

 This work is licensed under a
This work is licensed under a Abstract
The global epidemic of obesity is closely linked to the development of serious co-morbidities, including many forms of cancer. Epidemiological evidence consistently shows that obesity is associated with a similar or mildly increased incidence of prostate cancer but, more prominently, an increased risk for aggressive prostate cancer and prostate cancer-specific mortality. Studies in mice demonstrate that obesity induced by high-fat feeding increases prostate cancer progression; however, the mechanisms underpinning this relationship remain incompletely understood. Adipose tissue expansion in obesity leads to local tissue dysfunction and is associated with low-grade inflammation, alterations in endocrine function and changes in lipolysis that result in increased delivery of fatty acids to tissues of the body. The human prostate gland is covered anteriorly by the prominent peri-prostatic adipose tissue and laterally by smaller adipose tissue depots that lie directly adjacent to the prostatic surface. We discuss how the close association between dysfunctional adipose tissue and prostate epithelial cells might result in bi-directional communication to cause increased prostate cancer aggressiveness and progression. However, the literature indicates that several ‘mainstream’ hypotheses regarding obesity-related drivers of prostate cancer progression are not yet supported by a solid evidence base and, in particular, are not supported by experiments using human tissue. Understanding the links between obesity and prostate cancer will have major implications for the health policy for men with prostate cancer and the development of new therapeutic or preventative strategies.
Keywords: prostate cancer, obesity, lipid metabolism, inflammation, adipose tissue
Epidemiology of prostate cancer and obesity
Prostate cancer
Prostate cancer is an androgen-dependent malignancy that affects the aging male population, with the mean age at diagnosis being 66 years (1). Over the past decade, the incidence of prostate cancer has risen to be the second most commonly diagnosed cancer for males, representing 15% of male cancer diagnosis and 8% of all cancer cases (1, 2). Prostate cancer incidence varies more than 25-fold worldwide; the rates are highest in Australia/New Zealand and Northern America and in Western and Northern Europe (2). This is largely because of the increased availability of screening for prostate-specific antigen (PSA) in men without symptoms of the disease.
At diagnosis, most men present with early stage localised disease and commonly undergo radical prostatectomy surgery or external-beam radiotherapy. This therapy is curative for some men. However, for approximately one-third of cases, local recurrent and/or metastatic disease ensues, although the slow growing nature of prostate cancer means it often takes decades for this progression to become clinically relevant (3). Being a slow growing cancer, the 5-year survival rate from prostate cancer is a very high (∼98.9% based on SEER 2005–2011 data) (1). This is in contrast to other more aggressive cancers that have significantly lower 5-year survival rates (e.g., 17.4 and 64.9% in lung and colorectal cancer respectively) (1). Consequently, there are many men who live with prostate cancer for a decade or more, posing a significant burden on the healthcare system.
Despite the long latency of this disease progression, prostate cancer still accounts for a significant number of cancer deaths, being the fifth leading cause of death from cancer in men (6.6% of the total deaths in men; average age 80 years) (1, 2). There is less variation in mortality rates worldwide than is observed for incidence rates, with the number of deaths from prostate cancer being slightly greater in less developed regions. Mortality rates are generally high in predominantly black populations, very low in Asia and intermediate in the Americas and Oceania.
Obesity
Overweight and obesity are defined as the excessive accumulation of adipose tissue and stratification in these categories is often based on an individual's BMI, calculated as weight in kilograms divided by height in meters squared (kg/m2). Individuals with a BMI ranging from 18.5 to 24.9 are considered as having normal weight, those with a BMI 25–29.9 are considered overweight, and those with a BMI >30 are considered obese. Excess adiposity is an epidemic in developed countries, with two in three men now defined as overweight or obese (4). Compounding this upward trend is the increasing prevalence of obesity in children (5), leading to the prediction that by 2030 one-half of the world's population will be overweight or obese (6). In developed nations such as Australia, overweight and obesity is predicted to increase by between 0.4 and 0.8% per year, such that by 2025, 83% of males aged 20 years and older will be overweight or obese (7).
Linking prostate cancer prevalence and severity to obesity
Obesity is a major risk factor for many diseases, including cancer, accounting for approximately one-third of cancer-related deaths in 2012 (8). Obesity contributes to both the increased incidence and/or progression of many cancers including endometrial, bowel, (postmenopausal) breast and prostate cancer (9). While there is an association between adulthood obesity and similar (10, 11, 12) or increased risk (8, 13) of developing prostate cancer, the stronger and more compelling epidemiological evidence shows that obese men have a significantly higher risk of being diagnosed with aggressive, high-grade prostate cancer, compared to men of a healthy weight (10, 11, 14, 15, 16, 17). In contrast, obese men are less likely to be diagnosed with low-risk or localised prostate cancer (17, 18).
The impact of obesity on prostate cancer is illustrated in a meta-analysis of 17 studies encompassing 3 569 926 prostate cancer patients. Although the findings of the individual studies contributing to these meta-analyses differed dramatically, overall, obesity was not significantly correlated with the incidence of prostate cancer (relative risk (RR): 1.0; 95% CI, 0.95–1.06). However, obesity correlated with an increased risk of aggressive prostate cancer (RR: 1.14; 95% CI, 1.04–1.25) and risk of prostate cancer-associated mortality (RR: 1.24; 95% CI: 1.15–2.33) (12). This small, but statistically significant, result is meaningful considering the many millions of men affected by prostate cancer. Thus, obese men are more likely to die from prostate cancer-specific causes compared to men of a healthy weight. There is also evidence that obesity is an independent predictor of an increased risk of biochemical failure following radical prostatectomy and external-beam radiotherapy (14, 15). A separate meta-analysis of prostate cancer patients after primary treatment showed that a 5 kg/m2 increase in BMI (or 16 kg weight gain for a man of 1.8 m height) was associated with a 16% increase in biochemical recurrence (relapse of PSA) (19). Thus, obesity is a significant risk factor for aggressive prostate cancer, disease recurrence and prostate cancer-specific mortality. In light of the continuing increase in obesity prevalence, coupled with the worsening of prostate cancer outcome for obesity patients, this is an area of intense clinical significance.
An important consideration when assessing the epidemiology linking obesity to prostate cancer pathogenesis is the growing recognition that obesity is a heterogeneous condition. Among US adults, for example, ∼30% of individuals who are obese are considered ‘metabolically healthy,’ while ∼23% of normal-weight adults are defined as ‘metabolically unhealthy’(20). Thus, the body weight that is predictive of cancer progression and mortality is likely to be influenced by many co-variants including genetics, aerobic fitness and daily physical activity, age and an individual's metabolic and endocrine profile. The assessment of such phenotypic traits will be important in providing insights into the causal role of obesity-related metabolic abnormalities on prostate cancer progression.
Obesity and prostate cancer progression in rodent models
Studies in animals generally support a link between obesity and prostate cancer progression. The experimental approach mostly employed in such studies is to compare prostate tumour progression in mice fed a low-fat diet (LFD, ∼10% of energy derived from fat) with mice fed a hypercaloric, high-fat diet (HFD, ∼40–60% of energy derived from fat) for a period of 6–30 weeks. This high-fat feeding regime induces obesity and many of its associated metabolic and endocrine co-morbidities including increased visceral adipose tissue deposition, glucose intolerance, mild hyperinsulinemia, dyslipidemia and subclinical inflammation.
Xenograft studies that involve subcutaneous injection of immortalised human cancer cells into immune-deficient mice have generally shown, although not unanimously (21), that tumour mass and/or volume is increased in mice fed a HFD compared with mice fed a LFD (Table 1) (22, 23, 24, 25). Examination of transgenic mouse lines of accelerated prostate cancer including transgenic adenocarcinoma of mouse prostate (TRAMP) mice (24, 25, 26, 27), mice expressing human c-Myc in the prostate (Hi-Myc) (28, 29) and phosphatase and tensin homolog (Pten) haploinsufficient mice (30) have also demonstrated an increased rate of neoplastic progression toward adenocarcinoma with aggressive stromal invasion in HFD mice (Table 1). In contrast, calorie restriction in Hi-Myc mice fed a HFD prevented body weight gain and reduced the incidence and severity of invasive adenocarcinomas (28). Although equivalent transgenic models are not available in rats, two studies demonstrate that rats rendered obese by high-fat feeding have proliferative and hyperplastic changes in prostate morphology, which was associated with increased vasculature and extracellular matrix deposition (31, 32). Together, these findings of similar incidence of hyperplasia and low-grade prostatic intraepithelial neoplasia, but progression toward more aggressive prostate cancer in rodent obesity, reflect the general view in humans that obesity increases the risk of more aggressive disease in humans (see ‘Epidemiology prostate cancer and obesity’ above).
