

Abstract
Background
Platelets are essential for maintaining haemostasis and play a key role in the pathogenesis of cardiovascular disease. Upon ligation of platelet receptors through subendothelial matrix proteins, intracellular reactive oxygen species (ROS) are generated, further amplifying the platelet activation response. Thrombin, a potent platelet activator, can signal through GPIbα and protease-activated receptor (PAR) 1 and PAR4 on human platelets, and recently has been implicated in the generation of ROS. While ROS are known to have key roles in intra-platelet signalling and subsequent platelet activation, the precise receptors and signalling pathways involved in thrombin-induced ROS generation have yet to be fully elucidated.
Objective
To investigate the relative contribution of platelet GPIbα and PARs to thrombin-induced reactive oxygen species (ROS) generation.
Methods and results
Highly specific antagonists targeting PAR1 and PAR4, and the GPIbα-cleaving enzyme, Naja kaouthia (Nk) protease, were used in quantitative flow cytometry assays of thrombin-induced ROS production. Antagonists of PAR4 but not PAR1, inhibited thrombin-derived ROS generation. Removal of the GPIbα ligand binding region attenuated PAR4-induced and completely inhibited thrombin-induced ROS formation. Similarly, PAR4 deficiency in mice abolished thrombin-induced ROS generation. Additionally, GPIbα and PAR4-dependent ROS formation were shown to be mediated through focal adhesion kinase (FAK) and NADPH oxidase 1 (NOX1) proteins.
Conclusions
Both GPIbα and PAR4 are required for thrombin-induced ROS formation, suggesting a novel functional cooperation between GPIbα and PAR4. Our study identifies a novel role for PAR4 in mediating thrombin-induced ROS production that was not shared by PAR1. This suggests an independent signalling pathway in platelet activation that may be targeted therapeutically.
Abbreviations: ROS, reactive oxygen species; Nk, Naja kaouthia protease; CRP, collagen-related peptide; WT, wild type; PAR, protease-activated receptor
Keywords: Platelets, Reactive oxygen species, Thrombin, Receptors GPIbα, PAR1 and PAR4
Graphical abstract
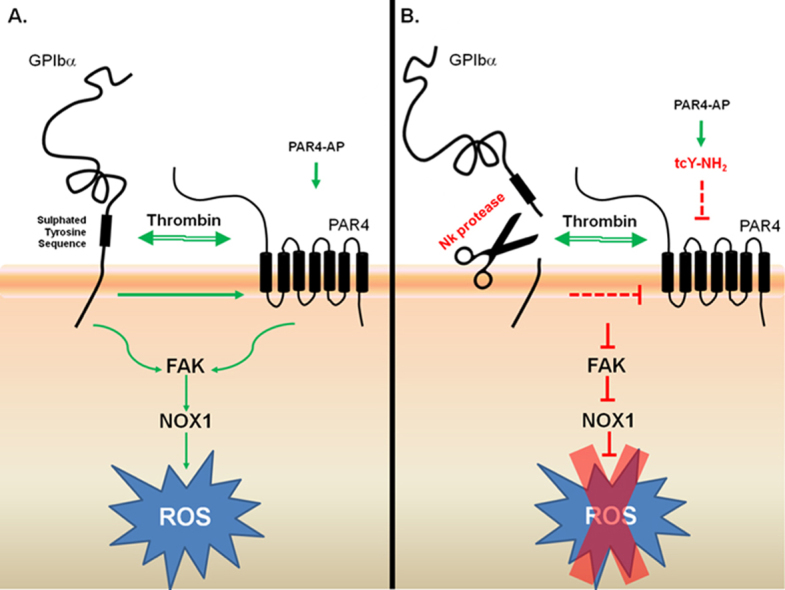
Both GPIbα and PAR4 are required for thrombin-induced ROS formation, suggesting a novel functional cooperation between GPIbα and PAR4. (A) Thrombin binds GPIbα, via the sulphated tyrosine sequence, and cleaves PAR4, activating the signalling molecules FAK and NOX1 and generating ROS. B. Nk protease cleaves GPIbα and inhibits thrombin-induced ROS generation, whilst hindering PAR4-induced ROS production. In the presence of the PAR4 antagonist, tcY-NH2, PAR4-AP-induced ROS production is abolished. GP: glycoprotein; PAR: protease activated receptor; FAK: focal adhesion kinase; NOX: NADPH oxidase; ROS: reactive oxygen species; Nk: Naja kouthia; AP: activating peptide.
Highlights
-
•
PAR4 plays an important role in platelet-derived ROS generation.
-
•
Thrombin-induced ROS generation in platelets require both GPIbα and PAR4.
-
•
Potential functional association between GPIbα and PAR4 receptors and mouse and human platelets.
-
•
GPIbα and PAR4-dependent ROS formation is mediated through FAK and NOX1 proteins.
1. Introduction
Platelets are essential for maintaining haemostasis and play a key role in the pathogenesis of cardiovascular disease [1]. Upon ligation of platelet receptors through subendothelial matrix proteins, intracellular reactive oxygen species (ROS) are generated and released, further amplifying the platelet activation response [2], [3], [4]. The main source of ROS is through NADPH oxidase (NOX) activation [5], and both NOX1 and NOX2 are expressed in human platelets [6]. We previously described a role for NOX1 in GPVI-dependent thrombus formation [7], suggesting platelet-derived ROS are a potential therapeutic target against cardiovascular disease. Recently, thrombin has been implicated in the generation of ROS [2], [3]. While ROS are known to have key roles in intra-platelet signalling [8] and subsequent platelet activation [4], the precise receptors and signalling pathways involved in thrombin-induced ROS generation have yet to be fully elucidated.
