


Abstract
Jumonji domain-containing protein 6 (JMJD6) is a nuclear protein involved in histone modification, transcription and RNA processing. Although JMJD6 is crucial for tissue development, the link between its molecular functions and its roles in any given differentiation process is unknown. We report that JMJD6 is required for adipogenic gene expression and differentiation in a manner independent of Jumonji C domain catalytic activity. JMJD6 knockdown led to a reduction of C/EBPβ and C/EBPδ protein expression without affecting mRNA levels in the early phase of differentiation. However, ectopic expression of C/EBPβ and C/EBPδ did not rescue differentiation. Further analysis demonstrated that JMJD6 was associated with the Pparγ2 and Cebpα loci and putative enhancers. JMJD6 was previously found associated with bromodomain and extra-terminal domain (BET) proteins, which can be targeted by the bromodomain inhibitor JQ1. JQ1 treatment prevented chromatin binding of JMJD6, Pparγ2 and Cebpα expression, and adipogenic differentiation, yet had no effect on C/EBPβ and C/EBPδ expression or chromatin binding. These results indicate dual roles for JMJD6 in promoting adipogenic gene expression program by post-transcriptional regulation of C/EBPβ and C/EBPδ and direct transcriptional activation of Pparγ2 and Cebpα during adipocyte differentiation.
INTRODUCTION
Jumonji C (JmjC) domain-containing proteins belong to the family of ferrous iron (Fe2+)- and 2-oxoglutarate (2-OG)-dependent dioxygenases that hydroxylate a broad range of substrates that includes proteins and nucleic acids (1,2). A number of JmjC domain-containing proteins act as epigenetic regulators involved in development as well as in diseases such as cancer, inflammation, metabolic and neurological disorders (3,4). Among the family of JmjC domain proteins, Jumonji domain containing protein 6 (JMJD6) is an evolutionarily conserved nuclear protein widely expressed in various tissues (5,6). Although knockout mice showed developmental delay and abnormalities in multiple tissues, the mechanism by which JMJD6 regulates tissue development has not been demonstrated (6–9).
JMJD6 regulates gene expression at different levels. It has been shown that JMJD6 alters covalent modifications on histones as well as on transcription factors and splicing proteins via its lysine hydroxylase or methyl-arginine demethylase activity (10–14), though the functional consequences of JMJD6-mediated modification of these proteins is not fully characterized. JMJD6 also controls the release of paused RNA polymerase II through its interaction with bromodomain containing 4 (BRD4) protein at distal regulatory elements (15). In addition, biochemical characterization of JMJD6 showed its capacity to bind RNA in vitro (16). Other evidence indicates that JMJD6 not only interacts with splicing factors but also associates with nascent RNAs derived from a reporter and from endogenous genes, and that the interaction modulates constitutive and alternative splicing (17,18). However, whether these regulatory functions in gene expression account for the roles of JMJD6 in tissue development has not yet been determined.
Adipocyte differentiation is a critical aspect of organismal development and plays a major role in adipose tissue homeostasis (19–22). An increase in adipocyte number (hyperplasia) has been linked to excess nutrient overload and obesity (23,24). On the contrary, impaired adipocyte differentiation is associated with lipodystrophy and systemic metabolic disorders such as insulin resistance, dyslipidemia and steatosis (25–27). The molecular events controlling adipocyte differentiation have been extensively studied in tissue culture systems and in animal models over the past 20 years. The integration of multiple signaling pathways leads to the activation of early adipogenic regulators including CCAAT/enhancer-binding protein β (Cebpβ) and CCAAT/enhancer-binding protein δ (Cebpδ), followed by the activation of the master regulators, peroxisome proliferator-activated receptor-γ (Pparγ) and CCAAT/enhancer-binding protein α (Cebpα) (28). PPARγ and C/EBPα in turn promote the expression of a large set of phenotypic genes in mature adipocytes (29,30). Recent advances in genome-wide analysis for DNase I hypersensitive sites (DHS) and the binding of transcription factors and cofactors have provided significant insights into the epigenetic control of gene expression during adipogenic differentiation (31–33).
In the present study, we investigated the roles of JMJD6 in gene expression regulation by using a well-characterized adipocyte differentiation model. The results showed that JMJD6 is essential for initiation of the transcriptional program that promotes the terminal differentiation of C3H10T1/2 mouse mesenchymal cells. The adipogenic function of JMJD6 is independent of its JmjC domain activity. Moreover, knockdown of JMDJ6 caused a deficiency in C/EBPβ and C/EBPδ protein expression without an effect on the mRNA levels, indicating a post-transcriptional regulatory function on C/EBPβ and C/EBPδ expression. Further analysis identified the binding of JMJD6 at the Pparγ2 and Cebpα genomic loci and nearby putative enhancers when Pparγ2 and Cebpα are expressed. Targeting the chromatin readers for JMJD6 with the bromodomain and extra-terminal domain (BET) protein inhibitor, JQ1, displaced JMJD6 from the loci and prevented gene expression, indicating a role for JMJD6 and BET proteins in regulating Pparγ2 and Cebpα gene transcription. Overall, our results indicate distinct functions of JMJD6 in facilitating adipogenic gene expression program via transcriptional and post-transcriptional mechanisms.
MATERIALS AND METHODS
Tissue culture
Mouse C3H10T1/2, 293T and BOSC23 cells were maintained in growth media consisting of Dulbecco's modified Eagle's medium (DMEM) high glucose medium (Invitrogen), 10% fetal calf serum (FCS, Sigma) and 100 U/ml penicillin/streptomycin (Invitrogen). For adipogenic induction, confluent cells were cultured in DMEM high glucose medium supplemented with 10% FCS, 10 μg/ml insulin, 0.5 mM IBMX, 1 μM dexamethasome and 5 μM Troglitazone (all reagents from Sigma) for 48 h. After induction, the differentiating cells were maintained in DMEM high glucose medium supplemented with 10% FCS and 5 μg/ml insulin. Media was changed every other day until harvest.
Plasmid DNA construction
pENTR/pTER+ vector and pLentiX2 DEST vector were gifts from Dr. Eric Campeau. The preparation of small hairpin RNA (shRNA) lentiviral constructs was performed as previously described (34). Briefly, JMJD6 shRNAs, GFP shRNA and scrambled sequence shRNA oligonucleotides were individually cloned into pENTR/pTER+ vector. The pENTR/pTER+ shRNA constructs were subsequently incubated with LR clonase II enzyme mix (Invitrogen) and pLentiX2 DEST vector containing a puromycin resistance gene to generate pLentiX2 DEST/shJmjd6, pLentiX2 DEST/shGFP and pLentiX2 DEST/shCtrl constructs. The sequences of shRNA oligonucleotides are listed in Supplementary Table S1. To generate the constructs for JMJD6 and HIF1AN protein expression, the coding sequence of JMJD6 and HIF1AN with a C-terminal FLAG tag sequence was amplified from mouse embryonic fibroblast cDNA by reverse transcriptase-polymerase chain reaction (RT-PCR) and cloned into pBABE retroviral vector containing a blasticidin resistance gene (35) and pcDNA3.1(-) vector (Invitrogen). The construct for the catalytic-inactive mutant JMJD6 (H187A/D189A) protein (14) was generated from the wild type JMJD6 construct by QuikChange Lightning site-directed mutagenesis kit (Agilent). The primers for cloning and mutagenesis are listed in Supplementary Table S1.
Virus preparation and transduction
Lentivirus preparation was performed as previously described (34). Briefly, the shRNA lentiviral constructs were co-transfected with pLP1, pLP2 and pVSVG packaging vectors into 293T cells with Lipofectamine 2000 reagent (Invitrogen). The constructs encoding wild type JMJD6 (JMJD6-wt) and the catalytic-inactive mutant JMJD6 (JMJD6-mut) were individually transfected into BOSC23 cells (36). The viral supernatant was harvested after 48 h incubation and filtered through 0.45 μm syringe filter (Millipore). To infect C3H10T1/2 cells, 2 ml of the filtered supernatant supplemented with 4 μg/ml of polybrene (Sigma) were used to infect one million cells for 48 h. The infected cells were then selected in media containing 2.5 μg/ml puromycin (Invitrogen) or 5 μg/ml of blasticidin (Invitrogen).
Oil Red O staining
Cells were washed once with PBS and fixed in 10% phosphate-buffered formalin (Fisher Scientific) overnight at room temperature. The next day, the cells were washed with 60% isopropanol and air dried completely. The cells were stained with 60% Oil-Red O (AMRESCO) for 10 min and washed extensively with running tap water. To quantify staining, Oil Red O was extracted with 100% isopropanol and measured at optical density 500 nm (OD500).
Gene expression analysis
Total RNA was isolated using TRIzol reagent (Invitrogen) and treated with DNaseI (Invitrogen) according to the manufacturer's instructions. cDNAs were prepared from 1 μg of total RNA with random hexamers (Roche) and the Superscript III reverse transcriptase kit (Invitrogen). Real-time quantitative PCR was performed on a StepOne Plus real-time PCR machine with Fast SYBR green master mix (Applied Biosystems). The relative gene expression levels were calculated as 2∧(Ct5S rRNA – Ctgene) and were normalized to the experimental control as indicated (37). The primers are listed in Supplementary Table S2.
Western blot analysis
Cells were washed twice with cold PBS and harvested in RIPA buffer (50 mM Tris-HCl, pH7.4, 150 mM NaCl, 1 mM EDTA, 1% NP-40 and 0.25% sodium deoxycholate) supplemented with protease inhibitor cocktail (Roche). Mouse adipose tissue extracts were prepared as described (38). Protein concentration was measured using Bio-Rad protein assay. The samples were mixed with 4X sodium dodecyl sulphate (SDS) loading buffer (240 mM Tris-HCl, pH6.8, 8% SDS, 40% glycerol, 0.01% bromophenol blue and 10% β-mercaptoethanol) and boiled at 95°C for 10 min. Samples were separated on 10% sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto PVDF membrane (Bio-Rad). The blots were blocked in 3% non-fat milk overnight at 4°C. After a sequential incubation with the primary antibodies and HRP-conjugated secondary antibodies, the blots were developed on X-ray films with ECL western blotting detection reagents (GE Healthcare Life Sciences). Primary antibodies against JMJD6 (sc-11366), JMJD6 (sc-28348), HIF1AN (sc-26219), PPARγ (sc-7273), C/EBPα (sc-61), C/EBPβ (sc-150), C/EBPδ (sc-151) and β-ACTIN (sc-81178) were purchased from Santa Cruz Biotechnology. PI3-kinase p85 (ABS233) antibody was purchased from Millipore. The phospho-C/EBPβ (#3084) antibody was purchased from Cell Signaling. The anti-FLAG rabbit antisera was described (39). The anti-BRD2 (A302–583A), anti-BRD3(A302–367A) and anti-BRD4 (A301–985A50) antibodies were purchased from Bethyl Laboratories. The secondary antibodies (NA9340 and NA9310) were purchased from GE Healthcare Life Sciences. ImageJ software was used for image quantification.