Table 1.
Effect of diet-induced obesity on prostate cancer progression in rodents
| Model | Diet | Cancer progression | Reference |
|---|---|---|---|
| Xenograft/Allograft models | |||
| PC3 cells xenograft into Swiss nu/nu mice | LFD vs HFD for 20–22 weeks | No difference in tumour mass 4–6 weeks after xenograft. | 21 |
| LNCaP cells xenograft into BALB/c-nu/nu mice | LFD (9.5% fat) vs HFD (59% fat) | Increase in tumour mass of HFD mice 14 weeks after xenograft. | 22 |
| LNCaP cells xenograft into nude mice | LFD (10% fat) vs HFD (57% fat) | Increased tumour volume and plasma PSA levels in HFD mice after 12 weeks. | 23 |
| TRAMP-C2 cells allograft into C57Bl/6 mice | LFD (10% fat) vs HFD (60% fat) starting at 4 weeks for 20 weeks | Increase in tumour volume, proliferation, angiogenesis and lymphangiogenesis in HFD mice 11 weeks after allograft. | 25 |
| TRAMP-C2 cells allograft into C57Bl/6 mice | LFD (9.4% fat) vs HFD (33% fat) starting at 6 weeks for 20 weeks | Increase in tumour mass and volume (20% NS) in HFD mice 10 weeks after allograft. | 24 |
| Transgenic mice models of prostate cancer | |||
| Pten haploinsufficient mice | LFD (12% fat) vs HFD (45% fat) starting at 4 weeks | Increased neoplastic progression, angiogenesis, inflammation and epithelial-mesenchymal transition in HFD mice after 3–6 months | 30 |
| TRAMP mice | LFD (9.4% fat) vs HFD (33% fat) starting at 6 weeks | No effects on tumour development, tumour differentiation, tumour grade, age of tumour palpation and age of death assessed after 36–40 weeks. Note, no difference in body mass. | 24 |
| TRAMP mice | LFD (16% fat) vs HFD (40% fat) starting at 3 weeks | Increased tumour formation rate and death rate in HFD mice after 28 weeks. | 26 |
| TRAMP mice | LFD (10% fat) vs HFD (45% fat) starting at 6 weeks | Lateral and dorsal PIN lesions worse in HFD mice after 10 weeks, but not 5 weeks. | 27 |
| TRAMP mice | LFD (10% fat) vs HFD (60% fat) starting at 4 weeks | Increased incidence of PDC, reduced PIN and increased lung and liver metastasis in HFD mice after 20 weeks | 25 |
| Hi-Myc mice | LFD (10% fat) vs HFD (60% fat) starting at 8 weeks | Similar incidence of hyperplasia and low-grade prostatic intraepithelial neoplasia in the ventral prostate at 3 and 6 months of age. Increased incidence of adenocarcinoma with aggressive stromal invasion in HFD mice at 6 months. | 28 |
| Hi-Myc mice | LFD (12% fat) vs HFD (42% fat) starting at 3 weeks | Increased transition from PIN to invasive adenocarcinoma in HFD mice assessed at 7 months. | 29 |
HFD, high-fat diet; Hi-Myc, mice express human c-Myc in the mouse prostate; LFD, low-fat diet; PDC, poorly differentiated carcinoma; PIN, prostatic intraepithelial neoplasia; Pten, phosphatase and tensin homolog; TRAMP, transgenic adenocarcinoma mouse prostate.
Changes in adipose tissue with obesity: links with prostate cancer progression
Despite the persuasive epidemiological evidence and proof-of-concept experiments in mice (Table 1), the biological mechanisms and associated cellular signalling pathways linking obesity and prostate cancer remain incompletely understood. Evidently, such mechanistic insight is important for the development of preventative or therapeutic strategies aimed at reducing prostate cancer burden in obesity.
Adipose tissue is a complex organ that consists of multiple cell types including adipocytes, adipocyte progenitor cells, mesenchymal stem cells, endothelial cells and various resident and infiltrating immune cells (33). It is unlikely that adipose tissue expansion per se is responsible for the development of obesity-associated complications, but rather, complications become evident when adipocyte hypertrophy occurs in the absence of appropriate neovascularisation (34). Under these circumstances, some argue that this leads to an inadequate supply of nutrients, growth factors and oxygen that in turn initiates a sequela of events including localised hypoxia, cellular death, inflammation, extracellular matrix remodelling and fibrosis and other stress responses (35). Together, this process of aberrant adipose tissue expansion is thought to cause the metabolic and endocrine dysregulation associated with obesity.
In this regard, numerous metabolic, endocrine and inflammatory changes that occur with obesity might promote prostate cancer progression (13). These include altered lipid metabolism and dyslipidemia, the development of pre-diabetes that is characterised by insulin resistance and mild hyperinsulinemia, altered secretion of adipokines, development of a subclinical pro-inflammatory state and alterations in several endocrine cascades including the growth hormone/insulin-like growth factor-1 axis, renin-angiotensin system and steroid hormones (Fig. 1). Herein, we will discuss the established and putative links between these obesity-generated defects and cancer progression in the knowledge that, in all likelihood, multiple hits from several obesogenic factors are likely to converge to activate a common set of signalling pathways to promote prostate cancer growth. We have also added the following text on p.8: ‘In our critical evaluation of the literature, we highlight the problematic interpretation of some experimental approaches and deficiencies in the field, which segues to the Perspectives section at the conclusion of the article.
Figure 1.

Linking obesity to prostate cancer. Changes in endocrine and metabolic function and the inflammatory milieu occur during the development of obesity. Changes associated with alterations in adipose tissue function are shown in the dark blue box and changes in the systemic metabolism and endocrine function are shown in the light blue box. Arrows indicate the direction of change in obese vs lean individuals. AngII, angiotensin II; AT11R, angiotensin II type 1 receptor; PEDF, pigment epithelium-derived factor; RTK, receptor tyrosine kinase
Obesity-driven inflammation and prostate cancer
Adipose tissue becomes increasingly infiltrated with macrophages, neutrophils, T- and B-cells, and mast cells during the development of obesity (34). In this regard, adipose tissue from obese mice and humans is infiltrated and accumulated with a large number of macrophages, which can ultimately comprise up to 40% of the cells in obese adipose tissue (36). Other immune cells are also recruited to the adipose tissue, especially lymphocytes, and produce numerous pro-inflammatory cytokines and chemokines including tumour necrosis factor (TNF)α, interleukin (IL)-6, IL-1β and monocyte chemoattractant protein (MCP)-1 (for review, see Olefsky and Glass (37)). It is important to note that this obesity-driven inflammatory state is characterised by small increases in blood cytokine levels, somewhere in the order of 0.5–5 pg/ml for TNFα and IL-6 respectively. Hence, low-grade or subclinical chronic inflammation may provide a permissive- or pro-tumorigenic endocrine and/or paracrine environment in obese individuals.
IL-6 is associated with prostate cancer progression (38) with several studies reporting high serum IL-6 levels in patients with advanced castrate-resistant and metastatic prostate cancer (39, 40, 41). It is possible that adipose tissue is an important source of IL-6 as systemic levels of IL-6 correlate positively with body weight and adipose-derived IL-6 accounts for up to 35% of circulating IL-6 (13). While historical data indicates that IL-6 is expressed in most prostate cancer cell lines and clinical specimens (42), a recent study showed that IL-6 production by prostate cancer tissues is negligible and that the growth promoting effects of IL-6 are paracrine (i.e., from adjacent adipose tissues; see ‘Peri-prostatic adipose tissue and local paracrine regulation of prostate cancer progression’) rather than autocrine (43). In support of this concept, peri-prostatic adipose tissue (PPAT) produces a significant amount of IL-6, which is likely to be an important local driver of prostate carcinogenesis (44).
