

Abstract
Background
Trigeminal neuralgia is accompanied by severe mechanical, thermal and chemical hypersensitivity of the orofacial area innervated by neurons of trigeminal ganglion (TG). We examined the role of the voltage-gated sodium channel subtype Nav1.9 in the development of trigeminal neuralgia.
Results
We found that Nav1.9 is required for the development of both thermal and mechanical hypersensitivity induced by constriction of the infraorbital nerve (CION). The CION model does not induce change on Nav1.9 mRNA expression in the ipsilateral TG neurons when evaluated 9 days after surgery.
Conclusions
These results demonstrate that Nav1.9 channels play a critical role in the development of orofacial neuropathic pain. New routes for the treatment of orofacial neuropathic pain focussing on regulation of the voltage-gated Nav1.9 sodium channel activity should be investigated.
Keywords: Nav1.9 sodium channels, Trigeminal ganglion (TG) neurons, Constriction of the infraorbital nerve, Trigeminal neuralgia, Neuropathic pain
Background
Trigeminal neuralgia is an excruciating pain syndrome characterized by severe facial pain that can be triggered by light touch of the orofacial surface area producing stabbing, shooting or burning sensations. Trigeminal neuralgia is accompanied by mechanical, thermal and/or chemical hypersensitivities of the orofacial area and is maintained by impaired signaling in sensory neurons of trigeminal ganglion (TG) [1]. However, little is known about the intracellular mechanisms or altered functioning of TG nociceptors underling the development of orofacial pain.
Voltage-gated sodium channels play a central role in nociception, being key determinants of neuronal excitability. Specific expression of tetrodotoxin-resistant (TTX-R) voltage-gated sodium channels Nav1.8 and Nav1.9 subtypes was found for a restricted population of peripheral nociceptors in TG [2–4], dorsal root ganglion (DRG) [5–9], nodose ganglion [10], unmyelinated afferent fibres and nerve terminals [8, 9, 11, 12]. While Nav1.8 channels carry the majority of inward current during the upstroke of the action potentials (AP) [13–15], Nav1.9 channels sustain repetitive firing and plateau potentials [16, 17] due to their characteristic activation at relatively negative membrane potentials as well as slow and incomplete inactivation, producing “persistent” Na+ flow at subthreshold voltages [18–20].
Studies of the role of Nav1.9 channels showed that SCN11A null mutant mice developed reduced hyperalgesia in different inflammatory pain models [formalin, carrageenan, complete freund’s adjuvant (CFA), prostaglandin E2] [21, 22] as well as diminished nociceptive sensitivity triggered by inflammatory mediators [bradykinin, serotonin, adenosine triphosphate (ATP)] [2]. In contrast, Nav1.9−/− mice showed unaltered pain-related behaviour in models of DRG-related neuropathic pain of various origins (partial sciatic nerve injury [2], chronic constriction injury (CCI) [23] and spinal nerve transaction [24]). Recent studies of human SCN11A gene mutations demonstrated that several mutations in this gene are associated with either, peripheral neuropathy and episodic chronic pain [25] or congenital inability to experience pain [26]. Despite the established role of Nav1.9 channels in pain perception, the contribution of this channel subtype to the development of orofacial pain remains unknown, and how cephalic neuropathic pain develops with loss of Nav1.9 channels has not been studied.
Using global Nav1.9 knockout mice, we show for the first time that loss of Nav1.9 channels is associated with the failure to develop orofacial pain in a model of trigeminal neuralgia.
Results
Basal orofacial sensitivity in Nav1.9+/+ and Nav1.9−/− mice
To examine if the loss of Nav1.9 channels could influence the sensitivity of the orofacial area, basal mechanical and thermal sensitivities were examined in Nav1.9+/+ and Nav1.9–/– mice. Mechanical orofacial sensitivity was evaluated as a functional reaction in response to 0.04 g von Frey filament applied to the animal forehead innervated by the trigeminal nerve (Fig. 1a). A filament of 0.04 g intensity was chosen since I have been found to evoke nociceptive responses in the orofacial area after unilateral constriction of the infraorbital nerve (CION), but present almost no effect in control mice [27]. No differences in basal responses to mechanical stimulation were observed between genotypes (n = 13–14 mice, p > 0.05; Fig. 1c). Groups did not differ with regard to basal thermal sensitivity, measured as a latency of response to radiant heat applied to the vibrissal pad surface (n = 13–14 mice, p > 0.05; Fig. 1b, d). Thus, loss of Nav1.9 channels does not affect basal orofacial sensitivity, either mechanical or thermal, in mice.
