

Abstract
In biological systems there is a balance between the production and neutralization of reactive oxygen species (ROS). This balance is maintained by the presence of natural antioxidants and antioxidant enzymes such as superoxide dismutase (SOD), catalase and glutathione peroxidase. The enhancement of lipid peroxidation or the decrease of antioxidant protection present in metabolic diseases or bad lifestyle can induce endothelial dysfunction and atherosclerosis. Clinical studies have shown that oxidative stress can increase ROS reducing the formation of antioxidant defences, especially in subjects with coronary artery disease (CAD). Some observation indicated that in the early stages of the disease there is a homeostatic up-regulation of the antioxidant enzyme system in response to increased free radicals to prevent vascular damage. As soon as free radicals get to chronically elevated levels, this compensation ceases. Therefore, SOD and the other enzymes may represent a good therapeutic target against ROS, but they are not useful markers for the diagnosis of CAD. In conclusion antioxidant enzymes are reduced in presence of metabolic disease and CAD. However the existence of genes that promote their enzymatic activity could contribute to create new drugs for the treatment of damage caused by metabolic diseases or lifestyle that increases the plasma ROS levels.
Keywords: Superoxide dismutase, Catalase, Glutathione peroxidase, Antioxidant enzyme, Coronary artery disease, Reactive oxygen species, Vascular inflammation
Core tip: This review shows that antioxidant enzymes are very important factors for the prevention and treatment of atherosclerotic disease, but more studies are required to understand whether they can be used as markers for diagnosis of coronary artery disease. The presence of polymorphic genes that increases the activity and expression of these enzymes can be considered important for the development of new therapeutic strategies. In our opinion further efforts should be directed especially on this last point, in order to find new therapies to increase the function of antioxidant enzymes in metabolic disease or other risk factors.
INTRODUCTION
Oxygen has played a crucial role in the evolution[1-2] inducing the aerobic organisms to develop an adaptation to its toxicity by the presence of antioxidant systems. Oxygen is always metabolized to produce oxygen derived free radicals[3] (superoxide O2-., hydroxyl OH-) and non-radical (hydrogen peroxide H2O2) all termed reactive oxygen species (ROS). In physiological condition there is a balance between the production and neutralization of ROS[4]. Small amount of ROS are constantly generated[5] and may often be useful for the immune system[6] and defense against microorganisms[5]. Conversely, high doses of ROS determine oxidative stress responsible for serious metabolic dysfunctions and damage to biological macromolecules[7]. The enhancement of lipid peroxidation or a decrease in antioxidant protection can frequently induce the reaction with the nucleophilic centers of the DNA, RNA and proteins leading to irreversible damage such as cytotoxicity, mutagenicity and carcinogenicity. For instance, intracellular O2-., hydroxyl radical (OH-) and H2O2 play an important role in endothelial dysfunction, hypertension and atherosclerosis, inducing the expression of ICAM-1 and monocyte adhesion in endothelial cells[8,9]. To minimize the damage caused by free radicals the organism utilize enzymatic and non-enzymatic antioxidant systems. Of the first group are the superoxide dismutase (SOD), glutathione peroxidase (GPX), catalase (CAT), glutathione (GSH), while the second group consists of vitamin A, ascorbic acid (vitamin C) and alpha-tocopherol (vitamin E)[10]. The fact that the activity of SOD is more intense in humans compared to other species could explain the longevity of our species suggesting that humans have better protection against ROS[11,12]. The imbalance between pro-oxidant and antioxidant systems can occur by an overproduction of ROS, (as the radical O2-., or OH-), or because of the drastic reduction of antioxidant systems. Among the main sources of generation of ROS are the mitochondrial electron transport chain[13], the system of NAPDH oxidase and nitric oxide synthase (Figure 1). It has been known for years that NAD(P)H oxidase is a major source of superoxide in vascular tissue[14,15] and in cardiac cells[16].
Figure 1.