Thrombin, a potent platelet activator, can signal through GPIbα and protease activated receptor (PAR)1 and PAR4 on platelets [9]. PAR1 and PAR4 are known as the major thrombin receptors on platelets [10]; however, thrombin additionally binds GPIbα initiating the phosphorylation of signalling molecules required for integrin αIIbβ3 activation [11], [12]. This signalling is inhibited by treatment of platelets with the purified snake venom-derived metalloproteinases, mocarhagin and Naja kaouthia (Nk) protease [12], [13], which cleave GPIbα between amino acids Glu282-Asp283 and Tyr276-Asp277, respectively, to remove the GPIbα ectodomain including a sulphated tyrosine sequence that binds thrombin [14], [15]. In human platelets both PAR1 and PAR4 initiate platelet activation through G-protein signalling, and PAR1 contains a thrombin-binding sequence, which is absent in PAR4, allowing thrombin to bind more readily (20–70-fold faster rate of activation than PAR4) and at lower concentrations [16], [17]. Human platelets possess both PAR1 and PAR4 receptors in similar numbers (approximately 1000 copies/platelet) [18], [19], however mouse platelets do not express PAR1 and activation occurs through PAR4 [20].
Distinct differences have been reported between PAR1 and PAR4 signalling in human platelets [21]. Calcium responses elicited through PAR1 are short and rapid, but prolonged and sustained following PAR4 stimulation [17]. PAR4, but not PAR1, is regulated by P2Y12-stimulated feedback [22], and desensitisation of PAR1 in platelets is overcome by signalling through PAR4 [23]. Furthermore, stimulation of PAR4 results in more robust procoagulant activity in comparison to PAR1 [24].
Both thrombin and the PAR1-specific agonist thrombin receptor-activating peptide as well as GPVI and FcγRIIa agonists stimulate ROS production in platelets [2], [7], [25]; however GPIbα- and PAR4-specific agonists have not been evaluated as to whether they induce ROS formation. To investigate the relative contributions of thrombin receptors to ROS generation, platelets were treated with highly specific PAR1 and PAR4 antagonists or, Nk protease, and subsequently ROS production was quantified by flow cytometry. Here, we provide evidence for functional roles for GPIbα and PAR4 in thrombin-induced ROS generation, independent of PAR1, and a potential synergy between GPIbα and PAR4. Furthermore, ROS produced via GPIbα and PAR4 activation are mediated through focal adhesion kinase (FAK) and NOX1.
2. Materials and methods
2.1. Materials
Anti-GPIbα (AK2) and anti-VWF (5D2) murine monoclonal antibodies have been previously described [26], [27]; the irrelevant isotype IgG2 was from BD Pharmingen (Oxford, UK). Rat anti-mouse GPIbα (Xia.G5) IgG2B and rat IgG2B isotype (both FITC conjugated) were obtained from Emfret (Würzburg, Germany). Cross-linked collagen related peptide (CRP) was obtained from Prof. Richard Farndale (Department of Biochemistry, Cambridge University, UK). The protein kinase C activator, phorbol myristoyl acetate (PMA), and the calcium ionophore, A23187, were from Sigma Aldrich (St. Louis, MO, USA). Thrombin was from Calbiochem (UK). PAR1 (PAR1-AP, SFLLRN-NH2) and PAR4 (PAR4-AP, AYPGKF-NH2) agonists were from Abgent Europe (Oxfordshire, UK). PAR4 antagonist, tcY-NH2, PAR1 antagonist, SCH79797, and PF-573228 (hereafter referred to as PF-228) were from Tocris Bioscience (R&D Systems Europe, UK). ML171 (2-acetylphenothiazine) and BMS200261 were purchased from Sigma Aldrich (St. Louis, MO, USA). Nk protease (a GPIbα-specific cleavage enzyme from the venom of cobra Naja kaouthia) was purified as previously described [28].
2.2. Preparation of washed human and mouse platelets
Blood collection from drug-free healthy volunteers was approved by the Medical Research Ethics Committee of the Royal College of Surgeons in Ireland (RCSI), ID number REC269, and the Australian Centre for Blood Diseases (ACBD), Monash University, Melbourne, Australia. Written informed consent was obtained from all donors prior to phlebotomy. Venous blood was drawn using acid citrate dextrose (ACD-15% v/v) as anticoagulant. In brief, platelet-rich plasma (PRP) was obtained by centrifugation of whole blood at 190g for 20 min without braking. Platelets were isolated from PRP by centrifugation for 8 min at 650g, resuspended and washed (3x) in CGS buffer (123 mM NaCl, 33.3 mM glucose, 14.7 mM trisodium citrate, pH 7.0). Platelets were resuspended to the required count in Ca2+-free HEPES Tyrode's buffer (5 mM HEPES, 5.5 mM glucose, 138 mM NaCl, 12 mM NaHCO3, 0.49 mM MgCl2, 2.6 mM KCl, 0.36 mM NaH2PO4, pH 7.4). Platelets were supplemented with 1.8 mM CaCl2 prior to experimentation.
