

Abstract
Mycobacterium tuberculosis, the agent of human tuberculosis has developed different virulence mechanisms and virulence-associated tools during its evolution to survive and multiply inside the host. Based on previous reports and by analogy with other bacteria, phospholipases C (PLC) of M. tuberculosis were thought to be among these tools. To get deeper insights into the function of PLCs, we investigated their putative involvement in the intracellular lifestyle of M. tuberculosis, with emphasis on phagosomal rupture and virulence, thereby re-visiting a research theme of longstanding interest. Through the construction and use of an M. tuberculosis H37Rv PLC-null mutant (ΔPLC) and control strains, we found that PLCs of M. tuberculosis were not required for induction of phagosomal rupture and only showed marginal, if any, impact on virulence of M. tuberculosis in the cellular and mouse infection models used in this study. In contrast, we found that PLC-encoding genes were strongly upregulated under phosphate starvation and that PLC-proficient M. tuberculosis strains survived better than ΔPLC mutants under conditions where phosphatidylcholine served as sole phosphate source, opening new perspectives for studies on the role of PLCs in the lifecycle of M. tuberculosis.
Most of the ~130 mycobacterial species1 are harmless to humans, whereas a few pose major threats to human health and life. Among the latter is Mycobacterium tuberculosis, the etiological agent of tuberculosis, which transmits efficiently among humans and globally accounts for 9 million new tuberculosis cases and 1.5 million deaths each year2. Many factors have been reported that contribute to the outstanding efficacy of M. tuberculosis to infect its host and circumvent eradication by the immune system3,4. Genome-based studies and advanced gene knock-out techniques have been instrumental for the identification of numerous virulence factors of M. tuberculosis that seem to be important for its lifestyle as key pathogen5,6. Comparative sequence analyses were also important for finding polymorphisms useful for molecular epidemiology7 and evolutionary studies8. Among the different approaches, genomic comparison of environmental, saprophytic mycobacteria with clinically relevant mycobacteria may provide important information.
One of the potential differences emerging from the comparison of non-pathogenic with pathogenic mycobacterial species is the presence of genes encoding phospholipase C (PLC) in the latter. For example, Mycobacterium abscessus, which represents an exceptional, emerging pathogen within the large group of otherwise mostly harmless fast-growing mycobacteria9,10,11,12, encodes a PLC involved in the intracellular survival of M. abscessus in amoebae13. Moreover, PLC-encoding genes are also present in several species of the slow-growing mycobacteria, which constitute a subgroup in the 16 S rDNA-based mycobacterial phylogeny14 and harbour the great majority of mycobacterial pathogens. Only few studies have addressed the impact of PLC on virulence of these pathogens. The most well known of these studies targeted PLCs of a clinical M. tuberculosis strain (MT103) and reported that PLC-knock-out mutants of this strain were attenuated at later stages of infection15. Together with reported cytotoxic effects of PLC16, these results were taken up by numerous review articles on mycobacterial pathogenicity5,17,18,19, leading to the widespread supposition that PLCs were important virulence factors of M. tuberculosis.
In the present study, we thus sought to gain deeper insights into the molecular mechanisms by which PLCs might contribute to virulence of M. tuberculosis. PLCs from selected species of other bacterial genera, such as Listeria monocytogenes or Clostridium perfringens, are known to play a significant role in helping the bacteria to escape from phagosomal containment inside host cells by acting together with pore-forming proteins such as listeriolysin or perfringolysin20,21,22.
M. tuberculosis was reported to produce membrane-damaging proteins associated with the ESX-1 secretion system, which are required for induction of phagosomal rupture and bacterial access to the cytosolic compartment of infected phagocytic cells4, However, it remains unknown if other bacterial factors, as for example PLCs, might also contribute to the M. tuberculosis-mediated disruption of the phagosomal membrane.
The first main objective of our study was thus to investigate whether the biological activities of ESX-1 and mycobacterial PLCs were linked. For this purpose, we constructed a PLC-deletion mutant in the M. tuberculosis H37Rv genetic background, and subjected it to dedicated cell-biological analyses in comparison with the wild-type (WT) M. tuberculosis H37Rv strain. To evaluate whether the PLC-deletion mutants had the ability to induce phagosomal rupture in host-macrophages, we used a recently developed fluorescence resonance energy transfer (FRET)-based method23,24. Results from the phagosomal rupture assay together with virulence tests in cellular and small animal infection models allowed us to revisit the role of PLCs of M. tuberculosis in the infection process, which to our surprise was found to be only marginal. These results open new perspectives for future research to elucidate the biological role of PLCs in M. tuberculosis and related slow-growing mycobacteria.
Results
Genome analysis and deletion of the plcABC operon in an M. tuberculosis H37Rv genetic background
Analysis of M. tuberculosis genome data from public databases shows that most M. tuberculosis strains harbour four PLC encoding genes. These genes, named plcA, plcB, plcC and plcD are located at two different genomic loci in M. tuberculosis, with plcA-B-C organised as an operon (rv2351c-rv2350c-rv2349c) at genome coordinates 2632–2627 kb (reverse strand) of strain H37Rv, and plcD, represented as a single gene (rv1755c), located about 640 kb upstream of plcA-C25. It is also known that PLC encoding genes are preferred integration sites (or hotspots) for the IS6110 insertion element, which may lead to the presence of two insertion elements in close proximity, favouring homologous recombination between the adjacent IS6110 elements and deletion of the intervening sequences26,27,28. The widely used reference strain M. tuberculosis H37Rv shows such IS6110-mediated truncation of the plcD gene26. However, despite plcD inactivation, M. tuberculosis H37Rv retains a fully virulent phenotype in mice29. We thus chose the H37Rv strain to construct a null PLC mutant, taking in consideration that truncation of plcD facilitated the construction of the PLC complete knock-out strain, as only the PlcA-B-C operon had to be deleted.
The M. tuberculosis H37Rv PLC null mutant (H37RvΔPLC) was constructed by using a previously described recombineering-based approach30. The different construction steps included the generation by 3-step-PCR31 of a linear DNA fragment containing an apramycin resistance cassette embedded in the flanking regions of the plcABC cluster (Fig. 1A), which was genetically transformed into the H37Rv strain. Selection of an appropriate clone that showed replacement of the plcABC cluster with the apramycin cassette was assessed by PCR and then confirmed by Southern blotting analysis (Fig. 1B,C). In addition, a H37RvΔPLC::plcABC complemented strain was obtained by integrating the plcABC gene cluster into the genome of H37RvΔPLC using the plasmid pPlcABC. This pYUB412-based vector32 contains the plcABC operon expressed under the control of its natural promoter.
Figure 1. Construction of M. tuberculosis H37RvplcABC KO (H37RvΔPLC).

(A) Schematic representation of genomic organization of plc genes in M. tuberculosis H37Rv wild type and H37RvΔPLC strains ; (B) AvrII restriction fragment profiles of M. tuberculosis WT and KO strains separated by agarose gel electrophoresis; (C) Pattern obtained from genomic DNAs digested with AvrII and hybridized with a probe specific for the plcC downstream region; Lanes: 1 (second lane from left), negative control (pYUB412 vector); 2, positive control pYUB412::plcABC; 3 and 4, M. tuberculosis H37Rv WT, 5 and 6, M. tuberculosis H37RvΔplcABC; 7, M, Smart Ladder (Eurobio).
As controls for selected experiments, we also included the previously described Myc2509ΔPLC mutant strain15, here referred to as MT103ΔPLC, and the isogenic MT103 parental M. tuberculosis strain.
