

ABSTRACT
Chlamydia trachomatis is an obligate intracellular bacterium that is a globally important human pathogen. The chlamydial plasmid is an attenuating virulence factor, but the molecular basis for attenuation is not understood. Chlamydiae replicate within a membrane-bound vacuole termed an inclusion, where they undergo a biphasic developmental growth cycle and differentiate from noninfectious into infectious organisms. Late in the developmental cycle, the fragile chlamydia-laden inclusion retains its integrity by surrounding itself with scaffolds of host cytoskeletal proteins. The ability of chlamydiae to developmentally free themselves from this cytoskeleton network is a fundamental virulence trait of the pathogen. Here, we show that plasmidless chlamydiae are incapable of disrupting their cytoskeletal entrapment and remain intracellular as stable mature inclusions that support high numbers of infectious organisms. By using deletion mutants of the eight plasmid-carried genes (Δpgp1 to Δpgp8), we show that Pgp4, a transcriptional regulator of multiple chromosomal genes, is required for exit. Exit of chlamydiae is dependent on protein synthesis and is inhibited by the compound C1, an inhibitor of the type III secretion system (T3S). Exit of plasmid-free and Δpgp4 organisms, which failed to lyse infected cells, was rescued by latrunculin B, an inhibitor of actin polymerization. Our findings describe a genetic mechanism of chlamydial exit from host cells that is dependent on an unknown pgp4-regulated chromosomal T3S effector gene.
IMPORTANCE
Chlamydia’s obligate intracellular life style requires both entry into and exit from host cells. Virulence factors that function in exiting are unknown. The chlamydial inclusion is stabilized late in the infection cycle by F-actin. A prerequisite of chlamydial exit is its ability to disassemble actin from the inclusion. We show that chlamydial plasmid-free organisms, and also a plasmid gene protein 4 (pgp4) null mutant, do not disassociate actin from the inclusion and fail to exit cells. We further provide evidence that Pgp4-regulated exit is dependent on the chlamydial type III secretion system. This study is the first to define a genetic mechanism that functions in chlamydial lytic exit from host cells. The findings also have practical implications for understanding why plasmid-free chlamydiae are highly attenuated and have the ability to elicit robust protective immune responses.
INTRODUCTION
Diseases caused by Chlamydia trachomatis bacteria are important global health problems. Ocular serovars (A to C) and genital serovars (D to K) are epithelium-tropic pathogens that infect the ocular and genital mucosae, respectively (1). Ocular infection can result in trachoma, the world’s leading cause of infectious blindness (2), whereas genital serovars are the leading cause of bacterial sexually transmitted disease (3) and a major risk factor in the transmission of HIV. The pathophysiology of chlamydial infections is poorly understood, but the organism’s highly conserved cryptic plasmid has recently emerged as a key virulence factor. Plasmidless organisms are highly attenuated in mouse (4) and nonhuman primate models of infections yet, paradoxically, generate superior levels of protective immunity against virulent plasmid-bearing organisms (5). The molecular basis for these infection characteristics is contributed by two of the plasmid’s eight open reading frames (ORFs): pgp3 and pgp4, respectively. A pgp3 deletion mutant is partially attenuated for mice (6), but the molecular basis for attenuation is unknown. Pgp4 is a transcriptional regulator of multiple chromosome genes (7) that are candidate virulence factors, but their roles in pathogenesis remain undefined.
A primary reason for chlamydia’s success as a pathogen is linked to its highly specialized and unique biphasic developmental cycle (8). The elementary body (EB) is a small (200 nm) metabolically inert extracellular form of the organism that initiates infection by attaching to and entering host cells. The internalized EB is confined to a membrane-bound vacuole termed an inclusion, which is rapidly modified, rendering it nonfusogenic with host lysosomes. The EB rapidly differentiates into a larger (800 nm) metabolically active noninfectious reticulate body (RB) that replicates by binary fission. The RB undergoes a secondary differentiation process back to the infectious EB, followed by host cell lysis and a release of EBs that reinitiate a new infectious cycle. The entire developmental cycle is completed between 36 and 48 h postinfection (p.i.).
An intriguing theme that has emerged in the study of chlamydial infection biology is the pathogen’s strategy to exploit the host cytoskeleton to facilitate entry, mobility of the cytosolic inclusion, and exit. EBs invade host cells by secreting the type III secretion system (T3S) effector translocated actin recruiting protein (TARP). TARP functions in invasion (9) by activating a Rac-dependent GTPase signaling cascade that rapidly recruits actin to the entry site (10). The EB-laden inclusion is then trafficked along microtubules, through a dynein-dependent mechanism, to the microtubule-organizing center (11). This migration positions the inclusion at the perinuclear region juxtapositioned against the trans-Golgi apparatus, where it acquires sphingomyelin (12) and cholesterol (13) from the host. The inclusion then undergoes a rapid expansion in size and number of chlamydial organisms. This expansive growth results in a fragile inclusion membrane that is stabilized by a RhoA GTPase-dependent mechanism that deposits scaffolds of F-actin and vimentin (14, 15) around the periphery of the inclusion. To exit host cells, chlamydiae must dismantle themselves from their entanglement in this cytoskeleton network. They achieve this, in part, by secretion of the chlamydial protease activity factor (CPAF) that cleaves vimentin but, interestingly, not actin (16). CPAF cleavage of vimentin is thought to relax the inclusion membrane, allowing for its optimum expansion late in the growth cycle. However, a central and unanswered question is how do chlamydiae dispose of actin late in the growth cycle to guarantee their successful exit and reinitiation of the infectious cycle? There is evidence that they accomplish this through a Rho GTPase actin-depolymerizing mechanism (17), but the chlamydial gene(s) that functions in this capacity is unknown.