Studies using platelets from PAR4 knock out (KO) mice were performed at the ACBD (Monash University, Melbourne, Australia). Blood was drawn by approved personnel, with approval from the corresponding local Ethics Committee. Platelets were prepared as described previously [7] with one noted exception: mice were overdosed with pentobarbitone (60 mg/kg) prior to blood collection. The generation and characterisation of the PAR4 KO mice have been previously described [29]. Age- and sex-matched wild-type (WT) littermates were used as controls.
2.3. Platelet aggregation
Platelet aggregation was preformed in a PAP 4-C aggregometer (Chrono-Log, PA, USA) using washed platelets (2.5×108/mL) under constant stirring at 1100 rpm at 37 °C. For all inhibitory studies throughout this study, platelets were preincubated with vehicle control or antagonists for 10 min at 37 °C before the addition of agonist.
2.4. Measurement of intracellular ROS
The measurement of intracellular ROS was performed as previously described [7], [30]. Briefly, washed platelets (2.5×108/mL) in HEPES Tyrode's were incubated for 30 min at 37 °C with 10 µM dihydrodichlorofluorescein diacetate (H2DCFDA, Cambridge Bioscience, UK), pre-treated with antagonists then stimulated with 1 µg/mL of CRP for 10 min at 37 °C. Samples were diluted 10-fold in HEPES Tyrode's (0.1% BSA) containing 10 µM H2DCFDA and analysed immediately. All flow cytometric analysis was performed on a FACSCanto™ II (in Ireland) or FACSCalibur™ (in Australia) and analysed using FACSDiva™ software (Becton Dickinson, San Jose, CA, USA). ROS stimulation indexes were calculated from the fluorescent geo-mean values expressed as fold change relative to unstimulated platelet sample set as 1. In some instances, to show effects of inhibitors, positive controls were set as 100% ROS production. It must be noted that H2DCFDA has some limitations [31]. The dye is preferentially oxidised by a variety of one-electron species including hydroxyl radicals, peroxyl radicals and peroxynitrite anions, and therefore does not offer a direct measurement of H2O2. Additionally, a degree of self-propagating signal amplification is involved once DCF radical is generated. Nevertheless, as shown in our previous study, fluorescent signal generated by H2DCFDA-loaded activated platelets could be abrogated by a specific NADPH oxidase inhibitor, ML171. This reduction of H2DCFDA-oxidising species corroborated with an impact on platelet functional response, specifically thromboxane production and thrombus forming ability [7].
2.5. Nk protease treatment of platelets
Washed platelets (1×109/mL) were resuspended in HEPES Tyrode's buffer with Ca2+ and incubated with or without 10 µg/mL of Nk protease for 30 min at 37 °C. Platelets were washed with CGS and centrifuged at 650g for 8 min with no brake and resuspended in HEPES Tyrode's buffer with Ca2+ at 2.5×108/mL.
2.6. Measurement of GPIbα cleavage
Washed platelets (2.5×108/mL) treated with or without Nk protease (10 µg/mL) were incubated with the PE-labelled GPIbα-specific antibody (2 µg/mL AN51) or isotype control (2 µg/mL) for 15 min at 37 °C, then diluted 100-fold in HEPES Tyrode's and measured for intact GPIbα content on a FACSCanto™.
2.7. Cell lines
COS-7 cells and COS-7 cells stably expressing the VWF-A1 domain containing an R543W mutation (a gain-of-function mutation found in Type 2B von Willebrand's Disease), hereafter designated as R543W cells, have been described previously [32]. The cell lines were grown and maintained in M199 medium supplemented with 2 mM glutamine, 10% (v/v) FBS and 1 µg/mL puromycin at 37 °C and 5% CO2 prior to use.
2.8. Analysis of COS-7 cells expressing VWF-A1 domain
The level of VWF-A1/R543W on transfected COS-7 cells was assessed by flow cytometry. Cells were harvested using TBS containing 10 mM EDTA, pelleted, washed and resuspended (2.5×107 cells/mL) in HEPES Tyrode's buffer. Cells were pre-incubated with either 10 µg/mL of mouse monoclonal antibodies anti-VWF (5D2) or 20 µg/mL anti-GPIbα (AK2, negative control) for 1 h at room temperature, washed, incubated for a further 30 min with anti-mouse FITC secondary antibody, washed, and resuspended in 200 µL HEPES Tyrode's for analysis on a FACSCalibur™.
2.9. Data analysis
All statistical analysis was performed using GraphPad Prism 5®. Results are shown as mean±SEM. Statistical significance of difference between means was determined using ANOVA, with post-hoc analysis by the Bonferroni test. A value of *P≤0.05 was considered to be statistically significant.
3. Results
3.1. Detection of ROS following PAR1 and PAR4 activation
Platelet-derived ROS generation through thrombin receptors was assessed using PAR1 and PAR4 agonists and thrombin. Fig. 1 shows aggregation and ROS generation of washed human platelets stimulated with increasing concentrations of thrombin, PAR1-AP and PAR4-AP. While thrombin at 0.2, 1 and 2 U/mL elicited maximal platelet aggregation responses, ROS generation was only measurable at 1–2 U/mL (Fig. 1A) consistent with other reports [2], [3], [33], suggesting a predominant role for PAR4 and not PAR1 in thrombin-dependent ROS production.
Fig. 1.