Evaluation of phospholipase C activity in mutant and WT M. tuberculosis strains
In a first step, we used a spectrophotometric assay to determine the PLC activity of whole-cell extracts from WT M. tuberculosis strains and the two PLC-deletion mutants. This assay is based on the detection of the hydrolysis of colourless p-nitrophenylphosphorylcholine (p-NPPC) to p-nitrophenol, which absorbs light at 410 nm and is yellow. As shown in Fig. 2, the PLC activity was decreased in H37RvΔPLC compared to the corresponding WT strain. However, a lower PLC activity was detected in M. tuberculosis H37Rv strain relative to the MT103 strain, which might be linked to the truncation of the fourth plc gene (plcD) in M. tuberculosis H37Rv. Complementation of M. tuberculosis H37RvΔPLC with plasmid pPlcABC restored PLC activity to the level of the H37Rv WT M. tuberculosis strain. As expected, the plcABC-unrelated M. tuberculosis H37Rv mutant ∆ESX1, which is lacking a functional ESX-1 secretion system due to the deletion of the region of difference RD123,24,33, showed a phospholipase C activity very similar to the H37Rv WT strain (Fig. 2A,B). Taken together, these results confirmed the loss of PCL activity in the H37RvΔPLC mutant.
Figure 2. Phospholipase C enzymatic assay.
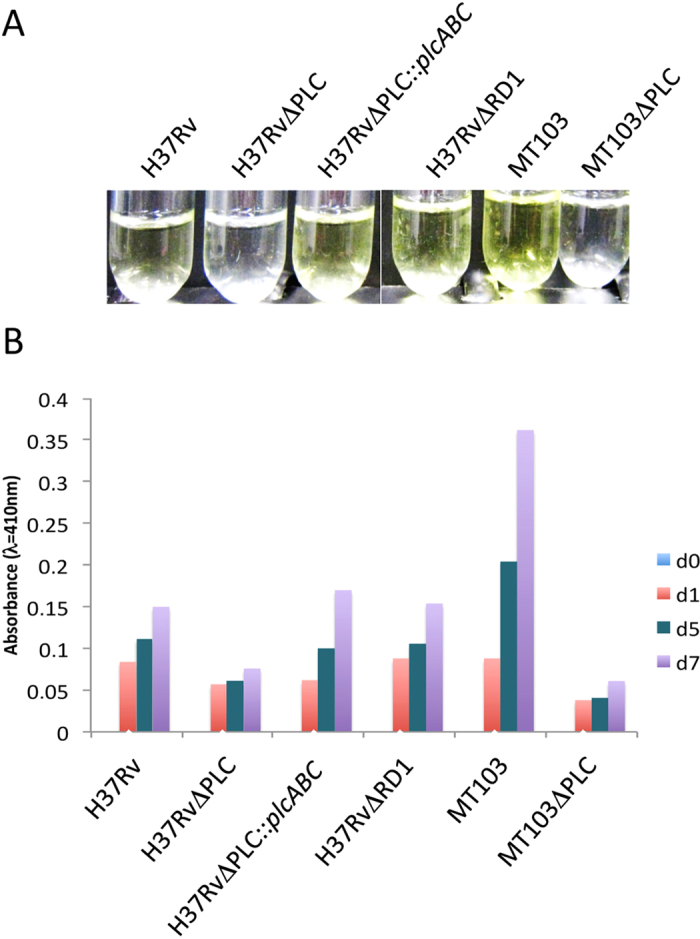
This assay is based on the detection of the hydrolysis of p-nitrophenylphosphorylcholine (p-NPPC) to p-nitrophenol. While the substrate, p-NPPC, is colourless, the product p-nitrophenol due to its ability to absorb light at 410 nm, is yellow. About 500 μg of total protein were used in the assay and measurements were performed in triplicates. (A) Crude extracts of 4 day-old cultures from different M. tuberculosis strains were incubated with 5 mmol.l−1 of p-nitrophenol phosphorylcholine; (B) Measurement of phospholipase activity of different M. tuberculosis strains over 3 timepoints. Measurements were performed in duplicates.
Phospholipase C is not involved in M. tuberculosis-induced phagosomal rupture
According to host-pathogen interaction data reported from a range of bacterial pathogens, phospholipase C activity is often required for egress of bacteria from phagosomal containment and cytosolic access20,21,22. We thus investigated the ability of PLC mutants H37RvΔPLC and MT103ΔPLC and WT strains to access the cytosol during infection of THP-1 human macrophage-like cells, by using a recent flow-cytometric phagosomal rupture screening method23. Briefly, this sensitive assay relies on the change in the emission spectrum of the cephalosporin-like FRET substrate CCF-4 upon cleavage by β-lactamases34,35, which serves as a readout for the detection of contact between β-lactamase-producing M. tuberculosis and the FRET substrate in different environments, including the host cytosol. As CCF-4 cannot enter an intact vacuole, the assay assesses whether cytosolic contact of M. tuberculosis occurs in the host cell during infection. Differentiated THP-1 cells were infected with the various M. tuberculosis strains at an MOI of 1:2, and the CCF-4 emission spectrum of cells was monitored over a three-day period. Both the WT and the ΔPLC-deletion M. tuberculosis strains were able to induce a switch in the emission spectrum from ~535 nm to ~450 nm, indicating that they were gaining access to the cytosol of the infected THP-1 cells (Fig. 3). In contrast, the attenuated ΔESX-1 (ΔRD1) M. tuberculosis strain, which is impaired in inducing phagosomal rupture in host cells23,24 and was included in the analysis as a negative control, was unable to induce a FRET inhibition, thereby validating the assay (Fig. 3, Supplementary Figure 1). From these results we concluded that PLC of M. tuberculosis was not required for inducing phagosomal rupture in THP-1 cells, neither in the H37Rv-, nor in the MT103 genetic backgrounds.
Figure 3. Phagosomal rupture by M. tuberculosis mutants.

Capacity of different M. tuberculosis strains and mutants to induce phagosomal rupture inTHP-1 cells, monitored by CCF-4 staining and flow cytometric analysis. Non infected (gray), infected (MOI = 1:10) with different strains of M. tuberculosis (purple) using a recently developed approach23. Results shown are representative of 2 independent experiments. The shift towards blue emission (447 nm) of CCF-4 is due to the inhibition of FRET and is proportional to the mycobacteria-induced phagosomal rupture/cytosolic access.
Virulence of M. tuberculosis H37RvΔPLC in the THP-1 infection model
As phagosomal rupture in M. tuberculosis is often linked to virulence36, we evaluated the survival and/or growth of the WT and ΔPLC-mutant M. tuberculosis strains in THP-1 cells. WT and mutant M. tuberculosis strains were used to infect THP-1 cells at an MOI of 1:20 (1 bacterium per ~ 20 cells), and the number of intracellular bacteria was determined immediately after phagocytosis (day 0) and 3, 5 and 7 days post infection. As shown in Fig. 4, the H37RvΔPLC mutant and the corresponding WT strain showed similar intracellular growth kinetics, resulting in a 1.5-Log increase in CFU number over a 7-day period. Consistent with previous observations from Raynaud and colleagues15, no differences were observed in the intracellular growth abilities of MT103ΔPLC and its isogenic parental strain. In contrast, the ΔESX-1 (ΔRD1) M. tuberculosis strain, showed attenuated growth relative to WT and ΔPLC strains (Fig. 4). These results indicate that PLCs, in contrast to the ESX-1 proteins, are not essential for M. tuberculosis intracellular survival and optimal growth in host macrophages.
Figure 4. Growth kinetics of M. tuberculosis strains in THP-1 derived macrophages.