Here, we show that the chlamydial plasmid plays an important role in exit from host cells. We demonstrate that chlamydial plasmid-free organisms, and a plasmid gene protein 4 null mutant (Δpgp4), do not disassociate actin from the inclusion and fail to exit cells.
RESULTS
Plaque-forming efficiency is plasmid and pgp4 dependent.
To better understand the role of the chlamydial plasmid in host-cell interactions, we initially compared the plaque-forming efficiencies of wild-type (L2), plasmid-deficient (L2R), and L2R cells transformed with the shuttle vector pBRCT (L2pBRCT), which possesses all eight plasmid ORFs in L929 cells (Fig. 1). In these and all subsequent experiments described here, the culture medium was not supplemented with cycloheximide. This was done to more closely mimic natural infection of mammalian host cells. Importantly, the addition of cycloheximide to culture medium significantly diminishes the lysis and exit phenotypes described here. There was a significant reduction (10-fold) in plaque-forming efficiency between the L2R and L2 strains (Fig. 1A). Transformation of L2R with pBRCT restored its plaque-forming phenotype to that of the wild-type L2 strain. One-step growth curves were created to compare the strains’ intracellular growth characteristics, as these could impact their plaque-forming efficiencies. L2, L2R, and L2RpBRCT strains grew at nearly identical rates postinfection and yielded similar infectious bursts at the completion of their growth cycles (Fig. 1B). Thus, the observed differences in plaque-forming efficiency between L2 and L2R cells were not due to differences in their ability to grow intracellularly. A more plausible explanation for our findings is that the plasmid plays a role in the lysis of infected cells, allowing release of infectious EBs that are then capable of initiating a new infection cycle which results in the formation of plaques. We observed similar results with Chlamydia muridarum and C. trachomatis serovar D plasmid-free strains (see Fig. S1 in the supplemental material), suggesting a conserved functional role for the plasmid in the lysis of chlamydia-infected cells.
FIG 1 .

Chlamydial plaque-forming efficiency is plasmid and pgp4 dependent. (A) PFU on L929 cell monolayers infected with a multiplicity of infection (MOI) of 0.0002 of wild-type (L2), plasmidless (L2R), or L2RpBRCT cells. (B) One-step growth curves were prepared for L929 cells infected with L2, L2R, and L2RpBRCT cells at an MOI of 5. Recoverable IFU were determined at various times postinfeciton. Each time point represents the mean rIFU from triplicate cultures. (C) PFU on L929 cell monolayers infected with an MOI of 0.0002 of L2, L2R, L2RpBRCT, or plasmid ORF deletion mutants. L2RpΔpgp4 had no effect on plaque formation, which indicated it has no or minimum impact on downstream gene expression; a finding consistent with other reports (18). (D) PFU on L929 cell monolayers infected with an MOI of 0.0002 of L2, L2R, L2RpBRCT, or plasmid ORF nonsense mutants. P values are shown above the bar graphs for the indicated comparisons. Plaques ranging in size from 2 to 4 mm were counted. The data represent the average results of three independent experiments.
Which plasmid gene controls cell lysis? We used the L2 strain to define the plasmid’s role in cell lysis, due to its predominant lytic phenotype and because we had previously generated a series of L2 plasmid deletion mutants. Of the eight plasmid ORFs, pgp1, pgp2, pgp6, and pgp8 are required for plasmid maintenance and are not amenable for the construction of stable transformants (7). Stable transformants of the remaining four ORFs, produced as either deletion mutants (L2RpΔpgp3, L2RpΔpgp4, L2RpΔpgp5, L2RpΔpgp7, and L2RpΔpgp34 [7]) or as single nucleotide polymorphism (SNP) nonsense mutations (L2Rppgp3T212A, L2Rppgp4A37T, and L2Rppgp3T212A/pgp4A37T) (see Fig. S2 in the supplemental material) were analyzed for their ability to form plaques. We found that the deletion mutants L2RpΔpgp3, L2RpΔpgp4, and L2RpΔpgp34 exhibited an L2R-like reduction in plaque-forming efficiency (Fig. 1C). Because the 3′ deletion in pgp3 has been previously shown to affect the downstream expression of pgp4 (18), it was not possible to assign a functional role for either gene in the plaque assay when we used deletion mutants. On the other hand, when the pgp3 and pgp4 nonsense mutants were tested, only the L2Rppgp4A37T mutant exhibited an L2R plaque-forming phenotype (Fig. 1D). These results demonstrate that the ability of chlamydiae to form plaques is pgp4 dependent.
Plasmid-deficient L2R and L2RpΔpgp4 do not exit host cells but remain highly infectious.