ROS generation through the activation of PARs. Washed human platelets (preloaded with 10 µM H2DCFDA for ROS experiments only) were stimulated with (A) 0.2–2 U/mL thrombin, (B) 10–50 µM PAR1-AP or (C) 150–250 µM PAR4-AP, and monitored for aggregation by light transmission aggregometry or ROS production by flow cytometry (n=3). (D) Platelet-derived ROS generation stimulated with PAR1-AP (50 µM), PAR4-AP (250 µM) and thrombin (1 U/mL) from the same platelet samples. Data are mean±SEM (n=3), ***P≤0.001.
Previous studies have demonstrated ROS production through PAR1 activation in platelets [3], but a role for PAR4 in platelet generated ROS has not been previously described. Here, PAR1 activation induced maximum platelet aggregation at concentrations as low as 10 µM but required higher concentrations (20–50 µM) to generate ROS (Fig. 1B). In contrast, PAR4-AP elicited a significant ROS response with the same concentrations (150–250 µM) that were required for optimum platelet aggregation (Fig. 1C). Additionally, at optimal concentrations, thrombin elicited a much higher response in comparison to PAR1 activation alone (Fig. 1D), suggesting a more predominant role for PAR4 or GPIbα in thrombin-induced ROS production.
3.2. Thrombin-induced ROS production requires PAR4, but not PAR1
Although both PAR1 and PAR4 agonists could generate ROS, the relative contribution of these receptors in thrombin-induced ROS production remained unclear. Specific blockade of PAR1 by SCH79797 (1 µM; Fig. 2A) did not inhibit ROS production induced by 2 U/mL thrombin, but significantly potentiated the level of ROS detected. This was confirmed with a second PAR1 antagonist, BMS200261 (1 µM; Supplement Fig. 1). In contrast, pre-treatment with the PAR4 antagonist, tcY-NH2 (400 µM), significantly inhibited ROS production (Fig. 2A). These data suggest a novel role for PAR4 in thrombin-induced ROS generation in platelets independent of PAR1. The specificity of these antagonists was confirmed by their effect on the response to the relevant PAR agonist peptide (data not shown).
Fig. 2.

Thrombin-induced ROS generation requires PAR4, but not PAR1. (A) H2DCFDA labelled human platelets were pre-treated with vehicle control (0.1% DMSO), PAR1 antagonist SCH79797 (1 µM) or PAR4 antagonist tcY-NH2 (400 µM), stimulated with thrombin (2 U/mL), and monitored for ROS production by flow cytometry. Data are mean±SEM (n=3), *P≤0.05, ***P≤0.001. (B) Washed platelets from WT and PAR4 KO mice (2.5×108/mL) were preloaded with 10 µM H2DCFDA and left unstimulated, or stimulated with thrombin (2 U/mL) and ROS production was measured by flow cytometry. Data are mean±SEM (n=3), *P<0.05 H2DCFDA labelled platelets from WT and PAR4 KO mice were left unstimulated, or stimulated with thrombin (2 U/mL) and ROS production was measured by flow cytometry. Data are mean±SEM (n=3), ***P≤0.001.
To confirm the role of PAR4 in thrombin-induced ROS formation, ROS production was assessed in platelets isolated from wild-type (WT) and PAR4-deficient mice (PAR4 KO). While basal ROS production in platelets was similar between the two mouse strains, stimulation with 2 U/mL thrombin induced significant ROS generation in WT mouse platelets, ROS formation was at background levels in platelets from PAR4 KO mice (Fig. 2B). Platelets derived from PAR4 KO mice readily generated ROS in response to the GPVI-specific agonist, collagen-related peptide (CRP; 10 µg/mL) or the combination of the protein kinase C activator, phorbol myristoyl acetate (PMA; 10 µM) and the calcium ionophore, A23187 (20 µM) (Supplement Fig. 2).
3.3. Thrombin-induced ROS production is GPIbα-dependent
In addition to PAR1 and PAR4, thrombin also binds GPIbα [9], although a role for GPIbα in ROS production has not been previously explored. To investigate the contribution of GPIbα in thrombin-induced ROS generation, human platelets were treated with Nk protease, that has been shown to specifically cleave GPIbα between amino acid residues Tyr276-Asp277, thus removing the GPIbα thrombin binding site [28]. Nk protease treatment (10 µg/mL for 30 min) consistently removed >99% of intact GPIbα (Supplement Fig. 3). Nk protease-treated platelets failed to produce ROS in response to thrombin (Fig. 3A) but generated ROS normally when treated with CRP (Fig. 3B). This suggests that GPIbα is required, along with PAR4 for thrombin-induced ROS production in human platelets. When stimulated with PAR4-AP, platelets showed significantly less ROS generation in the absence of the GPIbα N-terminal region (Fig. 3C), however the response to PAR1 agonist remained normal (Fig. 3D). Consistently, aggregation was ablated in Nk-treated platelets when stimulated with PAR4-AP (Fig. 3E), implying a functional association between GPIbα and PAR4.
Fig. 3.

GPIbα is required for thrombin-induced ROS generation. Human platelets +/− Nk protease treatment, and labelled with 10 µM H2DCFDA,were stimulated with (A) 2 U/mL thrombin (B) 10 µg/mL CRP, (C) 250 µM PAR4-AP or (D) 50 µM PAR1-AP, and ROS production was measured by flow cytometry. Data are mean±SEM (n=4). (E) Washed human platelets +/− Nk protease treatment were stimulated with 250 µM PAR4-AP for 3 min and monitored for platelet aggregation by light transmission aggregometry using a PAP4 platelet aggregometer. Data are mean±SEM (n=4). (F) H2DCFDA labelled mouse platelets +/− Nk protease treatment were stimulated with thrombin (2 U/mL) and monitored for ROS production by flow cytometry. Data are mean±SEM (n=3). *P≤0.05, **P≤0.01, ***P≤0.001. ns=statistical nonsignificance.
