salmonella infection upregulates the leaky protein claudin-2 in intestinal epithelial cells. PloS one 8, e58606, 10.1371/journal.pone.0058606 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mankertz J. et al. Functional crosstalk between Wnt signaling and Cdx-related transcriptional activation in the regulation of the claudin-2 promoter activity. Biochem Biophys Res Commun 314, 1001–1007, 10.1016/j.bbrc.2003.12.185 (2004). [DOI] [PubMed] [Google Scholar]
- Fujita H. et al. Tight junction proteins claudin-2 and -12 are critical for vitamin D-dependent Ca2+ absorption between enterocytes. Mol Biol Cell 19, 1912–1921, 10.1091/mbc.E07-09-0973 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu S. et al. Intestinal epithelial vitamin D receptor deletion leads to defective autophagy in colitis. Gut 2014-307436, 10.1136/gutjnl-2014-307436 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sentongo T. A. et al. Vitamin D status in children, adolescents, and young adults with Crohn disease. The American journal of clinical nutrition 76, 1077–1081 (2002). [DOI] [PubMed] [Google Scholar]
- Abreu M. T. et al. Measurement of vitamin D levels in inflammatory bowel disease patients reveals a subset of Crohn’s disease patients with elevated 1,25-dihydroxyvitamin D and low bone mineral density. Gut 53, 1129–1136, 10.1136/gut.2003.036657 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim W. C., Hanauer S. B. & Li Y. C. Mechanisms of disease: vitamin D and inflammatory bowel disease. Nat Clin Pract Gastroenterol Hepatol 2, 308–315, 10.1038/ncpgasthep0215(2005). [DOI] [PubMed] [Google Scholar]
- Wang T. T. et al. Direct and indirect induction by 1,25-dihydroxyvitamin D3 of the NOD2/CARD15-defensin beta2 innate immune pathway defective in Crohn disease. J Biol Chem 285, 2227–2231, 10.1074/jbc.C109.071225 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rufo P. A. & Bousvaros A. Current therapy of inflammatory bowel disease in children. Paediatr Drugs 8, 279–302, 10.2165/00148581-200608050-00002 (2006). [DOI] [PubMed] [Google Scholar]
- Simmons J. D., Mullighan C., Welsh K. I. & Jewell D. P. Vitamin D receptor gene polymorphism: association with Crohn’s disease susceptibility. Gut 47, 211–214, 10.1136/gut.47.2.211 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pei F. H. et al. Vitamin D receptor gene polymorphism and ulcerative colitis susceptibility in Han Chinese. Journal of digestive diseases 12, 90–98, 10.1111/j.1751-2980.2011.00483.x (2011). [DOI] [PubMed] [Google Scholar]
- Naderi N. et al. Association of vitamin D receptor gene polymorphisms in Iranian patients with inflammatory bowel disease. Journal of gastroenterology and hepatology 23, 1816–1822, 10.1111/j.1440-1746.2008.05525.x (2008). [DOI] [PubMed] [Google Scholar]
- Kim J. H. et al. Implication of intestinal VDR deficiency in inflammatory bowel disease. Biochimica et biophysica acta 1830, 2118–2128, 10.1016/j.bbagen.2012.09.020 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes D. J., McManus R., Neary P., O’Morain C. & O’Sullivan M. Common variation in the vitamin D receptor gene and risk of inflammatory bowel disease in an Irish case-control study. European journal of gastroenterology & hepatology 23, 807–812, 10.1097/MEG.0b013e328349283e (2011). [DOI] [PubMed] [Google Scholar]
- Capaldo C. T. et al. Tight function zonula occludens-3 regulates cyclin D1-dependent cell proliferation. Mol Biol Cell 22, 1677–1685, 10.1091/mbc.E10-08-0677 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov A. I. et al. Microtubules regulate disassembly of epithelial apical junctions. BMC cell biology 7, 12, 10.1186/1471-2121-7-12 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlberg C., Polly P. Gene regulation by vitamin D3. Critical reviews in eukaryotic gene expression 8, 19–42, 10.1615/CritRevEukarGeneExpr.v8.i1.20 (1998). [DOI] [PubMed] [Google Scholar]