To understand the mechanism of pgp4-linked plaque formation, we examined the interactions of L2, L2R, L2RpBRCT, L2RpΔpgp4, and L2RpΔpgp7 cells following infection of human cervical epithelial cells (HeLa 229). For these experiments, HeLa 229 cells were infected and observed at different times postinfection by phase microscopy for lysis and stained with iodine to detect glycogen production (Fig. 2). As previously described (7), L2, L2RpBRCT, and L2RpΔpgp7 inclusions accumulated glycogen, whereas inclusions of L2R and L2RpΔpgp4 did not (Fig. 2A). Notably, there was a marked difference in the ability of these strains to lyse cells. Cells infected with L2, L2RpBRCT, and L2RpΔpgp7 exhibited extensive cytopathology that resulted in complete lysis of the monolayer 72 h p.i. In contrast, infection with L2R or L2RpΔpgp4 did not lyse cells, despite extended incubation times (72 to 96 h p.i.). Remarkably, L2R and L2RpΔpgp4 organisms remained highly infectious despite being trapped within their intracellular environment (Fig. 2B). These findings clearly support a role for Pgp4 in chlamydial exit from host cells and provide a plausible explanation for why L2R and L2RpΔpgp4 exhibit significantly reduced plaque-forming efficiencies (Fig. 1).
FIG 2 .
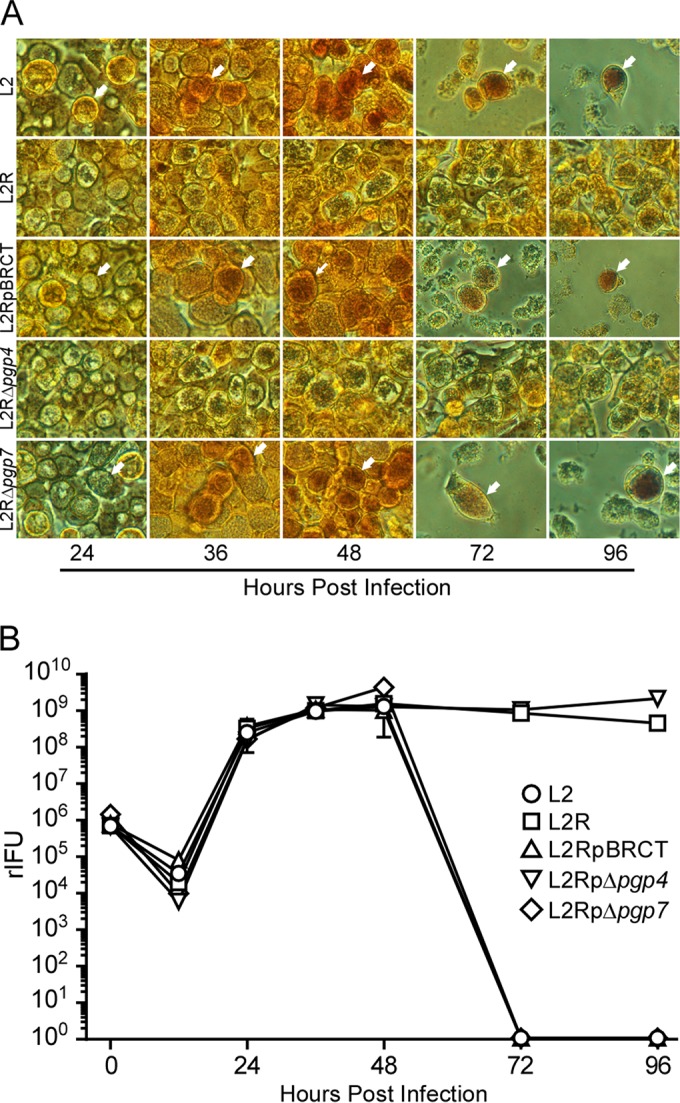
Plasmid-deficient and L2RpΔpgp4 chlamydiae do not exit host cells but remain highly infectious. (A) L2, L2R, L2RpBRCT, L2RpΔpgp4, or L2RpΔpgp7 was used to infect HeLa 229 cells at a multiplicity of infection (MOI) of 5. Cells were examined by phase microscopy (magnification, ×40) and stained with iodine to detect glycogen at various times postinfection. Inclusions positive for glycogen are marked with white arrows. (B) One-step growth curves were prepared with HeLa 229 cells infected with L2, L2R, L2RpBRCT, L2RpΔpgp4, or L2RpΔpgp7 cells at an MOI of 5. rIFU were determined at various times postinfection. Each time point represents the mean rIFU from triplicate cell cultures. Note the lack of lysis of cells infected with L2R or L2RpΔpgp4 and the sustained levels of rIFU from these infected cells at late time points (72 and 96 h p.i.). Results shown are from one of three independent experiments.
Chlamydial exit requires plasmid-mediated actin depolymerization.