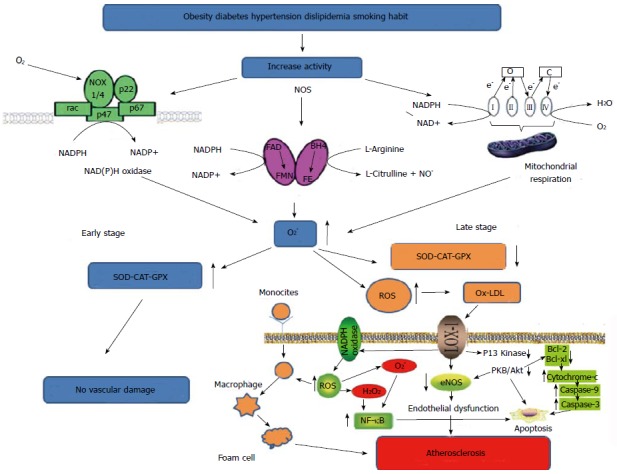
Biochemical events that favor the increase of reactive oxygen species. In the early stages of CAD, ROS do not cause damage due to the presence of an enzymatic compensatory mechanism. In late stage this mechanism is saturated and no longer allows an efficient defense, so that other biochemical events lead to vascular damage. ROS: Reactive oxygen species; SOD: Superoxide dismutase; CAT: Catalase; GPX: Glutathione peroxidase; CAD: Coronary artery disease; NF-κB: Nuclear factor-κB; NADPH: Nicotinamide adenine dinucleotide phosphate-oxidase; LOX-1: Lectin-like oxidized low-density lipoprotein receptor-1; eNOS: Endothelial nitric oxide synthase; FAD: Flavin adenine dinucelotide; BH4: Tetrahydrobiopterin; FE: Heme iron; FMN: Flavin mononucleotide.
NAD(P)H+2O2 → NAD(P)+H++2O2-.
And it has been demonstrated that its activity is increased by angiotensin II[17], thrombin, platelet-derived growth factor (PDGF), tumor necrosis factor-α (TNF-α) and lactosylceramide[18-20].
ENZYMATIC ANTIOXIDANT SYSTEM
SOD converts the highly reactive radical O2-. to the less reactive radical H2O2, which in turn can be destroyed by CAT or GPX, protecting the dehydratase (dehydratase hydroxyacid, aconitase, 6 phosphogluconate dehydratase, fumarase A and B). In humans, there are three forms of SOD: cytosolic (Cu, Zn-SOD), mitochondrial (Mn-SOD) and extracellular (EC-SOD)[21]. The respiratory chain in the mitochondria is the major source of oxygen radicals. Mn-SOD is of primary importance in removing O2-.[22] and is essential for life. Cu, Zn-SOD seems to play an important role in the first line of antioxidant defense catalyzing the dismutation of O2-. radicals to form H2O2 and molecular oxygen, however, knock-outs experiments have shown that it is not essential for life[23]. EC-SOD is a tetrameric glycoprotein containing zinc and copper that has a high affinity for heparin. In mammalian tissues it is regulated by cytokines[24].
CAT consists of 4 ferriprotoporfirinici groups per molecule, is known as the most efficient enzyme since it is never saturated by the presence of H2O2[7]. CAT reacts with H2O2 and with proton donors (ROOH) producing H2O. CAT protects the cells from the production of H2O2 playing an important role in the acquisition of tolerance to oxidative stress as an adaptive response of the cells[25].
GPX also catalyzes the reduction of a variety of hydroperoxides using GSH (ROOH and H2O2). The cells that contain low levels of GPX are much more susceptible to the toxicity of compounds such as adriamycin which produces hydroperoxides[26] and seems important as a line of defense against peroxynitrite.