Chromatin immunoprecipitation
Chromatin immunoprecipitation was performed as previous described (40). Briefly, cells were fixed in 1% formaldehyde at room temperature for 10 min and harvested as cell pellets. The cell pellets were re-suspended in cell lysis buffer (50 mM Tris-HCl, pH 8.0, 1% SDS, 10 mM EDTA, pH 8.0 and protease inhibitors) and sonicated in a Bioruptor (UCD-200, Diagenode) to obtain fragmented DNA sized around 500 bp. The samples were centrifuged at 12 000 x g for 10 min at 4°C to remove insoluble debris. The supernatant containing the chromatin was pre-cleared with 40 μl of a 50% slurry protein A beads (Protein A Sepharose CL-4B, GE Healthcare Life Sciences) supplemented with 0.2 mg/ml salmon sperm DNA (Invitrogen) and 0.5 mg/ml BSA (Invitrogen). 50 μg of DNA were immunoprecipitated with 4 μg of specific antibody or 20 μl of serum overnight at 4°C. The next day, the immunocomplexes were incubated with 60 μl of a 50% slurry of protein A beads for 1 h at 4°C. After extensively washing, the immunocomplexes were eluted in 1% SDS and 1% NaHCO3. The eluted samples were subjected to reverse cross-linking for 4 h at 65°C. The immunoprecipitated DNA fragments were purified using the QIAquick PCR purification kit (QIAGEN) and used as templates for quantitative PCR. The fold difference of immunoprecipitated DNA fraction relative to input was calculated as 2∧(Ctinput – CtChIP) (41). The primers are listed in Supplementary Table S3. The antibodies for ChIP assays are C/EBPβ (sc-150), C/EBPδ (sc-151), RNA polymerase II (sc-899) and rabbit IgG control (sc-2027) from Santa Cruz Biotechnology. The anti-BRD4 (A301–985A50) antibody was from Bethyl Laboratories. The anti-JMJD6 rabbit sera was made against a bacterial expressed GST-fusion with human JMJD6 (aa 2–414), which was cloned by PCR-amplification and subsequently inserted into pGEX-2T plasmid. The primers for cloning are listed in Supplementary Table S1. The specificity of the JMJD6 antisera was evaluated by western blot as shown in Supplementary Figure S1.
Immunoprecipitation
Cells were washed twice with cold PBS and lysed with a buffer containing 50 mM Tris-HCl, pH7.4, 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate and protease inhibitor cocktail (Roche) on ice for 30 min. The cell lysate were pre-cleared by 40 μl of a 50% slurry of protein A beads. 1 mg of the cell lysate was incubated with either 20 μl of anti-JMJD6 rabbit antisera or rabbit IgG control (sc-2027) for 2 h at 4°C. The samples were then incubated with 60 μl of a 50% slurry of protein A beads for 1 h at 4°C. The immunocomplexes were eluted by adding 2X SDS loading buffer and subsequently boiled at 95°C for 10 min before SDS-PAGE and subsequent western blotting.
Small interfering RNA knockdown
ON-TARGETplus mouse Cebpβ and Cepbδ siRNA SMART pools and the non-targeting control siRNA pool were purchased from Dharmacon GE Healthcare Life Sciences. The target sequences for Cepbβ mRNA were GAGCGACGAGUACAAGAUG, CCUUUAGACCCAUGGAAGU, GCACCCUGCGGAACUUGUU and GAAAAGAGGCGUAUGUAUA. The target sequences for Cepbδ mRNA were CGCGGAAGGAACACGGGAA, AGUUGUCGGCCGAGAACGA, GUAAGGAGAUGGACGCGUU and GGCACUGGACUGCGAGAGA. Briefly, cells were plated at a density of 1.5 × 105 cells per well in 12 well plates overnight and then transfected with siRNA pools using Lipofectamine 2000 in Opti-MEM medium (Invitrogen). After 4 h incubation, the medium was replaced with the normal growth medium for 20 h prior to adipogenic induction.
Plasmid and In vitro synthesized RNA transfection
pcDNA3-EGFP (#13031), pcDNA3.1(-) mouse C/EBPβ LAP (#12557) and pcDNA3.1(-) mouse C/EBPδ (#12559) were obtained from Addgene. For the preparation of in vitro synthesized RNA, pcDNA3.1(-) mouse C/EBPβ LAP, pcDNA3.1(-) mouse C/EBPδ and pcDNA3-EGFP plasmids were linearized with XbaI or HindIII and were in vitro transcribed with mMESSAGE mMACHINE T7 Ultra Transcription Kit (Ambion) according to the manufacturer's instructions. The capped and polyadenylated RNAs were further purified with RNA Clean and Concentrator Kit (Zymo Research). For transfection, 1.5 × 105 cells were plated in each well of 12-well plates overnight and transfected with either 2 μg of the plasmids or in vitro synthesized RNAs using Lipofectamine 2000 in Opti-MEM medium. After 4 h incubation, the media was replaced with the normal growth medium for 20 h prior to adipogenic induction.
Statistical analysis
At least three independent experiments were performed as indicated in the Figure legends. The values are presented as the mean ± SEM. Statistical analyses were performed using Student's t-test with two-tailed distribution and equal variance.
RESULTS
JMJD6 is expressed in adult adipose tissues and during the differentiation of C3H10T1/2 cells
To understand the biological roles of JMJD6 in adipocyte differentiation, we first examined the expression of JMJD6 in adult adipose tissues and in differentiating adipocytes. Western blot analysis showed that JMJD6 protein is expressed in both white and brown adipose tissues from 6-week-old male and female C57BL/6 mice (Figure 1A). For differentiating adipocytes, we used mouse multipotent mesenchymal C3H10T1/2 cells as a tissue culture model (42). C3H10T1/2 cells differentiate into adipocytes upon stimulation with an adipogenic induction media containing insulin, dexamethasone, 3-isobutyl-1-methylxanthine and Troglitazone. Induction of the master adipogenic regulators, PPARγ and C/EBPα, occurred on day 1 post-induction (Figure 1B). JMJD6 is present in differentiating C3H10T1/2 adipocytes even before the onset of the differentiation (Figure 1B). A transient increase of JMJD6 protein and mRNA expression was observed in the differentiating cells as compared to the levels at day 0 when the cells were undifferentiated (Figure 1C, D). Previous work has shown that JMJD6 proteins form oligomers (43–46). Consistent with western blot analysis and quantification of the monomer species (Figure 1B, C), examination of a time course of differentiating cells indicated that the oligomeric form was present at the onset of differentiation and its level increased by days 2–3 post-differentiation and decreased thereafter (Supplementary Figure S2). Based on these results, we concluded that JMJD6 is present in adult adipose tissues and expressed throughout the course of differentiation of C3H10T1/2 cells.
Figure 1.
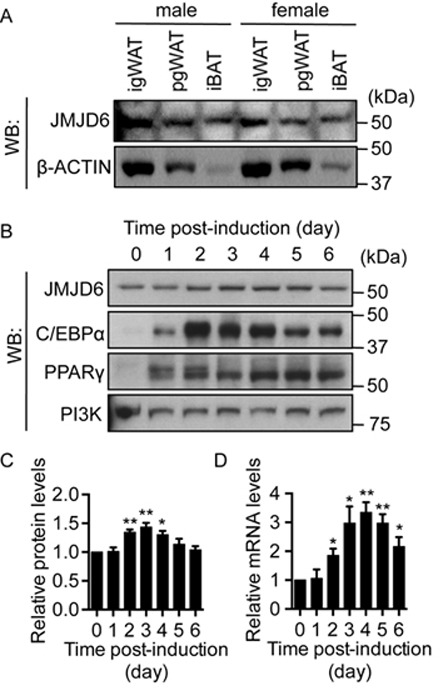
JMJD6 is expressed in adult adipose tissues and in differentiating C3H10T1/2 adipocytes. (A) Representative western blots for JMJD6 and β-ACTIN protein levels in adipose tissues from 6-week-old male and female C57BL/6 mice. igWAT: inguinal white adipose tissue; pgWAT: perigonadal white adipose tissue; iBAT: interscapular brown adipose tissue. (B) Representative western blots for JMJD6, C/EBPα and PPARγ protein levels in undifferentiated (day 0) and in differentiating (day 1 to 6) C3H10T1/2 cells. PI3K levels were probed as a loading control. (C) Quantification of JMJD6 protein levels from three independent western blots. The levels of JMJD6 were normalized to PI3K loading control and are presented as the relative expression levels to the day 0 sample (SEM, n = 3, *P < 0.05; **P < 0.01). (D)Jmjd6 mRNA levels in the differentiating C3H10T1/2 cells were determined by real-time qPCR. The mRNA levels were normalized to 5s rRNA levels and the sample at day 0 was set as 1 (SEM, n = 3, *P < 0.05; **P < 0.01).
