

Background: In plants, a key regulatory step in chlorophyll biosynthesis is catalyzed by two light-dependent isozymes.
Results: The two isozymes operate via the same reaction mechanism but differ in their catalytic efficiencies.
Conclusion: Different substrate affinities and conformational flexibilities modulate the catalytic reaction.
Significance: Detailed understanding of light-driven reactions in nature has a big impact on the development of artificial energy conversion systems.
Keywords: chlorophyll, enzyme kinetics, plant biochemistry, reductase, ultraviolet-visible spectroscopy (UV-Vis spectroscopy), NADPH:protochlorophyllide oxidoreductase, chlorophyll biosynthesis, low temperature absorption spectroscopy
Abstract
The enzyme protochlorophyllide oxidoreductase (POR, EC 1.3.1.33) has a key role in plant development. It catalyzes one of the later steps in chlorophyll synthesis, the light-induced reduction of protochlorophyllide (PChlide) into chlorophyllide (Chlide) in the presence of NADPH. Two isozymes of plant POR, POR A and POR B from barley, which differ in their function during plant life, are compared with respect to their substrate binding affinity, catalytic efficiency, and catalytic mechanism. POR B as compared with POR A shows an 5-fold higher binding affinity for PChlide and an about 6-fold higher catalytic efficiency measured as kcat/Km. Based on the reaction intermediates, which can be trapped at low temperatures the same reaction mechanism operates in both POR A and POR B. In contrast to results reported for POR enzymes from cyanobacteria, the initial light-driven step, which occurs at temperatures below 180 K already involves the full chemistry of the photoreduction and yields the reaction product, Chlide, in an enzyme-bound form. The subsequent dark reactions, which include cofactor (NADP+) release and cofactor (NADPH) rebinding, show different temperature dependences for POR A and POR B and suggest a higher conformational flexibility of POR B in the surrounding active center. Both the higher substrate binding affinity and well adapted enzyme dynamics are held responsible for the increased catalytic activity of POR B as compared with POR A.
Introduction
Chlorophyll is the main pigment of photosynthesis in plants, green algae, and cyanobacteria. The biosynthesis of chlorophyll therefore represents a fundamental metabolic process, which is strongly regulated in response to environmental cues (1, 2). The light-dependent protochlorophyllide oxidoreductase (POR)3 (EC 1.3.1.33) is the key regulatory enzyme of the chlorophyll synthesis pathway in all oxygen-producing photosynthetic organisms (3–7). In one of the last steps, POR catalyzes the light-driven reduction of protochlorophyllide (PChlide) into chlorophyllide (Chlide), from which chlorophyll finally derives. With reduced nicotinamide adenine dinucleotide phosphate (NADPH) as cofactor, the reduction involves the trans addition of hydrogen across the C(17)-C(18) double bond in ring D of PChlide (Fig. 1), whereas the pro-S hydride of the nicotinamide ring of NADPH is transferred to the C(17) position and, most likely, the proton of a conserved tyrosine residue to the C(18) position of the porphyrin molecule (8–9). Because of this light-driven H-transfer, which enables the exploitation of the abundant sun light as energy source for chemical synthesis, POR is also an attractive model for novel approaches in bio-inspired, green chemistry.
FIGURE 1.

The light-induced reduction of protochlorophyllide to chlorophyllide catalyzed by the enzyme NADPH:protochlorophyllide oxidoreductase in the presence of NADPH as cofactor.
Based on the comparison of the amino acid sequence with other sequences in the protein database POR is a member of the large family of enzymes known as short chain dehydrogenases/reductases that catalyze a variety of NADP(H)- or NAD(H)-dependent reactions (9–11). The common structural features include a glycine-rich GXXXGXG NADPH-binding motif as part of the so-called Rossman-fold, which is located close to the N-terminal region of the enzyme. In addition, tyrosine and lysine residues are arranged at the catalytic site. They are essential for the catalytic activity of POR and belong to the YXXXK substrate-binding motif, which is highly conserved across the short chain dehydrogenases/reductases superfamily of enzymes.
In barley (Hordeum vulgare) and perhaps in other plant species two distinct isozymes of POR, termed POR A and POR B exist (12–14), whereas in Arabidopsis (Arabidopsis thaliana) apart from POR A and POR B an additional third isozyme, POR C has been identified (15, 16). POR A and POR B differ remarkably in their expression pattern during plant development (17, 18), substrate specificities (19, 20), reaction efficiencies (21) and the mechanism by which their nuclear-encoded precursor proteins are imported into plastids (22–24). POR A is highly abundant in etiolated plants and mainly active during seedling de-etiolation. In the light, the expression of POR A is repressed resulting in its selective disappearance, whereas POR B becomes the predominant species. In contrast to POR A, POR B is not negatively regulated by light but constitutively expressed both in etiolated and light-adapted plants. Hence, in greened and adult plants, POR B is the only POR enzyme, which operates in chlorophyll synthesis (13, 14, 17, 18). With respect to their function, the two isozymes are proposed to cooperate in higher molecular weight light-harvesting complexes in the prolamellar body of etioplasts (19, 20). Based on a comparison of the amino acid sequences, POR A and POR B show a very high sequence homology (25) indicating that differences in the catalytic function can hardly be attributed to striking differences in their structure at first glance.
Thus far POR A and POR B have been mainly examined in comparative studies with respect to their function in light-harvesting complexes (19, 20, 26). Although POR B is the key enzyme in chlorophyll synthesis of light-adapted, green plants, details of the enzyme kinetics and catalytic mechanism are still unclear. In the present paper we have addressed this dearth of knowledge by comparing the two POR isozymes in parallel experiments with regard to their substrate affinities and catalytic efficiencies toward PChlide as a substrate (henceforth referred to as PChlide for reasons of simplicity). Furthermore, the catalytic mechanism is assessed by trapping transient intermediates in low temperature absorbance experiments. The results allow further conclusions on the catalytic dynamics of POR A and POR B as well as on differences in the catalytic mechanism between the two plant isozymes themselves and the enzyme from cyanobacteria, which does not exist in a multiple but only single form. That enzyme is of interest because POR from cyanobacteria can be seen as a phylogenetic ancestor of all other POR enzymes (27).