ASSOCIATION BETWEEN ANTIOXIDANT ENZYME AND VASCULAR DISEASE
The increased oxidative stress is associated with the pathogenesis of coronary artery disease (CAD)[27,28]. Clinical studies have shown that oxidative stress can increase ROS reducing the formation of antioxidant defenses[27,28]. Some authors have demonstrated that the reduction of activity of antioxidant enzymes such as CAT, SOD and GPX facilitates the oxidative aggression to the cells, especially in subjects with CAD[29]. The study showed that in the early stages of CAD, SOD and CAT levels increased to protect and prevent lipid peroxidation whereas they decreased significantly with the worsening of the disease[29]. These observations indicate that in the early stages of the disease there is a homeostatic up-regulation involving the antioxidant enzyme system in response to increased free radicals to prevent vascular damage. In later stages of the disease, when free radicals get to chronically elevated levels, this mechanism, that has reached the saturation, suddenly crashes showing a reduction of antioxidant enzyme activities. In our study[30] in which we examined a population of CAD patients with different numbers of affected vessels, we did not find a significant difference in SOD levels even if higher values were observed in patients with three or four numbers of injured vessels (Table 1). We can speculate that in presence of high oxygen free radical levels, the compensatory response is only partially related to the activity or expression of the EC-SOD enzyme linked to the damaged artery. However, our data do not disagree with the data of Gupta et al[29] concerning the patients in the late stages of the disease, when the vascular damage is already present; in fact they did not find any difference in SOD and CAT activity. The difference is mainly in the fact that the authors have also examined patients in the early stages of CAD, when the antioxidant enzyme system was not yet saturated. Therefore, EC-SOD do not appear a useful marker for the diagnosis of CAD or to stratify the patients population[30] (Table 1), but it may represent a good therapeutic target against ROS in CAD. In another study, Kotur-Stevuljevic et al[31] found that the enzymatic activity of erythrocyte SOD in patients with CAD was significantly decreased comparing to healthy volunteers. A significant difference was also evident among patients with stenosis less than 50% compared to those with stenosis higher than 50%. However, the activity of SOD in patients with stenosis less than 50% was not significantly different from the control group, showing that also a compensatory mechanism did not exist in this population. This means that the homeostatic enzyme response occurs when the vascular damage has not yet been produced. This phase could represent an important time point for therapeutic treatments that stimulate the enzyme system for a longer time or with natural scavenger to massively reduce the presence of ROS produced by metabolic diseases or from unhealthy lifestyle. From the clinical point of view it would be important to combine the analysis of SOD CAT, GPX with markers of vascular inflammation such as cytokines and C-reactive protein in cases where it is already established the presence of risk factors such as hypertension, diabetes or hypercholesterolemia, but in the absence of plaque formation. The intimal thickening, effect induced by endothelial activation, is an early event to consider. At this stage the cells begin to produce inflammatory cytokines that attract monocytes, adhesion molecules, receptors for oxidized lipoproteins and a massive increase of ROS. Of course, more studies are needed to determine the precise role of these enzymes in protecting the arteries from ROS damage, in order to clarify whether they can be inserted in both the prevention and the treatment of atherosclerotic disease.
Table 1.
Inflammatory parameters, and serum levels of extracellular superoxide dismutase and free radicals in a group of patients with coronary artery disease and healthy volunteers drawn from the study of Lubrano et al[30]
| Number of injured vassels | IL-6 (pg/mL) | TNF-α (pg/mL) | CRP (mg/dL) | Peroxy radicals (UC) | EC-SOD (U/mL) |
| Controls | 1.05 ± 0.2 | 2.5 ± 1.1 | 0.15 ± 0.03 | 197 ± 15.5 | 2.91 ± 0.4 |
| 1 | 2.7 ± 0.7 | 1.05 ± 0.3 | 0.83 ± 0.7 | 241 ± 30.7 | 2.9 ± 0.4 |
| 2 | 2.6 ± 1.7 | 1.34 ± 0.2 | 1.6 ± 0.1 | 246 ± 12.5 | 2.7 ± 0.6 |
| 3 | 3.3 ± 1a | 0.98 ± 0.4 | 1.1 ± 0.7 | 272 ± 20.2 | 3.8 ± 0.7 |
| 4 | 3.5 ± 0.6a | 3.9 ± 1.5 | 1.4 ± 1.1 | 273 ± 30.5 | 5.1 ± 1.3 |
P < 0.05. TNF-α: Tumor necrosis factor-α; EC-SOD: Extracellular superoxide dismutase; CAD: Coronary artery disease; CRP: C-reactive protein.
INFLUENCE OF THE MAIN RISK FACTORS IN THE MODULATION OF ANTIOXIDANT ENZYME
Obesity has been considered an important factor in causing various health problems, especially in vascular disease[32]. It has been observed that the adipose tissue secretes adipokines, responsible for the production of ROS, and independent factors, for the generation of systemic oxidative stress[33]. The persistence of obesity implies an increase of inflammatory cytokines and an excessive consumption of oxygen, which generates free radicals in the respiratory chain coupled to oxidative phosphorylation in mitochondria. In the long term, the accumulation of fat may deplete the sources of antioxidants and significantly decrease the activity of enzymes such as SOD, CAT and GPX and the presence of non-enzymatic factors such as vitamin E, vitamin C and β-carotene[33].