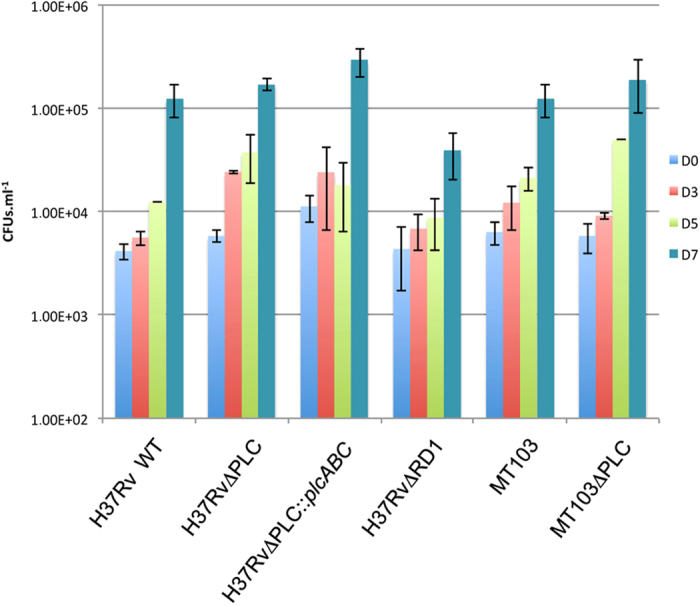
Number of colony forming units (CFU) obtained at different time points after infection. MOI was 1:20 (bacteria/cells). The figures show the means and the standard deviations obtained in 3 independent experiments.
Virulence of M. tuberculosis in mouse infection models
To further test whether PLC inactivation might result in a potential defect in in vivo growth ability that might not be detectable in macrophages cell lines, we evaluated the virulence properties of the H37RvΔPLC and WT strains in different mouse infection models.
Given the previously established suitability of the SCID (severe combined immune deficient) mouse infection model for distinguishing attenuated ΔESX-1 (ΔRD1) and virulent WT M. tuberculosis strains33,37,38, the potential impact of PLC-inactivation on virulence of M. tuberculosis was first assessed by testing the in vivo growth characteristics of ΔPLC and WT M. tuberculosis strains in SCID mice. Both the H37Rv ΔPLC mutant and the WT strain displayed an indistinguishable, high bacterial load in lungs and spleen of infected mice (Fig. 5A,B). This was also confirmed by visual inspection of the organs, which showed typical signs of massive infection (Supplementary Figure 2). Similarly, the MT103ΔPLC and WT strains both showed comparable, intense in vivo growth in SCID mice, as indicated by the presence of ~108 CFU in the organs after 3 weeks of infection, although it should be mentioned that for this latter strain couple, the CFU numbers determined for day 1 were somewhat higher for the ΔPLC mutant in comparison with the WT strain (Fig. 5A,B).
Figure 5. Virulence evaluation of M. tuberculosis strains in different mouse infection models.
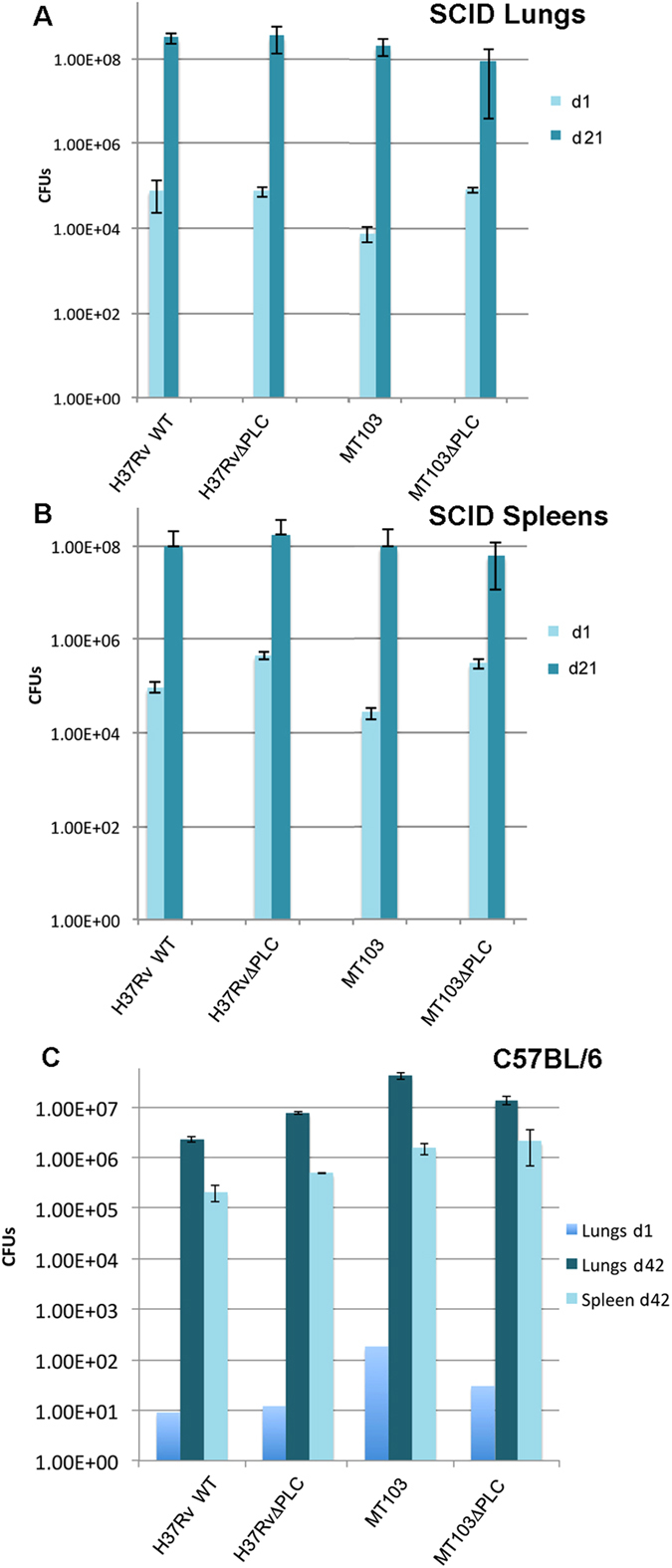
Number of colony forming units (CFU) 3 weeks days after intravenous infection with M. tuberculosis WT and mutant strains in (A) lungs; and (B) spleens of SCID mice. (C) Panel C shows the in vitro growth characteristics of the same panel of strains as above, in C57BL/6 mice 6 weeks after infection. Results shown are representative of 2 independent experiments.
To determine whether the findings obtained in the SCID mouse model were also relevant in immunocompetent mice, virulence studies with the H37Rv and MT103 ΔPLC and WT strain-pairs were also performed in an aerosol infection model of C57BL/6 mice, where the bacterial load in target organs was determined after 6 weeks of infection. As shown in Fig. 5C and Supplementary Figure 3, we did not observe a significant difference between WT and ΔPLC mutants in their in vivo growth properties. These findings, which are in overall agreement with results from the phagosomal rupture screen and the THP-1 infection assay, suggest that PLCs from M. tuberculosis might not represent very obvious virulence factors of M. tuberculosis.