Experimental Procedures
Expression and Purification of POR A and POR B from Barley (H. vulgare)
cDNA encoding the respective POR proteins (without transition peptides) was subcloned into the expression vector pRSET A (Invitrogen) via BamHI and HindIII restriction sites. The vector DNA (pRSET A-His-POR) was then transformed into competent Escherichia coli SoluBL21 host cells (amsbio). For protein expression, SoluBL21 E. coli cells were cultivated in M9 minimal medium containing 100 μg/ml of ampicillin at 24 °C for about 7 h. When the culture reached a value of OD600 ∼0.4 protein expression was induced by the addition of isopropyl β-d-1-thiogalactopyranoside to a final concentration of 1 mm. Then the cells were further incubated at 24 °C for ∼20 h. After harvesting by centrifugation the cell pellets were disrupted by cell lyses with lysozyme (1 mg/ml) and CelLytic ExpressTM (0.25 g/g wet cell paste). For purification, the cell lysate was subjected to affinity chromatography on nickel-agarose using a HisTrap FF column (GE Healthcare). The column was washed with 10 volumes of binding buffer (50 mm Tris-HCl, 0.5 m NaCl, 20 mm imidazole, 20% glycerol, 0.1% Triton X-100, 0.1% β-mercaptoethanol, pH 7.6.), whereas the POR protein was eluted with binding buffer containing 300 mm imidazole. The enzyme fractions obtained from this elution step were desalted on a HiPrep desalting column (GE Healthcare) with a buffer containing 50 mm Tris-HCl, 0.3 m NaCl, 20% glycerol, 0.1% Triton X-100, and 7 mm DTT, pH 7.6. The pooled fractions were concentrated in a centrifugal filter unit (Millipore) if necessary. After each preparation the homogeneity and purity of the isolated POR enzymes was verified by electrophoresis in 12.5% polyacrylamide gels with 0.1% SDS. Protein concentrations were determined by the Bradford assay using bovine serum albumin as a standard.
Expression and Purification of POR from Synechocystis
cDNA encoding the POR enzyme from Synechocystis sp. was also subcloned into expression vector pRSET A (Invitrogen) using BamHI and EcoRI cloning sites. The subsequent procedures including overexpression and purification of the enzyme were analogous to the experimental steps described above for the enzymes from barley (H. vulgare).
Preparation of PChlide
PChlide a was isolated from 5-day-old, dark grown oat seedlings (Avena sativa L.) as reported recently (28). Briefly, the coleoptiles were treated with 15 mm 5-aminolevulinic acid in 35 mm potassium phosphate buffer for 48 h. Following disruption of the coleoptiles by homogenization, PChlide was extracted into an ice-cold 10 mm Tricine buffer, pH 7.5, 75% acetone (v/v) mixture. After centrifugation, PChlide was transferred into diethyl ether followed by extraction into a 4:1 methanol, 0.01 m ammonia mixture. Finally, PChlide was transferred into diethyl ether from the water/methanol mixture and fractionated by HPLC on a reverse phase RP-18 column in a linear 20–80% acetonitrile gradient. The fractions containing highly pure PChlide were lyophilized and stored at 250 K until use. For the experiments PChlide was dissolved in methanol and diluted with reaction buffer to concentrations as required resulting in less than 1% methanol in the sample.
Kinetic Experiments
Kinetic assays for determination of the initial rates (v0), the Michaelis-Menten constant (Km), and the turnover number (kcat) were conducted in measuring buffer containing 50 mm Tris-HCl, 0.3 m NaCl, 20% glycerol, 0.1% Triton X-100, and 1 mm DTT, pH 7.6. The typical reaction mixture was composed of 10 μm POR, 100 μm NADPH, 1 mm DTT. PChlide was used at various concentrations as indicated under “Results.”
Initial reaction rates (v0) were measured by irradiating the ternary complex POR-NADPH-PChlide, which was pre-formed by incubation in the dark, with a HeNe laser 30–50 (Spindler and Hoyer) at λexc = 633 nm and a photon flux of 2.5 × 10−5 mol m−2 s−1. The reaction progress was followed by monitoring the absorbance spectra in the UV/visual region in dependence on the irradiation time using a Lambda 35 UV/visual spectrophotometer (PerkinElmer Instruments). In this set-up, the reaction rate corresponds to the decrease/increase in the PChlide/Chlide concentration. The concentrations were determined in each experiment using ϵ630 nm = 48,700 liters/mol cm−1 for PChlide and ϵ670 nm = 91,000 liters/mol cm−1 for Chlide. From plots of the initial rates versus the concentration of PChlide, the Km values were estimated according to the Michaelis-Menten equation,
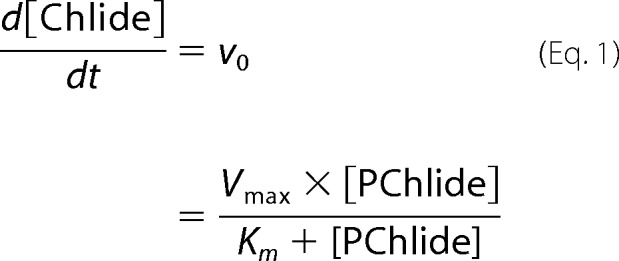 |
by means of a nonlinear least-squares curve fitting procedure in Matlab. The turnover number, kcat, was derived from maximum enzyme velocity (Vmax) according to,
 |
with [E0] − total concentration of the POR enzyme.
Microscale Thermophoresis (MST)
The affinity of PChlide to the POR enzymes was analyzed by microscale thermophoresis (29). The POR enzymes were labeled with the blue fluorescent dye NT-495-NHS (Nano Temper Technologies). The concentration of the labeled POR enzymes including the cofactor NADPH was kept constant at 83 nm for POR A and 166 nm for POR B, respectively, whereas the concentration of PChlide was varied in a 1:1 serial dilution starting from 5 μm. The samples, enzyme and substrate, were dissolved in measuring buffer (50 mm Tris-HCl, 0.3 m NaCl, 20% glycerol, 0.1% Triton X-100, 1 mm DTT, pH 7.6). After an incubation period of 15 min in the dark the MST measurements were carried out at 25 °C on a Monolith NT.115 instrument (Nano Temper Technologies). Thermophoresis was measured at 80% LED power and 20% MST power, respectively. From the binding curves, which yield the fraction of POR-bound PChlide as function of the titrated PChlide, the dissociation constants, KD, were determined using the equation,
 |
with
where [ES] is the concentration of the PChlide-bound POR-complex (enzyme-substrate complex) and [S0] the added PChlide concentration.
The solution of Equation 3 with substitutions of Equations 4 and 5 results in,
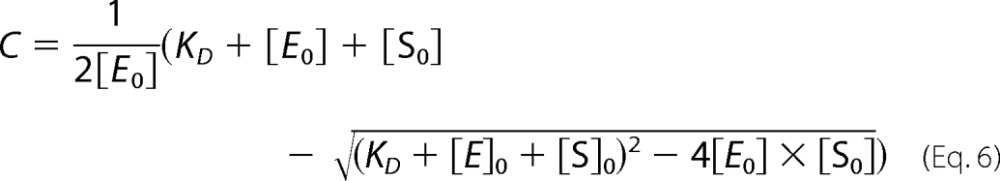 |
with the C− fraction of POR-bound PChlide.
From the fit of the binding curves to Equation 6 the dissociation constant, KD, was derived.
The change in the free enthalpy (ΔG0) upon binding of PChlide to the POR-NADPH complex was deduced from the dissociation constant, KD, by Equation 7.
Low Temperature Absorbance Measurements
Ternary POR-NADPH-PChlide complexes at 60/400/3.5 μm concentrations were incubated in the dark prior to each measurement. The measuring buffer was composed of 50 mm Tris-HCl, 60% glycerol, 0.3 m NaCl, 0.1% Triton X-100, 1 mm DTT, pH 7.6. The samples were frozen at 180 K using an OpstistatDN nitrogen bath cryostat (Oxford Instruments) and then irradiated by an LED cold-light source (Thorlabs, Inc.) at λexc = (632 ± 9) nm (400 microwatts) for 20 min to initiate POR activity. The absorbance spectra were registered at low temperatures between 180 and 305 K to characterize the intermediates trapped at the respective temperatures. The temperature of the samples was monitored with a thermocouple sensor (100 Ω platinum resistance thermometer) and the absorbance spectra were recorded with a Jasco V-630 UV/visual spectrophotometer (Jasco Germany GmbH). Normalization of the spectra and data analyses was performed by using Origin software.