Other inflammatory cytokines have been identified as potential mediators of inflammation and prostate cancer (for review, see Sfanos and De Marzo (45)). For example, increased circulating levels of macrophage inhibitory cytokine 1 (MIC-1), a member of the transforming growth factor-β (TGF-β) superfamily, predicts poor prostate cancer prognosis (46) by mediating the interaction between macrophages and prostate cancer development (47). In addition, other pro-inflammatory cytokines such as IL-8 (48, 49) and TNFα(50) correlate with prostate cancer aggressiveness and bone metastasis. It remains uncertain whether obesity is required for, or even associated with, pro-inflammatory cytokine/chemokine promotion of prostate tumourigenesis, because these interactions are seldom studied in the context of obesity. It is also unclear whether the major source of these elevated cytokine levels is the expanded adipose tissue or the prostate gland itself, and by extension, whether the effects on tumourigenesis are autocrine, paracrine or systemic.
It has long been recognised that inflammation is a predisposing factor for cancer initiation and progression, and indeed, chronic inflammation is now regarded as an enabling characteristic of human cancer (51). Chronic inflammation in the prostate gland can be induced by infections, dietary factors, hormonal changes and/or other unknown environmental exposures, leading to pathologies such as prostatitis and proliferative inflammatory atrophy (PIA) (52). These two chronic pathologies, mainly comprised of lymphocytes and macrophages (compared to neutrophils for acute inflammation), are associated with an increased risk of human prostate cancer development (53, 54). The impact of obesity on chronic or acute prostate cancer-associated inflammation in patients is unexplored. However, studies in mice have indicated a strong link between obesity, inflammation and increased cytokine production. For example, prostatic inflammation was observed in obese Hi-Myc mice, with a significant increase in T-lymphocytes and macrophages in the ventral prostate (28). This prostatic inflammation led to enhanced inflammatory (NFκB and cytokines) and growth factor (Akt/mTORC1 and STAT3) signalling, which in turn fuelled tumour progression (28). TRAMP mice also showed significant increases in serum IL-1α, IL-1β, IL-6 and TNF-α when fed a HFD vs LFD that was associated with accelerated tumourigenesis (26). However, the latter study did not determine whether the chronic inflammation underpinning the elevated cytokine secretion originated from the expanded adipose tissue, and/or extended to the prostate itself. Collectively, these data indicate that the inflamed adipose tissue of obese individuals provides a source of pro-inflammatory cytokines and possibly immune cells that infiltrate the prostate gland, although this concept is unexplored.
Adipokines and prostate cancer
Adipocytes are active endocrine cells responsible for the biosynthesis and secretion of a large number of hormones and cytokines that have a variety of biological functions (55). Screening studies in our laboratory have identified ∼170 adipokines including adiponectin, leptin, retinol binding protein (RBP)4, pigment-epithelium derived factor, visfatin, vascular endothelial growth factor (VEGF), TGF-β and various acute phase reactants (56). Protein secretion from adipocytes is modulated by a variety of processes including transcription, translation, post-translational modifications and secretion through classical and various non-classical pathways (57). Dysfunction of the expanded adipose tissue is postulated to significantly alter the adipokine secretion profile in obesity, in turn altering endocrine and paracrine/autocrine signalling that is postulated to impact both the systemic circulation and/or local tumour microenvironment in obese patients. However, studies in mice are not supportive of this hypothesis, (58) and a comprehensive examination of the adipocyte secretome in human obesity is not described. Nevertheless, it is well documented that some adipokines are differentially secreted in obesity; most prominently leptin secretion is increased and adiponectin decreased. Given that leptin stimulates prostate cancer cell growth and proliferation (59) and adiponectin is growth inhibitory (60), the concurrent changes in adipokine secretion support prostate cancer progression. A full interrogation of the adipokines produced by specific adipose tissue depots in obese vs lean men, including those with prostate cancer, is warranted.
Sex steroids, obesity and prostate cancer
Adiposity influences the synthesis and bioavailability of sex hormones. This is highly relevant in obese men with prostate cancer because it is a hormone-dependent cancer. Androgens are the major driver of prostate cancer growth and progression, and thus androgen-deprivation therapy is the standard of care for advanced disease. In addition to androgens, the prostate is also an estrogen-regulated organ and there is a body of evidence that estrogens induce major adverse and beneficial effects in the prostate gland through estrogen receptors ERα and ERβ respectively (61, 62).
In adult men, total testosterone levels have been reported to be inversely correlated with the BMI and subcutaneous and visceral fat content (63, 64). Furthermore, adipose-derived cytokines (such as TNFα and IL-6) inhibit the production of testosterone by negatively feeding back to the hypothalamus (65). The reduction in total testosterone levels in obese men is further compounded by the suppressed synthesis of sex-hormone binding globulin (SHBG) (66), a carrier protein that specifically binds circulating testosterone and reduces its bioavailability to peripheral tissues. Thus, both total and bioavailable testosterone is reduced in obese men. Despite the suppression of this hormone, which is the major driver of prostate cancer growth and development, a study of obese men with prostate cancer showed that increased adiposity, but not low testosterone, was associated with advanced disease and poor differentiation of prostate cancer (67). Thus, while obesity-associated changes in systemic testosterone availability are consistently observed, they may not be as important as local or adipose-derived factors in driving prostate cancer progression.
A second major endocrine change in obesity is an increase in estrogen production because adipose tissue expresses the estrogen metabolising enzyme, aromatase. Low testosterone in aging males is typically combined with higher estrogen levels, comparable to the level detected in postmenopausal women (68). Estradiol levels (and aromatase activity) are positively correlated with body fat mass and, more specifically, to subcutaneous abdominal fat, but not to visceral fat (69), such that serum estrone (E1) and 17β-estradiol (E2) were reported twofold elevated in a group of morbidly obese men (64). However, serum estrogen levels do not always correlate with tissue levels, and the changes in steroid biosynthesis and steroid receptor activity within the malignant prostate gland and/or the adjacent PPAT of obese men remain unknown.
Increased fatty acid availability and metabolic reprogramming as a driver of prostate cancer
All cells require the metabolic substrate to fuel adenosine triphosphate production to support cellular functions, to drive growth and permit proliferation. Distinct regulatory mechanisms have evolved to control cellular metabolism in proliferating vs non-proliferating cells. The Warburg effect describes how most solid tumours undergo metabolic reprogramming that results in increased glucose uptake, glycolysis and lactate production, even in the presence of sufficient oxygen (70). Prostate cancer is a slowly proliferating tumour and the rates of glucose uptake and glycolysis are relatively low, indicating distinct metabolic programming in this disease (57). This is due, at least in part, to a low abundance of the primary glucose transporter GLUT1 (71). In addition, the prostate uniquely produces and accumulates extraordinarily high citrate levels (10–50 000 nmol/g in prostate vs ∼200 nmol/g in muscle (72, 73)), and citrate is a powerful allosteric inhibitor of phosphofructokinase, a rate-limiting and regulatory enzyme of glycolysis (Fig. 2). Exemplifying this point is the poor utility of 2-deoxy-2-(18F)fluoro-D-glucose and positron emission tomography for prostate cancer detection or staging (74).
Figure 2.

Schematic outlining fatty acid metabolism in the prostate. Fatty acids derived from the systemic circulation and the resident peri-prostatic adipose tissue are transported into the prostate epithelial cell where they undergo several fates including (1) oxidation in the mitochondria, (2) bulk storage as triglycerides contained within lipid droplets, (3) incorporation into phospholipids and sterol lipids to facilitate membrane production and (4) conversion to signalling molecules. Fatty acids can inhibit glucose metabolism through several mechanisms. Fatty acids inhibit the activity of hexokinase, and products of fatty acid oxidation, including acetyl CoA and citrate, are powerful allosteric feedback signals that suppress the activity of rate-limiting enzymes of glucose metabolism. ATP, adenosine triphosphate; CD36, fatty acid translocate/CD36; G-6-P, glucose-6-phosphate; HK, hexokinase; PFK, phosphfructokinase.