A potential synergy between PAR4 and GPIbα in thrombin-induced ROS generation was confirmed using mouse platelets, which express PAR3 and PAR4 but not PAR1. When murine platelets were treated with Nk protease to prevent engagement of GPIbα by thrombin, ROS generation was significantly inhibited (Fig. 3F), indicating that GPIbα may act as an accessory receptor and aid the engagement of PAR4 by thrombin both in murine and human platelets.
3.4. ROS formation occurs through ligation and activation of GPIbα
To further understand the role of GPIbα in ROS formation, we used COS-7 cells stably transfected for cell surface expression of a recombinant von Willebrand Factor (VWF)-A1 domain containing a R543W gain-of-function point mutation as a GPIbα specific agonist (R543W cells). The A1 domain of VWF on R543W cells is in a constitutively open and active conformation on the cell membrane as a GPI-linked construct [32]. Surface expression was confirmed using the VWF-specific antibody, 5D2, with no binding to non-transfected (WT) cells (Supplement Fig. 4). R543W cells, but not WT cells, induced ROS in human platelets (Fig. 4A), which was inhibited by 5D2 (10 µg/mL), which blocks the GPIb binding site within VWF and the GPIbα-specific antibody AK2 (20 µg/mL), which blocks the VWF binding site on GPIb (Fig. 4B). These data demonstrate that engagement through GPIbα alone is sufficient to initiate platelet ROS formation.
Fig. 4.

ROS formation occurs through ligation and activation of GPIbα. (A) H2DCFDA labelled human platelets were stimulated with WT or R543W COS-7 cells (1:50; cell:platelet ratio) and monitored for ROS production by flow cytometry. Data are mean±SEM (n=3). (B) H2DCFDA labelled human platelets pre-treated with either the GPIbα-function blocking antibody, AK2 (20 µg/mL) or the VWF-function blocking antibody 5D2 (10 µg/mL) were stimulated with R543W cells (1:50; cell:platelet ratio) and monitored for ROS production. Data are mean±SEM (n=3). *P≤0.05, **P≤0.01, ***P≤0.001.
3.5. NOX1 is the main source of ROS downstream of GPIbα and PAR activation
We have previously shown NOX1 is the main NADPH oxidase isoform involved in CRP-dependent ROS generation through the platelet collagen receptor, GPVI [7]. Here, we explored the role of NOX1 in ROS formation induced by thrombin using the NOX1 inhibitor, ML171 (5 µM). ML171 significantly inhibited ROS generation when platelets were activated with thrombin, R543W cells or PAR4-AP (Fig. 5A). These results suggest that platelet-derived ROS generation is mainly NOX1-dependent regardless of the agonist employed.
Fig. 5.

ROS generation via GPIbα and PAR activation is mediated through FAK and NOX1. H2DCFDA labelled human platelets were pre-treated with the (A) NOX1 inhibitor, 5 µM ML171, or (B) FAK inhibitor, 1 µM PF-228, for 10 min, then stimulated with thrombin (1 U/mL), R543W cells (1:50; cell:platelet ratio) or PAR4-AP (250 µM) for 2 min and monitored for ROS production by flow cytometry. Data are mean±SEM (n=3–6). *P≤0.05, **P≤0.01, ***P≤0.001.
3.6. FAK is a prerequisite of GPIbα and PAR4-dependent ROS generation
We recently demonstrated that FAK is important for downstream signalling leading to ROS formation when platelets are stimulated with CRP [30]. The FAK inhibitor, PF-228 (1 µM), also inhibited ROS formation when platelets were stimulated with thrombin, R543W cells or PAR4-AP (Fig. 5B), suggesting that FAK also plays a functional role downstream of both GPIbα and PAR4 following thrombin-induced ROS production.
4. Discussion
Human PAR4 is a low affinity thrombin receptor initiating downstream signalling events following primary activation of platelets through PAR1 [34]. In this study, we identified a novel role for PAR4 in mediating thrombin-induced ROS production that was not shared by PAR1. Additionally, the potential for involvement of GPIbα in platelet-derived ROS generation was confirmed with a GPIbα-specific agonist. The combined data suggest GPIbα is required to facilitate the interaction of thrombin with PAR4 on the platelet membrane and implies a functional association between these two receptors in human and mouse platelets.
High concentrations of thrombin are typically required for PAR4 activation. In this study thrombin concentrations of 1 U/mL or greater, stimulated ROS formation consistent with an important role for PAR4. In line with previous studies [3], higher concentrations of PAR1-AP were required to induce ROS formation than were required to induce maximal platelet aggregation. In comparison, PAR4-AP concentrations required for maximum aggregation response induced significant ROS generation. While, a recent study associated platelet activation via PAR4 with the redox signalling molecule 12-LOX [35], to the best of our knowledge the present results are the first report of a role for PAR4 in platelet-derived ROS generation. Pharmacological antagonists acting at PAR1 (SCH79797) or PAR4 (tcY-NH2) confirmed a novel role for PAR4, but not PAR1, in thrombin-mediated ROS generation. Interestingly, SCH79797 actually potentiated thrombin-induced ROS generation. This suggests that in the absence of functional PAR1, ROS generation may be increased due to more thrombin being available to bind PAR4, and is consistent with PAR1 not being involved in thrombin-induced ROS generation.