Even in the case of diabetes there is convincing experimental evidence and clinical trials that have demonstrated that the onset of the disease is closely associated with oxidative stress[34,35]. Potential sources of ROS in diabetes can be justified by the increase in glucose oxidation and by the changes of redox balance through a cascade of ROS generated by mitochondria. This process has been associated with the onset of type 1 diabetes (DM1) caused by pancreatic beta cell apoptosis, and the onset of type 2 diabetes (DM2) caused by insulin resistance[36]. Some authors demonstrated that high glucose levels could stimulate cytochrome P450 activity by excessive nicotinamide adenine dinucleotide phosphate-oxidase (NADPH) produced by glucose metabolism[37]. In addition, ketosis, a hallmark of DM1, seems to increase the production of oxygen radicals in this patients[38]. The reduced enzymatic activity of CAT, SOD, GSH-PX, and glutathione reductase (GSH-Rx), as well as high levels of thiobarbituric acid (TBARS), an indirect measure of the production of ROS, that seem to be consistently high in diabetes[39] are important indices for interpreting the extent of the disease. Recent studies have shown that the levels of SOD and glutathione S-transferase activities were significantly lower in patients with T2DM compared to healthy subjects[40]. It is known that the use of vitamin E as a dietary supplement for patients with CAD entails a significant benefit in reducing the symptoms of angina pectoris[41]. In diabetic rats the beneficial effect of vitamin E showed the delay of onset of coronary atherosclerosis compared to untreated. The slowing of the development of the disease was due to a reduction in oxidative stress, and not secondary to a decrease in the glucose or cholesterol in plasma, for the fact that the respective plasma concentrations remained unchanged in the diabetic mice supplemented with vitamin E[42]. Other studies have supported these results, in fact, it was observed that a triple antioxidant therapy (Vitamin E, lipoic acid, and vitamin C) in diabetic volunteers attenuated oxidative stress reducing the formation of methemoglobin in vitro and in glycated hemoglobin in vivo[43]. Numerous clinical studies have shown a decrease in EC-SOD in African Americans with hypertension, in patients with vasospastic angina, calcific aortic stenosis and in patients with DM2, compared with control subjects[44,45]. Furthermore, it was observed that the standard dietary treatment for type 2 diabetic patients produces an increase of the SOD and GPX activity[46].
It is evident that the mechanism, that renders low-density lipoprotein a good substrate for the production of foam cells and the initiation of atherosclerotic events, is their oxidative modification[28]. As for other conditions, some investigators have shown that the overexpression of antioxidant enzymes can slow the progression of atherosclerosis[47]. Tests carried out on animals have found that the use of natural antioxidants supplements leads to the increase of enzyme activities[48], whereas the intake of excessive dietary lipids and therefore an excess of energy and cholesterol has a negative influence on antioxidant enzymes. A negative correlation between dietary cholesterol and the markers of antioxidant enzyme activity, including CAT and GPX, was observed. The authors have shown a significant improvement in erythrocyte antioxidant capacity, as increased activity for SOD, CAT and GPX in children with hypercholesterolemia who have followed a diet with reduced saturated fat and introduction of several fatty acids for 6 mo[49].
CLINICAL IMPLICATIONS
Increased expression of CuZn-SOD (SOD1) protects muscle cells from oxidative damage. It was observed that overexpression of SOD1 gene inhibits the DNA binding activity of activator protein-1 and NF-κB. Interesting prospects are given by the fact that the substitution of valine with alanine has been shown to induce an increase of 30%-40% in the activity Mn-SOD in the mitochondria with consequent reduction of the risk of CAD and acute myocardial infarction[50]. Even the overexpression of GPX reduces oxidation of the phospholipids, the formation of hydroperoxides of cholesterol, as well as pro-inflammatory lipid peroxides generated by LPO and COX, reducing oxidative stress and vascular atherosclerosis progression. From these observations we conclude that the antioxidant enzyme system is inversely associated with a high-fat diet, and as previously described, the increase in vitamin E, vitamin C, and β-carotene is associated with the strengthening of SOD, therefore the feeding is an important factor in the prevention and treatment of oxidative damage caused by ROS. In the near future it will be possible to study also the genetic polymorphism. The existence of a gene that promotes the enzymatic activity of SOD can contribute to create new drugs for the prevention of damage caused by metabolic diseases or lifestyle that increases the plasma levels of ROS. We believe that further studies should be performed to determine if there is a mechanism of compensation of the antioxidant enzyme system induced by the presence of ROS, and in this case to understand when it begins and what is its intensity. This fact is not very clear from previous studies, because if on one hand it seems to develop before vascular lesion, one the other hand it has never been observed in the presence of metabolic diseases, when the vascular damage has not yet happened.