Knocking down JMJD6 impaired adipogenic differentiation of C3H10T1/2 cells
To investigate the function of JMJD6 in adipocyte differentiation, we knocked down JMJD6 in C3H10T1/2 cells by introduction of small hairpin RNAs prior to the onset of differentiation. Two shRNAs specific for JMJD6 were used. shJ6–2 targets the 5′ UTR and the start of the 5′ coding sequence, whereas sh6–3 targets the 3′ UTR of Jmjd6 mRNA. The cells were induced to differentiate upon confluence. Adipogenic differentiation was evaluated at day 6 post-induction by quantifying the intracellular lipid content. Two distinct control shRNAs, a scramble sequence called shCtrl, and shGFP, had no effect on the adipogenic differentiation (Figure 2A, B; Supplementary Figure S3). A significant reduction in lipid accumulation in the JMJD6 knockdown cells compared to the scramble shRNA control cells was observed (Figure 2A, B), demonstrating that JMJD6 knockdown reduced the adipogenic potential of C3H10T1/2 cells. Moreover, the levels of the master regulators for terminal differentiation PPARγ and C/EBPα were significantly reduced in the day 6 post-induced JMJD6 knockdown cells (Figure 2C). Consistent with the reduction in protein levels, Pparγ and Cebpα mRNA expression was significantly reduced in the JMJD6 knockdown cells (Figure 2D). Other adipocyte-associated genes such as sterol regulatory element-binding transcription factor-1c (Srebp1c), fatty acid synthase (Fasn), adiponectin (AdipoQ) and fatty acid binding protein 4 (Fabp4) were also down-regulated (Figure 2D). These results indicated that the knockdown of JMJD6 led to a failure of differentiation of C3H10T1/2 cells due to the inability of the cells to activate the adipogenic gene expression program.
Figure 2.
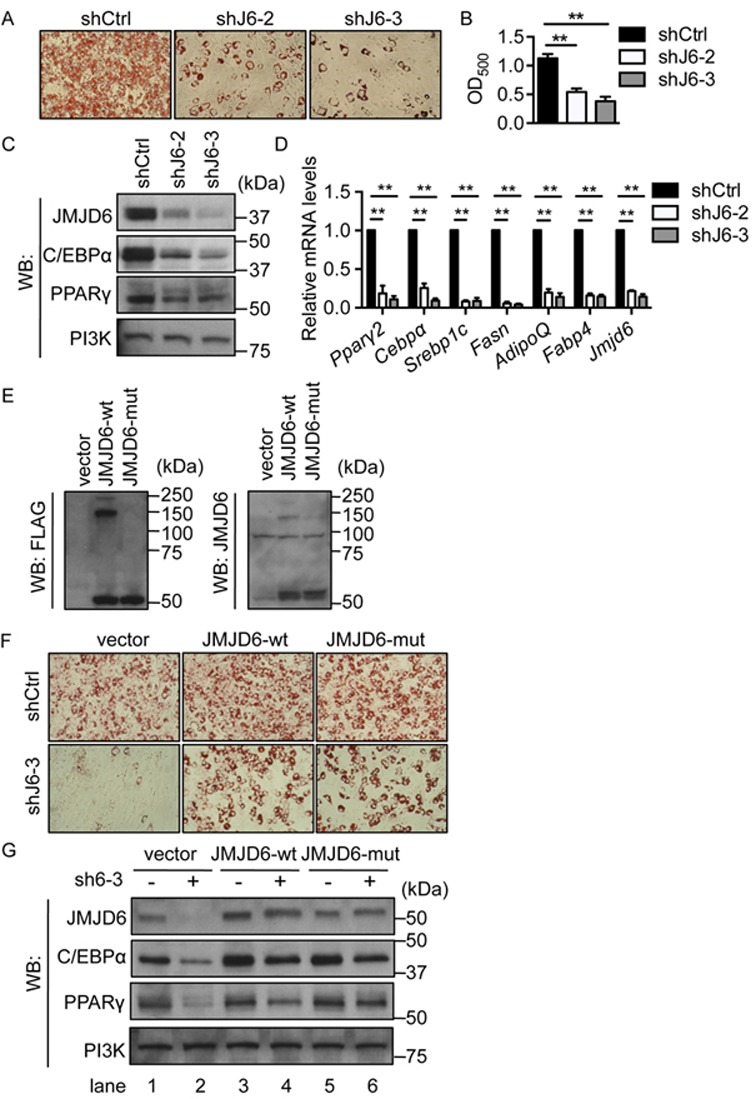
JMJD6 is required for the terminal differentiation of C3H10T1/2 cells independent of JmjC domain activity. (A) Representative Oil-Red O staining images of day 6 post-induced C3H10T1/2 cells. Endogenous JMJD6 was knocked down by either of two shRNAs (shJ6–2 or shJ6–3). A scramble shRNA construct (shCtrl) was used a control. (B) Quantification of Oil Red O staining. The values are presented as the average of optical density at 500 nm (OD500) from three independent experiments (SEM, n = 3, **P < 0.01). (C) Representative western blots for JMJD6, C/EBPα and PPARγ protein levels in the day 6 post-induced cells as indicated. PI3K levels were probed as a loading control. (D) The mRNA levels of adipocyte-associated genes in the day 6 post-differentiated cells. The individual mRNA levels were normalized to 5s rRNA levels. The normalized expression levels of the control cells were set as 1 (SEM, n = 3, *P < 0.05; **P < 0.01). (E) Representative western blots of the ectopically expressed wild type JMJD6 (JMJD6-wt) and catalytic-inactive mutant JMJD6 (JMJD6-mut) in C3H10T1/2 cells prior to differentiation. The blots were probed with either anti-FLAG antiserum or a JMJD6-specific antibody. (F) Representative Oil Red O staining images of day 6 post-differentiated cells as indicated. The C3H10T1/2 cells expressing either wild type JMJD6 (JMJD6-wt) or catalytic-inactive mutant (JMJD6-mut) or empty vector were subsequently transduced with lentiviral constructs containing either a scramble shRNA control (shCtrl) or a shRNA targeting the 3′ UTR of Jmjd6 mRNA (shJ6–3). (G) Representative western blots for JMJD6, C/EBPα and PPARγ protein levels in day 6 post-induced cells as indicated.
JMJD6 regulates adipogenesis independently of JmjC domain catalytic activity
To determine whether the catalytic activity of the JmjC domain in JMJD6 is required for adipocyte differentiation, we used a catalytically inactive mutant JMJD6 that includes substitution of histidine 187 and aspartate 189 with alanine (14). C3H10T1/2 cells were first transduced with the retroviral constructs encoding FLAG-tagged wild type JMJD6 (JMJD6-wt) or catalytically inactive mutant JMJD6 (JMJD6-mut) or with the empty vector. Ectopically expressed JMJD6 protein was evaluated by western blot analysis with an antiserum recognizing FLAG epitope and an antibody specific for JMJD6 (Figure 2E). Consistent with the literature indicating that JMJD6 oligomerization requires JmjC domain catalytic activity (45), the wild type JMJD6 protein oligomerized, but the over-expressed mutant JMJD6 was only present as a monomer (Figure 2E). The cells were subsequently transduced with the lentiviral vector containing shRNA to deplete endogenous JMJD6 protein. Upon adipogenic induction, a significant percentage of cells expressing FLAG-tagged wild type JMJD6 with endogenous JMJD6 depletion (JMJD6-wt/shJ6–3) differentiated into adipocytes (Figure 2F). Interestingly, the cells expressing the catalytically inactive mutant JMJD6 (JMJD6-mut/shJ6–3) differentiated into adipocytes to the same extent as the cells expressing wild type JMJD6 (JMJD6-wt/shJ6–3) (Figure 2F). Moreover, the protein levels of the reintroduced wild type and mutant JMJD6 proteins were similar to levels of endogenous JMJD6 in the control cells (Figure 2G, lane 4 and lane 6 compared to lane 1). The expression levels of PPARγ and C/EBPα protein were comparable in the wild type and in the mutant JMJD6 cells (Figure 2G, lane 4 and 6). These results suggest that the catalytic activity of the JmjC domain in the JMJD6 protein is dispensable for the adipogenic differentiation of C3H10T1/2 cells.
Down-regulation of C/EBPβ and C/EBPδ is associated with the differentiation deficiency in the JMJD6 knockdown cells
To determine the causes of the differentiation defect in the JMJD6 knockdown cells, we analyzed the activation of Pparγ and Cebpα genes, which is crucial for terminal differentiation. C/EBPβ and C/EBPδ are early adipogenic regulators that collaborate to activate Pparγ and Cebpα transcription during the early stage of differentiation (28,47). By examining the expression of C/EBPβ and C/EBPδ, we found that JMJD6 knockdown resulted in a reduction of C/EBPβ and C/EBPδ protein expression at 3 h post-induction (Figure 3A). However, we did not observe any effect of JMJD6 knockdown on Cebpβ and Cebpδ mRNA levels (Figure 3B), which suggests that JMJD6 is involved in the post-transcriptional control of C/EBPβ and C/EBPδ expression. Both Cebpβ and Cebpδ are intronless genes. Therefore, the known function of JMJD6 in RNA splicing would not be a direct mechanism that caused the reduction C/EBPβ and C/EBPδ protein expression. We examined the protein stability of C/EBPβ and C/EBPδ by cycloheximide treatment and western blot analysis. Protein samples were collected for up to 5 h in the presence of cycloheximide. No significant difference was detected in the degradation rate of C/EBPβ or C/EBPδ between the JMJD6 knockdown cells and the control cells (Supplementary Figure S4). Therefore, JMJD6 knockdown had no effect on C/EBPβ and C/EBPδ stability.
Figure 3.
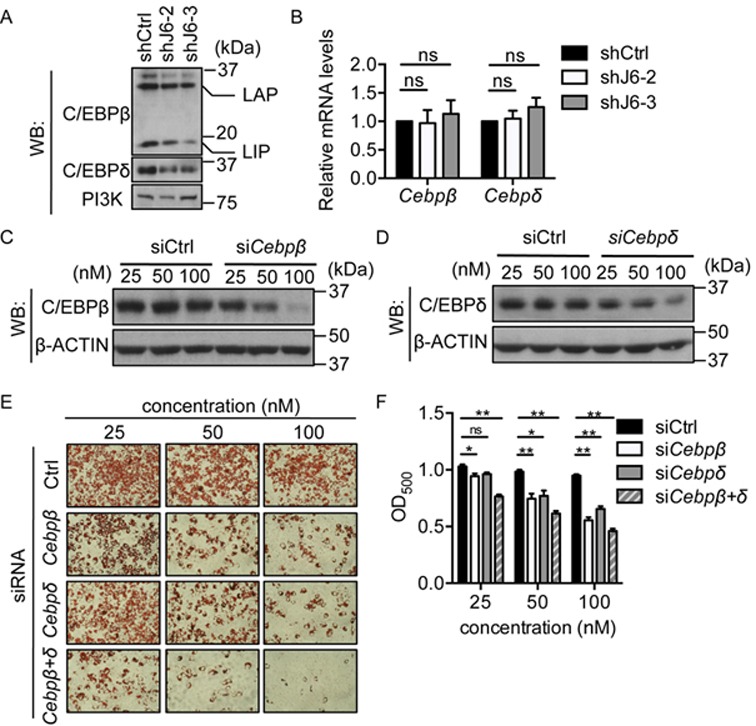
Down-regulation of C/EBPβ and C/EBPδ was associated with the differentiation deficiency in the JMJD6 knockdown cells. (A) Representative western blots for C/EBPβ and C/EBPδ protein levels in the 3 h post-induced control cells (shCtrl) and the JMJD6 knockdown cells (shJ6–2 and shJ6–3). PI3K levels were probed as a loading control. LAP and LIP represent different isoforms of C/EBPβ (B)Cebpβ and Cebpδ mRNA levels in the 3 h post-induced cells. The normalized expression levels of the control cells were set as 1. The values are presented as the average of relative expression levels with standard error (SEM, n = 3, ns: not significant). (C, D) Representative western blots for C/EBPβ and C/EBPδ protein levels in 3 h post-induced cells that were pre-treated with the indicated siRNAs for 24 h prior to adipogenic induction. β-ACTIN levels were probed as a loading control. (E) Representative Oil Red O staining images of day 6 post-induced C3H10T1/2 cells with siRNA treatment as indicated. (F) Quantification of Oil Red O staining. The values are presented as the average of optical density at 500 nm (OD500) from three independent experiments (SEM, n = 3, *P < 0.05, **P < 0.01).