For the determination of the activation energies (ΔG#), samples of the ternary POR complex with 30/200/5 μm POR/NADPH/PChlide concentrations were incubated in the dark, maintained at various temperatures between 160 and 200 K. At each selected temperature the initial rate (v0) was measured after initiating the POR-catalyzed reaction by irradiation at λexc =(632 ± 9) nm for different time intervals. From this data the rate constant k was estimated and the activation energy was then calculated by using the Arrhenius equation,
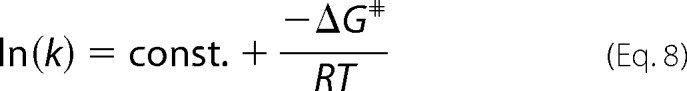 |
where ΔG# is the activation energy, k is the rate constant, R is the gas constant, and T is the absolute temperature.
Results
Binding of PChlide to POR A and POR B
The affinity of PChlide to isozymes POR A and POR B was first analyzed by MST. To this end the POR enzymes were labeled with the blue fluorescent dye NT-495 and the reconstituted POR-labeled NADPH complexes, the concentration of which was kept constant, were titrated with decreasing concentrations of PChlide in serial dilution. Fig. 2 shows the binding curves for POR A and POR B with the fraction of POR-bound PChlide plotted against the concentration of titrated PChlide. The binding constants were derived from the binding curves as equilibrium dissociation constants, KD, through the curve fitting procedure given under “Experimental Procedures.” This analysis yields dissociation constants for PChlide binding in the presence of NADPH of KD = 1.0 ± 0.014 μm in the case of POR A and KD = 0.19 ± 0.009 μm in the case of POR B. From the lower KD value for POR B it is apparent that this isozyme shows an about 5-fold higher binding affinity to PChlide as compared with POR A.
FIGURE 2.

Fraction of POR-bound PChlide in dependence on the concentration of added PChlide. The plots are shown for POR A (●) and POR B (■). The dissociation constant KD was estimated by fitting the binding curve with the quadratic solution for the fraction of the POR-NADPH complex that bound to PChlide, calculated from the law of mass action (Equation 6). The POR enzymes were labeled with the blue fluorescent dye NT-495. For the measurements POR-NADPH complexes (83 nm POR A and 166 nm POR B, as well as a 10-fold molar excess of NADPH) dissolved in measuring buffer (50 mm Tris-HCl, 0.3 m NaCl, 0.1% Triton X-100, 20% glycerol, 1 mm DTT, pH 7.6), were titrated with PChlide added in the concentration range from 1 nm to 5 μm. The experiments were repeated at least four times (n = 4). To exclude nonspecific binding a negative control containing only the labeled POR-NADPH complex in all capillaries was performed in parallel to the PChlide-bound POR-NADPH complex (▴). The corresponding data analysis indicates no binding at all.
To further characterize the affinity of the two POR isozymes to the substrate, the Michaelis-Menten constants, Km, were determined in a second set of experiments. Fig. 3 shows the dependence of the initial reaction rates on the PChlide concentration when the enzyme concentration was held constant. As can be seen, both, POR A and POR B follow the Michaelis-Menten kinetics with the initial rate, v0, being proportional to the PChlide concentration in the low concentration range and approaching its maximum, Vmax, at high PChlide concentrations thus resulting in a rectangular hyperbola. The fit of the rectangular hyperbolae to the Michaelis-Menten equation (Equation 1) yielded values of Km = 3.44 ± 0.3 μm for POR A and Km = 1.25 ± 0.05 μm for POR B. The value obtained for POR A is in gross agreement with the dissociation constant, KD, whereas Km determined for POR B differs to a slightly higher extent from KD (cf. Table 1). In any case, the Km values reproduce the trend observed for the KD constants, i.e. a lower value for POR B than POR A. From the same fit, values of Vmax = 0.05 μm s−1 were derived for POR A and Vmax = 0.1 μm s−1 for POR B, thus indicating that POR B reaches the maximum reaction rate at substrate saturation, Vmax, twice as fast as POR A.
FIGURE 3.

Dependence of the initial rate (vo) of the POR-catalyzed reaction on the PChlide concentration ([PChlide]). The plots are shown for POR A (●) and POR B (■). The assays were performed as described under “Experimental Procedures.” The Michaelis-Menten constant, Km, and the maximum reaction rate, Vmax, were estimated from the v0 versus [PChlide] plots by a nonlinear curve fitting procedure using the Michaelis-Menten equation (Equation 1) as fit function in Matlab.
TABLE 1.
Comparison of the binding parameters and kinetic constants between POR A and POR B
The dissociation constants, KD, are derived from MST measurements and the values of the kinetic parameters are obtained from plots of the initial rates, v0, against the PChlide concentration by using a nonlinear regression analysis for the Michaelis-Menten Equation 1. The estimates are the average of four independent experiments.
| Parameter | POR A (ternary complex) | POR B (ternary complex) |
|---|---|---|
| KD (μm) | 1.0 ± 0.014 | 0.19 ± 0.009 |
| Km (μm) | 3.44 ± 0.3 | 1.25 ± 0.07 |
| Vmax (μm s−1) | 0.05 ± 0.005 | 0.1 ± 0.015 |
| kcat (s−1) | 5.5 × 10−3 ± 5.5 × 10−4 | 1.16 × 10−2 ± 1.5 × 10−3 |
| kcat/Km (s−1 m−1) | 1.6 × 103 ± 1.6 × 102 | 9.3 × 103 ± 9.3 × 102 |
Catalytic Efficiency of POR A and POR B
To assess and compare the catalytic efficiencies of the two isozymes, POR A and POR B, the turnover number, kcat, and the specificity constant, kcat/Km, were used. kcat was determined from the maximum velocity, Vmax, under substrate saturation (Equation 2). Vmax and also Km, necessary for evaluation of the specificity constant, were derived from the plots in Fig. 3 by a nonlinear least squares fit as described in the preceding paragraph. The efficiency constants thus obtained are summarized in Table 1. As these data reveal, kcat is about two times higher for POR B than for POR A indicating a considerably enhanced rate for the conversion of PChlide when the reaction is catalyzed by POR B. This tendency is further confirmed by the specificity constant kcat/Km for the two isozymes. Compared with kcat, the ratio of kcat/Km does not only provide information on how fast the POR-catalyzed reaction is in the one or the other case, but also on how much of the substrate is required to reach Vmax. By using the respective values from Table 1 it is evident that the catalytic efficiency of POR B as compared with POR A is ∼6-fold higher. Thus, POR B is clearly the more efficient enzyme in the photoconversion of PChlide into Chlide.