Fig. 1.
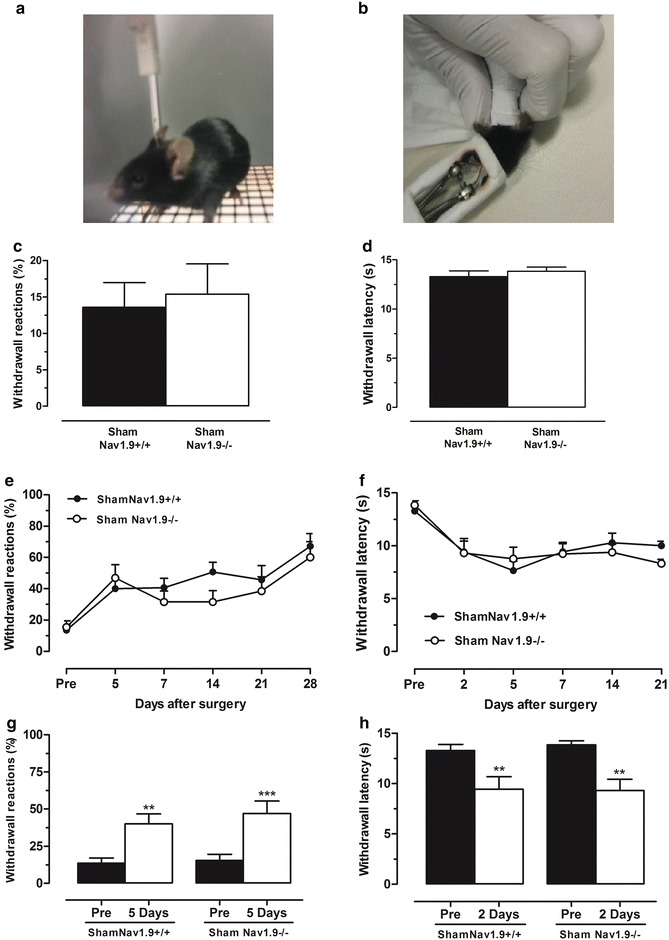
Loss of Nav1.9 does not influence orofacial sensitivity in mice. Mechanical orofacial sensitivity in response to 0.04 g von Frey filament applied to the forehead (a) and thermal orofacial sensitivity in response to radiant heat applied to a vibrissal pad (b) in sham-operated Nav1.9+/+ and Nav1.9−/− mice. Mechanical (c) and thermal (d) orofacial sensitivities baselines. Mechanical (e) and thermal (f) orofacial sensitivities evaluated before (pre) and at different time-points after surgery. Changes in mechanical (g) and thermal (h) orofacial sensitivities after sham-operation. Values represent mean ± SEM analysed for 13–14 mice. **p < 0.01, ***p < 0.001 compared to that before surgery (pre) (Student paired t test)
We next examined the responses of control and knockout mice to a sham surgical inflammation without nerve damage. No major differences in mechanical and thermal sensitivities were found between Nav1.9+/+ and Nav1.9–/– mice, submitted to this sham surgical procedure with no CION, at different time-points (on days 2, 5, 7, 14, 21 and 28 post-surgery; n = 13–14 mice; Fig. 1e, f). Although, the orofacial area was sensitised after the sham surgical procedure, the surgery-induced changes were similar between Nav1.9+/+ and Nav1.9−/− sham-operated mice for either threshold mechanical (by 26 %, p < 0.01 and by 31 %, p < 0.001 in Nav1.9+/+ and Nav1.9−/− mice, respectively; n = 13–14 mice; Fig. 1g) or threshold thermal sensitivities (by 29 % and by 31 %, n = 13–14 mice, p < 0.01 in Nav1.9+/+ and Nav1.9−/− mice, respectively; Fig. 1h).