Since JMJD6 knockdown did not completely abolish C/EBPβ and C/EBPδ expression, we examined whether suboptimal levels of C/EBPβ and C/EBPδ were sufficient to cause the differentiation deficiency. C3H10T1/2 cells were pre-treated with the siRNA targeting Cebpβ and Cebpδ mRNA 24 h prior to adipogenic induction. C/EBPβ and C/EBPδ protein levels were examined in 3 h post-induced cells (Figure 3C, D). At a concentration of 50 nM, the siRNA treatment caused only a modest decrease in C/EBPβ and C/EBPδ protein levels. However, the adipogenic differentiation of C3H10T1/2 was significantly blocked (Figure 3E, F). These results support the conclusion that the decease in C/EBPβ and C/EBPδ protein expression contributes to the differentiation deficiency in JMJD6 knockdown cells.
Reduced C/EBPβ protein levels correlated with the decrease of RNA Polymerase II recruitment at the Pparγ2 and Cebpα gene loci
A previous study suggested that C/EBPβ functions as a pioneer transcription factor for the activation of the transcriptional program during the early stages of adipocyte differentiation (31). Since phosphorylation of C/EBPβ modulates its transcriptional activity (48), we further examined the effect of JMJD6 knockdown on the levels of C/EBPβ and phosphorylated C/EBPβ at threonine 188 in the differentiating cells. We found that JMJD6 knockdown reduced C/EBPβ levels throughout the first 48 h of differentiation (Figure 4A), which is consistent with the previous results (Figure 3A). Moreover, the level of C/EBPβ phosphorylation was directly proportional to the level of total C/EBPβ both in shCtrl and shJ6–3 cells (Figure 4A and Supplementary Figure S5), indicating that JMJD6 was not specifically affecting C/EBPβ phosphorylation. We therefore explored the possibility that reduced C/EBPβ protein levels might directly contribute to the activation of the Pparγ2 and Cebpα genes by analyzing its binding to the corresponding promoter regions. As expected, ChIP assays showed that the binding of C/EBPβ was reduced at the Pparγ2 and Cebpα proximal promoters in the JMJD6 knockdown cells upon adipogenic induction (Figure 4B, C), which correlates with the inhibition of Pparγ2 and Cebpα expression. It has been shown that JMJD6 directly contributes to RNA polymerase II transcription through binding to distal enhancers (15). We therefore examined the binding of JMJD6 and RNA polymerase II at the Pparγ2 and Cebpα loci and their putative enhancers. For calling the putative enhancers, we searched published data sets for genomic regions that were enriched with DNAse I hypersensitivity (DHS), MED1, p300, and BRG1 binding, and H3K4me1/2 and H3K27Ac incorporation but with low levels of H3K4me3 incorporation (Supplementary Figure S6) (32,49). The −9.5 kb, +96 kb, and +100 kb regions of the Pparγ2 locus and +37 kb regions of the Cepbα were previously called as putative enhancers in immortalized brown preadipocytes (49). ChIP-qPCR results showed that the binding of JMJD6 increased upon differentiation across the Pparγ2 and Cebpα genomic loci (Supplementary Figure S7A, B). In addition, the binding of JMJD6 and RNA polymerase II at most of the sequences examined was decreased in the JMJD6 knockdown cells (Figure 4D–G). These data suggest that the failure of Pparγ2 and Cebpα gene activation caused by the JMJD6 knockdown is likely a consequence of the reduced binding of C/EBPβ, JMJD6 and RNA polymerase II at these genomic loci.
Figure 4.
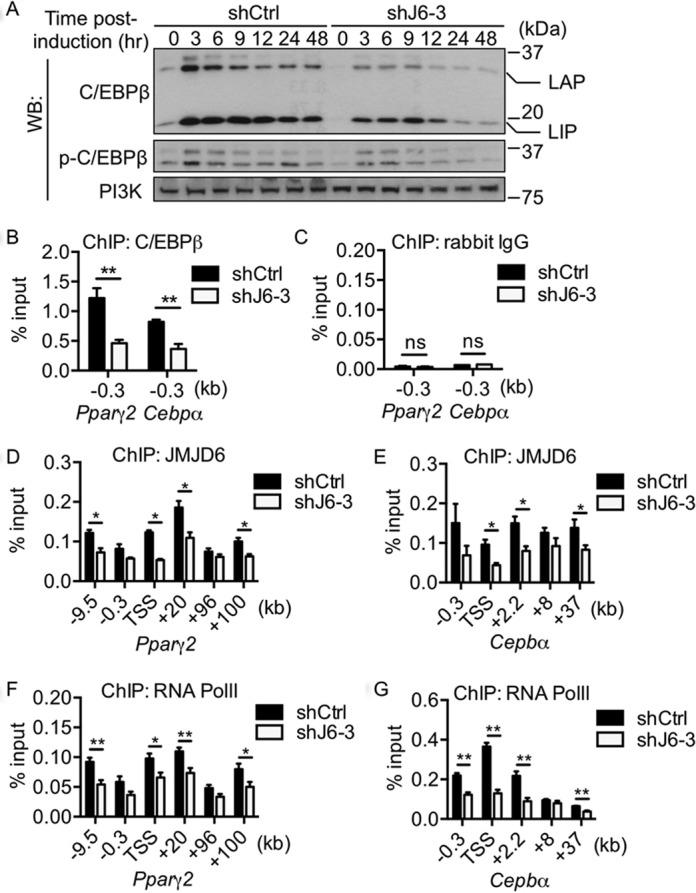
Reduced C/EBPβ protein levels correlated with the decrease in RNA polymerase II occupancy at the Pparγ2 and Cebpα loci and putative enhancers. (A) Representative western blots for the levels of C/EBPβ and phosphorylated C/EBPβ at threonine 188 in the control cells (shCtrl) and in the JMJD6 knockdown cells (shJ6–3) throughout the early phase of differentiation. PI3K levels were probed as a loading control. (B) ChIP of C/EBPβ at the Pparγ2 and Cebpα proximal promoters. (C) ChIP of rabbit IgG for negative control. (D, E) ChIP of JMJD6 and (F, G) ChIP of RNA polymerase II at the Pparγ2 and Cebpα loci and nearby putative enhancers. The values are presented as the average% input from at least three independent experiments (SEM, n≥3, *P < 0.05, **P < 0.01).
Ectopic expression of C/EBPβ and C/EBPδ is not sufficient to restore adipogenic differentiation in the JMJD6 knockdown cells
To determine whether the JMJD6-dependent defects in Pparγ2 and Cebpα gene activation were entirely related to the decrease in C/EBPβ and C/EBPδ protein levels, we attempted to rescue the adipogenic differentiation in the JMJD6 knockdown cells by ectopically expressing C/EBPβ and C/EBPδ. Transient transfection of the plasmids encoding C/EBPβ (LAP isoform) and C/EBPδ protein into the scramble control cells resulted in robust expression of C/EBPβ and C/EBPδ (Figure 5A). However, the JMJD6 knockdown cells failed to express these proteins (Figure 5A), even though the plasmid-derived mRNAs were present in the JMJD6 knockdown cells (Supplementary Figure S8A, B). These data suggest that the post-transcriptional mechanism by which JMJD6 regulates endogenous C/EBPβ and C/EBPδ also regulates the ectopic expression of these proteins. To bypass the post-transcriptional defects resulting from the knockdown of JMJD6, we introduced Cebpβ and Cebpδ mRNAs that were synthesized in vitro (Supplementary Figure S8C, D). This procedure generated equivalent protein expression both in the JMJD6 knockdown cells and in the control cells (Figure 5B). We then sought to determine whether the introduction of Cebpβ and Cebpδ mRNA could rescue the differentiation deficiency resulting from JMJD6 knockdown. For the mRNA transfection control, we chose in vitro transcribed EGFP mRNA because of the length similarity to Cebpβ and Cebpδ mRNA and the minimal effect of EGFP on the transcriptional program. As shown by Oil Red O staining, ectopic expression of Cebpβ and/or Cebpδ mRNA was not sufficient to rescue the differentiation deficiency (Figure 5C). The JMJD6 knockdown cells transfected with a combination of Cebpβ and Cebpδ mRNA failed to express endogenous Pparγ2 and Cebpα mRNA to the levels observed in the control cells (Figure 5D). Based on these results, we conclude that the JMJD6 knockdown cells must have multiple defects in Pparγ2 and Cepbα gene activation. Since JMJD6 was present at the Pparγ2 and Cepbα genomic loci (Figure 4D, E), we speculated that JMJD6 might directly contribute to the transcriptional activation of Pparγ2 and Cepbα.
Figure 5.