Reaction Pathway of POR A and POR B Probed at Low Temperatures
As shown by studies on POR enzymes from cyanobacteria, catalysis can be initiated by irradiation at cryogenic temperatures as low as 120 K. This allows the catalytic reaction steps to be followed by trapping reaction intermediates at low temperatures (6, 30–33). Therefore, to gain insight into the reaction mechanisms of POR A and POR B, the transient intermediates formed at low temperatures were characterized by their absorbance spectra. To this end the catalytically active ternary POR-NADPH-PChlide complex was cooled down to 180 K. After initiation of catalysis by irradiation with red light (λexc = 632 nm) the temperature was progressively increased in the dark and the absorbance spectra were taken at definite temperatures. The spectra thus obtained and the temperature dependence of the intermediates extracted from the absorbance spectra is shown in Figs. 4 and 5.
FIGURE 4.
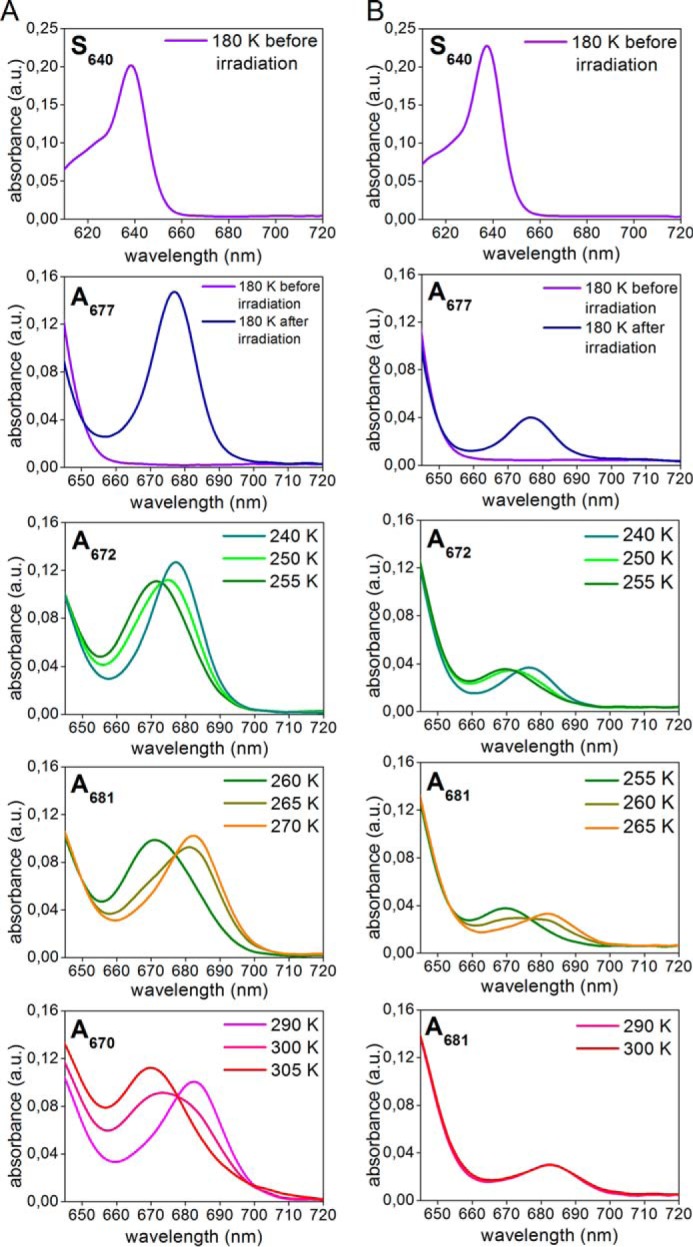
Intermediates in the reaction cycle of POR A and POR B as detected by low temperature absorbance spectroscopy. The ternary complexes of POR A and POR B (60 μm POR, 3.5 μm PChlide, 400 μm NADPH, 1 mm DTT in measuring buffer with 60% glycerol) were frozen at 180 K, irradiated by actinic light at λexc = 632 nm, and then the temperature was raised in the range between 180 and 305 K step by step. The absorbance spectra were monitored at the temperatures indicated in the insets. The left displays the absorbance spectra of POR A and the right those of POR B. For more details see ”Experimental Procedures.“
FIGURE 5.

Spectroscopic characteristics of the intermediates in the reaction cycle of POR A and POR B as obtained from exploratory factor analysis. A and B, absorbance spectra of the intermediates approached for the reaction pathway of POR A and POR B, respectively. C, temperature dependence of the intermediates in the reaction path of POR A (■) and POR B (□).
It is striking that upon binding of PChlide to both POR isozymes in the presence of the cofactor NADPH the absorbance shifts to the red by about 10 nm to give rise to an absorbance maximum at 640 nm. This effect is clearly seen at 180 K and reflects the formation of species S640, which represents the absorbance of enzyme-bound PChlide in the POR-NADPH-PChlide ternary complexes. In the room temperature spectra the red shift in absorbance is manifested solely by a broadening of the absorbance band between 630 and 650 nm (data not shown).
After photoexcitation of S640 at 180 K, the initial light-induced step involves the formation of a first intermediate with an absorbance maximum at 677 nm (A677). No intermediate form that precedes A677 can be observed even at temperatures below 180 K. The temperature dependence of the A677 intermediate reveals that it mainly occurs between 180 and 240 K in both POR A and POR B. At temperatures above 240 K the A677 absorbance band disappears accompanied by the simultaneous appearance of a new intermediate (A672) absorbing with a maximum at 672 nm. This first dark reaction, which requires temperatures close to or above the phase transition of proteins, is then followed a by second one. During the second dark process the A672 species is converted into a product peaking at 681 nm (A681). The temperature interval for the formation of A681 is between 255 and 270 K. This reaction step is shifted by about 5 °C to lower temperatures in POR B when compared with POR A. Until that point POR A and POR B do not differ significantly with respect to the sequence of the dark reactions as well as the spectral characteristics of the intermediates. The results of an SVD analysis confirm that three different reaction intermediates are required to accurately model the experimental datasets for both isozymes. In addition, spectral analysis using exploratory factor analysis (34) also identifies three intermediates with spectral characteristics and temperature dependence as shown in Fig. 5. They fully match the intermediate species monitored in the absorbance spectra (Fig. 4).
In POR A, the further increase in temperature above 290 K leads to the disappearance of the 681-nm absorbance band and simultaneous formation of a blue-shifted band at 670 nm, which represents the absorbance of free, unbound Chlide. Hence, this last reaction step indicates the release of Chlide, the final product of the enzymatic reaction, from the enzyme complex. The corresponding reaction is missing in POR B. There are no further spectral changes after formation of the A681 intermediate in the time period of the measurement.
Another difference between POR A and POR B refers to the yield of the first photoproduct (A677) formed after irradiation at 180 K (Fig. 4). Under the conditions of the low temperature experiments, which allow only a single catalytic turnover, it is evident that POR A generates a significantly larger amount of the first A677 intermediate than POR B.