Fatty acids are essential to support cancer cells, impacting directly on cell growth, proliferation, differentiation and motility (57, 75). Fatty acids impact these functions by meeting the bioenergetic requirements for cell proliferation; providing precursors for triglyceride synthesis, which are an important intracellular energy storage depot; providing the building blocks for phospholipid and sterol production that are incorporated into membranes to facilitate cell proliferation and growth; and acting as signalling molecules to impact a range of biochemical pathways in cancer proliferation and metastases (Fig. 2).
Fatty acids can be derived from triglycerides contained in circulating very low density lipoproteins and chylomicrons, fatty acids derived from adipose tissue lipolysis or de novo lipogenesis, which is the production of fatty acids from acetyl CoA that is mainly derived from glucose breakdown in the mitochondria. Lipogenic gene expression is increased in prostate cancer and targeted disruption of lipogenic proteins (e.g., ACLY, ACC, FASN or SCD1) can reduce prostate cancer progression (57, 75), highlighting the importance of fatty acids in prostate cancer progression.
Obesity is associated with increased systemic and local fatty acid availability owing largely to an increased fat mass and metabolic reprogramming. Adipose tissue lipolysis is the process of triglyceride breakdown to produce fatty acids and is increased under basal and postprandial conditions in overweight and obese men (76), resulting in excess fatty acid release relative to tissue needs. Obesity is also associated with increased fatty acid uptake by peripheral tissues, which results in excessive lipid storage in almost all tissues (77, 78). Thus, it is possible that increased fatty acid delivery contributes to more aggressive prostate cancer in obesity.
Although prostate cancer is proposed to be associated with a high dependence on fatty acid metabolism (57, 79), direct evidence supporting this claim was lacking until recently. Fatty acid oxidation is reported to be higher in immortalised prostate cancer cells (LNCaP, VCaP and PC3) compared with non-cancerous prostate epithelial cells (BPH-1 and WPMY-1) (80). The pharmacological blockade of either fatty acid oxidation (Etomoxir, carnitine palmitoyltransferase 1 inhibitor) or intracellular triglyceride hydrolysis (Orlistat, a lipase inhibitor) decreased proliferation and viability in immortalised and patient-derived prostate cancer cells (81), and treating mice with Etomoxir decreased xenograft growth, demonstrating the importance of fatty acid oxidation for cancer progression (81).
Studies in both solid tumours and haematopoietic cells have provided evidence for the dependence of cancer cells on fatty acid oxidation and, in turn, unveiled exciting therapeutic opportunities (82). The understanding of metabolism in prostate cancer is less advanced, and deciphering the changes in transcriptional regulation and cellular signalling that directs metabolic reprogramming in prostate cancer tumours will provide the requisite information to develop specific therapeutic interventions. Based on the current knowledge, and the evidence that fatty acid availability is increased in obesity (76), strategies aimed at reducing fatty acid uptake into prostate tumours may provide a novel therapeutic avenue for prostate cancer treatment.
Peri-prostatic adipose tissue and local paracrine regulation of prostate cancer progression
Although obesity-driven systemic effects are likely to be important in promoting prostate cancer, it is important to also consider the local prostatic milieu, because the microenvironment is a major determinant in directing local cancer growth, invasion and distant metastasis (51, 83). Adipocytes affect tumour characteristics in other cancers, most notably breast cancer, where adipocytes exist within tissue and bi-directional paracrine signalling can occur (84). The prostate may be subjected to similar regulation because it is covered anteriorly by PPAT, although distinct to breast, PPAT is located outside of the prostate gland (85). Clinical studies evaluating the prognostic value of PPAT report that PPAT quantity is increased in obesity and is a risk factor for both the diagnosis of prostate cancer and high-grade disease (86). In a study of 932 patients undergoing external radiotherapy or brachytherapy, PPAT quantity measured by computed tomography was associated with more aggressive (PSA >20 or Gleason score ≥8 or T3) disease (87). This finding was confirmed in pre-diagnosis populations where PPAT thickness was identified as a risk factor for prostate cancer detection upon biopsy and an independent predictive factor for high-grade prostate cancer (88).
In light of these associations, several studies have evaluated the tumour promoting potential of PPAT-secreted factors in vitro (89, 90). Immortalised prostate cancer cells were grown in the presence of PPAT-secreted factors collected in culture medium. The PPAT-secreted factors resulted in significantly increased proliferation (89) and motility (90) of the PC3 prostate cancer cell line, and these effects were exacerbated in cells treated from PPAT from obese vs lean men (89). Interestingly, the secreted products from the cells of the stromal vascular fraction (i.e., cells other than adipocytes) were less pro-tumorigenic than the whole PPAT secretome, indicating that factors from adipocytes and other cells act cooperatively in promoting a pro-tumorigenic environment (90). Another important insight from this work was that proliferation induced from PPAT conditioned medium was significantly increased over conditioned medium from either subcutaneous or visceral adipose tissue depots (89, 90), indicating that PPAT secretes a unique subset of molecules that promotes tumour aggressiveness. The composition of this secretome remains unresolved. Together, these studies indicate that PPAT secretes factors that are capable of altering the microenvironment of prostate tissue to promote tumour cell proliferation, survival and motility and that this secretome is more tumorigenic in obesity.
Based on the in vitro models described above, it appears that PPAT secretes factors to contribute to the prostate cancer microenvironment (85, 89, 91). However, direct support for this hypothesis is dependent on the demonstration of direct portal communication between these tissues. In addition to PPAT, there are adipocytes directly adjacent to the prostate gland itself (Fig. 3A), and it is tempting to speculate that these adipocytes signal in a paracrine manner to cancer foci. More importantly, in aggressive prostate cancer involving extra-prostatic extension, prostate cancer cells come into direct contact with adjacent adipocytes, increasing the likelihood of bi-directional paracrine interaction (Fig. 3B). It is also possible that once extension beyond the prostate occurs, signals produced by prostate cancer cells may alter adipocyte metabolism and endocrine function and promote inflammation (90). The pro-inflammatory milieu induced by the localised immune response to the presence of cancer, which often occurs in extra-prostatic tissues (Fig. 3B and C), would promote lipolysis in the adjacent adipocytes and in turn provide fatty acid substrate for tumour progression (see ‘Increased fatty acid availability and metabolic reprogramming as a driver of prostate cancer’).
Figure 3.