- Gombart A. F., Borregaard N. & Koeffler H. P. Human cathelicidin antimicrobial peptide (CAMP) gene is a direct target of the vitamin D receptor and is strongly up-regulated in myeloid cells by 1,25-dihydroxyvitamin D3. FASEB journal : official publication of the Federation of American Societies for Experimental Biology 19, 1067–1077, 10.1096/fj.04-3284com (2005). [DOI] [PubMed] [Google Scholar]
- Sun J. et al. Increased NF-kappaB activity in fibroblasts lacking the vitamin D receptor. American journal of physiology. Endocrinology and metabolism 291, E315–322, 10.1152/ajpendo.00590.2005 (2006). [DOI] [PubMed] [Google Scholar]
- Wu S., Xia Y., Liu X. & Sun J. Vitamin D receptor deletion leads to reduced level of IkappaBalpha protein through protein translation, protein-protein interaction, and post-translational modification. The international journal of biochemistry & cell biology 42, 329–336, 10.1016/j.biocel.2009.11.012 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charite J. et al. Transducing positional information to the Hox genes: critical interaction of cdx gene products with position-sensitive regulatory elements. Development 125, 4349–4358 (1998). [DOI] [PubMed] [Google Scholar]
- Duprey P. et al. A mouse gene homologous to the Drosophila gene caudal is expressed in epithelial cells from the embryonic intestine. Genes & development 2, 1647–1654, 10.1101/gad.2.12a.1647 (1988). [DOI] [PubMed] [Google Scholar]
- Suzuki T., Yoshinaga N. & Tanabe S. Interleukin-6 (IL-6) regulates claudin-2 expression and tight junction permeability in intestinal epithelium. J Biol Chem 286, 31263–31271, 10.1074/jbc.M111.238147 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toell A., Polly P. & Carlberg C. All natural DR3-type vitamin D response elements show a similar functionality in vitro. The Biochemical journal 352 Pt 2, 301–309 (2000). [PMC free article] [PubMed] [Google Scholar]
- Haussler M. R. et al. The nuclear vitamin D receptor: biological and molecular regulatory properties revealed. J. Bone and Mineral Research 13, 325–349, 10.1359/jbmr.1998.13.3.32 (1998). [DOI] [PubMed] [Google Scholar]
- Xue Y. & Fleet J. C. Intestinal vitamin D receptor is required for normal calcium and bone metabolism in mice. Gastroenterology 136, 1317–1327, e1311-1312, 10.1053/j.gastro.2008.12.051 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouillon R. et al. Vitamin D and human health: lessons from vitamin D receptor null mice. Endocr Rev 29, 726–776, 10.1210/er.2008-0004 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mankertz J. et al. TNFalpha up-regulates claudin-2 expression in epithelial HT-29/B6 cells via phosphatidylinositol-3-kinase signaling. Cell and tissue research 336, 67–77, 10.1007/s00441-009-0751-8 (2009). [DOI] [PubMed] [Google Scholar]
- Yamamoto T. et al. IL-1beta regulates expression of Cx32, occludin, and claudin-2 of rat hepatocytes via distinct signal transduction pathways. Experimental cell research 299, 427–441, 10.1016/j.yexcr.2004.06.011 (2004). [DOI] [PubMed] [Google Scholar]
- Ogura M. et al. Vitamin D3 modulates the expression of bile acid regulatory genes and represses inflammation in bile duct-ligated mice. J Pharmacol Exp Ther 328, 564–570, 10.1124/jpet.108.145987 (2009). [DOI] [PubMed] [Google Scholar]
- Kamen D. L. & Tangpricha V. Vitamin D and molecular actions on the immune system: modulation of innate and autoimmunity. J Mol Med 88, 441–450, 10.1007/s00109-010-0590-9 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterhouse J. C., Perez T. H. & Albert P. J. Reversing bacteria-induced vitamin D receptor dysfunction is key to autoimmune disease. Ann N Y Acad Sci 1173, 757–765, 10.1111/j.1749-6632.2009.04637.x (2009). [DOI] [PubMed] [Google Scholar]