Activation Energy for the First Light-induced Reaction Step
To gain further insight into the reaction mechanism the activation energies for the initial light-driven step were determined. For this purpose the rate constants for the formation of the first photoproduct, A677, were estimated over a range of temperatures from 160–200 K both for POR A and POR B. The data were plotted in an Arrhenius graph (ln(k) versus 1/T) and fitted to the Arrhenius equation (Fig. 6). Thus, activation energies (ΔG#) of 24.9 ± 2.1 kJ mol−1 for POR A and 35.9 ± 1.8 kJ mol−1 for POR B could be calculated implying a higher activation barrier that has to be crossed in POR B as compared with POR A to produce A677.
FIGURE 6.

Arrhenius plots displaying the logarithm of the rate constant (ln(k)) for the initial photoreaction of the ternary POR complex to the first photoproduct, A677, as a function of the inverse temperature (1/T). The plots are given for the reactions catalyzed by POR A (●) and POR B (■) in the temperature range from 160–200 K. From a fit of the data to the Arrhenius equation (Equation 8) the activation energies (ΔG#) are calculated to be 24.9 ± 2.1 kJ mol−1 for POR A and 35.9 ± 1.8 kJ mol−1 for POR B.
Comparison with the Reaction Intermediates in the Catalytic Cycle of POR from Cyanobacteria
To compare the reaction intermediates in the catalytic pathway of POR A and POR B with those already identified in the reaction cycle for POR from cyanobacteria (6, 30–33), the low temperature absorbance spectra were also monitored for the enzyme from Synechocystis under identical experimental conditions. The spectra of the intermediates resolved are summarized in Fig. 7. In the following, they are briefly discussed in respect of the results reported for Synechocystis POR (6, 30, 31). In full agreement with the Synechocystis studies, the first light-induced step yields a photoproduct, which is characterized by a broad absorbance band with a maximum at 698 nm (A698). In the subsequent dark reaction A698 is converted into another intermediate form, A677, with an absorbance band centered at 677 nm. In the last reaction step occurring above the glass transition temperature, A677 undergoes a blue-shift in its absorbance and forms the final product, A670, the absorbance features of which mirrors those of free, unbound Chlide and thus indicates the release of Chlide from the POR complex.
FIGURE 7.

Spectroscopic characterization of the reaction intermediates in the catalytic pathway of the POR enzyme from Synechocystis as detected by low temperature absorbance spectroscopy. The ternary complexes of Synechocystis POR (60 μm POR, 3.5 μm PChlide, 400 μm NADPH, 1 mm DTT in measuring buffer with 60% glycerol) were frozen at 180 K, irradiated by actinic light at λexc = 632 nm, and then the temperature was raised in the range between 180 and 305 K step by step. The absorbance spectra were monitored at the temperatures indicated in the insets. The figure reproduces the results reported in Refs. 6 and 30–32. It is shown to demonstrate that identical results are obtained under the experimental conditions used with the enzymes from barley.
Discussion
In the present work the two isozymes of POR, POR A and POR B, are compared with respect to their substrate-binding parameters, catalytic efficiency, and photochemical reaction cycle assessed at low temperatures. In this direct comparison, heterologously expressed plant enzymes from barley (H. vulgare L.) are used. Those enzymes allow a detailed spectroscopic and kinetic analysis of the enzyme-catalyzed reaction because disturbances, as for instance caused by aggregation or limited amounts of available enzymes, can be excluded.
The PChlide-binding affinities of the two POR isozymes are analyzed by a direct comparison of the dissociation constant, KD, and the Michaelis-Menten constant, Km. By using the MST technique, KD values of 1.0 ± 0.014 μm for POR A and 0.19 ± 0.009 μm for POR B have been calculated. The value of POR A is in gross agreement with the Km value, which is derived from kinetic measurements and determined to be 3.44 ± 0.3 μm. For POR B the KD constant differs to a slightly higher extent from the Km value, which is estimated as 1.25 ± 0.07 μm (Table 1). This discrepancy shows that the Michaelis-Menten constant does not represent a measure of the substrate affinity at least in case of POR B. The difference between Km and KD is not uncommon and applies for many enzymes. This can be explained by the fact that the rate constant for breakdown of the ES complex is not rate-limiting, i.e. not negligible relative to that for the formation of the ES complex or that other reaction steps such as the product release are rate-determining. Consequently, Km remains a complex function of various rate constants and cannot be considered as a measure of the substrate affinity even though the Michaelis-Menten equation and the characteristic saturation behavior of the enzyme still apply. However, irrespective of the situation for Km, the considerably smaller KD constant of POR B indicates a significantly higher (∼5-fold) affinity of this isozyme for PChlide than POR A. This result is in line with findings from in vitro reconstitution experiments of light-harvesting POR-PChlide-NADPH complexes. As shown in these studies by quantification of the acetone-extractable pigments, POR B as compared with POR A is more specific to PChlide by binding and photoconverting a higher amount of PChlide into Chlide (20). Moreover, the Km value of ∼3 μm obtained for POR A is of the same order as the values of 6.8 and 1.8 μm reported for Synechocystis POR and the POR enzyme from Thermosynechococcus, respectively (35, 36). On the other hand, there is a difference to the Km values of 0.27 and 0.47 μm estimated for plant PORs from pea and oat (37, 38). The reason for this discrepancy is most likely due to the use of “inhomogeneous” enzyme preparations in the assays. In the case of the enzyme from pea, dimers of POR fused with the high molecular affinity tag maltose-binding protein were assessed, whereas in the case of the oat enzyme, POR was directly extracted from etiolated seedlings and therefore most probably contains a mixture of POR A and POR B. With respect to the change in the free enthalpy (ΔG0) the binding of PChlide to the POR-NADPH complexes does not greatly differ between POR A and POR B. It is estimated from the KD as −34 and −38 kJ mol−1 for POR A and POR B, respectively.
The higher binding affinity of POR B to PChlide and therefore the higher concentration of the enzyme-substrate complex in the steady-state correlates with a considerably increased catalytic efficiency as compared with POR A. Both the turnover number kcat and the specificity constant kcat/Km commonly used for comparing enzyme activity are 2- and about 6-fold, respectively, larger for POR B than POR A. The 2-fold higher turnover number corresponds well with the previously reported 2-fold higher quantum efficiency of POR B versus POR A (21). One can speculate that the higher catalytic efficiency of POR B is related to its biological function. Although POR A appears mainly transiently in dark-grown seedlings and is the main enzyme during seedling de-etiolation, POR B becomes the predominant one in green plants, in which it is responsible for maximal PChlide photoreduction (13, 14, 18). Accordingly, it can be assumed that the high enzymatic activity of POR B guarantees the delivery of sufficient amounts of chlorophyll needed to meet the demands of plant growth. This role of POR B is of special importance for angiosperms, which only contain the light-dependent enzyme and lack the light-independent enzyme. Furthermore, it is known that free PChlide generates reactive oxygen species, primarily singlet oxygen, after light absorption and provokes photooxidative damage (39–42). Hence, the high binding constant and enhanced activity of POR B most likely prevents the accumulation of free PChlide and lowers the risk of photosensitized side reactions.