Expression of plcABC genes seems to linked to phosphate concentration
Previous studies on PLCs of P. aeruginosa have shown that induction of PLC expression was phosphate regulated, suggesting a putative role of PLCs for retrieval of phosphate from the environment20,39. To investigate whether PLCs of M. tuberculosis might present similar features, we established an in vitro growth model under phosphate-limiting conditions (Supplementary Figure 4). For monitoring promoter activities, a recombinant ΔPLC M. tuberculosis H37Rv strain expressing a translational 5′-plcA-egfp fusion under the natural plcABC promoter was constructed and named H37RvΔPLC::plcA-egfp. Results obtained from growth experiments with this strain showed that during the first 9 days fluorescence remained low, while starting from day 10 post-inoculation a strong increase in fluorescence was noted (Fig. 6A). By this time-point the phosphate concentration in the medium was below 0.3 mmol.l–1. The expression of the plcABC genes thus seems to be induced by low phosphate concentration, although an impact of other potential stress factors linked to the consumption and limitation of essential nutriments may not be excluded. To further investigate this point, an H37RvΔPLC::plcA-egfp strain that also expressed DsRed under a constitutive promoter was constructed. With the help of this strain promoter activity was studied at different phosphate ion concentrations, simultaneously controlling for the impact of cell density on fluorescence levels. Monitoring of green fluorescence relative to red fluorescence and absorbance levels showed that under low phosphate conditions green fluorescence increased strongly relative to the constant level of red fluorescence, confirming that the plcABC promoter was more strongly induced under low phosphate conditions (Fig. 6B). Finally, we also evaluated the GFP-fluorescence normalized to the cell density measured in OD, and again observed that at low phosphate concentration the promoter activity of the plcABC operon was increased (Fig. 6C). Starvation of phosphate ions thus seems to represent a stress that the bacteria try to counterbalance by induction of PLC production.
Figure 6. plcA-egfp fusion gene expression of M. tuberculosis H37RvΔPLC::Pr_plcA-egfp during growth in phosphate limiting conditions.

(A) The curve shows the phosphate concentration in samples over time of in vitro growth. Histogram represents increase of the culture fluorescence intensity due to GFP expression over time. Results shown are representative of 2 independent experiments. Note that towards the end of the experiment the phosphate concentration slightly increased, which is plausibly due to lysis of some of the older bacterial cells. (B) Measurement of fluorescence divided into GFP and red fluorescence in a culture of H37RvΔPLC::Pr_plcA-egfp complemented with a DsRed expressing plasmid. DS-red is expressed via a constitutive promoter while GFP expression is dependent on plcA promoter activity. (C) Promoter activity of M. tuberculosis H37RvΔPLC::Pr_plcA-egfp in presence of decreasing phosphate concentration due to in vitro growth of culture during 7 days at 37 °C under shaking conditions. Measures show the ratio between fluorescence and absorbance, the first reflecting GFP expression levels and the latter reflecting cell density. (D) Survival of M. tuberculosis H37Rv WT and mutant strains in broth that provides phosphatidylcholine as the sole phosphate source. Results shown represent two different experiments. For each experiment the different strains tested were plated and counted in triplicate.
Finally, we also conducted experiments wherein the WT, ΔPLC and ΔPLC::plcABC H37Rv M. tuberculosis strains were grown in liquid medium supplemented with phosphatidylcholine as the sole phosphate source. As shown in Fig. 6D, the WT M. tuberculosis H37Rv strain and the complemented strain survived better under these conditions compared to the ΔPLC mutant. It should be emphasized, however, that none of the strains was able to actively grow under these experimental settings.
Discussion
PLCs are widely distributed enzymes in living organisms. PLCs hydrolyze phospholipids such as phosphatidylcholine or sphingomyelin at the phosphodiester bond. In bacteria, these enzymes have been reported to function in a wide variety of cellular tasks during infection, including membrane lysis, intracellular signalling, lipid metabolism and/or pathogenicity-associated activity40,41. In our study, we focused on the PLCs of M. tuberculosis, which belong to the superfamily of haemolytic phosphocholine-specific PLCs for which PLC of P. aeruginosa is the paradigm member42. Our initial objective was to evaluate whether these enzymes were involved in the process of phagosomal rupture induced by M. tuberculosis during infection of macrophages. In L. monocytogenes, or C. perfringens, PLCs play important roles together with pore forming listeriolysin or perfringolysin, respectively, to lyse the phagosomal membrane and allow the bacteria to gain access to the cytosol and promote cell-to-cell spread22,43. Concerning the infection with M. tuberculosis, the scenario seems more complex. While it was long thought that M. tuberculosis resists degradation in the phagosome by inhibiting the fusion with lysosomes, favoring intra-phagosomal survival and multiplication44, more recent studies by van der Wel and colleagues, using cryo-electron microscopy, provided evidence of cytosolic presence of M. tuberculosis at later stages of infection45. Similarly, cytosolic access of virulent M. tuberculosis strains was recently also reported by using a FRET-based read-out, combined with automated confocal microscopy24 or flow cytometry23. We here used the latter method to test the M. tuberculosis ΔPLC and WT strains for their ability to cause phagosomal rupture in comparison with a ΔESX-1 (ΔRD1) negative control and found that the M. tuberculosis ΔPLC mutants and WT strains all showed very similar abilities to gain cytosolic access.
Given the result that PLCs of M. tuberculosis were not required for inducing phagosomal rupture and cytosolic contact of M. tuberculosis, which are attributes usually linked to mycobacterial pathogenicity36, we subjected the ΔPLC mutants and WT strains to virulence analyses in in vitro/ex vivo and in vivo models. In the obtained data, we could only detect minor, not significant virulence differences between the ΔPLC and WT M. tuberculosis strains of two genetic backgrounds, i.e. MT103 and H37Rv, in the 3 models used. These results, which were different from those of previous work reporting that PLCs were involved in virulence of M. tuberculosis15, remained puzzling.
Review of the available literature suggests that the number of functional PLC-encoding genes in different strains of the M. tuberculosis complex is highly variable and ranges from 0 to 4 copies. In many M. tuberculosis strains, including H37Rv, the plcD gene, which represents a hotspot for IS6110 insertion, is inactivated or deleted27,28,46. Similarly, extensive IS6110 insertion is also observed for the plcABC locus, but to a lesser extent47. Moreover, in a study on genetic polymorphisms affecting the four PLC encoding genes in M. tuberculosis isolates, Viana-Niero and coworkers found that 19 of 25 clinical isolates showed loss of parts of genes or complete genes from the plcABC and/or plcD loci, whereby five isolates retrieved from patients with active tuberculosis had all 4 plc genes interrupted48. PLC-encoding loci are also variable in different lineages of the M. tuberculosis complex; PlcA/B/C, which are also known as the “mtp40” mycobacterial protein(s), are missing from the M. bovis lineage due to the RD5 deletion, and also are absent from certain other tubercle bacilli27,49. In this respect it is also noteworthy that M. bovis strains with an IS6110 insertion in the remaining plcD gene were described. Interestingly, these strains without a functional PLC encoding gene were responsible for causing tuberculosis lesions in cattle for which no differences in the organ distribution relative to other M. bovis strains were noticed50. These findings are also in agreement with results from a high-density transposon screen, wherein PLC-encoding genes have not been identified as essential for in vivo growth of M. tuberculosis in the mouse model51. Taken together, these reports and our experimental findings with two different ΔPLC mutants of M. tuberculosis cast doubt on an essential role of PLC in virulence of tubercle bacilli. PLCs of M. tuberculosis might play a less important role in the infectious lifecycle of M. tuberculosis than previously thought.