To understand the mechanism of Pgp4-mediated exit, we focused on the host cytoskeletal proteins actin and vimentin, which are recruited to the inclusion during the middle to late stage of the developmental cycle (14). It has been proposed that these proteins provide structural support to the rapidly expanding and fragile inclusion membrane late in the developmental cycle (14). Predictably, the inclusion membranes of L2R and L2RpΔpgp4 cells stained positive for actin (Fig. 3A), and the number of inclusions staining positive increased at later times postinfection (Fig. 3B). A similar staining pattern was also observed for vimentin (Fig. 4). We hypothesized that if L2R and L2RpΔpgp4 were incapable of removing these proteins from the inclusion that prevented their exit, it should be possible to rescue them by treatment with latrunculin B or Withaferin A, inhibitors of actin and vimentin polymerization, respectively. For these experiments, cells were incubated for 48 h p.i., and the medium was removed and replaced with medium containing latrunculin B or Withaferin A. The treated infected cells were incubated an additional 8 h, and the culture medium was collected and assayed for recoverable inclusion-forming units (rIFU). Latrunculin B treatment rescued L2R and L2RpΔpgp4 infectivity. An approximate 100-fold increase in rIFU was found in the supernatants of latrunculin B-treated cultures (Fig. 3C). In contrast, Withaferin A treatment, which is known to disassemble vimentin while leaving F-actin networks intact, was ineffective in rescuing infectivity (Fig. 4C). Collectively, these findings implicate actin as the primary cytoskeletal protein responsible for providing the structural stability to the chlamydial inclusion. Furthermore, they clearly support a role for Pgp4 in controlling the disposition of actin from the inclusion membrane late in the growth cycle, which provides a novel escape strategy allowing chlamydiae to exit cells.
FIG 3 .

The inability of L2R and L2RpΔpgp4 strains to exit cells can be rescued by latrunculin B. Arrows identify F-actin (A), the inclusion (I), and nucleus (N). (A) HeLa 229 cells were infected with L2R or L2RpΔpgp4 at a multiplicity of infection (MOI) of 5 or 2.5, respectively, fixed, and stained for actin with rhodamine-conjugated phalloidin and DAPI at various times postinfection. Note the assembly of actin at the inclusion membrane. (B) HeLa 229 cells were infected with L2R or L2RpΔpgp4 at an MOI of 5 or 2.5, respectively, and the numbers of actin-positive inclusions were quantified at various times postinfection. (C) HeLa 229 cells infected with wild-type L2, L2R, L2RpBRCT, or L2RpΔpgp4 at an MOI of 5 for 48 to 52 h were treated for 8 h with latrunculin B (500 nM) or dimethyl sulfoxide (DMSO). Following treatment, rIFU in the supernatants were assayed. HeLa cell viability was not affected by treatment with 500 nM latrunculin B for 8 hours, as determined in a lactate dehydrogenase assay. The results shown are representative of three independent experiments.
FIG 4 .
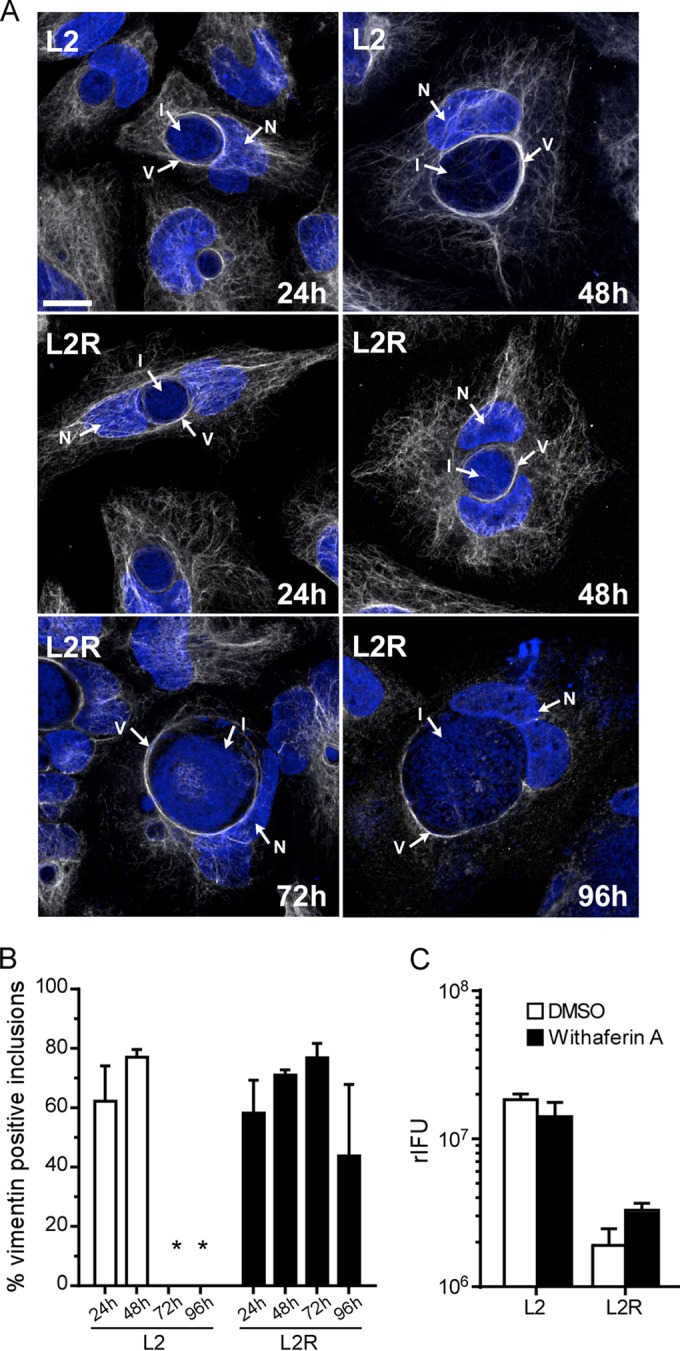
Vimentin is not essential for chlamydial exit from host cells. Arrows identify vimentin (V), the inclusion (I), and nucleus (N). (A) HeLa 229 cells were infected with L2 or L2R at a multiplicity of infection (MOI) of 5, fixed and stained with anti-vimentin antibody, followed by indirect immunofluorescence and DAPI staining at various times postinfection. (A) L2 and L2R stained positive for vimentin (arrows), and the positive staining persisted in L2R cells at late time points (72 and 96 h p.i.). (B) Assembly of vimentin filaments increased with time. The asterisks indicate lysis of the cell monolayers at 72 and 96 h p.i. (C) HeLa 229 cells were infected with L2 or L2R at an MOI of 5 for 50 h and then treated with Withaferin A (2 µM; Chromadex) or dimethyl sulfoxide (DMSO) for 8 h. The effect of treatment was determined by measuring rIFU in cell culture supernatants. Note that unlike latrunculin B, Withaferin A did not rescue L2R infectivity. The results shown are representative of three independent experiments.