Very recently, reconstituted enzyme complexes of POR A, POR B, and POR C from A. thaliana were studied with respect to their low temperature fluorescence spectra and catalytic activity (26). Whereas the fluorescence spectra indicate formation of highly aggregated POR complexes under the experimental conditions used, POR A and POR B did not differ in their catalytic activity. This result is in disagreement with those from the barley isozymes reported in this contribution. The disagreement might be explained by the following reasons. (i) The Arabidopsis isozymes reflect the behavior of aggregated POR-NADPH-PChlide complexes, the photochemistry of which could be affected by PChlide-PChlide interactions and/or energy transfer processes between PChlide and Chlide. In contrast the barley isozymes were dissolved in a buffer containing a detergent above the critical micellar concentration resulting in monomeric enzyme forms. It remains to be seen whether competitive energy transfer processes in the aggregated POR complexes of Arabidopsis override the reaction chemistry observed for the monomeric enzymes in this study. (ii) On the other hand, differences between the Arabidopsis and Hordeum isozymes are not surprising, given that the lineages that led to Arabidopsis (eudicots) and barley (monocots) separated about 200 million years ago (43). Furthermore, based on the evolutionary relationship of POR enzymes from diverse organisms as reported in Ref. 27, POR A and POR B isozymes from barley appear to trace back to ancient gene duplication within grasses. Therefore, differences between the POR A and POR B barley isozymes are not unexpected in the frame of subfunctionalization or other diversification events. Thus, it cannot be ruled out that the differences between the Arabidopsis and Hordeum isozymes are due to differences in the organism from which the isozymes originate. (iii) In this context it is worth mentioning that differences in the structure of the Arabidopsis and barley POR B have been shown very recently (26, 44). The structural difference is localized at the enzyme surface, which enables POR B to form high molecular light harvesting complexes in barley but not in Arabidopsis. However, so far it is unclear whether such structural differences also affect the geometry at the catalytic site of POR B from the two organisms. In general, the reason for the difference in the catalytic efficiency between the Arabidopsis and Hordeum POR A and POR B isozymes requires further investigation.
In the low temperature experiments the 180 K spectra of ternary POR A and POR B complexes, represented by the S640 species, exhibit a red shift in the PChlide absorbance from 630 to 640 nm (Fig. 4). Such a red shift is well known from the POR enzymes of cyanobacteria and suggested to represent the formation of the finally photoactive state as a consequence of conformational rearrangements upon binding of PChlide and NADPH within the catalytic site (6, 30, 37, 42).
The first photoproduct produced after photoexcitation at 180 K is A677 in both POR A and POR B. No other intermediate than A677 has been observed even at temperatures below 180 K. Due to its spectral characteristics A677 can be attributed to Chlide, the final reaction product, which, however, remains still bound in the POR-NADP+-Chlide complex. This first reaction step differs significantly from that observed in POR from cyanobacteria (6, 30–32). In the latter enzymes the first photochemical step creates an intermediate, A696, in which PChlide has received the hydride (H−) from NADPH and forms an intermolecular charge transfer state with NADP+ (42). The H+ transfer then occurs in the subsequent dark reaction and produces A681 that obviously corresponds to the A677 species seen as the primary photoproduct in POR A and POR B. Evidently, a charge-transfer intermediate similar to A696 is not to be observed in the two Hordeum PORs. The comparative analysis of the low temperature intermediates trapped in Synechocystis POR and the two barley isozymes confirms this conclusion. The results show that a charge-transfer intermediate (A698) can be identified in the case of Synechocystis POR, whereas it is definitely missing in the low temperature spectra of POR A and POR B (Figs. 4 and 7). That means that either the activation barrier separating A698 from A681 is too small in the two isozymes studied so that an intermediate similar to A698 cannot be trapped at low temperatures, or that in general the H−/H+ transfer reaction occurs in a concerted process that directly produces Chlide as reaction product. In any case it seems that the active site has optimally evolved for the reaction chemistry in the two barley isozymes compared with the Synechocystis enzyme.
As is further evident from Fig. 4, a higher amount of the first photoproduct, A677, is generated in POR A as compared with POR B. This finding agrees with the lower activation energy estimated with 24.9 ± 2.1 kJ mol−1 for POR A as opposed to 35.9 ± 1.8 kJ mol−1 for POR B. However, on second glance it seems to be in contrast with the higher reaction quantum yield reported for POR B as compared with POR A (21). The reason for this is that in the low temperature experiments the reaction pathway is followed for only one reaction cycle, whereas the results on the quantum yields were achieved in steady-state measurements, i.e. in multiple turnover assays at room temperature. Under those conditions substrate binding and enzyme flexibility also affect the catalytic efficiency (see below).
In both POR A and POR B the initial light-induced reaction is followed by three and two, respectively, dark reactions, which take place above the glass transition temperature. Therefore, they most likely require the rearrangement of protein domains/sites near or at the catalytic center to modulate the product binding interactions and to release the cofactor and product. The absorbance shifts, characteristic of the different dark reaction stages, are very similar to those identified in low temperature binding studies of Chlide to POR in the presence of NADPH and NADP+ (32). Taking the assignment that has been made there, the dark reactions can be attributed to the release of NADP+ (A677 → A672), its replacement by NADPH (A672 → A681), and the expulsion of Chlide from the active site (A681 → A670). However, in case of POR B it appears that Chlide remains trapped in the binding pocket under the experimental conditions used. It is most likely that this effect reflects a higher binding affinity of Chlide to POR B than to POR A, similar to the distinct binding affinities of PChlide.
Furthermore, a difference in the temperature dependence of the second dark step, which involves the conversion A672 → A681 and goes along with the liberation of NADP+ and rebinding of NADPH, was observed. This reaction is shifted to lower temperatures in POR B as compared with POR A (Fig. 5). Because the reaction only occurs above the glass-transition temperature where protein dynamics plays a prominent role, the temperature shift suggests higher protein flexibility in the POR B enzyme, which finally facilitates the replacement of NADP+ by NADPH within the active site.
Apart from possible differences in protein dynamics, the low temperature experiments clearly indicate that both the initial photoactive POR-NADPH-PChlide complex and the reaction intermediates, which are formed in the course of PChlide reduction, are spectroscopically identical in POR A and POR B. Beyond that, the reaction sequence in the appearance of the different intermediate species is also the same in the two isozymes. These findings imply that the same mechanism of catalysis is likely to operate in POR A and POR B. Thus, the higher catalytic efficiency of POR B as compared with POR A is not the result of differences in the catalytic mechanism. It is first of all due to the higher binding affinity of PChlide to POR B and most probably to a higher conformational flexibility of POR B, as a result of which the cofactor release and cofactor rebinding steps are modulated. In addition, enzyme dynamics may also play a role for PChlide binding in the ternary enzyme-substrate complex as it was recently observed for POR enzymes from cyanobacteria (45). Thus, even if protein dynamics might not directly affect the catalytic rate itself it seems to be an integral part in the catalytic function of POR B that could explain the higher catalytic activity when compared with POR A.
In conclusion, the results of the present study reveal significant differences between the two isozymes of barley POR, POR A and POR B. In accordance with its prominent role for chlorophyll biosynthesis in light-adapted plants, POR B shows an about 6-fold higher catalytic efficiency as compared with POR A. As is apparent from the intermediates trapped at low temperatures, the two isozymes do not differ in their reaction mechanism. Therefore, the higher catalytic efficiency of POR B obviously results from the higher binding affinity of PChlide to the enzyme and, most likely, from dynamic effects, which control conformational fluctuations of protein sites around the catalytic center.