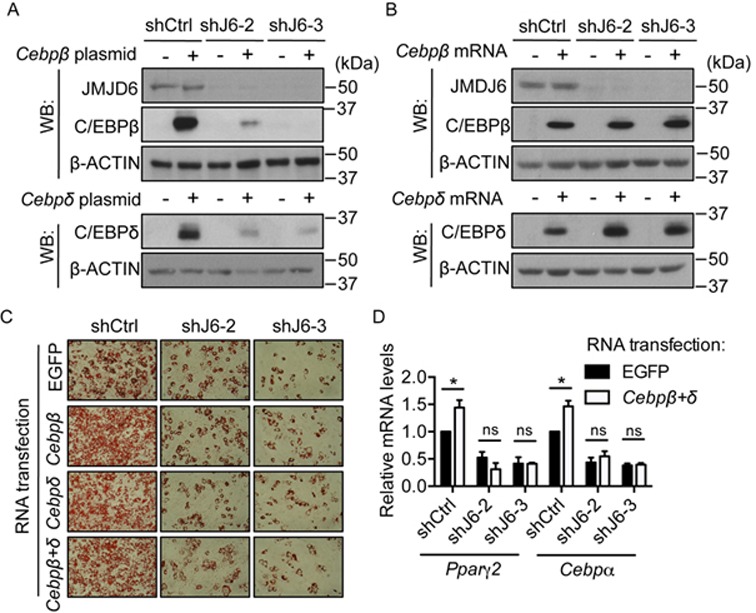
Ectopic expression of C/EBPβ and C/EBPδ was not sufficient to rescue the differentiation deficiency caused by JMJD6 knockdown. (A) Representative western blots for the levels of C/EBPβ, C/EBPδ and JMJD6 protein expression in the control cells (shCtrl) and in the JMJD6 knockdown cells (shJ6–2 and shJ6–3) 24 h after transfection with the plasmids encoding C/EBPβ(LAP) and C/EBPδ protein. An empty vector was used as a transfection control. (B) C/EBPβ, C/EBPδ and JMJD6 protein expression in the cells 24 h after transfection with in vitro synthesized Cebpβ and Cebpδ mRNA. In vitro synthesized EGFP mRNA was used as a transfection control. (C) Representative Oil Red O staining images of the day 6 post-induced cells as indicated. (D)Pparγ2 and Cebpα mRNA levels in the day 6 post-induced cells that were transfected with EGFP mRNA or a combination of Cebpβ and Cebpδ mRNA (Cebpβ+δ) as indicated. The values are presented as the average of relative expression levels with standard error (SEM, n = 3, *P < 0.05; **P < 0.01, ns: not significant).
Targeting the chromatin reader for JMJD6 blocked the adipogenic transcriptional program
JMJD6 has no characterized DNA binding domain (50) and also lacks DNA binding ability in vitro (16). Studies have shown that JMJD6 physically interacts with the chromatin reader proteins BRD4 and BRD2 (15,51). To determine the mechanisms by which JMJD6 binds to the genomic loci and directly controls Pparγ2 and Cepbα gene activation, we targeted the chromatin reader for JMJD6 by the BET protein-specific inhibitor JQ1 (52). We found that JQ1 treatment significantly inhibited adipogenic differentiation at a 1 μM dose (Figure 6A, B). By analyzing the expression of adipogenic regulators, we found that JQ1 treatment blocked PPARγ and C/EBPα protein expression (Figure 6C). Unlike in the JMJD6 knockdown cells, JQ1 had no effect on C/EBPβ and C/EBPδ levels at either 3 or 24 h post-induction (Figure 6C). Consistent with the protein analysis, JQ1 treatment inhibited the expression of Pparγ2 and Cebpα mRNAs without or with minimal effect on Cebpβ and Cebpδ mRNA expression, respectively (Figure 6D). The data indicate that the inhibition of adipogenic differentiation by JQ1 occurs via a mechanism that affects PPARγ2 and C/EBPα expression without affecting expression of the C/EBPβ and C/EBPδ. Though JQ1 affects neither the expression of JMJD6 (Figure 6C) nor the interaction of JMJD6 with BRD4 (Supplementary Figure S9), ChIP experiments showed that JQ1 treatment reduced the binding of JMJD6, BRD4 and RNA polymerase II at the Pparγ2 and Cepbα loci and their putative enhancers (Figure 7A–F), thereby providing a mechanism for the inhibition of Pparγ2 and Cebpα gene expression. JQ1 treatment did not affect the binding of C/EBPβ and C/EBPδ at the Pparγ2 and Cebpα promoter regions (Figure 7G, H). These results indicate that BRD4-mediated JMJD6 chromatin binding at the Pparγ2 and Cepbα genomic loci contributes to gene activation during differentiation.
Figure 6.
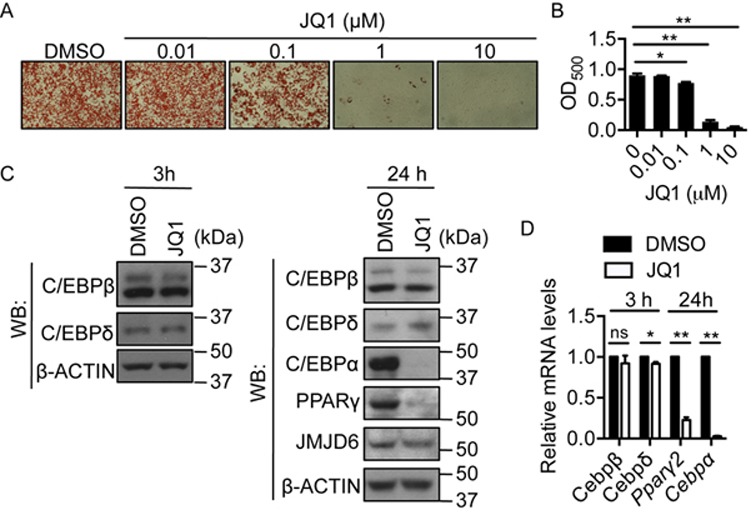
Targeting the chromatin reader for JMJD6 impaired adipogenic differentiation. (A) Representative Oil Red O staining images of day 6 post-induced C3H10T1/2 cells treated with JQ1 or DMSO. The treatment was performed in the first 48 h during the adipogenic induction. (B) Quantification of Oil Red O staining. The values are presented as the average of optical density at 500 nm (OD500) from three independent experiments (SEM, n = 3, *P < 0.05, **P < 0.01). (C) Representative western blots for the indicated proteins in the 3 h and 24 h post-induced cells in the presence of 1 μM JQ1 or DMSO as a vehicle control. (D) The mRNA levels of the indicated genes in the 3 h and 24 h post-induced cells in the presence of 1 μM JQ1 or DMSO. The values are presented as the average of relative expression levels from three independent experiments with standard error (SEM, n = 3, *P < 0.05; **P < 0.01, ns: not significant).
Figure 7.
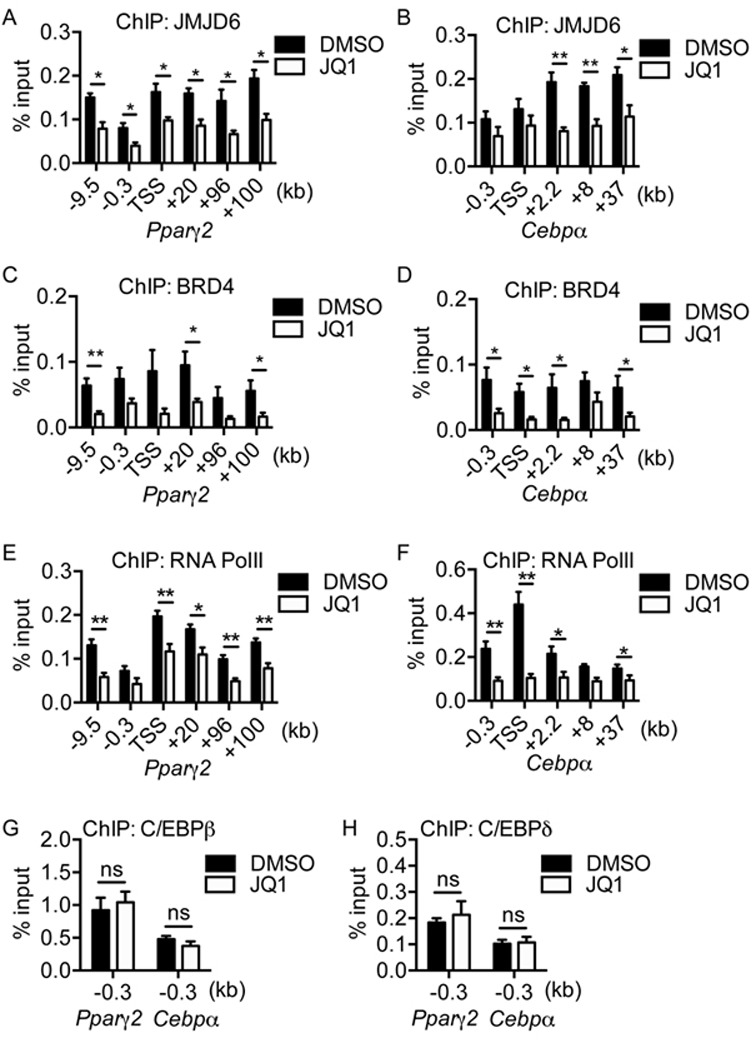
JQ1 treatment perturbed the binding of JMJD6 and RNA polymerase II at the Pparγ2 and Cebpα loci and putative enhancers. (A, B) ChIP of JMJD6, (C, D) ChIP of BRD4, and (E, F) ChIP of RNA polymerase II at the Pparγ2 and Cebpα loci and nearby putative enhancers in 24 h post-induced C3H10T1/2 cells in the presence of 1μM JQ1 or DMSO. (G) ChIP of C/EBPβ and (H) ChIP of C/EBPδ at the Pparγ2 and Cebpα proximal promoters in 24 h post-induced C3H10T1/2 cells. The values are presented as the average% input from four independent experiments (SEM, n = 4, *P < 0.05, **P < 0.01, ns: not significant). For negative control, ChIP of rabbit IgG was performed under the same conditions. The average % input from rabbit IgG ChIPs was below 0.05% in all cases (data not shown).
Collectively, the data support the conclusion that JMDJ6 acts as both a post-transcriptional and a transcriptional regulator during adipogenesis. A schematic diagram summarizing our findings is presented in Figure 8.
Figure 8.