ACKNOWLEDGMENTS
The authors are grateful to Lucrecia Mota Garcia and Laura Sabatino for their English editing support.
Footnotes
Conflict-of-interest statement: There is no conflict of interest associated with any of the authors that contributed their efforts in this manuscript.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: June 20, 2015
First decision: July 27, 2015
Article in press: November 4, 2015
P- Reviewer: Gomes A, Hassan M S- Editor: Tian YL L- Editor: A E- Editor: Wu HL
References
- 1.Falkowski PG. Evolution. Tracing oxygen’s imprint on earth’s metabolic evolution. Science. 2006;311:1724–1725. doi: 10.1126/science.1125937. [DOI] [PubMed] [Google Scholar]
- 2.Farrugia G, Balzan R. Oxidative stress and programmed cell death in yeast. Front Oncol. 2012;2:64. doi: 10.3389/fonc.2012.00064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Raymond J, Segrè D. The effect of oxygen on biochemical networks and the evolution of complex life. Science. 2006;311:1764–1767. doi: 10.1126/science.1118439. [DOI] [PubMed] [Google Scholar]
- 4.Turrens JF, Boveris A. Generation of superoxide anion by the NADH dehydrogenase of bovine heart mitochondria. Biochem J. 1980;191:421–427. doi: 10.1042/bj1910421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lee YJ, Galoforo SS, Berns CM, Chen JC, Davis BH, Sim JE, Corry PM, Spitz DR. Glucose deprivation-induced cytotoxicity and alterations in mitogen-activated protein kinase activation are mediated by oxidative stress in multidrug-resistant human breast carcinoma cells. J Biol Chem. 1998;273:5294–5299. doi: 10.1074/jbc.273.9.5294. [DOI] [PubMed] [Google Scholar]
- 6.Sun J, Chen Y, Li M, Ge Z. Role of antioxidant enzymes on ionizing radiation resistance. Free Radic Biol Med. 1998;24:586–593. doi: 10.1016/s0891-5849(97)00291-8. [DOI] [PubMed] [Google Scholar]
- 7.Lledías F, Rangel P, Hansberg W. Oxidation of catalase by singlet oxygen. J Biol Chem. 1998;273:10630–10637. doi: 10.1074/jbc.273.17.10630. [DOI] [PubMed] [Google Scholar]
- 8.Bradley JR, Johnson DR, Pober JS. Four different classes of inhibitors of receptor-mediated endocytosis decrease tumor necrosis factor-induced gene expression in human endothelial cells. J Immunol. 1993;150:5544–5555. [PubMed] [Google Scholar]
- 9.Sellak H, Franzini E, Hakim J, Pasquier C. Reactive oxygen species rapidly increase endothelial ICAM-1 ability to bind neutrophils without detectable upregulation. Blood. 1994;83:2669–2677. [PubMed] [Google Scholar]
- 10.Mataix J, Quiles JL, Huertas JR, Battino M, Mañas M. Tissue specific interactions of exercise, dietary fatty acids, and vitamin E in lipid peroxidation. Free Radic Biol Med. 1998;24:511–521. doi: 10.1016/s0891-5849(97)00288-8. [DOI] [PubMed] [Google Scholar]
- 11.Tolmasoff JM, Ono T, Cutler RG. Superoxide dismutase: correlation with life-span and specific metabolic rate in primate species. Proc Natl Acad Sci USA. 1980;77:2777–2781. doi: 10.1073/pnas.77.5.2777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lopez-Torres M, Perez-Campo R, Rojas C, Cadenas S, Barja G. Maximum life span in vertebrates: relationship with liver antioxidant enzymes, glutathione system, ascorbate, urate, sensitivity to peroxidation, true malondialdehyde, in vivo H2O2, and basal and maximum aerobic capacity. Mech Ageing Dev. 1993;70:177–199. doi: 10.1016/0047-6374(93)90047-u. [DOI] [PubMed] [Google Scholar]