Moreover, with respect to the initial photochemistry the two studied isozymes differ from the enzymes of cyanobacteria. The initial photoreaction, which occurs below 180 K, yields already fully reduced Chlide, even if Chlide remains at first still bound to the enzyme and is only released in the following dark reaction steps at increasing temperatures. The photoreduction in two separate reaction steps including the (i) light-induced hydride transfer from NADPH to PChlide followed by (ii) the proton transfer reaction in the subsequent dark reaction as it is reported for the enzymes from cyanobacteria (31, 32, 45) is not realized in the two barley isozymes.
Author Contributions
A. G. and N. A. designed, performed, and analyzed the experiments of the paper. P. F. Z. revised the manuscript critically. B. D. and G. H. designed research, acquired funding, drafted, and revisited the paper.
Acknowledgments
Andrea Hartmann, Leibniz Institute for Natural Product Research and Infection Biology, is gratefully acknowledged for help with the microscale thermophoresis technique. We also thank Prof. Dr. G. Theißen, Friedrich Schiller University Jena, for helpful discussions on the evolution of the light-dependent protochlorophyllide oxidoreductases.
This work was supported by a grant from the Deutsche Forschungsgemeinschaft, Jena School for Microbial Communication (JSMC) (to A. G., G. H., and B. D.). The authors declare that they have no conflicts of interest with the contents of this article.
- POR
- protochlorophyllide oxidoreductase
- PChlide
- protochlorophyllide
- Chlide
- chlorophyllide
- MST
- microscale thermophoresis
- Tricine
- N-[2-hydroxy-1,1-bis(hydroxymethyl)ethyl]glycine.
References
- 1.Beale S. I. (1999) Enzymes of chlorophyll biosynthesis. Photosynth. Res. 60, 43–73 [Google Scholar]
- 2.Yin L., and Bauer C. E. (2013) Controlling the delicate balance of tetrapyrrole biosynthesis. Phil. Trans. R. Soc. B 368, 20120262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lebedev N., and Timko M. P. (1998) Protochlorophyllide photoreduction. Photosynth. Res. 58, 5–23 [Google Scholar]
- 4.Masuda T., and Takamiya K. (2004) Novel insights into enzymology, regulation and physiological functions of light-dependent protochlorophyllide oxidoreductase in angiosperms. Photosynth. Res. 81, 1–29 [DOI] [PubMed] [Google Scholar]
- 5.Reinbothe C., El Bakkouri M., Buhr F., Muraki N., Nomata J., Kurisu G., Fujita Y., and Reinbothe S. (2010) Chlorophyll biosynthesis: spotlight on protochlorophyllide reduction. Trends Plant Sci. 15, 614–624 [DOI] [PubMed] [Google Scholar]
- 6.Heyes D. J., and Hunter C. N. (2005) Making light work of enzyme catalysis: protochlorophyllide oxidoreductase. Trends Biochem. Sci. 30, 642–649 [DOI] [PubMed] [Google Scholar]
- 7.Scrutton N. S., Groot L.-M., and Heyes D. J. (2012) Excited state dynamics and catalytic mechanism of the light-driven enzyme protochlorophyllide oxidoreductase. Phys. Chem. Chem. Phys. 14, 8818–8824 [DOI] [PubMed] [Google Scholar]
- 8.Begley T. P., and Young H. (1989) Protochlorophyllide reductase: determination of the regiochemistry and the stereochemistry of the reduction of protochlorophyllide to chlorophyllide. J. Am. Chem. Soc. 111, 3095–3096 [Google Scholar]
- 9.Wilks H. M., and Timko M. P. (1995) A light-dependent complementation system for analysis of NADPH:protochlorophyllide oxidoreductase: identification and mutagenesis of two conserved residues that are essential for enzyme activity. Proc. Natl. Acad. Sci. U.S.A. 92, 724–728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Baker M. E. (1994) Protochlorophyllide reductase is homologous to human carbonyl reductase and pig 20 β-hydrosteroid dehydrogenase. Biochem. J. 300, 605–607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Townley H. E., Sessions R. B., Clarke A. R., Dafforn T. R., and Griffiths W. T. (2001) Protochlorophyllide oxidoreductase: a homology model examined by site-directed mutagenesis. Proteins 44, 329–335 [DOI] [PubMed] [Google Scholar]
- 12.Armstrong G. A., Runge S., Frick G., Sperling U., and Apel K. (1995) Identification of NADPH:protochlorophyllide oxidoreductases A and B. Plant Physiol. 108, 1505–1517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Holtorf H., Reinbothe S., Reinbothe C., Bereza B., and Apel K. (1995) Two routes of chlorophyllide synthesis that are differently regulated by light in barley (Hordeum vulgare L.). Proc. Natl. Acad. Sci. U.S.A. 92, 3254–3258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Reinbothe S., Reinbothe C., Lebedev N., and Apel K. (1996) PORA and PORB, two light-dependent protochlorophyllide reducing enzymes of angiosperm chlorophyll biosynthesis. Plant Cell 8, 763–769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Su Q., Frick G., Armstrong G., and Apel K. (2001) PORC of Arabidopsis thaliana: a third light- and NADPH-dependent protochlorophyllide oxidoreductase that is differentially regulated by light. Plant Mol. Biol. 47, 805–813 [DOI] [PubMed] [Google Scholar]
- 16.Pattanayak G. K., and Tripathy B. C. (2002) Catalytic function of a novel protochlorophyllide oxidoreductase C of Arabidopsis thaliana. Biochem. Biophys. Res. Commun. 291, 921–924 [DOI] [PubMed] [Google Scholar]
- 17.Holtorf H., and Apel K. (1996) Transcripts of the two NADPH:protochlorophyllide oxidoreductase genes PorA and PorB are differently degraded in etiolated barley. Plant Mol. Biol. 31, 387–392 [DOI] [PubMed] [Google Scholar]
- 18.Reinbothe S., Reinbothe C., Holtorf H., and Apel K. (1995) Two NADPH:protochlorophyllide oxidoreductases in barley: evidence for the selective disappearance of PORA during the light-induced greening of etiolated seedlings. Plant Cell 7, 1933–1940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Reinbothe C. Lebedev N., and Reinbothe S. (1999) A protochlorophyllide light-harvesting complex involved in de-etiolation of higher plants. Nature 397, 80–84 [Google Scholar]
- 20.Reinbothe C., Buhr F., Pollmann S., and Reinbothe S. (2003) In vitro reconstitution of light-harvesting POR-protochlorophyllide complex with protochlororphyllides a and b. J. Biol. Chem. 278, 807–815 [DOI] [PubMed] [Google Scholar]
- 21.Hanf R., Fey S., Schmitt M., Hermann G., Dietzek B., and Popp J. (2012) Catalytic efficiency of a photoenzyme: an adaptation to natural light conditions. ChemPhysChem 13, 2013–2015 [DOI] [PubMed] [Google Scholar]