Plasmid-mediated exit is dependent on chlamydial protein synthesis and is blocked by a type III secretion inhibitor.
HeLa 229 cells infected with L2, L2R, L2RpBRCT, or L2RpΔpgp4 were treated with chloramphenicol, an inhibitor of prokaryotic protein synthesis, to determine if chlamydial exit was protein dependent. We assayed for the rIFU in the supernatant of treated cells as the measure of exit. Chloramphenicol treatment of L2 and L2RpBRCT late in the developmental cycle significantly reduced their ability to exit cells (Fig. 5A) but had no effect on L2R or L2RpΔpgp4 exit. Moreover, treatment with compound C1 [N'-(3,5-dibromo-2-hydroxybenzylidene)-4-nitrobenzohydrazide], a small-molecule inhibitor of the chlamydial T3S apparatus, similarly prevented L2 and L2RpBRCT exit (Fig. 5B). These findings are consistent with a role for T3S in exit, but other possibilities cannot be formally excluded. Collectively, these findings support a role for a Pgp4 transcriptionally regulated late-cycle chromosomal gene(s) as an effector that targets the active removal of actin from the inclusion.
FIG 5 .

Plasmid-mediated exit is dependent on chlamydial protein synthesis and is blocked by a T3S inhibitor. (A) HeLa 229 cells were infected with L2, L2R, L2RpBRCT, or L2RpΔpgp4 at a multiplicity of infection (MOI) of 5 for 36 h and then treated with chloramphenicol (100 µg/ml) or dimethyl sulfoxide (DMSO) for 18 h. (B) HeLa 229 cells were infected with L2, L2R, L2RpBRCT, or L2RΔpgp4 at an MOI of 5 for 36 h and then treated with compound C1 (50 µM) for 18 h. The effect of treatment was determined by measuring the rIFU in the supernatants of infected cells. Results shown are from one of three independent experiments.
A CPAF null mutant does not exit host cells.
CPAF has been implicated in proteolytic modification of cytoskeletal proteins that leads to cell lysis (14). It was therefore important to study its potential role in actin depolymerization in our experimental system. HeLa cells were infected with an L2 CPAF null mutant (RST17) and its isogenic strain (RST5) and examined by phase microscopy at various times postinfection for cell lysis. Similar to the L2R and L2RpΔpgp4 strains, the CPAF null mutant also failed to lyse infected HeLa 229 cells when cultured in Dulbecco’s modified Eagle’s medium containing 10% fetal bovine serum (FBS; DMEM-10) without cycloheximide (Fig. 6). Because CPAF was present in both L2R and L2RpΔpgp4 strains, it was essential for us to exclude a direct role for CPAF in the proteolysis of actin as a confounding mechanism for our findings.
FIG 6 .

A C. trachomatis L2 CPAF null mutant does not exit host cells. CPAF isogenic (RST5) and CPAF null (RST17) strains were used to infect HeLa 229 cells at a multiplicity of infection of 1. Cells were examined by phase microscopy (magnification, ×400) at various times postinfection. Note the similar appearance and development of inclusions in both the isogenic and null strains at 24 and 48 h p.i. At later times (72 and 96 h p.i.), the CPAF isogenic strain lysed infected cells, as shown by the lack of discernible inclusions and the appearance of host cell debris. In contrast, the CPAF null-infected cells exhibited large mature inclusions at these later times postinfection and were morphologically similar to L2R-infected cells (see Fig. 2). The nonlytic phenotype of the CPAF null and L2R strains was dependent on culturing infected cells in DMEM-10 without cycloheximide.
Host cytoskeletal proteins are not cleaved in L2- or L2R-infected cells.