Loss of Nav1.9 channels alleviates CION-induced orofacial hypersensitivity
Constriction of the infraorbital nerve [25] led to a development of long-lasting mechanical hypersensitivity in Nav1.9+/+. This hypersensitivity developed 7 days after surgery, reached a peak at 2 weeks, and remained persistent until 3 weeks (Fig. 2a). CION-induced mechanical hypersensitivity was robust, showing a significantly increased reaction in response to von Frey filament applied to the forehead of Nav1.9+/+ and Nav1.9−/− mice. The increase in the response frequency was 26 % (p < 0.05), 35 % (p < 0.01) and 31 % (p < 0.01) on day 7, 14 and 21 after CION respectively compared to sham-operated mice (n = 13–14 mice; Fig. 2a). Strikingly, in Nav1.9−/− mice, CION was not accompanied by the development of hypersensitivity of the orofacial area. The responses of Nav1.9−/− mice were similar to those observed in the littermates of the sham-operated group over all periods tested. Thus, loss of Nav1.9 channels abolished the development of CION-induced mechanical hypersensitivity.
Fig. 2.
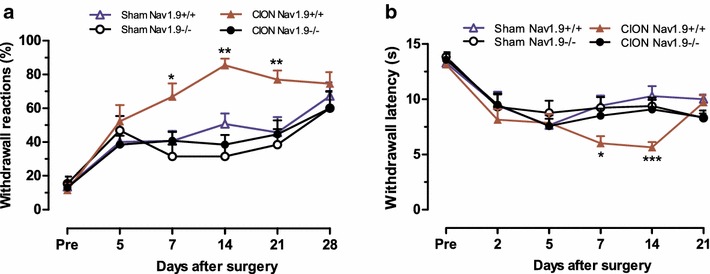
Orofacial neuropathic hypersensitivity was attenuated in Nav1.9 KO mice. Changes in mechanical orofacial sensitivity in response to 0.04 g von Frey filament applied to the forehead (a) and thermal orofacial sensitivity in response to radiant heat applied to a ipsilateral vibrissal pad (b) in Nav1.9+/+ and Nav1.9−/− mice before surgery (pre) and at different time-point after CION. Values represent mean ± SEM. analysed for 13–14 mice. *p < 0.05, **p < 0.01, ***p < 0.001 compared to the correspondent time-point in sham Nav1.9+/+ mice (Two-way ANOVA followed by Bonferroni’s test)
Loss of Nav1.9 channels also abolished the development of CION-induced thermal hypersensitivity. Consistent with previous studies [28], CION produced a long-lasting thermal hypersensitivity in Nav1.9+/+ mice, which developed within 1 week after surgery. The CION-induced decrease in thermal latency in Nav1.9+/+ mice was 10 % (p < 0.05) on day 7 and 12 % (p < 0.001) on day 14 post-surgery. However, the latency was not changed in Nav1.9−/− mice following CION over all period tested (n = 13–14 mice; Fig. 2b). Together, these results indicate that the Nav1.9 channel is required for the development of neuropathic orofacial pain.
Expression of Nav1.9 mRNA in TG on Sham and CION Nav1.9+/+ mice
As the Nav1.9−/− mice did not develop mechanical or thermal hypersensitivity after CION, the next step was to evaluate if the CION model induced any change in the expression of Nav1.9 mRNA in TG neurons. Sample from ipsilateral TG was processed on day 9 after surgery, for extraction of mRNA. As showed on Fig. 3, no Nav1.9 mRNA expression difference was observed between the CION (1.06 ± 0.42) and Sham (1.12 ± 0.28) groups in Nav1.9+/+ mice (p = 0.9026).
Fig. 3.