- 13.Narayanan D, Xi Q, Pfeffer LM, Jaggar JH. Mitochondria control functional CaV1.2 expression in smooth muscle cells of cerebral arteries. Circ Res. 2010;107:631–641. doi: 10.1161/CIRCRESAHA.110.224345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rajagopalan S, Kurz S, Münzel T, Tarpey M, Freeman BA, Griendling KK, Harrison DG. Angiotensin II-mediated hypertension in the rat increases vascular superoxide production via membrane NADH/NADPH oxidase activation. Contribution to alterations of vasomotor tone. J Clin Invest. 1996;97:1916–1923. doi: 10.1172/JCI118623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pagano PJ, Ito Y, Tornheim K, Gallop PM, Tauber AI, Cohen RA. An NADPH oxidase superoxide-generating system in the rabbit aorta. Am J Physiol. 1995;268:H2274–H2280. doi: 10.1152/ajpheart.1995.268.6.H2274. [DOI] [PubMed] [Google Scholar]
- 16.Mohazzab-H KM, Kaminski PM, Wolin MS. Lactate and PO2 modulate superoxide anion production in bovine cardiac myocytes: potential role of NADH oxidase. Circulation. 1997;96:614–620. doi: 10.1161/01.cir.96.2.614. [DOI] [PubMed] [Google Scholar]
- 17.Griendling KK, Minieri CA, Ollerenshaw JD, Alexander RW. Angiotensin II stimulates NADH and NADPH oxidase activity in cultured vascular smooth muscle cells. Circ Res. 1994;74:1141–1148. doi: 10.1161/01.res.74.6.1141. [DOI] [PubMed] [Google Scholar]
- 18.De Keulenaer GW, Alexander RW, Ushio-Fukai M, Ishizaka N, Griendling KK. Tumour necrosis factor alpha activates a p22phox-based NADH oxidase in vascular smooth muscle. Biochem J. 1998;329(Pt 3):653–657. doi: 10.1042/bj3290653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Marumo T, Schini-Kerth VB, Fisslthaler B, Busse R. Platelet-derived growth factor-stimulated superoxide anion production modulates activation of transcription factor NF-kappaB and expression of monocyte chemoattractant protein 1 in human aortic smooth muscle cells. Circulation. 1997;96:2361–2367. doi: 10.1161/01.cir.96.7.2361. [DOI] [PubMed] [Google Scholar]
- 20.Patterson C, Ruef J, Madamanchi NR, Barry-Lane P, Hu Z, Horaist C, Ballinger CA, Brasier AR, Bode C, Runge MS. Stimulation of a vascular smooth muscle cell NAD(P)H oxidase by thrombin. Evidence that p47(phox) may participate in forming this oxidase in vitro and in vivo. J Biol Chem. 1999;274:19814–19822. doi: 10.1074/jbc.274.28.19814. [DOI] [PubMed] [Google Scholar]
- 21.Majima HJ, Oberley TD, Furukawa K, Mattson MP, Yen HC, Szweda LI, St Clair DK. Prevention of mitochondrial injury by manganese superoxide dismutase reveals a primary mechanism for alkaline-induced cell death. J Biol Chem. 1998;273:8217–8224. doi: 10.1074/jbc.273.14.8217. [DOI] [PubMed] [Google Scholar]
- 22.Guan Y, Hickey MJ, Borgstahl GE, Hallewell RA, Lepock JR, O’Connor D, Hsieh Y, Nick HS, Silverman DN, Tainer JA. Crystal structure of Y34F mutant human mitochondrial manganese superoxide dismutase and the functional role of tyrosine 34. Biochemistry. 1998;37:4722–4730. doi: 10.1021/bi972394l. [DOI] [PubMed] [Google Scholar]
- 23.Reaume AG, Elliott JL, Hoffman EK, Kowall NW, Ferrante RJ, Siwek DF, Wilcox HM, Flood DG, Beal MF, Brown RH, et al. Motor neurons in Cu/Zn superoxide dismutase-deficient mice develop normally but exhibit enhanced cell death after axonal injury. Nat Genet. 1996;13:43–47. doi: 10.1038/ng0596-43. [DOI] [PubMed] [Google Scholar]
- 24.Buschfort C, Muller MR, Seeber S, Rajewsky MF, Thomale J. DNA excision repair profiles of normal and leukemic human lymphocytes: functional analysis at the single-cell level. Cancer Res. 1997;57:651–658. [PubMed] [Google Scholar]