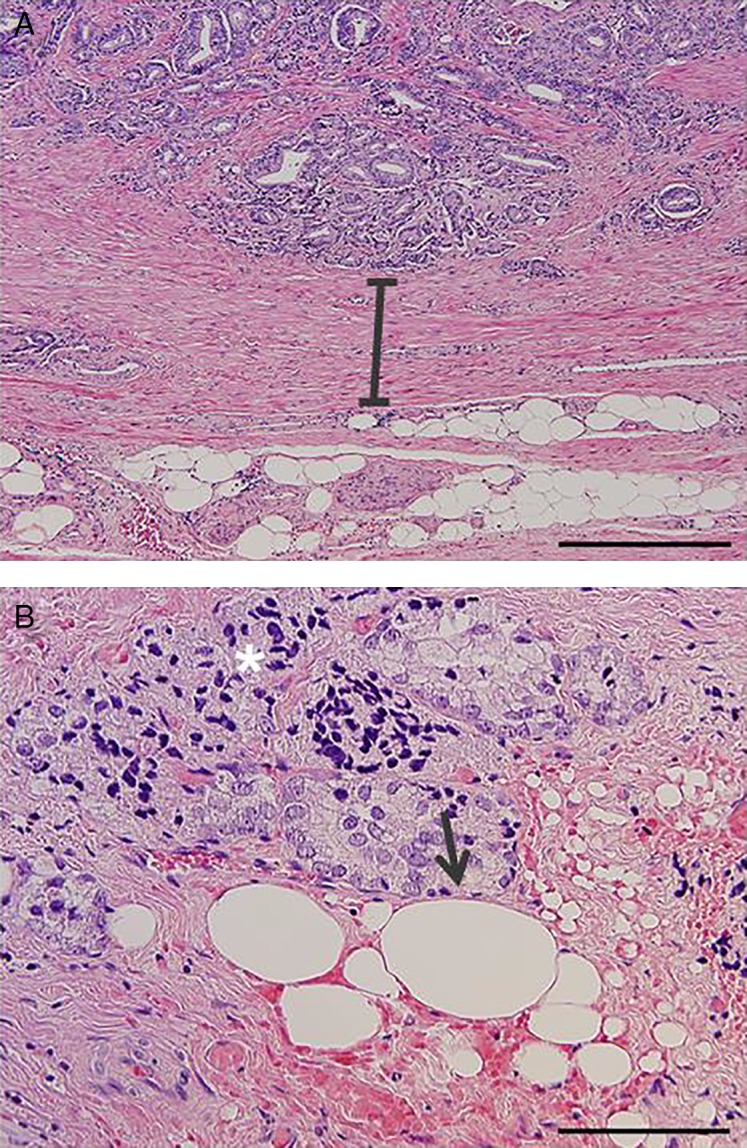
(A) Hematoxylin and eosin stain (H&E) image showing that human prostate tissue is physically separated from adjacent adipocytes by a concentric rim of condensed fibromuscular stroma at the periphery of the prostate. Adipocytes are located in the extra-prostatic region and not within the prostate gland itself. (B) H&E image showing evidence of prostate cancer cells infiltrating extra-prostatic tissue. Arrow indicates direct cell–cell contact between cancer cells and adipocytes. Asterisks indicate regions of chronic inflammation associated with the cancer cell infiltrate. Scale =400 μm (A) and 200 μm (B).
Perspectives
Despite the persuasive epidemiological evidence that obesity is associated with more prevalent high-grade prostate cancer and increased cancer-specific mortality, the biological mechanisms and associated cellular signalling pathways linking obesity and prostate cancer remain incompletely understood. The literature indicates that several mainstream hypotheses regarding obesity-related drivers of prostate cancer progression are not yet supported by a solid evidence base and, in particular, are not supported by experiments using human tissue. In this regard, many studies have been conducted in experimental models that bear limited relevance to human disease progression. For example, co-culturing of isolated adipocytes and immortalized prostate cancer cells does not faithfully reproduce the anatomical relationship between adipose tissue and the prostatic tumour (Fig. 3). In human studies, there is a need to define the obesity co-morbidities that most closely associate with prostate cancer aggressiveness, in the knowledge that obesity itself is unlikely to promote prostate cancer. Other outstanding issues remain, including an understanding of whether locally produced factors from PPAT impacts prostate cancer progression and, if so, what the major PPAT-secreted factors are. In addition, the relationship between lipid metabolism and cancer progression is in its infancy, and a thorough understanding of metabolism in human prostate malignancy is required before we advance to identifying new therapeutic and/or public health strategies (e.g., exercise or diet). Next, whether adipose-derived inflammation is pronounced enough to drive prostate cancer progression is unclear, as is the role of most inflammatory molecules in prostate cancer per se. Finally, altered sex steroid production, specifically reduced testosterone and elevated estrogen levels, has long been assumed to be a major driver of prostate cancer progression in obesity, but the evidence in humans is inconsistent. It is critical to determine the intra-tissue levels of steroid production to address this unresolved question.
In conclusion, while there is a gathering body of research in this field, our knowledge of the inter-relationship between obesity and prostate cancer development and/or progression is limited and less advanced than for other solid tumour types. Understanding the endocrine and metabolic pathways that play roles in disease progression and malignancy is important for identifying potential therapeutic avenues to slow prostate growth and metastasis.
Acknowledgements
The authors wish to acknowledge intellectual input from Ashlee Clark and Sam Norden.
Declaration of interest
There are no conflicts of interest that could be perceived as prejudicing the impartiality of this review.
Funding
This work was supported by the Prostate Cancer Foundation of Australia (Concept Grant, NCG 3313) and Diabetes Australia Research Trust. M J Watt is supported by a research fellowship from the National Health and Medical Research Council of Australia (ID: APP1077703).
References
- 1.Howlader N, et al SEER Cancer Statistics Review, 1975–2012, National Cancer Institute. Bethesda, MD. http://seer.cancer.gov/csr/1975_2012/, 2015.
- 2.Ferlay J, Soerjomataram I, Ervik M. Cancer Incidence and Mortality Worldwide: IARC CancerBase No. 11 (Internet). 2013: Lyon, France: International Agency for research on Cancer.
- 3.Shao YH, Demissie K, Shih W, Mehta AR, Stein MN, Roberts CB, Dipaola RS, Lu-Yao GL. Contemporary risk profile of prostate cancer in the United States. Journal of National Cancer Institute. 2009;101:1280–1283. doi: 10.1093/jnci/djp262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ng M, Fleming T, Robinson M, Thomson B, Graetz N, Margono C, Mullany EC, Biryukov S, Abbafati C, Abera SF, et al. Global, regional, and national prevalence of overweight and obesity in children and adults during 1980–2013: a systematic analysis for the Global Burden of Disease Study 2013. Lancet. 2014;384:766–781. doi: 10.1016/S0140-6736(14)60460-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lobstein T, Jackson-Leach R, Moodie ML, Hall KD, Gortmaker SL, Swinburn BA, James WP, Wang Y, McPherson K, et al. Child and adolescent obesity: part of a bigger picture. Lancet. 2015;385:2510–2520. doi: 10.1016/S0140-6736(14)61746-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kelly T, Yang W, Chen CS, Reynolds K, He J. Global burden of obesity in 2005 and projections to 2030. International Journal of Obesity. 2008;32:1431–1437. doi: 10.1038/ijo.2008.102. [DOI] [PubMed] [Google Scholar]
- 7.Haby MM, Markwick A, Peeters A, Shaw J, Vos T. Future predictions of body mass index and overweight prevalence in Australia, 2005–2025. Health Promotion International. 2012;27:250–260. doi: 10.1093/heapro/dar036. [DOI] [PubMed] [Google Scholar]
- 8.Wilson KM, Giovannucci EL, Mucci LA. Lifestyle and dietary factors in the prevention of lethal prostate cancer. Asian Journal of Andrology. 2012;14:365–374. doi: 10.1038/aja.2011.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Calle EE, Kaaks R. Overweight, obesity and cancer: epidemiological evidence and proposed mechanisms. Nature Reviews. Cancer. 2004;4:579–591. doi: 10.1038/nrc1408. [DOI] [PubMed] [Google Scholar]
- 10.Moller H, Roswall N, Van Hemelrijck M, Larsen SB, Cuzick J, Holmberg L, Overvad K, Tjønneland A, et al. Prostate cancer incidence, clinical stage and survival in relation to obesity: a prospective cohort study in Denmark. International Journal of Cancer. 2015;136:1940–1947. doi: 10.1002/ijc.29238. [DOI] [PubMed] [Google Scholar]
- 11.Wright ME, Chang SC, Schatzkin A, Albanes D, Kipnis V, Mouw T, Hurwitz P, Hollenbeck A, Leitzmann MF, et al. Prospective study of adiposity and weight change in relation to prostate cancer incidence and mortality. Cancer. 2007;109:675–684. doi: 10.1002/cncr.22443. [DOI] [PubMed] [Google Scholar]
- 12.Zhang X, Zhou G, Sun B, Zhao G, Liu D, Sun J, Liu C, Guo H. Impact of obesity upon prostate cancer-associated mortality: A meta-analysis of 17 cohort studies. Oncology Letters. 2015;9:1307–1312. doi: 10.3892/ol.2014.2841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Khandekar MJ, Cohen P, Spiegelman BM. Molecular mechanisms of cancer development in obesity. Nature Reviews. Cancer. 2011;11:886–895. doi: 10.1038/nrc3174. [DOI] [PubMed] [Google Scholar]