Previous studies have shown a physical association of the GPIb-IX-V complex with other important signalling surface receptor on platelets, GPVI and FcγRIIa [32], [36], [37], [38]. While GPVI- and FcγRIIa-dependent signalling results in ROS formation [33], [39], the role of GPIbα itself in platelet ROS generation is not clear. In this study, when the N-terminal domain of GPIbα was removed by Nk protease, the thrombin-induced ROS response was completely abolished and the platelets also responded poorly to PAR4-AP. These results suggest a possible functional relationship between GPIbα and PAR4 on the platelet surface. PAR4-AP-induced platelet aggregation was also reduced following removal of the GPIbα thrombin-binding domain, but not abolished, suggesting GPIbα is required for optimal PAR4 activation, further supporting a potential link between these two receptors. Thrombin-induced ROS generation in WT mouse platelets was also decreased by Nk protease treatment, supporting a functional cooperation between GPIbα and PAR4 in mouse platelets. In contrast to human platelets, PAR3 is also known to enhance PAR4 activation [40]. To date, there is no reported connection between PAR4 and GPIbα. There is evidence that GPIbα acts as a co-factor for PAR1 but not PAR4 activation [41], and can amplify PAR1 responses [42]. A possible explanation for this discrepancy is that the GPIbα association with PAR4 is masked by that with PAR1 with respect to platelet aggregation but evident with ROS generation at higher thrombin concentrations, which do not involve PAR1. The capacity of GPIbα to generate ROS was confirmed using the GPIbα specific agonist, R543W cells.
Downstream of thrombin binding and GPIbα/PAR4 activation, ROS generation was found to require both FAK and NOX1. These data suggest that agonist-dependent ROS production in platelets is mainly derived through NOX1 activation, consistent with our previous report for GPVI-dependent ROS formation [7]. In addition, these data also confirm that common signalling proteins such as FAK are involved in both GPIbα- and PAR4-dependent ROS production. Whether or not PAR4 directly activates NOX1 and/or like GPIb-IX and GPVI directly interacts with TRAF4 [25], was not determined in this study. PAR4 could also potentially utilise GPIb-associated NOX. The detailed mechanisms for PAR4-dependent activation of downstream NOX1 and FAK remain to be elucidated.
It is clear from this study that both GPIbα and PAR4 play a prominent role in thrombin-induced platelet ROS generation. In the functional absence of either receptor on human platelets, thrombin cannot elicit ROS production, implying a mechanism that involves both receptors. Anti-PAR1 drugs, such as Vorapaxar, are associated with severe bleeding risks [43], [44]. Shifting focus to the GPIbα-PAR4 axis could provide an alternate anti-platelet therapeutic strategy [45].
Contribution of authors
P.M. and M.C.B. designed the study. N.C. performed experiments and acquired data. N.C., P.M. and M.C.B. interpreted the results. P.M. supervised data analysis. N.C. and P.M. composed the majority of the manuscript. J.R.H. provided PAR4 KO mice and assisted in experimental design. J.F.A. performed experiments and acquired data. E.E.G. and R.K.A. provided essential reagents, supervised data analysis and interpreted the results. P.M. prepared the structure of the manuscript and the final figures. All authors read the manuscript.
Acknowledgements
We would like to acknowledge Science Foundation Ireland (Grant no. 09/IN.1/B2601), Biomedical Diagnostics Institute, Royal College of Surgeons in Ireland, Australian Centre for Blood Diseases, Monash University and Curtin University Faculty of Health Sciences and Curtin Health Innovation Research Institute for their financial, infrastructure and technical support.
Footnotes
Supplementary data associated with this article can be found in the online version at doi:10.1016/j.redox.2015.10.009.