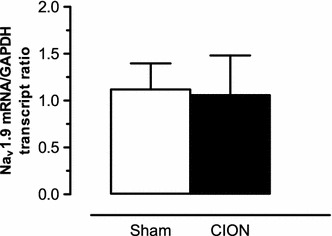
Expression of Nav1.9 mRNA in TG on Sham and CION Nav1.9+/+ mice 9 days after surgery. Values represent mean ± SEM analysed for 6–7 mice (Student paired t test)
Discussion
Considerable evidence suggests participation of Nav1.9 channels in the perception of pain. Both gain of function and loss of function pain conditions have been associated with point mutations in Nav1.9 [25, 26]. Two of those mutations led to a reduction in the current threshold and increased firing frequency in response to suprathreshold stimuli [25]. Mutations in SCN11A (R225C and A808G), reported in patients experienced episodic chronic pain, caused an increase in the Nav1.9 channels-mediated current density and hyperexcitability of nociceptive DRG neurons without changes in the resting membrane potential [29]. Other gain of function mutations lead Nav1.9 channels to display excessive activity at resting voltages, causing sustained depolarization of nociceptors, impaired generation of action potentials and resulting in a pain-free state [26]. The principal mechanism by which Nav1.9 channels contribute to painful states includes sensitization of nociceptors triggered by generation of persistent Na+ currents at subthreshold voltages that promotes depolarization near the resting membrane potential, reduces the current threshold required to trigger an action potential, and results in hyperexcitability of sensory neurons [7, 16, 18–20, 30]. Here using Nav1.9−/− mice, we show that Nav1.9 channels are also involved in sustained (tonic) firing of nociceptive TG neurons and are required for the development of orofacial neuropathic pain.
The role of the Nav1.9 channel in maintaining peripheral inflammatory pain has been demonstrated in different studies [21, 22, 30]. Inflammatory pain of different origins (induced by formalin, carrageenan, CFA, capsaicin, ATP, IL-1β or prostaglandin-E2) was substantially reduced in Nav1.9−/− mice [21, 22, 30], while neuropathic pain, produced by CCI or spinal nerve transection, still developed, showing mechanical allodynia of the hind paw of Nav1.9−/− mice [24, 31]. Using CION-induced model of orofacial neuropathic pain [28, 32], we demonstrate here the absence of both mechanical and thermal hypersensitivity of the orofacial area in Nav1.9−/− mice after CION. Loss of Nav1.9 channels did not change basal orofacial sensitivity (mechanical or thermal), indicating no major effects of Nav1.9 deletion on normal nociceptive pathways. Our findings of a crucial role for Nav1.9 channels in the development of neuropathic orofacial pain are in contrast with reports showing no critical involvement of these channels in the development of neuropathic pain in other somatic neuropathic pain models [24, 31]. This may reflect distinct mechanisms of neuropathic pain.
Nav1.9 channels enhance sustained repetitive (tonic) firing in TG neurons. Previous studies showing that Nav1.9 channels maintain sustained repetitive firing in DRG nociceptors and modulate patterns of firing discharge, including plateau potentials, active hyperpolarizing responses, and sustained oscillatory bursting discharges [16, 18, 33]. It is intriguing that gain of function Nav1.9 mutations in humans may result in enhanced pain or a complete loss of pain sensation [25, 26]. In transgenic mice, inflammatory pain is associated with enhanced Nav1.9 persistent sodium channel activity [30]. In the present study, neuropathic pain is dependent on the activity of Nav1.9 in a model of trigeminal neuralgia. A critical role for Nav1.9 in regulating pain thresholds is thus clear, but the precise mechanisms that contribute to the different pain phenotypes remains to be established.
Conclusions
Our study demonstrates for the first time that the Nav1.9 channel plays a critical role in the development of neuropathic orofacial pain associated with trigeminal neuralgia. Understanding how orbital nerve damage leads to myofascial pain through the modulation of Nav1.9 activity may provide new approaches to the treatment of trigeminal neuralgia.
Methods
Animals
C57Bl/6 mice (2–3 month-old) were used in accordance with the protocols approved by the UK Home Office and UCL ethics committee under a Home Office project license. All efforts were made to minimise animal suffering and to reduce the number of animals used. Experiments were conducted using both male and female wild type littermate and global Nav1.9 knockout mice bred from heterozygous (Nav1.9+/−) animals. The generation and characterization of the Nav1.9 null mutant line was previously described [34]. Mouse colonies were genotyped by PCR using ear biopsy samples. Only homozygous Nav1.9-null (Nav1.9−/−) and wild type (Nav1.9+/+) mice were used in these studies. Genomic DNA was extracted using a lysis buffer containing (in mM) 67 Tris, 16 (NH4)2SO4, 6.7 MgCl2, 100 β-mercaptoethanol, and 0.5 % Triton X-100 and 0.05 mg/ml proteinase K. The primers used were: 5′-AACAGTCTTACGCTGTTCCGATG-3′ (sense), 5′-ATGTGGCACTGGGCTTGAACTC-3′ (antisense), 5′-CTCGTCGTGACCCATGGCGAT-3′ (Neomycin FW). PCR was performed in one reaction and resulted in a 276 bp fragment product for WT and a 600 bp band for knockout mice.