- 25.Hunt CR, Sim JE, Sullivan SJ, Featherstone T, Golden W, Von Kapp-Herr C, Hock RA, Gomez RA, Parsian AJ, Spitz DR. Genomic instability and catalase gene amplification induced by chronic exposure to oxidative stress. Cancer Res. 1998;58:3986–3992. [PubMed] [Google Scholar]
- 26.Taylor SD, Davenport LD, Speranza MJ, Mullenbach GT, Lynch RE. Glutathione peroxidase protects cultured mammalian cells from the toxicity of adriamycin and paraquat. Arch Biochem Biophys. 1993;305:600–605. doi: 10.1006/abbi.1993.1467. [DOI] [PubMed] [Google Scholar]
- 27.Antoniades C, Tousoulis D, Tentolouris C, Toutouzas P, Stefanadis C. Oxidative stress, antioxidant vitamins, and atherosclerosis. From basic research to clinical practice. Herz. 2003;28:628–638. doi: 10.1007/s00059-003-2417-8. [DOI] [PubMed] [Google Scholar]
- 28.Stocker R, Keaney JF. Role of oxidative modifications in atherosclerosis. Physiol Rev. 2004;84:1381–1478. doi: 10.1152/physrev.00047.2003. [DOI] [PubMed] [Google Scholar]
- 29.Gupta S, Sodhi S, Mahajan V. Correlation of antioxidants with lipid peroxidation and lipid profile in patients suffering from coronary artery disease. Expert Opin Ther Targets. 2009;13:889–894. doi: 10.1517/14728220903099668. [DOI] [PubMed] [Google Scholar]
- 30.Lubrano V, Di Cecco P, Zucchelli GC. Role of superoxide dismutase in vascular inflammation and in coronary artery disease. Clin Exp Med. 2006;6:84–88. doi: 10.1007/s10238-006-0100-0. [DOI] [PubMed] [Google Scholar]
- 31.Kotur-Stevuljevic J, Memon L, Stefanovic A, Spasic S, Spasojevic-Kalimanovska V, Bogavac-Stanojevic N, Kalimanovska-Ostric D, Jelić-Ivanovic Z, Zunic G. Correlation of oxidative stress parameters and inflammatory markers in coronary artery disease patients. Clin Biochem. 2007;40:181–187. doi: 10.1016/j.clinbiochem.2006.09.007. [DOI] [PubMed] [Google Scholar]
- 32.Lastra G, Manrique CM, Hayden MR. The role of beta-cell dysfunction in the cardiometabolic syndrome. J Cardiometab Syndr. 2006;1:41–46. doi: 10.1111/j.0197-3118.2006.05458.x. [DOI] [PubMed] [Google Scholar]
- 33.Fernández-Sánchez A, Madrigal-Santillán E, Bautista M, Esquivel-Soto J, Morales-González A, Esquivel-Chirino C, Durante-Montiel I, Sánchez-Rivera G, Valadez-Vega C, Morales-González JA. Inflammation, oxidative stress, and obesity. Int J Mol Sci. 2011;12:3117–3132. doi: 10.3390/ijms12053117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rösen P, Nawroth PP, King G, Möller W, Tritschler HJ, Packer L. The role of oxidative stress in the onset and progression of diabetes and its complications: a summary of a Congress Series sponsored by UNESCO-MCBN, the American Diabetes Association and the German Diabetes Society. Diabetes Metab Res Rev. 2001;17:189–212. doi: 10.1002/dmrr.196. [DOI] [PubMed] [Google Scholar]
- 35.Sayed MR, Iman MM, Dawlat AS. Biochemical changes in experimental diabetes before and after treatment with mangifera indica and psidium guava extracts. Int J Pharm Biomed Sci. 2011;2:29–41. [Google Scholar]
- 36.West IC. Radicals and oxidative stress in diabetes. Diabet Med. 2000;17:171–180. doi: 10.1046/j.1464-5491.2000.00259.x. [DOI] [PubMed] [Google Scholar]
- 37.Jain SK. Hyperglycemia can cause membrane lipid peroxidation and osmotic fragility in human red blood cells. J Biol Chem. 1989;264:21340–21345. [PubMed] [Google Scholar]