Appendix A. Supplementary material
Supplementary material
References
- 1.Berndt M.C., Metharom P., Andrews R.K. Primary haemostasis: newer insights. Haemophilia. 2014;20(Suppl. 4):15–22. doi: 10.1111/hae.12427. [DOI] [PubMed] [Google Scholar]
- 2.Bakdash N., Williams M.S. Spatially distinct production of reactive oxygen species regulates platelet activation. Free. Radic. Biol. Med. 2008;45(2):158–166. doi: 10.1016/j.freeradbiomed.2008.03.021. [DOI] [PubMed] [Google Scholar]
- 3.Begonja A.J. Platelet NAD(P)H-oxidase-generated ROS production regulates alphaIIbbeta3-integrin activation independent of the NO/cGMP pathway. Blood. 2005;106(8):2757–2760. doi: 10.1182/blood-2005-03-1047. [DOI] [PubMed] [Google Scholar]
- 4.Krotz F. NAD(P)H oxidase-dependent platelet superoxide anion release increases platelet recruitment. Blood. 2002;100(3):917–924. doi: 10.1182/blood.v100.3.917. [DOI] [PubMed] [Google Scholar]
- 5.Seno T. Involvement of NADH/NADPH oxidase in human platelet ROS production. Thromb. Res. 2001;103(5):399–409. doi: 10.1016/s0049-3848(01)00341-3. [DOI] [PubMed] [Google Scholar]
- 6.Vara D., Campanella M., Pula G. The novel NOX inhibitor 2-acetylphenothiazine impairs collagen-dependent thrombus formation in a GPVI-dependent manner. Br. J. Pharmacol. 2013;168(1):212–224. doi: 10.1111/j.1476-5381.2012.02130.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Walsh T.G. The role of Nox1 and Nox2 in GPVI-dependent platelet activation and thrombus formation. Redox Biol. 2014;2:178–186. doi: 10.1016/j.redox.2013.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Finkel T. Redox-dependent signal transduction. FEBS Lett. 2000;476(1–2):52–54. doi: 10.1016/s0014-5793(00)01669-0. [DOI] [PubMed] [Google Scholar]
- 9.De Candia E. Mechanisms of platelet activation by thrombin: a short history. Thromb. Res. 2012;129(3):250–256. doi: 10.1016/j.thromres.2011.11.001. [DOI] [PubMed] [Google Scholar]
- 10.Coughlin S.R. Thrombin signalling and protease-activated receptors. Nature. 2000;407(6801):258–264. doi: 10.1038/35025229. [DOI] [PubMed] [Google Scholar]
- 11.Adam F., Guillin M.C., Jandrot-Perrus M. Glycoprotein Ib-mediated platelet activation. A signalling pathway triggered by thrombin. Eur. J. Biochem. 2003;270(14):2959–2970. doi: 10.1046/j.1432-1033.2003.03670.x. [DOI] [PubMed] [Google Scholar]
- 12.Dubois C. Thrombin binding to GPIbalpha induces integrin alphaIIbbeta3 dependent platelet adhesion to fibrin in ex vivo flowing whole blood. Thromb. Haemost. 2004;91(2):233–237. doi: 10.1160/TH03-03-0126. [DOI] [PubMed] [Google Scholar]
- 13.Andrews R.K. Structure-activity relationships of snake toxins targeting platelet receptors, glycoprotein Ib-IX-V and glycoprotein VI. Curr. Med. Chem. Cardiovasc. Hematol. Agents. 2003;1(2):143–149. doi: 10.2174/1568016033477559. [DOI] [PubMed] [Google Scholar]
- 14.Lova P. Thrombin induces platelet activation in the absence of functional protease activated receptors 1 and 4 and glycoprotein Ib-IX-V. Cell Signal. 2010;22(11):1681–1687. doi: 10.1016/j.cellsig.2010.06.008. [DOI] [PubMed] [Google Scholar]
- 15.Ravanat C. A central role of GPIb-IX in the procoagulant function of platelets that is independent of the 45-kDa GPIbalpha N-terminal extracellular domain. Blood. 2010;116(7):1157–1164. doi: 10.1182/blood-2010-01-266080. [DOI] [PubMed] [Google Scholar]
- 16.Andersen H. Protease-activated receptor 1 is the primary mediator of thrombin-stimulated platelet procoagulant activity. Proc. Natl. Acad. Sci. USA. 1999;96(20):11189–11193. doi: 10.1073/pnas.96.20.11189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Covic L., Gresser A.L., Kuliopulos A. Biphasic kinetics of activation and signaling for PAR1 and PAR4 thrombin receptors in platelets. Biochemistry. 2000;39(18):5458–5467. doi: 10.1021/bi9927078. [DOI] [PubMed] [Google Scholar]
- 18.Dupont A. An intronic polymorphism in the PAR-1 gene is associated with platelet receptor density and the response to SFLLRN. Blood. 2003;101(5):1833–1840. doi: 10.1182/blood-2002-07-2149. [DOI] [PubMed] [Google Scholar]
- 19.Burkhart J.M. The first comprehensive and quantitative analysis of human platelet protein composition allows the comparative analysis of structural and functional pathways. Blood. 2012;120(15):e73–e82. doi: 10.1182/blood-2012-04-416594. [DOI] [PubMed] [Google Scholar]
- 20.Kahn M.L. A dual thrombin receptor system for platelet activation. Nature. 1998;394(6694):690–694. doi: 10.1038/29325. [DOI] [PubMed] [Google Scholar]
- 21.Sidhu T.S., French S.L., Hamilton J.R. Differential signaling by protease-activated receptors: implications for therapeutic targeting. Int. J. Mol. Sci. 2014;15(4):6169–6183. doi: 10.3390/ijms15046169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Holinstat M. PAR4, but not PAR1, signals human platelet aggregation via Ca2+ mobilization and synergistic P2Y12 receptor activation. J. Biol. Chem. 2006;281(36):26665–26674. doi: 10.1074/jbc.M602174200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Falker K. Protease-activated receptor 1 (PAR1) signalling desensitization is counteracted via PAR4 signalling in human platelets. Biochem. J. 2011;436(2):469–480. doi: 10.1042/BJ20101360. [DOI] [PubMed] [Google Scholar]