L2- and L2R-infected HeLa cells were lysed with either RIPA buffer or 8 M urea at various times postinfection and analyzed by Western blotting using a panel of antibodies against host cytoskeletal and chlamydial proteins (Fig. 7). Both lysis methods were employed, as it was recently shown that the complete inactivation of CPAF’s protease activity is not achieved following lysis of infected cells by using standard RIPA buffer but requires the direct lysis of cells in 8 M urea or hot SDS (19). We found that in RIPA-solubilized infected L2 and L2R cells, both of which expressed CPAF, there was an obvious degradation of keratin 18 and vimentin, but not of actin. In contrast, when L2- and L2R-infected cells where directly lysed in 8 M urea, there was no proteolytic cleavage of keratin 18 or vimentin. Collectively, these results argue against a direct role for CPAF, or a plasmid-regulated protease, in the cleavage of actin that is prerequisite for chlamydial lytic exit. Thus, it is most likely that CPAF plays an indirect but crucial role in plasmid-regulated host cell lysis.
FIG 7 .

Host cytoskeletal proteins are not degraded in L2- or L2R-infected cells. HeLa 229 cells were infected with L2 or L2R at a multiplicity of infection of 3. Lysates of chlamydia-infected HeLa 229 cells were prepared in RIPA buffer or 8 M urea at various times postinfection. Lysates were electrophoresed on 10% Criterion precast gels and probed with antibodies to keratin 18, vimentin, actin, CPAF, major outer membrane protein (MOMP), or glyceraldehyde-3-phosphate dehydrogenase (GAPDH). Note both L2 and L2R express CPAF. When infected cells were solubilized in RIPA buffer, keratin 18 and vimentin were cleaved. In contrast, neither protein was cleaved following lysis in 8 M urea. Notably, actin was not cleaved following lysis in RIPA or urea. We found no evidence for actin proteolysis in either strain, despite the fact that the strains express CPAF. We also observed higher levels of MOMP and less CPAF in L2-infected cells, but we do not know the significance of these findings.
DISCUSSION
Exit is an essential pathobiological event in the interaction of C. trachomatis with its host cell. Here, we present novel findings demonstrating that the chlamydial plasmid controls lytic exit. We further demonstrated by using deletion mutants of the eight plasmid-carried genes, that Pgp4, a transcriptional regulator of multiple chromosomal genes, is required for exit. We have provided evidence that Pgp4-regulated exit is dependent on the chlamydial T3S system (Fig. 5). This study is the first to define a genetic mechanism that functions in chlamydial lytic exit from host cells. A plasmid-regulated chromosomal gene(s) that is expressed late in the developmental cycle is a logical T3S effector candidate(s) that functions in the destabilization of actin filaments from the inclusion membrane. In fact, a rather restricted subset of genes meeting these criteria has been identified (7, 20–22), such as CTL0236, CTL0237, and CTLO238, which are part of a proposed T3S operon (20). It therefore should be feasible, using recently develop chlamydial genetic tools (23–25), to generate null mutants of these genes to define their possible effector functions in actin destabilization. Although our studies did not not implicate a potential mechanism for a T3S effector, they did exclude a proteolytic function (Fig. 7). Overall, this study supports the general conclusion and pathogenic strategy that chlamydial organisms manipulate the host cytoskeletal network throughout their developmental growth cycle for successful parasitism of mammalian host cells.
Chlamydiae exit by either a lytic or nonlytic inclusion extrusion mechanism (17). More aggressive strains or species, such as C. trachomatis strain LGV and C. muridarum, tend to be more lytic, whereas non-LGV genital and ocular strains tend to be less lytic and exit predominantly by extrusion (26). The ability of the LGV strain to form plaques was found to be predominantly, but not exclusively, plasmid dependent (Fig. 1). Small numbers of plaques were produced by both L2R and L2RΔpgp4 organisms. These findings implicate a role for different exit mechanisms. Perhaps the plasmid-independent mechanism was due to inclusion extrusion rather than cell lysis. Why do these strains exhibit different exit mechanisms? We hypothesize that the chlamydial plasmid plays an important role in the exit of all strains via its ability to depolymerize actin from the inclusion membrane, but differences in the structural integrity of the late inclusion membrane among strains dictates whether exit occurs predominately by lysis or extrusion. How might this occur? We speculate that it is the result of chlamydial phospholipase proteins that act on the inclusion membrane. The chlamydial plasticity zone (PZ) is the location of virulence genes that are known to differ among strains and function in strain-specific infection tropism and pathogenesis (27). Multiple phospholipase genes are located in the PZ. The number and structure of phospholipase genes are variable among chlamydial strains (27, 28) and are expressed mid- to late cycle (21). Aggressive strains, like LGV, with more or more highly active phospholipases, may have a greater propensity to weaken the integrity of the mature inclusion membrane. Following actin destabilization, the membrane ruptures, releasing chlamydiae into the cytosol and leading to lysis of host cells. In contrast, strains with lower phospholipase activity (non-LGV strains) have more stable inclusion membranes that resist spontaneous intracellular lysis. These inclusions are then extruded by the host cell intact by a myosin-dependent mechanism (29). Interestingly, differences in cell exit mechanisms strongly correlate with the in vivo invasiveness and host cell tropism of these strains, suggesting that the mechanisms are important determinants of pathogenesis. Based on our findings, and consistent with the in vivo growth environment results, a more accurate measurement of exit strategies among strains should be conducted using cell culture medium not supplemented with cycloheximide.