- 22.Reinbothe S., Runge S., Reinbothe C., van Cleve B., and Apel K. (1995) Substrate-dependent transport of the NADP:protochlorophyllide oxidoreductase into isolated plastids. Plant Cell 7, 161–172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Reinbothe S., Reinbothe C., Runge S., and Apel K. (1995) Enzymatic product formation impairs both the chloroplast receptor binding function as well as translocon competence of the NADPH:protochlorophyllide oxidoreductase, a nuclear-encoded plastid protein. J. Cell Biol. 129, 299–308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Reinbothe C., Lebedev N., Apel K., and Reinbothe S. (1997) Regulation of chloroplast protein import through a protochlorophyllide-responsive transit peptide. Proc. Natl. Acad. Sci. U.S.A. 94, 8890–8894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Buhr F., El Bakkouri M., Valdez O., Pollmann S., Lebedev N., Reinbothe S., and Reinbothe C. (2008) Photoprotective role of NADPH:protochlorophyllide oxidoreductase A. Proc. Natl. Acad. Sci. U.S.A. 105, 12629–12634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gabruk M., Stecka A., Stral̸zka W., Kruk J., Stral̸zka K., and Mysliwa-Kurdziel B. (2015) Photoactive protochlorophyllide-enzyme complexes reconstituted with PORA, PORB and PORC proteins of A. thaliana: fluorescence and catalytic properties. PLoS ONE 10, e0116990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yang J., and Cheng Q. (2004) Origin and evolution of the light-dependent protochlorophyllide oxidoreductase (LPOR) genes. Plant Biol. 6, 537–544 [DOI] [PubMed] [Google Scholar]
- 28.Colindres-Rojas M., Wolf M. M., Gross R., Seidel S., Dietzek B., Schmitt M., Popp J., Hermann G., and Diller R. (2011) Excited-state dynamics of protochlorophyllide revealed by subpicosecond infrared spectroscopy. Biophys. J. 100, 260–267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Seidel S. A., Dijkman P. M., Lea W. A., van den Bogaart G., Jerabek-Willemsen M., Lazic A., Joseph J. S., Srinivasan P., Baaske P., Simeonov A., Katritch I., Melo F. A., Ladbury J. E., Schreiber G., Watts A., Braun D., and Duhr S. (2013) Microscale thermophoresis quantifies biomolecular interactions under previously challenging conditions. Methods 59, 301–315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Heyes D. J., Ruban A. V., Wilks H. M., and Hunter C. N. (2002) Enzymology below 200 K: the kinetics and thermodynamics of the photochemistry catalysed by protochlorophyllide oxidoreductase. Proc. Natl. Acad. Sci. U.S.A. 99, 11145–11150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Heyes D. J., Ruban A. V., and Hunter C. N. (2003) Protochlorophyllide oxidoreductase: “dark” reactions of a light-driven enzyme. Biochemistry 42, 523–528 [DOI] [PubMed] [Google Scholar]
- 32.Heyes D. J., and Hunter C. N. (2004) Identification and characterization of the product release steps within the catalytic cycle of protochlorophyllide oxidoreductase. Biochemistry 43, 8265–8271 [DOI] [PubMed] [Google Scholar]
- 33.Menon B. R., Waltho J. P., Scrutton N. S., and Heyes D. J. (2009) Cryogenic and laser photoexcitation studies identify multiple roles for active site residues in the light-driven enzyme protochlorophyllide oxidoreductase. J. Biol. Chem. 284, 18160–18166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yong A. G., and Pearce S. (2013) Beginners guide to factor analysis: focusing on exploratory factor analysis. Tutor. Quant. Methods Psychol. 9, 79–94 [Google Scholar]
- 35.Heyes D. J., Martin G. E., Reid R. J., Hunter C. N., and Wilks H. M. (2000) NADPH:protochlorophyllide oxidoreductase from Synechocystis: overexpression, purification and preliminary characterization. FEBS Lett. 483, 47–51 [DOI] [PubMed] [Google Scholar]
- 36.McFarlane M. J., Hunter C. N., and Heyes D. J. (2005) Kinetic characterization of the light-driven protochlorophyllide oxidoreductase (POR) from Thermosynechococcus elongates. Photochem. Photobiol. Sci. 4, 1055–1059 [DOI] [PubMed] [Google Scholar]
- 37.Martin G. E., Timko M. P., and Wilks H. M. (1997) Purification and kinetic analysis of pea (Pisum sativum L.) NADPH:protochlorophyllide oxidoreductase expressed as a fusion with maltose-binding protein in Escherichia coli. Biochem. J. 325, 139–145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Klement H., Helfrich M., Oster U., Schoch S., and Rüdiger W. (1999) Pigment-free NADPH:protochlorophyllide oxidoreductase from Avena sativa L. Eur. J. Biochem. 265, 862–974 [DOI] [PubMed] [Google Scholar]
- 39.Dietzek B., Tschierlei S., Hermann G., Yartsev A., Pascher T., Sundström V., Schmitt M., and Popp J. (2009) Protochlorophyllide a: a comprehensive photophysical picture. ChemPhysChem 10, 144–150 [DOI] [PubMed] [Google Scholar]
- 40.Sytina O. A., van Stokkum I. H., Heyes D. J., Hunter C. N., van Grondelle R., and Groot M. L. (2010) Protochlorophyllide excited-state dynamics in organic solvents studied by time-resolved visible and mid-infrared spectroscopy. J. Phys. Chem. B 114, 4335–4344 [DOI] [PubMed] [Google Scholar]
- 41.Hanf R., Fey S., Dietzek B., Schmitt M., Reinbothe C., Reinbothe S., Hermann G., and Popp J. (2011) Protein-induced excited-state dynamics of protochlorophyllide. J. Phys. Chem. A 115, 7873–7881 [DOI] [PubMed] [Google Scholar]
- 42.Heyes D. J., Heathcote P., Rigby S. E., Palacios M. A., van Grondelle R., and Hunter C. N. (2006) The first catalytic step of the light-driven enzyme protochlorophyllide oxidoreductase proceeds via a charge transfer complex. J. Biol. Chem. 281, 26847–26853 [DOI] [PubMed] [Google Scholar]
- 43.Wolfe K. H., Gouy M., Yang Y-W., Sharp P. M., and Li W.-H. (1989) Date of the monocot-dicot divergence estimated from chloroplast DNA sequence data. Proc. Natl. Acad. Sci. U.S.A. 86, 6201–6205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yuan M., Zhang D.-W., Zhang Z.-W., Chen Y.-E., Yuan S., Guo Y.-R., and Lin H.-H. (2012) Assembly of NADPH:protochlorophyllide oxidoreductase complex is needed for effective greening of barley seedlings. J. Plant Physiol. 169, 1311–1316 [DOI] [PubMed] [Google Scholar]
- 45.Heyes D. J., Menon B. R., Sakuma M., and Scrutton N. S. (2008) Conformational events during ternary enzyme-substrate complex formation are rate limiting in the catalytic cycle of the light-driven enzyme protochlorophyllide oxidoreductase. Biochemistry 47, 10991–10998 [DOI] [PubMed] [Google Scholar]