- 24.Duvernay M. Protease-activated receptor (PAR) 1 and PAR4 differentially regulate factor V expression from human platelets. Mol. Pharmacol. 2013;83(4):781–792. doi: 10.1124/mol.112.083477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Arthur J.F. TNF receptor-associated factor 4 (TRAF4) is a novel binding partner of glycoprotein Ib and glycoprotein VI in human platelets. J. Thromb. Haemost. 2011;9(1):163–172. doi: 10.1111/j.1538-7836.2010.04091.x. [DOI] [PubMed] [Google Scholar]
- 26.Chong B.H. Heparin-induced thrombocytopenia: mechanism of interaction of the heparin-dependent antibody with platelets. Br. J. Haematol. 1989;73(2):235–240. doi: 10.1111/j.1365-2141.1989.tb00258.x. [DOI] [PubMed] [Google Scholar]
- 27.De Luca M. Structure and function of the von Willebrand factor A1 domain: analysis with monoclonal antibodies reveals distinct binding sites involved in recognition of the platelet membrane glycoprotein Ib-IX-V complex and ristocetin-dependent activation. Blood. 2000;95(1):164–172. [PubMed] [Google Scholar]
- 28.Wijeyewickrema L.C. Fractionation of snake venom metalloproteinases by metal ion affinity: a purified cobra metalloproteinase, Nk, from Naja kaouthia binds Ni2+-agarose. Toxicon. 2007;50(8):1064–1072. doi: 10.1016/j.toxicon.2007.07.006. [DOI] [PubMed] [Google Scholar]
- 29.Hamilton J.R., Cornelissen I., Coughlin S.R. Impaired hemostasis and protection against thrombosis in protease-activated receptor 4-deficient mice is due to lack of thrombin signaling in platelets. J. Thromb. Haemost. 2004;2(8):1429–1435. doi: 10.1111/j.1538-7836.2004.00783.x. [DOI] [PubMed] [Google Scholar]
- 30.Carrim N. Role of focal adhesion tyrosine kinases in GPVI-dependent platelet activation and reactive oxygen species formation. Plos One. 2014;9(11):e113679. doi: 10.1371/journal.pone.0113679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kalyanaraman B. Measuring reactive oxygen and nitrogen species with fluorescent probes: challenges and limitations. Free. Radic. Biol. Med. 2012;52(1):1–6. doi: 10.1016/j.freeradbiomed.2011.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gardiner E.E. GPIbalpha-selective activation of platelets induces platelet signaling events comparable to GPVI activation events. Platelets. 2010;21(4):244–252. doi: 10.3109/09537101003695339. [DOI] [PubMed] [Google Scholar]
- 33.Arthur J.F. ITAM receptor-mediated generation of reactive oxygen species in human platelets occurs via Syk-dependent and Syk-independent pathways. J. Thromb. Haemost. 2012;10(6):1133–1141. doi: 10.1111/j.1538-7836.2012.04734.x. [DOI] [PubMed] [Google Scholar]
- 34.Kahn M.L. Protease-activated receptors 1 and 4 mediate activation of human platelets by thrombin. J. Clin. Invest. 1999;103(6):879–887. doi: 10.1172/JCI6042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yeung J. 12-Lipoxygenase activity plays an important role in PAR4 and GPVI-mediated platelet reactivity. Thromb. Haemost. 2013;110(3):569–581. doi: 10.1160/TH13-01-0014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Arthur J.F. Glycoprotein VI is associated with GPIb-IX-V on the membrane of resting and activated platelets. Thromb. Haemost. 2005;93(4):716–723. doi: 10.1160/TH04-09-0584. [DOI] [PubMed] [Google Scholar]
- 37.Baker J. GPIb potentiates GPVI-induced responses in human platelets. Platelets. 2004;15(4):207–214. doi: 10.1080/09537100410001701010. [DOI] [PubMed] [Google Scholar]
- 38.Sullam P.M. Physical proximity and functional interplay of the glycoprotein Ib-IX-V complex and the Fc receptor FcγRIIA on the platelet plasma membrane. J. Biol. Chem. 1998;273(9):5331–5336. doi: 10.1074/jbc.273.9.5331. [DOI] [PubMed] [Google Scholar]
- 39.Arthur J.F. Platelet receptor redox regulation. Platelets. 2008;19(1):1–8. doi: 10.1080/09537100701817224. [DOI] [PubMed] [Google Scholar]
- 40.Nakanishi-Matsui M. PAR3 is a cofactor for PAR4 activation by thrombin. Nature. 2000;404(6778):609–613. doi: 10.1038/35007085. [DOI] [PubMed] [Google Scholar]
- 41.De Candia E. Binding of thrombin to glycoprotein Ib accelerates the hydrolysis of Par-1 on intact platelets. J. Biol. Chem. 2001;276(7):4692–4698. doi: 10.1074/jbc.M008160200. [DOI] [PubMed] [Google Scholar]
- 42.Adam F. Thrombin-induced platelet PAR4 activation: role of glycoprotein Ib and ADP. J. Thromb. Haemost. 2003;1(4):798–804. doi: 10.1046/j.1538-7836.2003.00138.x. [DOI] [PubMed] [Google Scholar]
- 43.Tricoci P. Thrombin-receptor antagonist vorapaxar in acute coronary syndromes. N Engl. J. Med. 2012;366(1):20–33. doi: 10.1056/NEJMoa1109719. [DOI] [PubMed] [Google Scholar]
- 44.Morrow D.A. Efficacy and safety of vorapaxar in patients with prior ischemic stroke. Stroke. 2013;44(3):691–698. doi: 10.1161/STROKEAHA.111.000433. [DOI] [PubMed] [Google Scholar]
- 45.Metharom P. Current state and novel approaches of antiplatelet therapy. Arter. Thromb. Vasc. Biol. 2015;35(6):1327–1338. doi: 10.1161/ATVBAHA.114.303413. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material