CPAF has been implicated in the proteolysis of vimentin (14) and therefore might have a role in modifying the cytoskeletal structure surrounding the inclusion that affects exit. We made the observation that, similar to L2R, the LGV CPAF null mutant (16) also failed to lyse infected HeLa 229 cells when cultured in the absence of cycloheximide (Fig. 6). However, we believe that the inability of CPAF null organisms to lyse cells is not a direct effect of the protein proteolytic function in modifying the cytoskeletal structure encapsulating the inclusion. The reasons for this conclusion are that L2R expressed normal levels of CPAF, yet we observed no proteolysis of actin or other cytoskeletal proteins in L2R-infected cells. Moreover, CPAF is expressed and localizes to the cytosol at midcycle (21, 30); thus, its modification of inclusion-stabilizing actin at this relatively early time point would theoretically lead to premature lysis. Collectively, our findings implicate a role for both the plasmid and CPAF in host cell lysis but suggest CPAF’s role is indirect rather than direct. What might this role be? CPAF cleaves the developmentally late-expressed cysteine-rich outer membrane protein OmcB (31). Thus, CPAF could also target other outer membrane proteins, specifically, a T3S apparatus or its cognate effectors that affect their function.
Plasmid genes are known virulence factors for many pathogenic bacteria. Chlamydiae are no exception, as plasmidless strains are highly attenuated (5). The inability to exit host cells in vivo could in part provide an explanation for this strong attenuating phenotype. For example, nonlytic natural infections would be incapable of efficiently spreading infection to other cells within the same host or transmitting infection between hosts. As shown here, nonlytic strain-infected cells are capable of supporting a complete chlamydial growth cycle, yielding large numbers of infectious organisms as well as chlamydia-secreted antigens. Might these stable isolated depots of diverse high antigenic mass serve as atypical targets for dendritic cells that are superior immunogens for the selective induction of protective antichlamydial T cell immunity with unique mucosal homing or retention properties? Our previous findings demonstrating that ocular infection of macaques with plasmid-free trachoma organisms elicited an unexpected CD8+ T cell-mediated protective immunity distinguished by a rapid recall of local T cell cytokines are consistent with this possibility.
MATERIALS AND METHODS
Chlamydia strains.
C. trachomatis L2/434/Bu, the plasmid-deficient strain L2R (25667R), an L2 CPAF null strain (Rst17), its L2 isogenic strain (Rst5), strain D/UW-3/Cx, and C. muridarum (Weiss) were grown in HeLa 229 cells in DMEM-10 supplemented with high glucose, 10% FBS, and gentamicin (1 µg/ml). EBs were purified by density gradient centrifugation (32).
Generation of Chlamydia transformants.
C. trachomatis transformants L2RpBRCT, L2RpΔpgp3, L2RpΔpgp4, L2RpΔpgp5, L2RpΔpgp7, and L2RpΔpgp34 were described previously (7). Nonsense mutants L2Rppgp3T212A, L2Rppgp4A37T, and L2Rppgp3T212A/pgp4A37T were similarly constructed and transformed into L2R cells.
Plaque assays and one-step growth curves.
Plaque assays and one-step growth curves were performed in L929 cells and HeLa 229 cells, respectively (33), and the culture medium was not supplemented with cycloheximide.
Cytoskeleton staining.
HeLa 229 cells were grown on 12-mm coverslips in 24-well plates. At various times postinfection, cells were fixed with 2.5% paraformaldehyde in phosphate-buffered saline for 15 min at 37°C. Following permeabilization with 0.1% Triton X-100, F-actin was detected with rhodamine-conjugated phalloidin (7.5 U/ml; Life Technologies). Vimentin was detected by indirect immunofluorescence using mouse antivimentin antibody (Sigma-Aldrich) followed by Alexa Fluor 568-conjugated anti-mouse secondary antibody (Life Technologies). Coverslips were mounted onto glass slides using ProLong Gold antifade reagent containing 4',6-diamidino-2-phenylindole (DAPI; Molecular Probes). Fluorescent images were obtained using a Zeiss LSM 710 confocal microscope.
Chlamydial exit inhibition assays.
The exit inhibition assays consisted of three parts. (i) First, actin destabilization was evaluated in HeLa 229 cells infected with chlamydiae for 48 to 52 h, rinsed with warm Hanks balanced salt solution (HBSS), and treated for 8 h with latrunculin B (500 nM; Sigma-Aldrich). (ii) Chlamydial protein synthesis was measured in HeLa 229 cells infected with chlamydiae for 36 h, rinsed with warm HBSS, and treated with chloramphenicol (100 μg/ml; Sigma-Aldrich) for an additional 18 h (iii) Finally, the T3S was evaluated in HeLa 229 cells infected with chlamydiae for 36 h, rinsed once with warm HBSS, and treated with 50 µM compound C1 for 18 h. The effect of the different treatments on chlamydial exit was determined by assaying rIFU in the supernatants of treated and untreated control cell cultures.