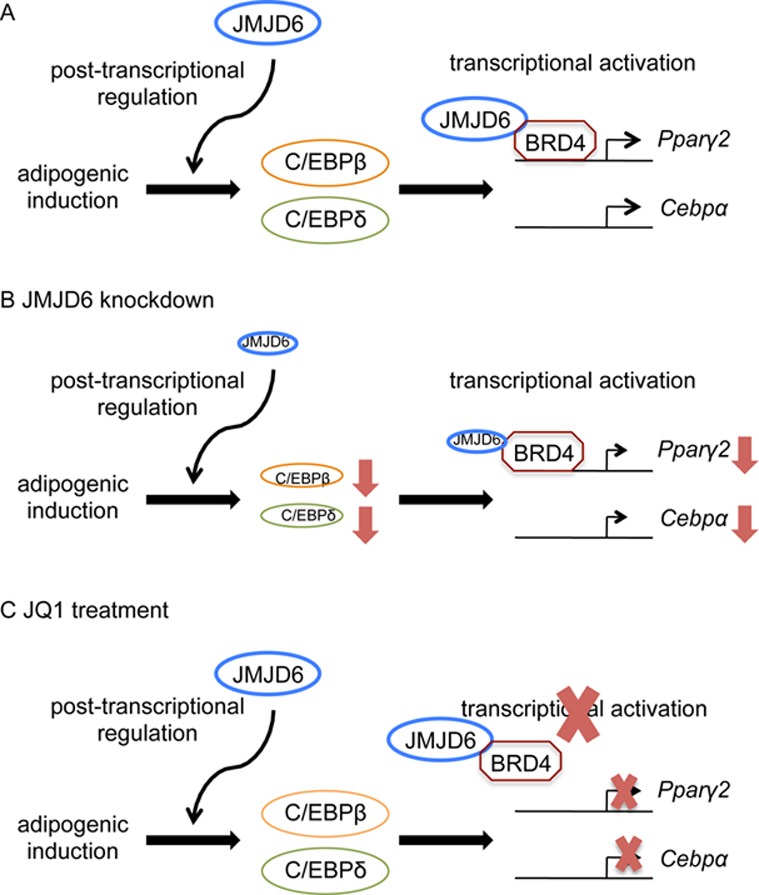
Schematic diagram of the dual functions of JMJD6 in the control of adipogenic regulator expression. (A) JMJD6 participates in the post-transcriptional regulation of C/EBPβ and C/EBPδ and transcriptional activation of Ppaγ2 and Cebpα genes. (B) Upon JMJD6 knockdown, dual functions of JMJD6 are compromised, and that results in a reduction of C/EBPβ and C/EBPδ protein levels and the mRNA levels from the Ppaγ2 and Cebpα genes. (C) Treatment of JQ1 blocks the transcriptional activation function of JMJD6 without an effect on C/EBPβ and C/EBPδ protein levels.
DISCUSSION
JMJD6 has multiple mechanisms of action
The effect of JMJD6 knockdown on C/EBPβ and C/EBPδ expression suggests a novel role of JMJD6 in the post-translational control of gene expression. We had determined that JMJD6 knockdown had no effect on C/EBPβ and C/EBPδ protein stability, and since both Cebpβ and Cebpδ are intronless genes, the reduction of C/EBPβ and C/EBPδ expression cannot be due to a defect in splicing of these two transcripts. Moreover, the general translation machinery is unlikely affected, because the in vitro synthesized Cebpβ and Cebpδ mRNA can be translated in the JMJD6 knockdown cells. Therefore, we concluded that JMJD6 promotes C/EBPβ and C/EBPδ expression at one or more steps between RNA synthesis and translation. The known JMJD6 interacting proteins are mostly involved in RNA processing (14). In addition, structural and biochemical characterization of JMJD6 also suggests a broad RNA binding capability (16). Perhaps JMJD6 is a part of Cebpβ and Cebpδ messenger ribonucleoprotein particles (mRNPs) and is involved in the assembly, processing, remodeling and localization of these mRNPs. Alternatively, JMJD6 might facilitate mRNA binding to ribosomes, which would be supported by the known interaction with ribosomal proteins and translation initiation factors (14,46). Taken together, we propose that JMJD6 plays multiple roles in post-transcriptional regulation of gene expression, regulating splicing on intron containing genes and as well as a second function related to other aspects of RNA processing. To get insight on how JMJD6 is involved in the various steps of mRNA metabolism in the context of adipogenesis, it would be interesting to identify and characterize the interacting protein partners as well as the associated RNA species with unbiased approaches such as immunoprecipitation followed by mass spectrometry or deep sequencing.
Though our studies demonstrated that JMJD6 dependent regulation of C/EBPβ and C/EBPδ expression contributes to the activation of the lineage determinants Pparγ2 and Cebpα, the work also indicates that this function is not sufficient to drive Pparγ2 and Cebpα expression and adipogenic differentiation. Thus there are additional roles for JMJD6 in promoting adipogenesis. Our ChIP results showed that JQ1 treatment caused the loss of JMJD6 binding at the Pparγ2 and Cebpα loci in a manner independent of controlling C/EBPβ and C/EBPδ expression. Therefore, JMJD6 likely directly contributes to the transcriptional activation of Pparγ2 and Cebpα genes, perhaps in cooperation with the known JMJD6 interacting protein, BRD4. Consistent with this hypothesis, treatment of cells with JQ1 caused a decrease in BRD4 and RNA polymerase II occupancy at the examined loci. Though previous work by others connected JMJD6 to the release of RNA polymerase II pausing (15), it appears that in the context of Pparγ2 and Cebpα activation, JMJD6 may also promote RNA polymerase II recruitment.
The decrease of RNA polymerase II binding at the putative enhancers near the Pparγ2 and Cebpα loci indicates that both JMJD6 knockdown and JQ1 treatment might not only inhibit the transcription of Pparγ2 and Cebpα genes but also the enhancer-derived RNAs (eRNAs). Noncoding eRNA transcripts have been identified in a number of cell types including adipocytes (53,54). Although the function of eRNAs are not fully understood, several lines of evidence suggest the roles of eRNA in promoting enhancer-promoter chromatin looping (55) and RNA polymerase II recruitment at promoters and gene bodies (56,57). Therefore the contributions of JMJD6 to transcription might occur via at both promoter and enhancer sequences.
Studies examining the effects of JQ1 on RNA polymerase II transcription have indicated that JQ1 selectively binds to the BRD4 bromodomain, and prevents the genomic binding by BRD4 and its associated factors, pTEFb and Mediator (58,59). Our study indicates that JMJD6 is one of the BRD4-associated proteins in the differentiating cells (Supplementary Figure S9), and the binding of JMJD6 at the genomic loci is prevented by JQ1 (Figure 7). However, the inhibitory effects of JQ1 on the adipogenic transcription program might be pleiotrophic, because BRD4 interacts with multiple transcription factors and chromatin regulators in addition to acetylated histones through its bromodomain (60). Furthermore, it is important to note that JQ1 has high affinity for the bromodomains of BRD2, BRD3, BRD4 and BRDT (52). Except for testes/oocyte-specific BRDT, BRD2, BRD3, BRD4 might all contribute the gene transcription program during adipogenic differentiation, possibly because these BET proteins facilitate transcriptional elongation through acetylated nucleosomes (61,62). Further complicating matters, BRD2 shows anti-adipogenic function in a hypomorphic mouse model as well as in 3T3-L1 preadipocytes (63,64). It is still unclear how BRD2 represses Pparγ2 gene expression, but the evidence indicated that BRD2 associates with PPARγ and represses the ability of PPARγ to activate downstream target genes (63). There may therefore be a regulatory balance between positive and negatively acting BET proteins. Although we did not identify any interaction of JMJD6 with BRD2 and BRD3 at the time point we assayed in the differentiating cells by co-immunoprecipitation (Supplementary Figure S9), since JMJD6 potentially interacts with both BRD2 and BRD4 (15,51), it is possible that JMJD6 collaborates with these distinct chromatin readers in a temporal-and spatial-dependent manner that differentially regulates the adipogenic transcriptional program.
JMJD6 function during adipogenesis is independent of the JmjC domain activity
JMJD6 was originally identified as an arginine demethylase (10) but subsequent work from different groups has been contradictory; some studies support the original findings while others do not and suggest that JMJD6 instead acts as a lysine hydroxylase (12–15,45,65). Our study demonstrated that the adipogenic function of JMJD6 is independent of JmjC domain activity. A recent study also reported a JmjC domain independent function of JMJD6 in the splicing of a reporter gene (17). Thus while JMJD6 has the potential to post-translationally modify chromatin and/or proteins involved in transcription, splicing, or any other cellular process, any such activity is not required for adipogenic differentiation, or, at best, contributes to the process in a non-essential manner. Given the diverse roles described for JMJD6, perhaps it functions as a scaffold protein that facilitates the localization and functions of other regulatory proteins.
The idea of JMJD6 function that is independent of JmjC domain catalytic activity raises the question of what JMJD6 domains mediate its functions. In addition to the JmjC domain, JMJD6 has a polyserine domain, multiple nuclear localization sequences, a nuclear export sequences, an AT-hook DNA binding motif, and a sumoylation site (50,66). To date, only the polyserine domain and nuclear localization sequences have been functionally evaluated (5,46,66). We attempted to identify regions of JMJD6 that are necessary for adipogenic differentiation by ectopically expressing a series of truncated JMJD6 proteins in C3H10T1/2 cells. However, the expression levels of these truncated JMJD6 proteins were either very low or not detectable (Supplementary Figure S10). These findings are consistent with the work of others who have also reported that truncations and deletions in JMJD6 decrease protein stability or form protein aggregates when they are ectopically expressed (44). Therefore, the domain(s) of JMJD6 necessary for the adipogenic function is still unclear. A systematic structure-function study using point mutations or small deletions would be necessary to identify the JmjC domain-independent functions of JMJD6.
SUMMARY
In conclusion, our work demonstrated that JMJD6 has multiple roles in promoting adipogenic differentiation via post-transcriptional regulation of C/EBPβ and C/EBPδ regulatory proteins and direct transcriptional regulation of the genes encoding the PPARγ2 and C/EBPα regulatory proteins (Figure 8). JMJD6 is therefore a critical regulator of the lineage determining transcription factors that control adipogenic differentiation.
Supplementary Material
Acknowledgments
We thank Dr Q. Wu for technical assistance, Dr E. Campeau for providing plasmids and R. Barutcu and Dr T. Padilla-Benavides for critical reading of the manuscript.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institutes of Health (NIH) [DK084278 to S.S. and A.N.I., GM56244 to A.N.I., DK32520 to UMass Medical School Diabetes and Endocrine Research Center]. Funding for open access charge: NIH [GM56244].