However, it is intriguing that despite the apparently marginal role of PLCs in virulence of M. tuberculosis, most strains have conserved one or more copies of PLC-encoding genes in their genomes, similar to certain non-tuberculous (NTM) mycobacteria. There are only few mycobacterial species that harbour genes encoding PLCs. Database analyses show that for the group of slow-growing mycobacteria, PLC-encoding genes are present in the genomes of smooth tubercle bacilli52, members of the M. tuberculosis complex, members of the Mycobacterium kansasii-Mycobacterium gastri cluster, Mycobacterium asiaticum, and members of the Mycobacterium marinum-Mycobacterium ulcerans cluster. PLCs are absent from the genomes of Mycobacterium leprae, and members of the Mycobacterium avium-intercellulare complex. In the more distantly related rapid growing mycobacteria, only M. abscessus is known to carry a PLC, which shows 37% amino-acid identity with PLCs from M. tuberculosis and seems to be the result of a specific horizontal gene transfer (HGT) into M. abscessus13,53. In contrast, the PLCs in M. tuberculosis and other slow-growing mycobacteria seem to share a common origin with PLCs from different Gordonia species, with which they show about 60% amino acid identity. It seems thus likely that a common progenitor of the phylogenetic subgroup of slow-growing mycobacteria comprising M. kansasii, M. gastri, M. asiaticum, M. marinum and M. tuberculosis has acquired the PLC-encoding genes during evolution through HGT from more distantly related actinobacteria.
This feature prompted us to search for potential alternative biological functions of PLCs in slow growing mycobacteria, not necessarily linked with virulence. As one hypothesis, PLCs of M. tuberculosis might help in the acquisition of phosphate. Cleavage of phospholipids by PLCs results in the generation of two molecular entities, a glycerol part and a residue containing a phosphate group, which might serve as a potential source of phosphate for the bacterium. The finding that expression of the plcA-egfp fusion was inversely correlated with the phosphate concentration in the medium suggests that the specific promoter activity might be downregulated under phosphate-sufficient environmental conditions. This assumption is in agreement with previous observations with PLCs from P. aeruginosa, for which an impact of phosphate concentration on plc gene regulation was noted20,39. Moreover, it has previously been reported that during infection of THP-1 cells by M. tuberculosis, the expression of the plcABC operon was upregulated for the first 24 h of infection15. Given the results obtained with our GFP-fusion assay, it is thus tempting to speculate that this upregulation might be related to a limited phosphate concentration inside the phagosome. Limitation of phosphate during phagosomal containment was also postulated by results from large-scale transcriptome studies, which found genes encoding phosphate transporters upregulated during infection18,54. It is plausible that M. tuberculosis can vary its supply in phosphate between inorganic phosphate, which is the preferred source of phosphorus for many bacteria18, and acquisition of organic phosphates through the action of phosphatases and/or phospolipases. A similar scenario was recently suggested for SpmT (Rv0888) of M. tuberculosis, which harbours a surface-exposed C-terminal sphingomyelinase domain and a putative N-terminal channel domain that mediates glucose and phosphocholine uptake across the outer membrane55. However, at present it remains unknown if the PLCs of M. tuberculosis may contribute to the phosphate supply of the bacterium in a similar way. Our results point to such a possibility, although more in depth studies are needed to clarify this question.
In conclusion, our study calls into question the impact of PLCs on virulence of M. tuberculosis, and provides new hints on putative alternative functions of PLCs in M. tuberculosis.
Methods
Bacterial strains and culture conditions
Escherichia coli DH10B and Top10 (Invitrogen) strains, used for cloning procedures, were grown on LB agar medium and/or LB broth. M. smegmatis mc2155 and M. tuberculosis strains were obtained from stock held at the Institut Pasteur. The M. tuberculosis MT103 strain and the corresponding Myc2509ΔPLC mutant strain15 were a gift of Prof. Gicquel, Institut Pasteur.
Mycobacterial strains were cultured in Middlebrook 7H9 broth supplemented with ADC (Difco) and 0.05% Tween 80 or on Middlebrook 7H11 medium supplemented with OADC (Difco). When required, antibiotics were included for selection purposes at following concentrations: Hygromycin (200 μg.ml–1) and Zeocin (25 μg.ml–1) for E. coli; Hygromycin (50 μg.ml–1), Apramycin (50 μg.ml–1), Kanamycin (25 μg.ml–1), Zeocin (25 μg.ml–1), for mycobacteria.
For the PlcABC-promoter induction assay, phosphate-free Sauton medium was prepared as follows: L-asparagine 4 g/L, MgSO4-7 H2O 0.5 g/L; ammonium iron III citrate 0.05 g/L; citric acid 2 g/L; ZnSO4 1% 0.1 ml/L; glycerol 60 ml/L. pH was adjusted between 7.2–7.3 by a buffer solution of ammonium hydroxide.
Construction of a plcABC deletion mutant in M. tuberculosis H37Rv
The M. tuberculosis H37Rv ΔplcABC mutant was constructed by allelic replacement using the recombineering method30. The allelic exchange substrate plcABC::Apra was obtained by a three step PCR approach56. Briefly, two 500–bp fragments corresponding to the plcABC upstream and downstream regions were amplified by PCR from the M. tuberculosis H37Rv genomic DNA and linked to a third PCR fragment encoding the apramycin resistance cassette, to generate the 2 kb-fragment plcABC::Apra. The plcABC::Apra fragment was thus used to transform a M. tuberculosis H37Rv recombinant strain containing the pJV53 vector. The pJV53 plasmid encodes the recombination proteins gp60 and gp6157, whose expression is induced by incubation with30 0.2% acetamide for 24 h. The H37Rv-pJV53 acetamide-activated transformants were selected on solid medium for resistance to Kanamycin and Apramycin. The obtained Kanamycin and Apramycin resistant clones were thus tested for the plcABC deletion by PCR. One out of 116 tested clones revealed an amplification profile consistent with the replacement of the plcABC cluster with the apramycin cassette, and was thus subjected to Southern blot analyses. Genomic DNAs from M. tuberculosis strains were digested with AvrII, separated by gel electrophoresis and transferred onto Hybond-C-Extra nitrocellulose (GE). Hybridization was performed with [α-32 P] dCTP-labeled PCR-probe, specific for the plcABC downstream region, in 6x SSC, 0.5% SDS, 0.01 M EDTA, 5x Denhardt’s solution, 100 μg.ml−1 salmon-sperm DNA, at 68 °C. After washing, membranes were exposed to phosphorimager screens, which were scanned in a STORM phosphorimager31,57.
Construction of M. tuberculosis H37RvΔPLC complemented strains
Two different integrative pYUB412-based plasmids (pPlcABC and pYUB412-Pr_plcA-egfp) harbouring the plcABC operon and the plcA gene, respectively, were constructed. To obtain the pPlcABC plasmid, the plcABC operon and its natural promoter region, were amplified by PCR and cloned into the pYUB412 vector backbone. Similarly, to construct the pYUB412-Pr_plcA-egfp plasmid, the plcA gene and the plcABC promoter region were amplified by PCR using modified primers (Supplementary Table 1), which allow the introduction of additional HindIII and NheI recognition sequences in the amplified fragment obtained. The resulting PCR product was digested and ligated into the HindIII-NheI-digested pYUB412::egfp (a pYUB412 derivative cosmid that allows the expression of transcriptional eGFP fusion protein constructs32).
Both pPlcABC and pYUB412-Pr_plcA-egfp constructs were used to transform the M. tuberculosis H37RvΔPLC mutant strain. Transformed clones were selected on solid medium for resistance to hygromycin.
PCR amplification and DNA Sequencing
PCR reactions to obtain fragments used in cloning procedures or in screening of transformed clones were carried out with Pwo (Roche) or similar high fidelity DNA polymerases, respectively, as previously reported58. Sequences of primers used in amplification reactions are listed in Table S1. All amplified PCR products and plasmids were sequenced by using the Big Dye cycle sequencing Kit (Applied Biosystems) in an automated DNA sequencer (Applied Biosystems, 3130xl genetic analyser).