Infraorbital nerve constriction
To produce the painful condition of trigeminal neuralgia, we used a model of unilateral CION [25] according to a method previously described for rats [28, 35] with some modifications. Briefly, mice were anesthetized with an intramuscular (i.m.) injection of a mixture of ketamine (Fort Dodge Animal Health LTD, UK) and medetomidine (Orion Pharma, UK) in the doses of 50 and 0.5 mg/kg, respectively. One incision was made below the right eye 1 mm caudal to the myofascial (vibrissal) pads. The superior lip elevator and anterior superficial masseter muscles were dissected to expose the rostral end of the infraorbital nerve, as it emerges from the infraorbital fissure. Extra care was taken to prevent facial nerve damage. Two silk ligatures (No. 8.0, Ethicon) were loosely tied at a distance of 2 mm around the infraorbital nerve, producing a development of orofacial hypersensitivity and preventing infraorbital nerve destruction [35]. The incision was closed with polypropylene sutures (No. 6.0, Ethicon). After surgery, all animals were injected i.m. with atipamezole (5 mg/kg, Orion Pharma) and were maintained in a warmed area until full recovery from anaesthesia. Animals operated on an identical manner with no ligature applied to the nerve were used as control (sham).
Behavioral tests
Measurement of mechanical hyperalgesia
Mice were acclimatized in an individual animal enclosure (4″ × 4″) chamber for at least 1 h. Sham-operated or CION-injured animals were submitted to a repeated mechanical stimulus (0.04 g von Frey filament) applied to the forehead surface innervated by the trigeminal nerve (Fig. 1a). Each trial was repeated 10 times at 30-s interval at least. The occurrence of head reactions (attack/escape or head withdrawal) was expressed for each trial as the mean of percentage reactions that represents an index of mechanical nociceptive sensitivity [27]. To determine basal mechanical sensitivity, the mechanical stimulus was applied to the forehead of animals 1 day before surgery. A development of mechanical hyperalgesia following CION was estimated at different days after surgery.
Measurement of thermal hyperalgesia
Thermal hyperalgesia of the orofacial area was measured as previously described [28, 36]. Each animal was held in front of a radiant heat source positioned 1 cm from the vibrissal pad surface (Fig. 1b). The intensity of thermal stimulus applied to the vibrissal pad skin was adjusted to temperature of ~50 °C (15 s cut off). Once the animal started to withdraw its head or vigorously flick a snout, the heat beam was turned off. The time between the start of the beam and a functional response was defined as a latency of response. A reduction in the response latency reflected thermal hyperalgesia. To determine basal thermal sensitivity, a heat stimulus was applied to the ipsilateral side of the snout 1 day before surgery. Basal latency of responses was typically from 10 to 15 s. The development of thermal hyperalgesia following CION was measured at different days after surgery.
Nav1.9 expression analysis real-time PCR
After animal euthanasia (with a CO2 chamber following by cervical dislocation), TG was quickly removed and placed in buffer RTL and RNA was isolated using RNeasy mini kit (Quiagen) according to the manufacturer instructions. The RNA was quantified by Nanodrop and converted to cDNA using iScript™ reverse transcription supermix for qRT-PCR (BioRad) according to the manufacturer instructions. PCR amplifications were performed using a Mastercycler ep realplex (Eppendorf). All samples were run in triplicate in a final volume of 20 µl containing 10 ng of cDNA, 1 µM of primers and SsoAdvanced™ universal SYBR® Green supermix (BioRad), according to manufacturer protocol. Prior to PCR, an 8 min enzyme activation step was done at 95 °C. The PCR protocol consisted of 10 s denaturation at 95 °C, 5 s at annealing temperature at 63 °C, 10 s elongation at 72 °C for 40 cycles. The annealing temperature was confirmed by melting curve. The primers sequences used were the following: TTCCACTCTACGTACCTTCCGAGT (forward) and ATTCCCATGAAGAGCTGCTGACCA (reverse) for Nav1.9; TGCGACTTCAACAGCAACTC (forward) and CTTGCTCAGTGTCCTTGCTG (reverse) for GAPDH.