- 38.Jain SK, Kannan K, Lim G. Ketosis (acetoacetate) can generate oxygen radicals and cause increased lipid peroxidation and growth inhibition in human endothelial cells. Free Radic Biol Med. 1998;25:1083–1088. doi: 10.1016/s0891-5849(98)00140-3. [DOI] [PubMed] [Google Scholar]
- 39.Johansen JS, Harris AK, Rychly DJ, Ergul A. Oxidative stress and the use of antioxidants in diabetes: linking basic science to clinical practice. Cardiovasc Diabetol. 2005;4:5. doi: 10.1186/1475-2840-4-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Verma S, Sagar N, Vats P, Shukla KN, Abbas M, Banerjee M. Antioxidant enzyme levelsas markers for type 2 diabetes mellitus. Int J Bioassays. 2013;2:685–690. [Google Scholar]
- 41.Shute EV. Proposed study of vitamin E therapy. Can Med Assoc J. 1972;106:1057. [PMC free article] [PubMed] [Google Scholar]
- 42.Otero P, Bonet B, Herrera E, Rabano A. Development of atherosclerosis in the diabetic BALB/c mice. Prevention with Vitamin E administration. Atherosclerosis. 2005;182:259–265. doi: 10.1016/j.atherosclerosis.2005.02.024. [DOI] [PubMed] [Google Scholar]
- 43.Coleman MD, Fernandes S, Khanderia L. A preliminary evaluation of a novel method to monitor a triple antioxidant combination (vitamins E, C and α-lipoic acid) in diabetic volunteers using in vitro methaemoglobin formation. Environ Toxicol Pharmacol. 2003;14:69–75. doi: 10.1016/S1382-6689(03)00027-9. [DOI] [PubMed] [Google Scholar]
- 44.Yamashita K, Takahiro K, Kamezaki F, Adachi T, Tasaki H. Decreased plasma extracellular superoxide dismutase level in patients with vasospastic angina. Atherosclerosis. 2007;191:147–152. doi: 10.1016/j.atherosclerosis.2006.03.008. [DOI] [PubMed] [Google Scholar]
- 45.Liao M, Liu Z, Bao J, Zhao Z, Hu J, Feng X, Feng R, Lu Q, Mei Z, Liu Y, et al. A proteomic study of the aortic media in human thoracic aortic dissection: implication for oxidative stress. J Thorac Cardiovasc Surg. 2008;136:65–72, 72.e1-3. doi: 10.1016/j.jtcvs.2007.11.017. [DOI] [PubMed] [Google Scholar]
- 46.Sekeroğlu MR, Sahin H, Dülger H, Algün E. The effect of dietary treatment on erythrocyte lipid peroxidation, superoxide dismutase, glutathione peroxidase, and serum lipid peroxidation in patients with type 2 diabetes mellitus. Clin Biochem. 2000;33:669–674. doi: 10.1016/s0009-9120(00)00190-9. [DOI] [PubMed] [Google Scholar]
- 47.Blankenberg S, Rupprecht HJ, Bickel C, Torzewski M, Hafner G, Tiret L, Smieja M, Cambien F, Meyer J, Lackner KJ. Glutathione peroxidase 1 activity and cardiovascular events in patients with coronary artery disease. N Engl J Med. 2003;349:1605–1613. doi: 10.1056/NEJMoa030535. [DOI] [PubMed] [Google Scholar]
- 48.Shih CK, Chang JH, Yang SH, Chou TW, Cheng HH. beta-Carotene and canthaxanthin alter the pro-oxidation and antioxidation balance in rats fed a high-cholesterol and high-fat diet. Br J Nutr. 2008;99:59–66. doi: 10.1017/S0007114507781497. [DOI] [PubMed] [Google Scholar]
- 49.Codoñer-Franch P, Bataller Alberola A, Domingo Camarasa JV, Escribano Moya MC, Valls Bellés V. Influence of dietary lipids on the erythrocyte antioxidant status of hypercholesterolaemic children. Eur J Pediatr. 2009;168:321–327. doi: 10.1007/s00431-008-0762-6. [DOI] [PubMed] [Google Scholar]
- 50.Fujimoto H, Taguchi J, Imai Y, Ayabe S, Hashimoto H, Kobayashi H, Ogasawara K, Aizawa T, Yamakado M, Nagai R, et al. Manganese superoxide dismutase polymorphism affects the oxidized low-density lipoprotein-induced apoptosis of macrophages and coronary artery disease. Eur Heart J. 2008;29:1267–1274. doi: 10.1093/eurheartj/ehm500. [DOI] [PubMed] [Google Scholar]