Transformation of mycobacterial strains
To obtain mycobacterial competent cells, M. tuberculosis H37Rv and M. smegmatis Mc2155 were recovered from cultures at exponential growth, and washed three times in 10% glycerol. Aliquots (100 μl) of freshly prepared electro-competent cells in glycerol 10% were transformed with 100 - 200 ng of vector DNA in 0.2-cm cuvettes (2.0 kV; 25 μF : 1000 Ohms) at room temperature58. Transformant clones were selected by incubation on solid medium supplemented with the corresponding antibiotic for 2–3 weeks at 37 °C.
Phospholipase C assay
PLC activities were measured using the p-nitrophenylphosphorylcholine (p-NPPC) substrate (Sigma-Aldrich). Activity is defined as the ability of an enzyme to catalyse p-NPPC into p-nitrophenyl (p-NP) of yellow chromogenic nature. Briefly, 0.5 mg of total protein was incubated in 3 ml of 10 mM Tris HCl (pH = 7.2), containing 5 mM of p-NPPC and 1.5% of sorbitol16. The reaction mix was incubated at 37 °C under shaking at 100 rpm. The reaction was stopped after 0, 1, 2, 3, 4, or 7 days by NaOH at 0.1 N (final concentration). The release of p-NP was measured at 410 nm. Buffer without proteins served as blank reference. For initial experimental setup, a control assay was performed with 40 U of purified PLC from B. cereus (Invitrogen).
Phosphate assay by colorimetric method
Phosphate ions in presence of L(+) ascorbic acid (Merck) and ammonium molybdate tetra-hydrate (Sigma) form a complex showing a blue/green colour. Briefly, comparator samples containing between a standard range of 0 and 6.10−5 mol. L−1 of phosphate (KH2PO4) were prepared in 15 mL glass tubes. After adding 1 mL of stock solution of ascorbic acid at 0.1 mol.L−1 and ammonium molybdate tetra-hydrate at 0.2 mol.L−1, the react volume was adjusted to 10 ml final volume with Milli-Q (Millipore) purified water. All tubes were incubated at 80 °C in a water bath during 10 minutes and slowly cooled down to room temperature on the bench. Then all samples and comparator samples were diluted (d = 1/2) before measurement at 750 nm.
Infection of THP-1 derived macrophages with M. tuberculosis
THP-1 cells were grown at 37 °C with 5% CO2. Cells were maintained in RPMI 1640 + glutamax (Life technologies) and 10% of foetal bovine serum. THP-1 cells were seeded in 96 well plates at 7.5 × 104 cells per well, and differentiated by incubation with 10 ng.ml−1 of PMA for 2 days. Before infection, the medium was removed and the wells were washed 3 times with PBS. Bacterial strains were added at a multiplicity of infection (MOI) = 1: 20 (1 bacterium: 20 macrophages). After 2 hours (day 0) or 3, 5 and 7 days post-infection, cells were lysed in PBS 0,01% of Triton X-100. The number of viable intracellular mycobacteria was determined/counted by plating serial dilutions of macrophage lysates on solid medium.
Phagosomal rupture assay by flow cytometry analysis
Differentiated THP-1 cells were infected at MOI of 0.5 and were stained at day 3 post-infection with 8 μM CCF-4 (Invitrogen) in EM buffer (120 mM NaCl, 7 mM KCl, 1.8 mM CaCl2, 0.8 mM MgCl2, 5 mM glucose and 25 mM Hepes, pH 7.3) complemented with 2.5 μM probenecid, during 1 h at room temperature. Cells were then washed once in PBS and stained with anti-CD11b-APC (BD) monoclonal antibody in PBS containing 3% fetal calf serum and 0.1% NaN3 and fixed with 4% paraformaldehyde overnight at 4 °C. Cells were analyzed in a CyAn cytometer by use of Summit software (Beckman Coulter, France). Data were analyzed with FlowJo software (Treestar, OR).
M. tuberculosis virulence studies in mice
Six-week-old female CB17/Ico SCID mice (Charles River) were infected intravenously via the lateral tail vein with 200 μl of bacterial suspension of 1 × 106 CFU.ml−1. For aerosol infection, a customized apparatus was used following a previously established procedure59 Six-week-old female C57BL/6B mice (Charles-River) were infected with a suspension containing 5 × 105 bacteria.ml−1 to obtain an inhaled dose of ca. 100 CFU. At selected time points after infection, mice were killed and organs homogenised using a tissue Lyser apparatus from Quiagen and 2.5 mm diameter glass beads to determine CFU numbers as previously reported52,59.
All animal studies were approved by the Institut Pasteur Safety Committee (Protocol 11.245; experimentation authorization number 75–1469), in accordance with European and French guidelines (Directive 86/609/CEE and Decree 87–848 of 19 October 1987), and implicating approval from local ethical committees (CETEA 2013–0036).
plcABC-promoter induction assay
The H37RvΔPLC::Pr_plcA-egfp strain was complemented with a DsRed expressing plasmid. The resulting strain was named H37RvΔPLC::Pr_plcA-egfp::Pr-hsp60-DsRed. In this construct DsRed is expressed via a constitutive promoter while GFP expression is dependent on plcA promoter activity. Briefly, for these experiments bacteria were grown in 7H9 + ADC + Hygromycin 50 μg.ml−1/Zeocin 25 μg.ml−1 medium until 0.4–0.6 OD. Bacteria of this preculture were inoculated in fresh Sauton medium containing 0.05% Tween80 and Hygromycin 50 μg.ml−1/Zeocin 25 μg.ml−1 at 0.05 OD. After 7 days of culture, bacteria were harvested and centrifuged during 5 min at 5000 g. After 3 washing steps, using 5 ml of fresh Sauton medium lacking PO43–, bacteria were inoculated at 0.05 final OD in Sauton medium containing phosphate or not. In addition, the bacterial quantities were monitored by plating aliquots of each strain onto agar plates (in triplicate) for CFU determination. The GFP (Ex = 475 nm/Em = 504 nm) and DsRed fluorescence (Ex = 558/Em = 583) was monitored using a microplate reader (BGM Labtech) and analysed by Omega software. To avoid an increase of fluorescence due to the growth, we chose to normalize fluorescence values relative to absorbance. These ratio measurements were converted in percentage, with the day 0, as reference point.
Mycobacterial phosphatidylcholine survival assay
M. tuberculosis H37Rv WT, ΔplcABC and complement strains were inoculated into phosphate-free Sauton medium supplemented with phosphatidylcholine (3.6 mM) (Sigma) as the sole phosphate source, considering that 1 mole of phosphate was equal to 1 mole of phosphatidylcholine. Addition of phosphatidylcholine rendered the medium turbid, which precluded the use of OD measurement. CFU counting was used as an alternative for quantification of bacteria, as previously reported60.
Statistical analyses
Potential statistical differences in bacterial loads were evaluated by ANOVA test with Tukey correction, after conversion of CFU numbers in Log10 CFU values. Statistical significance was considered to be a P value ≤ 0.05.
Additional Information
How to cite this article: Le Chevalier F. et al. Revisiting the role of phospholipases C in virulence and the lifecycle of Mycobacterium tuberculosis. Sci. Rep. 5, 16918; doi: 10.1038/srep16918 (2015).