Nav1.9 and GAPDH cDNA relative amounts were calculated function of the samples cycle threshold (Ct) using a standard concentration curve constructed with a serial dilution (10 times) from 100 to 0.01 ng of cDNA mix. For each sample, the relative amount of Nav1.9 cDNA was normalized by the GAPDH cDNA amount.
Statistical analysis
Data were analyzed using GraphPad Prism 5. The results are presented as mean ± SEM with n referring to the number of animals tested. Multiple groups were compared using two-way analysis of variance [37] followed by Bonferroni post hoc test or Student paired t test as indicated.
Values of p less than 0.05 was considered as statistically significant for either test used.
Authors’ contributions
APL: research concept, animal surgery and behavioural studies, data analysis and interpretation, drafting of the manuscript; OK: research concept, drafting of the manuscript; SS-V: animal surgery and pictures; JNW: research concept, supervision of studies, critical revision of the manuscript. All authors read and approved the final manuscript.
Acknowledgements
This work was supported by Brazilian National Research Council Grant (CNPq), Brazilian Government Scholarship Programme Science without Border (A.P.L.), Welcome Trust, the MRC and Arthritis UK Grants (J.N.W.). The authors thank Dr. Michael S. Minett for advice in behavioural studies.
Competing interests
The authors declare that they have no competing interests.
Abbreviations
- AP
action potential
- ATP
adenosine triphosphate
- CCI
chronic constriction injury
- CFA
complete freund adjuvant
- CION
constriction of the infraorbital nerve
- DRG
dorsal root ganglion
- IL-1β
interleukin-1 beta
- mRNA
messenger ribonucleic acid
- Nav1.8
voltage-gated sodium channel 1.8 subtype
- Nav1.9
voltage-gated sodium channel 1.9 subtype
- qRT-PCR
quantitative reverse transcription polymerase chain reaction
- SCN11A
gene coding voltage-gated sodium channel 1.9 subtype
- TG
trigeminal ganglion
- TTX-R
tetrodotoxin-resistant
Footnotes
Ana Paula Luiz and Olga Kopach contributed equally to this work
Contributor Information
Ana Paula Luiz, Phone: +44 207 679 0793, Email: a.luiz@ucl.ac.uk.
Olga Kopach, Email: o.kopach@ucl.ac.uk.
Sonia Santana-Varela, Email: s.santana@ucl.ac.uk.
John N. Wood, Email: j.wood@ucl.ac.uk
References
- 1.Wilcox SL, et al. Trigeminal nerve anatomy in neuropathic and non-neuropathic orofacial pain patients. J Pain. 2013;14(8):865–872. doi: 10.1016/j.jpain.2013.02.014. [DOI] [PubMed] [Google Scholar]
- 2.Amaya F, et al. Diversity of expression of the sensory neuron-specific TTX-resistant voltage-gated sodium ion channels SNS and SNS2. Mol Cell Neurosci. 2000;15(4):331–342. doi: 10.1006/mcne.1999.0828. [DOI] [PubMed] [Google Scholar]
- 3.Padilla F, et al. Expression and localization of the Nav1.9 sodium channel in enteric neurons and in trigeminal sensory endings: implication for intestinal reflex function and orofacial pain. Mol Cell Neurosci. 2007;35(1):138–152. doi: 10.1016/j.mcn.2007.02.008. [DOI] [PubMed] [Google Scholar]
- 4.Scroggs RS. The distribution of low-threshold TTX-resistant Na+ currents in rat trigeminal ganglion cells. Neuroscience. 2012;222:205–214. doi: 10.1016/j.neuroscience.2012.07.012. [DOI] [PubMed] [Google Scholar]
- 5.Akopian AN, Sivilotti L, Wood JN. A tetrodotoxin-resistant voltage-gated sodium channel expressed by sensory neurons. Nature. 1996;379(6562):257–262. doi: 10.1038/379257a0. [DOI] [PubMed] [Google Scholar]
- 6.Black JA, et al. Changes in the expression of tetrodotoxin-sensitive sodium channels within dorsal root ganglia neurons in inflammatory pain. Pain. 2004;108(3):237–247. doi: 10.1016/j.pain.2003.12.035. [DOI] [PubMed] [Google Scholar]