- 14.Ma J, Li H, Giovannucci E, Mucci L, Qiu W, Nguyen PL, Gaziano JM, Pollak M, Stampfer MJ. Prediagnostic body-mass index, plasma C-peptide concentration, and prostate cancer-specific mortality in men with prostate cancer: a long-term survival analysis. Lancet Oncology. 2008;9:1039–1047. doi: 10.1016/S1470-2045(08)70235-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Allott EH, Masko EM, Freedland SJ. Obesity and prostate cancer: weighing the evidence. European Urology. 2013;63:800–809. doi: 10.1016/j.eururo.2012.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cao Y, Ma J. Body mass index, prostate cancer-specific mortality, and biochemical recurrence: a systematic review and meta-analysis. Cancer Prevention Research. 2011;4:486–501. doi: 10.1158/1940-6207.CAPR-10-0229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vidal AC, Howard LE, Moreira DM, Castro-Santamaria R, Andriole GL, Jr, Freedland SJ. Obesity increases the risk for high-grade prostate cancer: results from the REDUCE study. Cancer Epidemiology, Biomarkers & Prevention. 2014;23:2936–2942. doi: 10.1158/1055-9965.EPI-14-0795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hsing AW, Sakoda LC, Chua S., Jr Obesity, metabolic syndrome, and prostate cancer. American Journal of Clinical Nutrition. 2007;86:s843–s857. doi: 10.1093/ajcn/86.3.843S. [DOI] [PubMed] [Google Scholar]
- 19.Hu MB, Xu H, Bai PD, Jiang HW, Ding Q. Obesity has multifaceted impact on biochemical recurrence of prostate cancer: a dose-response meta-analysis of 36,927 patients. Medical Oncology. 2014;31:829. doi: 10.1007/s12032-013-0829-8. [DOI] [PubMed] [Google Scholar]
- 20.Wildman RP, Gu D, Muntner P, Wu X, Reynolds K, Duan X, Chen CS, Huang G, Bazzano LA, He J. Trends in overweight and obesity in Chinese adults: between 1991 and 1999-2000. Obesity. 2008;16:1448–1453. doi: 10.1038/oby.2008.208. [DOI] [PubMed] [Google Scholar]
- 21.Moiola CP, De Luca P, Zalazar F, Cotignola J, Rodríguez-Seguí SA, Gardner K, Meiss R, Vallecorsa P, Pignataro O, Mazza O. Prostate tumor growth is impaired by CtBP1 depletion in high-fat diet-fed mice. Clinical Cancer Research. 2014;20:4086–4095. doi: 10.1158/1078-0432.CCR-14-0322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huang M, Narita S, Numakura K, Tsuruta H, Saito M, Inoue T, Horikawa Y, Tsuchiya N, Habuchi T, et al. A high-fat diet enhances proliferation of prostate cancer cells and activates MCP-1/CCR2 signaling. Prostate. 2012;72:1779–1788. doi: 10.1002/pros.22531. [DOI] [PubMed] [Google Scholar]
- 23.Narita S, Tsuchiya N, Saito M, Inoue T, Kumazawa T, Yuasa T, Nakamura A, Habuchi T. Candidate genes involved in enhanced growth of human prostate cancer under high fat feeding identified by microarray analysis. Prostate. 2008;68:321–335. doi: 10.1002/pros.20681. [DOI] [PubMed] [Google Scholar]
- 24.Bonorden MJ, Grossmann ME, Ewing SA, Rogozina OP, Ray A, Nkhata KJ, Liao DJ, Grande JP, Cleary MP. Growth and progression of TRAMP prosate tumors in relationship to diet and obesity. Prostate Cancer. 2012;2012:543970. doi: 10.1155/2012/543970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cho HJ, Kwon GT, Park H, Song H, Lee KW, Kin JI, Park JHY. A high-fat diet containing lard accelerates prostate cancer progression and reduces survival rate in mice: possible contribution of adipose tissue-derived cytokines. Nutrients. 2015;7:2539–2561. doi: 10.3390/nu7042539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xu H, Hu MB, Bai PD, Zhu WH, Liu SH, Hou JY, Xiong ZQ. Proinflammatory cytokines in prostate cancer development and progression promoted by high-fat diet. BioMed Research International. 2015;2015:249741. doi: 10.1155/2015/249741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chang SN, Han J, Abdelkader TS, Kim TH, Lee JM, Song J, Kim KS, Park JH, Park JH. High animal fat intake enhances prostate cancer progression and reduces glutathione peroxidase 3 expression in early stages of TRAMP mice. Prostate. 2014;74:1266–1277. doi: 10.1002/pros.22843. [DOI] [PubMed] [Google Scholar]
- 28.Blando J, Moore T, Hursting S, Jiang G, Saha A, Beltran L, Shen J, Repass J, Strom S, DiGiovanni J. Dietary energy balance modulates prostate cancer progression in Hi-Myc mice. Cancer Prevention Research. 2011;4:2002–2014. doi: 10.1158/1940-6207.CAPR-11-0182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kobayashi N, Barnard RJ, Said J, Hong-Gonzalez J, Corman DM, Ku M, Doan NB, Gui D, Elashoff D, Cohen P, et al. Effect of low-fat diet on development of prostate cancer and Akt phosphorylation in the Hi-Myc transgenic mouse model. Cancer Research. 2008;68:3066–3073. doi: 10.1158/0008-5472.CAN-07-5616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu J, Ramakrishnan SK, Khuder SS, Kaw MK, Muturi HT, Lester SG, Lee SJ, Fedorova LV, Kim AJ, Mohamed IE, et al. High-calorie diet exacerbates prostate neoplasia in mice with haploinsufficiency of Pten tumor suppressor gene. Molecular Metabolism. 2015;4:186–198. doi: 10.1016/j.molmet.2014.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Silva SA, Gobbo MG, Pinto-Fochi ME, Rafacho A, Taboga SR, Almeida EA, Góes RM, Ribeiro DL. Prostate hyperplasia caused by long-term obesity is characterized by high deposition of extracellular matrix and increased content of MMP-9 and VEGF. International Journal of Experimental Pathology. 2015;96:21–30. doi: 10.1111/iep.12107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ribeiro DL, Pinto ME, Rafacho A, Bosqueiro JR, Maeda SY, Anselmo-Franci JA, Taboga SR, Góes RM. High-fat diet obesity associated with insulin resistance increases cell proliferation, estrogen receptor, and PI3K proteins in rat ventral prostate. Journal of Andrology. 2012;33:854–865. doi: 10.2164/jandrol.111.016089. [DOI] [PubMed] [Google Scholar]
- 33.Khandekar MJ, Cohen P, Spiegelman BM. Molecular mechanisms of cancer development in obesity. Nature Reviews. Cancer. 2011;11:886–895. doi: 10.1038/nrc3174. [DOI] [PubMed] [Google Scholar]
- 34.Rutkowski JM, Davis KE, Scherer PE. Mechanisms of obesity and related pathologies: the macro- and microcirculation of adipose tissue. FEBS Journal. 2009;276:5738–5746. doi: 10.1111/j.1742-4658.2009.07303.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cao Y. Angiogenesis modulates adipogenesis and obesity. Journal of Clinical Investigation. 2007;117:2362–2368. doi: 10.1172/JCI32239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW., Jr Obesity is associated with macrophage accumulation in adipose tissue. Journal of Clinical Investigation. 2003;112:1796–1808. doi: 10.1172/JCI200319246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Olefsky JM, Glass CK. Macrophages, inflammation, and insulin resistance. Annual Review of Physiology. 2010;72:219–246. doi: 10.1146/annurev-physiol-021909-135846. [DOI] [PubMed] [Google Scholar]
- 38.Culig Z. Proinflammatory cytokine interleukin-6 in prostate carcinogenesis. American Journal of Clinical and Experimental Urology. 2014;2:231–238. [PMC free article] [PubMed] [Google Scholar]