Conflict of interest statement. None declared.
REFERENCES
- 1.Klose R.J., Kallin E.M., Zhang Y. JmjC-domain-containing proteins and histone demethylation. Nat. Rev. Genet. 2006;7:715–727. doi: 10.1038/nrg1945. [DOI] [PubMed] [Google Scholar]
- 2.Loenarz C., Schofield C.J. Physiological and biochemical aspects of hydroxylations and demethylations catalyzed by human 2-oxoglutarate oxygenases. Trends Biochem. Sci. 2011;36:7–18. doi: 10.1016/j.tibs.2010.07.002. [DOI] [PubMed] [Google Scholar]
- 3.Johansson C., Tumber A., Che K., Cain P., Nowak R., Gileadi C., Oppermann U. The roles of Jumonji-type oxygenases in human disease. Epigenomics. 2014;6:89–120. doi: 10.2217/epi.13.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cloos P.A.C., Christensen J., Agger K., Helin K. Erasing the methyl mark: histone demethylases at the center of cellular differentiation and disease. Genes Dev. 2008;22:1115–1140. doi: 10.1101/gad.1652908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cui P., Qin B., Liu N., Pan G., Pei D. Nuclear localization of the phosphatidylserine receptor protein via multiple nuclear localization signals. Exp. Cell Res. 2004;293:154–163. doi: 10.1016/j.yexcr.2003.09.023. [DOI] [PubMed] [Google Scholar]
- 6.Böse J., Gruber A.D., Helming L., Schiebe S., Wegener I., Hafner M., Beales M., Köntgen F., Lengeling A. The phosphatidylserine receptor has essential functions during embryogenesis but not in apoptotic cell removal. J. Biol. 2004;3:15. doi: 10.1186/jbiol10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li M.O., Sarkisian M.R., Mehal W.Z., Rakic P., Flavell R.A. Phosphatidylserine receptor is required for clearance of apoptotic cells. Science. 2003;302:1560–1563. doi: 10.1126/science.1087621. [DOI] [PubMed] [Google Scholar]
- 8.Kunisaki Y., Masuko S., Noda M., Inayoshi A., Sanui T., Harada M., Sasazuki T., Fukui Y. Defective fetal liver erythropoiesis and T lymphopoiesis in mice lacking the phosphatidylserine receptor. Blood. 2004;103:3362–3364. doi: 10.1182/blood-2003-09-3245. [DOI] [PubMed] [Google Scholar]
- 9.Schneider J.E., Böse J., Bamforth S.D., Gruber A.D., Broadbent C., Clarke K., Neubauer S., Lengeling A., Bhattacharya S. Identification of cardiac malformations in mice lacking Ptdsr using a novel high-throughput magnetic resonance imaging technique. BMC Dev. Biol. 2004;4:16. doi: 10.1186/1471-213X-4-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chang B., Chen Y., Zhao Y., Bruick R.K. JMJD6 is a histone arginine demethylase. Science. 2007;318:444–447. doi: 10.1126/science.1145801. [DOI] [PubMed] [Google Scholar]
- 11.Unoki M., Masuda A., Dohmae N., Arita K., Yoshimatsu M., Iwai Y., Fukui Y., Ueda K., Hamamoto R., Shirakawa M., et al. Lysyl 5-hydroxylation, a novel histone modification, by jumonji domain containing 6 (JMJD6) J. Biol. Chem. 2013;288:6053–6062. doi: 10.1074/jbc.M112.433284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Poulard C., Rambaud J., Hussein N., Corbo L., le Romancer M. JMJD6 regulates ERα methylation on arginine. PLoS One. 2014;9:e87982. doi: 10.1371/journal.pone.0087982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang F., He L., Huangyang P., Liang J., Si W., Yan R., Han X., Liu S., Gui B., Li W., et al. JMJD6 promotes colon carcinogenesis through negative regulation of p53 by hydroxylation. PLoS Biol. 2014;12:e1001819. doi: 10.1371/journal.pbio.1001819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Webby C.J., Wolf A., Gromak N., Dreger M., Kramer H., Kessler B., Nielsen M.L., Schmitz C., Butler D.S., Yates J.R., et al. Jmjd6 catalyses lysyl-hydroxylation of U2AF65, a protein associated with RNA splicing. Science. 2009;5936:90–93. doi: 10.1126/science.1175865. [DOI] [PubMed] [Google Scholar]
- 15.Liu W., Ma Q., Wong K., Li W., Ohgi K., Zhang J., Aggarwal A.K., Rosenfeld M.G. Brd4 and JMJD6-associated anti-pause enhancers in regulation of transcriptional pause release. Cell. 2013;155:1581–1595. doi: 10.1016/j.cell.2013.10.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hong X., Zang J., White J., Wang C., Pan C., Zhao R., Murphy R.C., Dai S., Henson P., Kappler J.W., et al. Interaction of JMJD6 with single-stranded RNA. Proc. Natl. Acad. Sci. U.S.A. 2010;107:14568–14572. doi: 10.1073/pnas.1008832107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Heim A., Grimm C., Udo M., Simon H., Mackeen M.M., Merl J., Hauck S.M., Kessler B.M., Schofield C.J., Wolf A., et al. Jumonji domain containing protein 6 (Jmjd6) modulates splicing and specifically interacts with arginine-serine-rich (RS) domains of SR- and SR-like proteins. Nucleic Acids Res. 2014;42:7833–7850. doi: 10.1093/nar/gku488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Boeckel J., Guarani V., Koyanagi M., Roexe T., Lengeling A., Schermuly R.T., Gellert P., Braun T., Zeiher A., Dimmeler S. Jumonji domain-containing protein 6 (Jmjd6) is required for angiogenic sprouting and regulates splicing of VEGF-receptor 1. Proc. Natl. Acad. Sci. U.S.A. 2011;108:3276–3281. doi: 10.1073/pnas.1008098108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Spalding K.L., Arner E., Westermark P.O., Bernard S., Buchholz B. a, Bergmann O., Blomqvist L., Hoffstedt J., Näslund E., Britton T., et al. Dynamics of fat cell turnover in humans. Nature. 2008;453:783–787. doi: 10.1038/nature06902. [DOI] [PubMed] [Google Scholar]
- 20.Rosen E.D., MacDougald O.A. Adipocyte differentiation from the inside out. Nat. Rev. Mol. Cell Biol. 2006;7:885–896. doi: 10.1038/nrm2066. [DOI] [PubMed] [Google Scholar]
- 21.Gesta S., Tseng Y.-H., Kahn C.R. Developmental origin of fat: tracking obesity to its source. Cell. 2007;131:242–256. doi: 10.1016/j.cell.2007.10.004. [DOI] [PubMed] [Google Scholar]
- 22.Cristancho A.G., Lazar M.A. Forming functional fat: a growing understanding of adipocyte differentiation. Nat. Rev. Mol. Cell Biol. 2011;12:722–734. doi: 10.1038/nrm3198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sethi J.K., Vidal-Puig A.J. Thematic review series: adipocyte biology. Adipose tissue function and plasticity orchestrate nutritional adaptation. J. Lipid Res. 2007;48:1253–1262. doi: 10.1194/jlr.R700005-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang Q.A., Tao C., Gupta R.K., Scherer P.E. Tracking adipogenesis during white adipose tissue development, expansion and regeneration. Nat. Med. 2013;19:1338–44. doi: 10.1038/nm.3324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Garg A. Acquired and Inherited Lipodystrophies. N. Engl. J. Med. 2004;350:1220–1234. doi: 10.1056/NEJMra025261. [DOI] [PubMed] [Google Scholar]
- 26.Gustafson B., Gogg S., Hedjazifar S., Jenndahl L., Hammarstedt A., Smith U. Inflammation and impaired adipogenesis in hypertrophic obesity in man. Am. J. Physiol. Endocrinol. Metab. 2009;297:999–1003. doi: 10.1152/ajpendo.00377.2009. [DOI] [PubMed] [Google Scholar]
- 27.Bays H.E., Toth P.P., Kris-Etherton P.M., Abate N., Aronne L.J., Brown W.V., Gonzalez-Campoy J.M., Jones S.R., Kumar R., La Forge R., et al. Obesity, adiposity, and dyslipidemia: a consensus statement from the National Lipid Association. J. Clin. Lipidol. 2013;7:304–383. doi: 10.1016/j.jacl.2013.04.001. [DOI] [PubMed] [Google Scholar]
- 28.Farmer S.R. Transcriptional control of adipocyte formation. Cell Metab. 2006;4:263–273. doi: 10.1016/j.cmet.2006.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lefterova M.I., Zhang Y., Steger D.J., Schupp M., Schug J., Cristancho A., Feng D., Zhuo D., Stoeckert C.J., Liu X.S., et al. PPARgamma and C/EBP factors orchestrate adipocyte biology via adjacent binding on a genome-wide scale. Genes Dev. 2008;22:2941–2952. doi: 10.1101/gad.1709008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nielsen R., Pedersen T., Hagenbeek D., Moulos P., Siersbæk R., Megens E., Denissov S., Børgesen M., Francoijs K.J., Mandrup S., et al. Genome-wide profiling of PPARγ: RXR and RNA polymerase II occupancy reveals temporal activation of distinct metabolic pathways and changes in RXR dimer composition duirng adipogenesis. Genes Dev. 2008;22:2953–2967. doi: 10.1101/gad.501108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Siersbæk R., Nielsen R., John S., Sung M.-H., Baek S., Loft A., Hager G.L., Mandrup S. Extensive chromatin remodelling and establishment of transcription factor ‘hotspots’ during early adipogenesis. EMBO J. 2011;30:1459–1472. doi: 10.1038/emboj.2011.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Siersbæk R., Rabiee A., Nielsen R., Sidoli S., Traynor S., Loft A., La Cour Poulsen L., Rogowska-Wrzesinska A., Jensen O.N., Mandrup S. Transcription factor cooperativity in early adipogenic hotspots and super-enhancers. Cell Rep. 2014;7:1443–1455. doi: 10.1016/j.celrep.2014.04.042. [DOI] [PubMed] [Google Scholar]