- 7.Coste B, Crest M, Delmas P. Pharmacological dissection and distribution of NaN/Nav1.9, T-type Ca2+ currents, and mechanically activated cation currents in different populations of DRG neurons. J Gen Physiol. 2007;129(1):57–77. doi: 10.1085/jgp.200609665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dib-Hajj S, et al. NaN/Nav1.9: a sodium channel with unique properties. Trends Neurosci. 2002;25(5):253–259. doi: 10.1016/S0166-2236(02)02150-1. [DOI] [PubMed] [Google Scholar]
- 9.Dib-Hajj SD, et al. NaN, a novel voltage-gated Na channel, is expressed preferentially in peripheral sensory neurons and down-regulated after axotomy. Proc Natl Acad Sci USA. 1998;95(15):8963–8968. doi: 10.1073/pnas.95.15.8963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Matsumoto S, et al. Effect of 8-bromo-cAMP on the tetrodotoxin-resistant sodium (Nav 1.8) current in small-diameter nodose ganglion neurons. Neuropharmacology. 2007;52(3):904–924. doi: 10.1016/j.neuropharm.2006.10.008. [DOI] [PubMed] [Google Scholar]
- 11.Fang X, et al. Intense isolectin-B4 binding in rat dorsal root ganglion neurons distinguishes C-fiber nociceptors with broad action potentials and high Nav1.9 expression. J Neurosci. 2006;26(27):7281–7292. doi: 10.1523/JNEUROSCI.1072-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Persson AK, et al. Sodium–calcium exchanger and multiple sodium channel isoforms in intra-epidermal nerve terminals. Mol Pain. 2010;6:84. doi: 10.1186/1744-8069-6-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Blair NT, Bean BP. Roles of tetrodotoxin (TTX)-sensitive Na+ current, TTX-resistant Na+ current, and Ca2+ current in the action potentials of nociceptive sensory neurons. J Neurosci. 2002;22(23):10277–10290. doi: 10.1523/JNEUROSCI.22-23-10277.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cummins TR, et al. Glial-derived neurotrophic factor upregulates expression of functional SNS and NaN sodium channels and their currents in axotomized dorsal root ganglion neurons. J Neurosci. 2000;20(23):8754–8761. doi: 10.1523/JNEUROSCI.20-23-08754.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Suwanchai A, et al. NaV 1.8, but not NaV 1.9, is upregulated in the inflamed dental pulp tissue of human primary teeth. Int Endod J. 2012;45(4):372–378. doi: 10.1111/j.1365-2591.2011.01986.x. [DOI] [PubMed] [Google Scholar]
- 16.Maingret F, et al. Inflammatory mediators increase Nav1.9 current and excitability in nociceptors through a coincident detection mechanism. J Gen Physiol. 2008;131(3):211–225. doi: 10.1085/jgp.200709935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Osorio N, Korogod S, Delmas P. Specialized functions of Nav1.5 and Nav1.9 channels in electrogenesis of myenteric neurons in intact mouse ganglia. J Neurosci. 2014;34(15):5233–5244. doi: 10.1523/JNEUROSCI.0057-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Coste B, et al. Gating and modulation of presumptive NaV1.9 channels in enteric and spinal sensory neurons. Mol Cell Neurosci. 2004;26(1):123–134. doi: 10.1016/j.mcn.2004.01.015. [DOI] [PubMed] [Google Scholar]
- 19.Cummins TR, et al. A novel persistent tetrodotoxin-resistant sodium current in SNS-null and wild-type small primary sensory neurons. J Neurosci. 1999;19(24):RC43. doi: 10.1523/JNEUROSCI.19-24-j0001.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Maruyama H, et al. Electrophysiological characterization of the tetrodotoxin-resistant Na+ channel, Na(v)1.9, in mouse dorsal root ganglion neurons. Pflugers Arch. 2004;449(1):76–87. doi: 10.1007/s00424-004-1315-0. [DOI] [PubMed] [Google Scholar]