- 39.Wise GJ, Marella VK, Talluri G, Shirazian D. Cytokine variations in patients with hormone treated prostate cancer. Journal of Urology. 2000;164:722–725. doi: 10.1016/S0022-5347(05)67289-8. [DOI] [PubMed] [Google Scholar]
- 40.Adler HL, McCurdy MA, Kattan MW, Timme TL, Scardino PT, Thompson TC. Elevated levels of circulating interleukin-6 and transforming growth factor-β1 in patients with metastatic prostatic carcinoma. Journal of Urology. 1999;161:182–187. doi: 10.1016/S0022-5347(01)62092-5. [DOI] [PubMed] [Google Scholar]
- 41.Nguyen DP, Li J, Tewari AK. Inflammation and prostate cancer: the role of interleukin 6 (IL-6) BJU International. 2014;113:986–992. doi: 10.1111/bju.12452. [DOI] [PubMed] [Google Scholar]
- 42.Giri D, Ozen M, Ittmann M. Interleukin-6 is an autocrine growth factor in human prostate cancer. American Journal of Pathology. 2001;159:2159–2165. doi: 10.1016/S0002-9440(10)63067-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yu SH, Zheng Q, Esopi D, Macgregor-Das A, Luo J, Antonarakis ES, Drake CG, Vessella R, Morrissey C, De Marzo AM, Sfanos KS. A paracrine role for IL6 in prostate cancer patients: lack of production by primary or metastatic tumor cells. Cancer Immunology Research. 2015;3:1175–1184. doi: 10.1158/2326-6066.CIR-15-0013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Finley DS, Calvert VS, Inokuchi J, Lau A, Narula N, Petricoin EF, Zaldivar F, Santos R, Tyson DR, Ornstein DK. Periprostatic adipose tissue as a modulator of prostate cancer aggressiveness. Journal of Urology. 2009;182:1621–1627. doi: 10.1016/j.juro.2009.06.015. [DOI] [PubMed] [Google Scholar]
- 45.Sfanos KS, De Marzo AM. Prostate cancer and inflammation: the evidence. Histopathology. 2012;60:199–215. doi: 10.1111/j.1365-2559.2011.04033.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Brown DA, Lindmark F, Stattin P, Bälter K, Adami HO, Zheng SL, Xu J, Isaacs WB, Grönberg H, Breit SN, et al. Macrophage inhibitory cytokine 1: a new prognostic marker in prostate cancer. Clinical Cancer Research. 2009;15:6658–6664. doi: 10.1158/1078-0432.CCR-08-3126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Karan D, Holzbeierlein J, Thrasher JB. Macrophage inhibitory cytokine-1: possible bridge molecule of inflammation and prostate cancer. Cancer Research. 2009;69:2–5. doi: 10.1158/0008-5472.CAN-08-1230. [DOI] [PubMed] [Google Scholar]
- 48.Lehrer S, Diamond EJ, Mamkine B, Stone NN, Stock RG. Serum interleukin-8 is elevated in men with prostate cancer and bone metastases. Technology in Cancer Research & Ttreatment. 2004;3:411. doi: 10.1177/153303460400300501. [DOI] [PubMed] [Google Scholar]
- 49.Neveu B, Moreel X, Deschênes-Rompré MP, Bergeron A, LaRue H, Ayari C, Fradet Y, Fradet V. IL-8 secretion in primary cultures of prostate cells is associated with prostate cancer aggressiveness. Research and Reports in Urology. 2014;6:27–34. doi: 10.2147/RRU.S58643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chadha KC, Miller A, Nair BB, Schwartz SA, Trump DL, Underwood W. New serum biomarkers for prostate cancer diagnosis. Clinical Cancer Investigation Journal. 2014;3:72–79. doi: 10.4103/2278-0513.125802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 52.De Marzo AM, Platz EA, Sutcliffe S, Xu J, Grönberg H, Drake CG, Nakai Y, Isaacs WB, Nelson WG. Inflammation in prostate carcinogenesis. Nature Reviews. Cancer. 2007;7:256–269. doi: 10.1038/nrc2090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nelson WG, Demarzo AM, Yegnasubramanian S. The diet as a cause of human prostate cancer. Cancer Treatment and Research. 2014;159:51–68. doi: 10.1007/978-3-642-38007-5_4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nelson WG, Demarzo AM, Yegnasubramanian S. Prostatitis, sexually transmitted diseases, and prostate cancer: the California Men's Health Study. PLoS ONE. 2010;5:e8736. doi: 10.1371/journal.pone.0008736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Galic S, Oakhill JS, Steinberg GR. Adipose tissue as an endocrine organ. Molecular and Cellular Endocrinology. 2010;316:129–139. doi: 10.1016/j.mce.2009.08.018. [DOI] [PubMed] [Google Scholar]
- 56.Crowe S, Wu LE, Economou C, Turpin SM, Matzaris M, Hoehn KL, Hevener AL, James DE, Duh EJ, Watt MJ. Pigment epithelium-derived factor contributes to insulin resistance in obesity. Cell Metabolism. 2009;10:40–47. doi: 10.1016/j.cmet.2009.06.001. [DOI] [PubMed] [Google Scholar]
- 57.Zadra G, Photopoulos C, Loda M. The fat side of prostate cancer. Biochimica et Biophysica Acta. 2013;1831:1518–1532. doi: 10.1016/j.bbalip.2013.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hocking SL, Wu LE, Guilhaus M, Chisholm DJ, James DE. Intrinsic depot-specific differences in the secretome of adipose tissue, preadipocytes, and adipose tissue-derived microvascular endothelial cells. Diabetes. 2010;59:3008–3016. doi: 10.2337/db10-0483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Stattin P, Söderberg S, Hallmans G, Bylund A, Kaaks R, Stenman UH, Bergh A, Olsson T. Leptin is associated with increased prostate cancer risk: a nested case-referent study. Journal of Clinical Endocrinology and Metabolism. 2001;86:1518–1532. doi: 10.2337/db10-0483. [DOI] [PubMed] [Google Scholar]
- 60.Bub JD, Miyazaki T, Iwamoto Y. Adiponectin as a growth inhibitor in prostate cancer cells. Biochemical and Biophysical Research Communications. 2006;340:1158–1166. doi: 10.1016/j.bbrc.2005.12.103. [DOI] [PubMed] [Google Scholar]
- 61.McPherson SJ, Hussain S, Balanathan P, Hedwards SL, Niranjan B, Grant M, Chandrasiri UP, Toivanen R, Wang Y, Taylor RA, et al. Estrogen receptor-β activated apoptosis in benign hyperplasia and cancer of the prostate is androgen independent and TNFα mediated. PNAS. 2010;107:3123–3128. doi: 10.1073/pnas.0905524107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ellem SJ, Risbridger GP. Treating prostate cancer: a rationale for targeting local oestrogens. Nature Reviews. Cancer. 2007;7:621–627. doi: 10.1038/nrc2174. [DOI] [PubMed] [Google Scholar]
- 63.Chavarro JE, Toth TL, Wright DL, Meeker JD, Hauser R. Body mass index in relation to semen quality, sperm DNA integrity, and serum reproductive hormone levels among men attending an infertility clinic. Fertility and Sterility. 2010;93:2222–2231. doi: 10.1016/j.fertnstert.2009.01.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Schneider G, Kirschner MA, Berkowitz R, Ertel NH. Increased estrogen production in obese men. Journal of Clinical Endocrinology and Metabolism. 1979;48:633–638. doi: 10.1210/jcem-48-4-633. [DOI] [PubMed] [Google Scholar]
- 65.Davidson LM, Millar K, Jones C, Fatum M, Coward K. Deleterious effects of obesity upon the hormonal and molecular mechanisms controlling spermatogenesis and male fertility. Human Fertility. 2015;18:184–193. doi: 10.3109/14647273.2015.1070438. [DOI] [PubMed] [Google Scholar]