- 33.Steger D.J., Grant G.R., Schupp M., Tomaru T., Lefterova M.I., Schug J., Manduchi E., Stoeckert C.J., Lazar M. a. Propagation of adipogenic signals through an epigenomic transition state. Genes Dev. 2010;24:1035–1044. doi: 10.1101/gad.1907110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Campeau E., Ruhl V.E., Rodier F., Smith C.L., Rahmberg B.L., Fuss J.O., Campisi J., Yaswen P., Cooper P.K., Kaufman P.D. A versatile viral system for expression and depletion of proteins in mammalian cells. PLoS One. 2009;4:e6529. doi: 10.1371/journal.pone.0006529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Guidi C.J., Veal T.M., Jones S.N., Imbalzano A.N. Transcriptional compensation for loss of an allele of the Ini1 tumor suppressor. J. Biol. Chem. 2004;279:4180–4185. doi: 10.1074/jbc.M312043200. [DOI] [PubMed] [Google Scholar]
- 36.Pear W.S., Nolan G.P., Scott M.L., Baltimore D. Production of high-titer helper-free retroviruses by transient transfection. Proc. Natl. Acad. Sci. U.S.A. 1993;90:8392–8396. doi: 10.1073/pnas.90.18.8392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Livak K.J., Schmittgen T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 38.Hung C.-M., Calejman C.M., Sanchez-Gurmaches J., Li H., Clish C.B., Hettmer S., Wagers A.J., Guertin D.A. Rictor/mTORC2 loss in the Myf5 lineage reprograms brown fat metabolism and protects mice against obesity and metabolic disease. Cell Rep. 2014;8:256–271. doi: 10.1016/j.celrep.2014.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.De Serna I.L., Ohkawa Y., Berkes C.A., Bergstrom D.A., Dacwag C.S., Tapscott S.J., Imbalzano A.N. MyoD targets chromatin remodeling complexes to the myogenin locus prior to forming a stable DNA-bound complex. Mol. Cell. Biol. 2005;25:3997–4009. doi: 10.1128/MCB.25.10.3997-4009.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hernández-Hernández J.M., Mallappa C., Nasipak B.T., Oesterreich S., Imbalzano A.N. The Scaffold attachment factor b1 (Safb1) regulates myogenic differentiation by facilitating the transition of myogenic gene chromatin from a repressed to an activated state. Nucleic Acids Res. 2013;41:5704–5716. doi: 10.1093/nar/gkt285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Carey M.F., Peterson C.L., Smale S.T. Chromatin immunoprecipitation (ChIP) Cold Spring Harb. Protoc. 2009;9 doi: 10.1101/pdb.prot5279. doi:10.1101/pdb.prot5279. [DOI] [PubMed] [Google Scholar]
- 42.Pinney D.F., Emerson C.R. 10T1/2 cells: An in vitro model for molecular genetic analysis of mesodermal determination and differentiation. Environ. Health Perspect. 1989;80:221–227. doi: 10.1289/ehp.8980221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tibrewal N., Liu T., Li H., Birge R.B. Characterization of the biochemical and biophysical properties of the phosphatidylserine receptor (PS-R) gene product. Mol. Cell. Biochem. 2007;304:119–125. doi: 10.1007/s11010-007-9492-8. [DOI] [PubMed] [Google Scholar]
- 44.Hahn P., Wegener I., Burrells A., Böse J., Wolf A., Erck C., Butler D., Schofield C.J., Böttger A., Lengeling A. Analysis of Jmjd6 cellular localization and testing for its involvement in histone demethylation. PLoS One. 2010;5:e13769. doi: 10.1371/journal.pone.0013769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Han G., Li J., Wang Y., Li X., Mao H., Liu Y., Chen C.D. The hydroxylation activity of Jmjd6 is required for its homo-oligomerization. J. Cell. Biochem. 2012;113:1663–1670. doi: 10.1002/jcb.24035. [DOI] [PubMed] [Google Scholar]
- 46.Wolf A., Mantri M., Heim A., Müller U., Erika F., Mackeen M.M., Schermelleh L., Dadie G., Leonhardt H., Vénien-Bryan C., et al. The polyserine domain of the lysyl-5 hydroxylase Jmjd6 mediates subnuclear localization. Biochem. J. 2013;453:357–370. doi: 10.1042/BJ20130529. [DOI] [PubMed] [Google Scholar]
- 47.Salma N., Xiao H., Imbalzano A.N. Temporal recruitment of CCAAT/enhancer-binding proteins to early and late adipogenic promoters in vivo. J. Mol. Endocrinol. 2006;36:139–151. doi: 10.1677/jme.1.01918. [DOI] [PubMed] [Google Scholar]
- 48.Park B., Qiang L., Farmer S.R. Phosphorylation of C/EBPβ at a consensus extracellular signal-regulated kinase/glycogen synthase kinase 3 site is required for the induction of adiponectin gene expression during the differentiation of mouse fibroblasts into adipocytes. Mol. Cell. Biol. 2004;24:8671–8680. doi: 10.1128/MCB.24.19.8671-8680.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lee J.E., Wang C., Xu S., Cho Y.W., Wang L., Feng X., Baldridge A., Sartorelli V., Zhuang L., Peng W., et al. H3K4 mono- And di-methyltransferase MLL4 is required for enhancer activation during cell differentiation. Elife. 2013;24:e01503. doi: 10.7554/eLife.01503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hahn P., Böse J., Edler S., Lengeling A. Genomic structure and expression of Jmjd6 and evolutionary analysis in the context of related JmjC domain containing proteins. BMC Genomics. 2008;9:293. doi: 10.1186/1471-2164-9-293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rahman S., Sowa M.E., Ottinger M., Smith J.A., Shi Y., Harper J.W., Howley P.M. The Brd4 extraterminal domain confers transcription activation independent of pTEFb by recruiting multiple proteins, including NSD3. Mol. Cell. Biol. 2011;31:2641–2652. doi: 10.1128/MCB.01341-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Filippakopoulos P., Qi J., Picaud S., Shen Y., Smith W.B., Fedorov O., Morse E.M., Keates T., Hickman T.T., Felletar I., et al. Selective inhibition of BET bromodomains. Nature. 2010;468:1067–1073. doi: 10.1038/nature09504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Step S.E., Lim H.-W., Marinis J.M., Prokesch A., Steger D.J., You S.-H., Won K.-J., Lazar M.A. Anti-diabetic rosiglitazone remodels the adipocyte transcriptome by redistributing transcription to PPARγ-driven enhancers. Genes Dev. 2014;28:1018–1028. doi: 10.1101/gad.237628.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lam M.T.Y., Li W., Rosenfeld M.G., Glass C.K. Enhancer RNAs and regulated transcriptional programs. Trends Biochem. Sci. 2014;39:170–182. doi: 10.1016/j.tibs.2014.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li W., Notani D., Ma Q., Tanasa B., Nunez E., Chen A.Y., Merkurjev D., Zhang J., Ohgi K., Song X., et al. Functional roles of enhancer RNAs for oestrogen-dependent transcriptional activation. Nature. 2013;498:516–520. doi: 10.1038/nature12210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mousavi K., Zare H., Dell'orso S., Grontved L., Gutierrez-Cruz G., Derfoul A., Hager G.L., Sartorelli V. eRNAs promote transcription by establishing chromatin accessibility at defined genomic loci. Mol. Cell. 2013;51:606–617. doi: 10.1016/j.molcel.2013.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schaukowitch K., Joo J.-Y., Liu X., Watts J.K., Martinez C., Kim T.-K. Enhancer RNA facilitates NELF release from immediate early genes. Mol. Cell. 2014;56:29–42. doi: 10.1016/j.molcel.2014.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lovén J., Hoke H., Lin C.Y., Lau A., Orlando D. a, Vakoc C.R., Bradner J.E., Lee T.I., Young R.A. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell. 2013;153:320–334. doi: 10.1016/j.cell.2013.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Patel M.C., Debrosse M., Smith M., Dey A., Huynh W., Sarai N., Heightman T.D., Tamura T., Ozato K. BRD4 coordinates recruitment of pause release factor P-TEFb and the pausing complex NELF/DSIF to regulate transcription elongation of interferon-stimulated genes. Mol. Cell. Biol. 2013;33:2497–2507. doi: 10.1128/MCB.01180-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wu S.-Y., Lee A.-Y., Lai H.-T., Zhang H., Chiang C.-M. Phospho switch triggers Brd4 chromatin binding and activator recruitment for gene-specific targeting. Mol. Cell. 2013;49:843–857. doi: 10.1016/j.molcel.2012.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.LeRoy G., Rickards B., Flint S.J. The double bromodomain proteins Brd2 and Brd3 couple histone acetylation to transcription. Mol. Cell. 2008;30:51–60. doi: 10.1016/j.molcel.2008.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kanno T., Kanno Y., LeRoy G., Campos E., Sun H.-W., Brooks S.R., Vahedi G., Heightman T.D., Garcia B. a, Reinberg D., et al. BRD4 assists elongation of both coding and enhancer RNAs by interacting with acetylated histones. Nat. Struct. Mol. Biol. 2014;21:1047–1057. doi: 10.1038/nsmb.2912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wang F., Liu H., Blanton W.P., Belkina A., Lebrasseur N.K., Denis G. V. Brd2 disruption in mice causes severe obesity without Type 2 diabetes. Biochem. J. 2010;425:71–83. doi: 10.1042/BJ20090928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zang K., Wang J., Dong M., Sun R., Wang Y., Huang Y., Liu X., Li Y., Wang F., Yu M. Brd2 inhibits adipogenesis via the ERK1/2 signaling pathway in 3T3-L1 adipocytes. PLoS One. 2013;8:e78536. doi: 10.1371/journal.pone.0078536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mantri M., Webby C.J., Loik N.D., Hamed R.B., Nielsen M.L., McDonough M.A., McCullagh J.S.O., Böttger A., Schofield C.J., Wolf A. Self-hydroxylation of the splicing factor lysyl hydroxylase, JMJD6. Med. Chem. Commun. 2012;3:80–85. [Google Scholar]
- 66.Cikala M., Alexandrova O., David C.N., Pröschel M., Stiening B., Cramer P., Böttger A. The phosphatidylserine receptor from Hydra is a nuclear protein with potential Fe (II) dependent oxygenase activity. BMC Cell Biol. 2004;5:26. doi: 10.1186/1471-2121-5-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.