- 21.Lolignier S, et al. Nav1.9 channel contributes to mechanical and heat pain hypersensitivity induced by subacute and chronic inflammation. PLoS One. 2011;6(8):e23083. doi: 10.1371/journal.pone.0023083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Priest BT, et al. Contribution of the tetrodotoxin-resistant voltage-gated sodium channel NaV1.9 to sensory transmission and nociceptive behavior. Proc Natl Acad Sci USA. 2005;102(26):9382–9387. doi: 10.1073/pnas.0501549102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hillsley K, et al. Dissecting the role of sodium currents in visceral sensory neurons in a model of chronic hyperexcitability using Nav1.8 and Nav1.9 null mice. J Physiol. 2006;576(Pt 1):257–267. doi: 10.1113/jphysiol.2006.113597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Minett MS, et al. Pain without nociceptors? Nav1.7-independent pain mechanisms. Cell Rep. 2014;6(2):301–312. doi: 10.1016/j.celrep.2013.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huang J, et al. Gain-of-function mutations in sodium channel Na(v)1.9 in painful neuropathy. Brain. 2014;137(Pt 6):1627–1642. doi: 10.1093/brain/awu079. [DOI] [PubMed] [Google Scholar]
- 26.Leipold E, et al. A de novo gain-of-function mutation in SCN11A causes loss of pain perception. Nat Genet. 2013;45(11):1399–1404. doi: 10.1038/ng.2767. [DOI] [PubMed] [Google Scholar]
- 27.Luiz AP, et al. Contribution and interaction of kinin receptors and dynorphin A in a model of trigeminal neuropathic pain in mice. Neuroscience. 2015;300:189–200. doi: 10.1016/j.neuroscience.2015.05.015. [DOI] [PubMed] [Google Scholar]
- 28.Luiz AP, et al. Kinin B(1) and B(2) receptors contribute to orofacial heat hyperalgesia induced by infraorbital nerve constriction injury in mice and rats. Neuropeptides. 2010;44(2):87–92. doi: 10.1016/j.npep.2009.10.005. [DOI] [PubMed] [Google Scholar]
- 29.Zhang XY, et al. Gain-of-function mutations in SCN11A cause familial episodic pain. Am J Hum Genet. 2013;93(5):957–966. doi: 10.1016/j.ajhg.2013.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Amaya F, et al. The voltage-gated sodium channel Na(v)1.9 is an effector of peripheral inflammatory pain hypersensitivity. J Neurosci. 2006;26(50):12852–12860. doi: 10.1523/JNEUROSCI.4015-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Leo S, D’Hooge R, Meert T. Exploring the role of nociceptor-specific sodium channels in pain transmission using Nav1.8 and Nav1.9 knockout mice. Behav Brain Res. 2010;208(1):149–157. doi: 10.1016/j.bbr.2009.11.023. [DOI] [PubMed] [Google Scholar]
- 32.Krzyzanowska A, Avendano C. Behavioral testing in rodent models of orofacial neuropathic and inflammatory pain. Brain Behav. 2012;2(5):678–697. doi: 10.1002/brb3.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Baker MD, et al. GTP-induced tetrodotoxin-resistant Na+ current regulates excitability in mouse and rat small diameter sensory neurones. J Physiol. 2003;548(Pt 2):373–382. doi: 10.1113/jphysiol.2003.039131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ostman JA, et al. GTP up-regulated persistent Na+ current and enhanced nociceptor excitability require NaV1.9. J Physiol. 2008;586(4):1077–1087. doi: 10.1113/jphysiol.2007.147942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vos BP, Strassman AM, Maciewicz RJ. Behavioral evidence of trigeminal neuropathic pain following chronic constriction injury to the rat’s infraorbital nerve. J Neurosci. 1994;14(5 Pt 1):2708–2723. doi: 10.1523/JNEUROSCI.14-05-02708.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Almeida TF, Roizenblatt S, Tufik S. Afferent pain pathways: a neuroanatomical review. Brain Res. 2004;1000(1–2):40–56. doi: 10.1016/j.brainres.2003.10.073. [DOI] [PubMed] [Google Scholar]
- 37.Bergamaschi G, et al. Saporin, a ribosome-inactivating protein used to prepare immunotoxins, induces cell death via apoptosis. Br J Haematol. 1996;93(4):789–794. doi: 10.1046/j.1365-2141.1996.d01-1730.x. [DOI] [PubMed] [Google Scholar]