- 66.Hajshafiha M, Ghareaghaji R, Salemi S, Sadegh-Asadi N, Sadeghi-Bazargani H. Association of body mass index with some fertility markers among male partners of infertile couples. International Journal of General Medicine. 2013;6:447–451. doi: 10.2147/IJGM.S41341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Jentzmik F, Schnoeller TJ, Cronauer MV, Steinestel J, Steffens S, Zengerling F, Al Ghazal A, Schrader MG, Steinestel K, Schrader AJ. Corpulence is the crucial factor: association of testosterone and/or obesity with prostate cancer stage. International Journal of Urology. 2014;21:980–986. doi: 10.1111/iju.12494. [DOI] [PubMed] [Google Scholar]
- 68.Colangelo LA, Ouyang P, Liu K, Kopp P, Golden SH, Dobs AS, Szklo M, Vaidya D, Cushman M, Gapstur SM. Association of endogenous sex hormones with diabetes and impaired fasting glucose in men: multi-ethnic study of atherosclerosis. Diabetes Care. 2009;32:1049–1051. doi: 10.2337/dc08-2216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Vermeulen A, Kaufman JM, Goemaere S, van Pottelberg I. Estradiol in elderly men. Aging Male. 2002;5:98–102. doi: 10.1080/tam.5.2.98.102. [DOI] [PubMed] [Google Scholar]
- 70.Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029–1033. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chandler JD, Williams ED, Slavin JL, Best JD, Rogers S. Expression and localization of GLUT1 and GLUT12 in prostate carcinoma. Cancer. 2003;97:2035–2042. doi: 10.1002/cncr.11293. [DOI] [PubMed] [Google Scholar]
- 72.Bavenholm PN, Pigon J, Saha AK, Ruderman NB, Efendic S. Fatty acid oxidation and the regulation of malonyl-CoA in human muscle. Diabetes. 2000;49:1078–1083. doi: 10.2337/diabetes.49.7.1078. [DOI] [PubMed] [Google Scholar]
- 73.Costello LC, Franklin RB, Narayan P. Citrate in the diagnosis of prostate cancer. Prostate. 1999;38:237–245. doi: 10.1002/(SICI)1097-0045(19990215)38:3%3c237::AID-PROS8%3e3.0.CO;2-O. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Effert PJ, Bares R, Handt S, Wolff JM, Büll U, Jakse G. Metabolic imaging of untreated prostate cancer by positron emission tomography with 18fluorine-labeled deoxyglucose. Journal of Urology. 1996;155:994–998. doi: 10.1016/S0022-5347(01)66366-3. [DOI] [PubMed] [Google Scholar]
- 75.Zadra G, Priolo C, Patnaik A, Loda M. New strategies in prostate cancer: targeting lipogenic pathways and the energy sensor AMPK. Clinical Cancer Research. 2010;16:3322–3328. doi: 10.1158/1078-0432.CCR-09-1955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Koutsari C, Jensen MD. Thematic review series: Patient-Oriented Research. Free fatty acid metabolism in human obesity. Journal of Lipid Research. 2006;47:1643–1650. doi: 10.1194/jlr.R600011-JLR200. [DOI] [PubMed] [Google Scholar]
- 77.Boren J, Taskinen MR, Olofsson SO, Levin M. Ectopic lipid storage and insulin resistance: a harmful relationship. Journal of Internal Medicine. 2013;274:25–40. doi: 10.1111/joim.12071. [DOI] [PubMed] [Google Scholar]
- 78.Glatz JF, Luiken JJ, Bonen A. Membrane fatty acid transporters as regulators of lipid metabolism: implications for metabolic disease. Physiological Reviews. 2010;90:367–417. doi: 10.1152/physrev.00003.2009. [DOI] [PubMed] [Google Scholar]
- 79.Liu Y. Fatty acid oxidation is a dominant bioenergetic pathway in prostate cancer. Prostate Cancer and Prostatic Diseases. 2006;9:230–234. doi: 10.1038/sj.pcan.4500879. [DOI] [PubMed] [Google Scholar]
- 80.Schlaepfer IR, Glodé LM, Hitz CA, Pac CT, Boyle KE, Maroni P, Deep G, Agarwal R, Lucia SM, Cramer SD. Inhibition of Lipid Oxidation Increases Glucose Metabolism and Enhances 2-Deoxy-2-((18)F)Fluoro-D-Glucose Uptake in Prostate Cancer Mouse Xenografts. Molecular Imaging and Biology. 2015;17:529–538. doi: 10.1007/s11307-014-0814-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Schlaepfer IR, Rider L, Rodrigues LU, Gijón MA, Pac CT, Romero L, Cimic A, Sirintrapun SJ, Glodé LM, Eckel RH. Lipid catabolism via CPT1 as a therapeutic target for prostate cancer. Molecular Cancer Therapeutics. 2014;13:2361–2371. doi: 10.1158/1535-7163.MCT-14-0183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Carracedo A, Cantley LC, Pandolfi PP. Cancer metabolism: fatty acid oxidation in the limelight. Nature Reviews. Cancer. 2013;13:227–232. doi: 10.1038/nrc3483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Taylor RA, Risbridger GP. Prostatic tumor stroma: a key player in cancer progression. Current Cancer Drug Targets. 2008;8:490–497. doi: 10.2174/156800908785699351. [DOI] [PubMed] [Google Scholar]
- 84.Tan J, Buache E, Chenard MP, Dali-Youcef N, Rio MC. Adipocyte is a non-trivial, dynamic partner of breast cancer cells. International Journal of Developmental Biology. 2011;55:851–859. doi: 10.1387/ijdb.113365jt. [DOI] [PubMed] [Google Scholar]
- 85.Ribeiro R, Monteiro C, Catalán V, Hu P, Cunha V, Rodríguez A, Gómez-Ambrosi J, Fraga A, Príncipe A, Lobato C, et al. Obesity and prostate cancer: gene expression signature of human periprostatic adipose tissue. BMC Medicine. 2012;10:108. doi: 10.1186/1741-7015-10-108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bhindi B, Trottier G, Elharram M, Fernandes KA, Lockwood G, Toi A, Hersey KM, Finelli A, Evans A, van der Kwast TH, Fleshner NE. Measurement of peri-prostatic fat thickness using transrectal ultrasonography (TRUS): a new risk factor for prostate cancer. BJU International. 2012;110:980–986. doi: 10.1111/j.1464-410X.2012.10957.x. [DOI] [PubMed] [Google Scholar]
- 87.van Roermund JG, Hinnen KA, Tolman CJ, Bol GH, Witjes JA, Bosch JL, Kiemeney LA, van Vulpen M. Periprostatic fat correlates with tumour aggressiveness in prostate cancer patients. BJU International. 2011;107:1775–1779. doi: 10.1111/j.1464-410X.2010.09811.x. [DOI] [PubMed] [Google Scholar]
- 88.Woo S, Cho JY, Kim SY, Kim SH. Periprostatic fat thickness on MRI: correlation with Gleason score in prostate cancer. AJR. American Journal of Roentgenology. 2015;204:W43–W47. doi: 10.2214/AJR.14.12689. [DOI] [PubMed] [Google Scholar]
- 89.Venkatasubramanian PN, Brendler CB, Plunkett BA, Crawford SE, Fitchev PS, Morgan G, Cornwell ML, McGuire MS, Wyrwicz AM, Doll JA, et al. Periprostatic adipose tissue from obese prostate cancer patients promotes tumor and endothelial cell proliferation: A functional and MR imaging pilot study. Prostate. 2014;74:326–335. doi: 10.1002/pros.22756. [DOI] [PubMed] [Google Scholar]
- 90.Ribeiro R, Monteiro C, Cunha V, Oliveira MJ, Freitas M, Fraga A, Príncipe P, Lobato C, Lobo F, Morais A, et al. Human periprostatic adipose tissue promotes prostate cancer aggressiveness in vitro. Journal of Experimental & Clinical Cancer Research. 2012;31:32. doi: 10.1186/1756-9966-31-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Finley DS, Calvert VS, Inokuchi J, Lau A, Narula N, Petricoin EF, Zaldivar F, Santos R, Tyson DR, Ornstein DK. Periprostatic adipose tissue as a modulator of prostate cancer aggressiveness. Journal of Urology. 2009;182:1621–1627. doi: 10.1016/j.juro.2009.06.015. [DOI] [PubMed] [Google Scholar]