- Liu P. T., Krutzik S. R. & Modlin R. L. Therapeutic implications of the TLR and VDR partnership. Trends Mol Med 13, 117–124, 10.1016/j.molmed.2007.01.006 (2007). [DOI] [PubMed] [Google Scholar]
- Kong J. et al. Novel role of the vitamin D receptor in maintaining the integrity of the intestinal mucosal barrier. American journal of physiology. Gastrointestinal and liver physiology 294, G208–216, 10.1152/ajpgi.00398.2007 (2008). [DOI] [PubMed] [Google Scholar]
- Lagishetty V. et al. Vitamin d deficiency in mice impairs colonic antibacterial activity and predisposes to colitis. Endocrinology 151, 2423–2432, 10.1210/en.2010-0089 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams J. S. & Hewison M. Unexpected actions of vitamin D: new perspectives on the regulation of innate and adaptive immunity. Nat Clin Pract Endocrinol Metab 4, 80–90, 10.1038/ncpendmet0716 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu S., Bruce D., Froicu M., Weaver V. & Cantorna M. T. Failure of T cell homing, reduced CD4/CD8alphaalpha intraepithelial lymphocytes, and inflammation in the gut of vitamin D receptor KO mice. Proc Natl Acad Sci U S A 105, 20834–20839, 10.1073/pnas.0808700106 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen L. & Turner J. R. Role of epithelial cells in initiation and propagation of intestinal inflammation. Eliminating the static: tight junction dynamics exposed. American journal of physiology. Gastrointestinal and liver physiology 290, G577–582, 10.1152/ajpgi.00439.2005 (2006). [DOI] [PubMed] [Google Scholar]
- Laukoetter M. G. et al. JAM-A regulates permeability and inflammation in the intestine in vivo. J Exp Med 204, 3067–3076, 10.1084/jem.20071416 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blikslager A. T., Moeser A. J., Gookin J. L., Jones S. L. & Odle J. Restoration of barrier function in injured intestinal mucosa. Physiol Rev 87, 545–564, 10.1152/physrev.00012.2006 (2007). [DOI] [PubMed] [Google Scholar]
- Farhadi A., Banan A., Fields J. & Keshavarzian A. Intestinal barrier: an interface between health and disease. Journal of gastroenterology and hepatologyc 18, 479–497, 10.1046/j.1440-1746.2003.03032.x (2003). [DOI] [PubMed] [Google Scholar]
- Rajapaksa T. E., Stover-Hamer M., Fernandez X., Eckelhoefer H. A. & Lo D. D. Claudin 4-targeted protein incorporated into PLGA nanoparticles can mediate M cell targeted delivery. J Control Release 142, 196–205, 10.1016/j.jconrel.2009.10.033 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling J., Liao H., Clark R., Wong M. S. & Lo D. D. Structural constraints for the binding of short peptides to claudin-4 revealed by surface plasmon resonance. J Biol Chem 283, 30585–30595, 10.1074/jbc.M803548200 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y. G., Wu S. & Sun J. Vitamin D, Vitamin D Receptor, and Tissue Barriers. Tissue barriers 1, e23118, 10.4161/tisb.23118 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu R. et al. Chronic effects of a Salmonella type III secretion effector protein AvrA in vivo. PloS one 5, e10505, 10.1371/journal.pone.0010505 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Cromphaut S. J. et al. Duodenal calcium absorption in vitamin D receptor-knockout mice: functional and molecular aspects. Proc Natl Acad Sci U S A 98, 13324–13329, 10.1073/pnas.231474698 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan Y. et al. beta-Catenin activity negatively regulates bacteria-induced inflammation. Lab Invest 87, 613–624, 10.1038/labinvest.3700545 (2007). [DOI] [PubMed] [Google Scholar]
- Sun J. et al. Crosstalk between NF-kappaB and beta-catenin pathways in bacterial-colonized intestinal epithelial cells. American journal of physiology. Gastrointestinal and liver physiology 289, G129–137, 10.1152/ajpgi.00515.2004 (2005). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.