Supplementary Material
Acknowledgments
We are grateful to Graham F. Hatfull for providing the mycobacterial recombineering vector pJV53. Mycobacterial knock-out strain Myc2509ΔPLC and the M. tuberculosis MT103 parental strain used as controls, were a gift from Brigitte Gicquel; strain M. tuberculosis H37RvΔRD1 was a gift from William R. Jacobs, Jr. We thank Karim Sébastien for expert assistance in animal care in biosafety-A3 facilities. Funding: We acknowledge the support by the Agence National de Recherche (ANR-14-JAMR-001-02) and the Fondation pour la Recherche Médicale FRM (DEQ20130326471). R.B. is a member of the LabEx consortium IBEID at the Institut Pasteur. E.C.B. was supported by a stipend from the Pasteur–Paris University (PPU) International PhD program and the Institut Carnot Pasteur Maladies Infectieuses. F.L-C was supported by the French Region Ile-de- France (Domaine d’Intérêt Majeur Maladies Infectieuses et Emergentes) PhD program.
Footnotes
Author Contributions F.L.C., A.C., J.L.H. and R.B. conceived the project. F.L.C., A.C., W.F., A.P., E.C.B. and L.M. performed the experiments. F.L.C., A.C., D.B., J.L.H. and R.B. analyses the data and interpreted results. F.L.C. and R.B. wrote the paper. A.C., W.F., A.P., E.C.B., D.B., L.M. and J.L.H. commented and edited the different versions of the paper and all authors approved the manuscript.
References
- Magee J. G. & Ward A. C. in Bergey’s Manual of Systematic Bacteriology, The Actinobacteria Vol. 5 (eds Goodfellow M. et al.) 312–375 (Springer, 2012). [Google Scholar]
- WHO. Global tuberculosis report 2014. (World Health Organization, Geneva, 2014).
- Cambier C. J., Falkow S. & Ramakrishnan L. Host evasion and exploitation schemes of Mycobacterium tuberculosis. Cell. 159, 1497–1509, 1410.1016/j.cell.2014.1411.1024 (2014). [DOI] [PubMed] [Google Scholar]
- Majlessi L., Prados-Rosales R., Casadevall A. & Brosch R. Release of mycobacterial antigens. Immunol Rev. 264, 25–45, 10.1111/imr.12251 (2015). [DOI] [PubMed] [Google Scholar]
- Le Chevalier F., Cascioferro A., Majlessi L., Herrmann J. L. & Brosch R. Mycobacterium tuberculosis evolutionary pathogenesis and its putative impact on drug development. Future Microbiol. 9, 969–85, 10.2217/fmb.2214.2270 (2014). [DOI] [PubMed] [Google Scholar]
- Warner D. F., Koch A. & Mizrahi V. Diversity and disease pathogenesis in Mycobacterium tuberculosis. Trends Microbiol. 23, 14–21, 10.1016/j.tim.2014.1010.1005 (2015). [DOI] [PubMed] [Google Scholar]
- Takiff H. E. & Feo O. Clinical value of whole-genome sequencing of Mycobacterium tuberculosis. Lancet Infect Dis. 15, 1077–1090, 1010.1016/S1473-3099(1015)00071-00077 (2015). [DOI] [PubMed] [Google Scholar]
- Boritsch E. C. et al. A glimpse into the past and predictions for the future: the molecular evolution of the tuberculosis agent. Mol Microbiol. 93, 835–852, 10.1111/mmi.12720 (2014). [DOI] [PubMed] [Google Scholar]
- Roux A. L. et al. Comparing Mycobacterium massiliense and Mycobacterium abscessus lung infections in cystic fibrosis patients. J Cyst Fibros. 14, 63–69, 10.1016/j.jcf.2014.1007.1004 (2015). [DOI] [PubMed] [Google Scholar]
- Bryant J. M. et al. Whole-genome sequencing to identify transmission of Mycobacterium abscessus between patients with cystic fibrosis: a retrospective cohort study. Lancet 381, 1551–1560, 10.1016/S0140-6736(13)60632-7 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aitken M. L. et al. Respiratory outbreak of Mycobacterium abscessus subspecies massiliense in a lung transplant and cystic fibrosis center. Am J Respir Crit Care Med 185, 231–232, 10.1164/ajrccm.185.2.231 (2012). [DOI] [PubMed] [Google Scholar]
- Pawlik A. et al. Identification and characterization of the genetic changes responsible for the characteristic smooth-to-rough morphotype alterations of clinically persistent Mycobacterium abscessus. Mol. Microbiology 90, 612–629, 10.1111/mmi.12387 (2013). [DOI] [PubMed] [Google Scholar]
- Bakala N’Goma J. C. et al. Mycobacterium abscessus phospholipase C expression is induced during coculture within amoebae and enhances M. abscessus virulence in Mice. Infect Immun. 83, 780–791, 710.1128/IAI.02032-02014 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Springer B., Stockman L., Teschner K., Roberts G. D. & Bottger E. C. Two-laboratory collaborative study on identification of mycobacteria: molecular versus phenotypic methods. J Clin Microbiol 34, 296–303 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raynaud C. et al. Phospholipases C are involved in the virulence of Mycobacterium tuberculosis. Mol Microbiol 45, 203–217 (2002). [DOI] [PubMed] [Google Scholar]
- Bakala N’goma J., C., Schue M., Carriere F., Geerlof A. & Canaan S. Evidence for the cytotoxic effects of Mycobacterium tuberculosis phospholipase C towards macrophages. Biochim Biophys Acta. 1801, 1305–1313, 1310.1016/j.bbalip.2010.1308.1007 (2010). [DOI] [PubMed] [Google Scholar]
- Hingley-Wilson S. M., Sambandamurthy V. K. & Jacobs W. R. Jr. Survival perspectives from the world’s most successful pathogen, Mycobacterium tuberculosis. Nat Immunol 4, 949–955 (2003). [DOI] [PubMed] [Google Scholar]
- Niederweis M. Nutrient acquisition by mycobacteria. Microbiology. 154, 679–692 (2008). [DOI] [PubMed] [Google Scholar]
- Ligon L. S., Hayden J. D. & Braunstein M. The ins and outs of Mycobacterium tuberculosis protein export. Tuberculosis (Edinb). 92, 121–132, 110.1016/j.tube.2011.1011.1005 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Titball R. W. Bacterial phospholipases C. Microbiol. Rev. 57, 347–366 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Awad M. M., Ellemor D. M., Boyd R. L., Emmins J. J. & Rood J. I. Synergistic effects of alpha-toxin and perfringolysin O in Clostridium perfringens-mediated gas gangrene. Infect Immun. 69, 7904–7910 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cossart P. Illuminating the landscape of host-pathogen interactions with the bacterium Listeria monocytogenes. Proc Natl Acad Sci USA 108, 19484–19491, 19410.11073/pnas.1112371108 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simeone R. et al. Cytosolic access of Mycobacterium tuberculosis: Critical impact of phagosomal acidification control and demonstration of ccurrence in vivo. PLoS Pathog. 11, e1004650. 1004610.1001371/journal.ppat.1004650 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simeone R. et al. Phagosomal rupture by Mycobacterium tuberculosis results in toxicity and host cell death. PLoS Pathog 8, e1002507 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole S. T. et al. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 393, 537–544 (1998). [DOI] [PubMed] [Google Scholar]
- Brosch R. et al. Genomic analysis reveals variation between Mycobacterium tuberculosis H37Rv and the attenuated M. tuberculosis H37Ra strain. Infect Immun 67, 5768–5774 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon S. V. et al. Identification of variable regions in the genomes of tubercle bacilli using bacterial artificial chromosome arrays. Mol Microbiol 32, 643–656 (1999). [DOI] [PubMed] [Google Scholar]
- Ho T. B., Robertson B. D., Taylor G. M., Shaw R. J. & Young D. B. Comparison of Mycobacterium tuberculosis genomes reveals frequent deletions in a 20 kb variable region in clinical isolates. Yeast. 17, 272–282 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manca C. et al. Mycobacterium tuberculosis CDC1551 induces a more vigorous host response in vivo and in vitro, but is not more virulent than other clinical isolates. J Immunol 162, 6740–6746 (1999). [PubMed] [Google Scholar]
- van Kessel J. C. & Hatfull G. F. Recombineering in Mycobacterium tuberculosis. Nat Methods 4, 147–152 (2007). [DOI] [PubMed] [Google Scholar]
- Chaveroche M. K., Ghigo J. M. & d’Enfert C. A rapid method for efficient gene replacement in the filamentous fungus Aspergillus nidulans. Nucleic Acids Res. 28, E97. (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bange F. C., Collins F. M. & Jacobs W. R. Jr. Survival of mice infected with Mycobacterium smegmatis containing large DNA fragments from Mycobacterium tuberculosis. Tuber Lung Dis 79, 171–180 (1999). [DOI] [PubMed] [Google Scholar]
- Hsu T. et al. The primary mechanism of attenuation of bacillus Calmette-Guerin is a loss of secreted lytic function required for invasion of lung interstitial tissue. Proc Natl Acad Sci USA 100, 12420–12425 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charpentier X. & Oswald E. Identification of the secretion and translocation domain of the enteropathogenic and enterohemorrhagic Escherichia coli effector Cif, using TEM-1 beta-lactamase as a new fluorescence-based reporter. J Bacteriol. 186, 5486–5495 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nothelfer K., Dias Rodrigues C., Bobard A., Phalipon A. & Enninga J. Monitoring Shigella flexneri vacuolar escape by flow cytometry. Virulence. 2, 54–57, 10.4161/viru.4162.4161.14666 (2011). [DOI] [PubMed] [Google Scholar]
- Houben D. et al. ESX-1-mediated translocation to the cytosol controls virulence of mycobacteria. Cell Microbiol 14, 1287–1298, 10.1111/j.1462-5822.2012.01799.x (2012). [DOI] [PubMed] [Google Scholar]
- Pym A. S., Brodin P., Brosch R., Huerre M. & Cole S. T. Loss of RD1 contributed to the attenuation of the live tuberculosis vaccines Mycobacterium bovis BCG and Mycobacterium microti. Mol Microbiol 46, 709–717 (2002). [DOI] [PubMed] [Google Scholar]
- Bottai D. et al. Increased protective efficacy of recombinant BCG strains expressing virulence-neutral proteins of the ESX-1 secretion system. Vaccine. 33, 2710–2718, 2710.1016/j.vaccine.2015.2703.2083 (2015). [DOI] [PubMed] [Google Scholar]
- Shortridge V. D., Lazdunski A. & Vasil M. L. Osmoprotectants and phosphate regulate expression of phospholipase C in Pseudomonas aeruginosa. Mol Microbiol. 6, 863–871 (1992). [DOI] [PubMed] [Google Scholar]
- Schmiel D. H. & Miller V. L. Bacterial phospholipases and pathogenesis. Microbes Infect. 1, 1103–1112 (1999). [DOI] [PubMed] [Google Scholar]
- Dedieu L., Serveau-Avesque C., Kremer L. & Canaan S. Mycobacterial lipolytic enzymes: a gold mine for tuberculosis research. Biochimie. 95, 66–73, 10.1016/j.biochi.2012.1007.1008. (2013). [DOI] [PubMed] [Google Scholar]
- Stonehouse M. J. et al. A novel class of microbial phosphocholine-specific phospholipases C. Mol Microbiol. 46, 661–676 (2002). [DOI] [PubMed] [Google Scholar]
- O’Brien D. K. & Melville S. B. Effects of Clostridium perfringens alpha-toxin (PLC) and perfringolysin O (PFO) on cytotoxicity to macrophages, on escape from the phagosomes of macrophages, and on persistence of C. perfringens in host tissues. Infect Immun. 72, 5204–5215 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong J. A. & Hart P. D. Response of cultured macrophages to Mycobacterium tuberculosis, with observations on fusion of lysosomes with phagosomes. J Exp Med. 134, 713–740. (1971). [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Wel N. et al. M. tuberculosis and M. leprae translocate from the phagolysosome to the cytosol in myeloid cells. Cell 129, 1287–1298 (2007). [DOI] [PubMed] [Google Scholar]
- Tsolaki A. G. et al. Functional and evolutionary genomics of Mycobacterium tuberculosis: insights from genomic deletions in 100 strains. Proc Natl Acad Sci USA 101, 4865–4870 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong Y. et al. Distribution of insertion- and deletion-associated genetic polymorphisms among four Mycobacterium tuberculosis phospholipase C genes and associations with extrathoracic tuberculosis: a population-based study. J Clin Microbiol 43, 6048–6053. (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viana-Niero C., de Haas P. E., van Soolingen D. & Leao S. C. Analysis of genetic polymorphisms affecting the four phospholipase C (plc) genes in Mycobacterium tuberculosis complex clinical isolates. Microbiology 150, 967–978 (2004). [DOI] [PubMed] [Google Scholar]
- Weil A., Plikaytis B. B., Butler W. R., Woodley C. L. & Shinnick T. M. The mtp40 gene is not present in all strains of Mycobacterium tuberculosis. J Clin Microbiol. 34, 2309–2311 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viana-Niero C. et al. Identification of an IS6110 insertion site in plcD, the unique phospholipase C gene of Mycobacterium bovis. J Med Microbiol 55, 451–457 (2006). [DOI] [PubMed] [Google Scholar]
- Sassetti C. M. & Rubin E. J. Genetic requirements for mycobacterial survival during infection. Proc Natl Acad Sci USA 100, 12989–12994. (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Supply P. et al. Genomic analysis of smooth tubercle bacilli provides insights into ancestry and pathoadaptation of Mycobacterium tuberculosis. Nat Genet 45, 172–179 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ripoll F. et al. Non mycobacterial virulence genes in the genome of the emerging pathogen Mycobacterium abscessus. PLoS One 4, e5660, 10.1371/journal.pone.0005660 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnappinger D. et al. Transcriptional Adaptation of Mycobacterium tuberculosis within Macrophages: Insights into the Phagosomal Environment. J Exp Med 198, 693–704. (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speer A. et al. Surface hydrolysis of sphingomyelin by the outer membrane protein Rv0888 supports replication of Mycobacterium tuberculosis in macrophages. Mol Microbiol 3, 13073 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derbise A., Lesic B., Dacheux D., Ghigo J. M. & Carniel E. A rapid and simple method for inactivating chromosomal genes in Yersinia. FEMS Immunol Med Microbiol. 38, 113–116. (2003). [DOI] [PubMed] [Google Scholar]
- van Kessel J. C., Marinelli L. J. & Hatfull G. F. Recombineering mycobacteria and their phages. Nat Rev Microbiol. 6, 851–857, 810.1038/nrmicro2014 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodin P. et al. Functional analysis of early secreted antigenic target-6, the dominant T-cell antigen of Mycobacterium tuberculosis, reveals key residues involved in secretion, complex formation, virulence, and immunogenicity. J Biol Chem 280, 33953–33959 (2005). [DOI] [PubMed] [Google Scholar]
- Majlessi L. et al. Influence of ESAT-6 secretion system 1 (RD1) of Mycobacterium tuberculosis on the interaction between mycobacteria and the host immune system. J Immunol 174, 3570–3579 (2005). [DOI] [PubMed] [Google Scholar]
- Munoz-Elias E. J. & McKinney J. D. Mycobacterium tuberculosis isocitrate lyase 1 and 2 are jointly required for in vivo growth and virulence. Nat Med. 11, 638–644 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.