Immunoblotting.
Lysates of chlamydial-infected HeLa 229 cells were processed as previously described (34). Briefly, HeLa cells were grown and infected in 6-well tissue culture plates. Cells were (i) harvested by trypsinization, washed with ice-cold HBSS, and lysed on ice for 10 min in RIPA buffer (50 mM Tris [pH 7.5], 150 mM NaCl, 0.1% SDS, 0.5% sodium deoxycholate, 1% NP-40) supplemented with protease inhibitors (Roche) or (ii) solubilized directly in 8 M urea for 10 min on ice. Equal volumes of cell lysates were mixed with Laemmli sample buffer, boiled for 10 min, and electrophoresed on 10% Criterion precast gels (Bio-Rad). Proteins were transferred to nitrocellulose membranes and subjected to immunoblot analysis.
Statistical analyses.
GraphPad Prism 6.0 software was used for data analysis. Statistical significance was determined by using unpaired Student’s t tests for two groups or one-way analysis of variance for three or more groups. P values of <0.05 were considered statistically significant.
SUPPLEMENTAL MATERIAL
Plaque-forming efficiencies of C. muridarum and C. trachomatis serovar D. (A) PFU on L929 cell monolayers infected with a multiplicity of infection (MOI) of 0.0002 of C. muridarum P+, C. muridarum P−, or C. muridarum pBRCm. (B) PFU on L929 cell monolayers infected with an MOI of 0.0002 of C. trachomatis DP+ or DP−. The data shown represent the averages of three independent experiments. Download
A schematic showing the ORF and promoter locations for pgp3 and pgp4. Dashed lines indicate the deleted region for each construct. The vertical lines indicate the location of nonsense mutations, with the transversion mutation shown. Download
ACKNOWLEDGMENTS
We thank Grant McClarty, Michael Patton, and John Collins for critical reading and helpful suggestions in manuscript preparation. We thank Raphael Valdivia for the CPAF null (Rst17) and isogenic (Rst5) strains, Guangming Zhong for the anti-CPAF antibody, and Huizhou Fan for the T3S inhibitor C1. We thank Austin Athman, Anita Mora, and Hua Su for assistance with graphic arts and Kelly Haynes for manuscript preparation and editing.
This work was supported by the Intramural Research Program of the National Institute of Allergy and Infectious Diseases, National Institutes of Health.
Footnotes
Citation Yang C, Starr T, Song L, Carlson JH, Sturdevant GL, Beare PA, Whitmire WM, Caldwell HD. 2015. Chlamydial lytic exit from host cells is plasmid regulated. mBio 6(6):e01648-15. doi:10.1128/mBio.01648-15.
REFERENCES
- 1.Schachter J, Sweet RL, Grossman M, Landers D, Robbie M, Bishop E. 1986. Experience with the routine use of erythromycin for chlamydial infections in pregnancy. N Engl J Med 314:276–279. doi: 10.1056/NEJM198601303140503. [DOI] [PubMed] [Google Scholar]
- 2.Whitcher JP, Srinivasan M, Upadhyay MP. 2001. Corneal blindness: a global perspective. Bull World Health Organ 79:214–221. [PMC free article] [PubMed] [Google Scholar]
- 3.Brunham RC, Rey-Ladino J. 2005. Immunology of Chlamydia infection: implications for a Chlamydia trachomatis vaccine. Nat Rev Immunol 5:149–161. doi: 10.1038/nri1551. [DOI] [PubMed] [Google Scholar]
- 4.O’Connell CM, Ingalls RR, Andrews CW, Scurlock AM, Darville T. 2007. Plasmid-deficient Chlamydia muridarum fail to induce immune pathology and protect against oviduct disease. J Immunol 179:4027–4034. doi: 10.4049/jimmunol.179.6.4027. [DOI] [PubMed] [Google Scholar]
- 5.Kari L, Whitmire WM, Olivares-Zavaleta N, Goheen MM, Taylor LD, Carlson JH, Sturdevant GL, Lu C, Bakios LE, Randall LB, Parnell MJ, Zhong G, Caldwell HD. 2011. A live-attenuated chlamydial vaccine protects against trachoma in nonhuman primates. J Exp Med 208:2217–2223. doi: 10.1084/jem.20111266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu Y, Huang Y, Yang Z, Sun Y, Gong S, Hou S, Chen C, Li Z, Liu Q, Wu Y, Baseman J, Zhong G. 2014. Plasmid-encoded Pgp3 is a major virulence factor for Chlamydia muridarum to induce hydrosalpinx in mice. Infect Immun 82:5327–5335. doi: 10.1128/IAI.02576-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Song L, Carlson JH, Whitmire WM, Kari L, Virtaneva K, Sturdevant DE, Watkins H, Zhou B, Sturdevant GL, Porcella SF, McClarty G, Caldwell HD. 2013. Chlamydia trachomatis plasmid-encoded Pgp4 is a transcriptional regulator of virulence-associated genes. Infect Immun 81:636–644. doi: 10.1128/IAI.01305-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Moulder JW. 1991. Interaction of Chlamydiae and host cells in vitro. Microbiol Res 55:143–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Clifton DR, Fields KA, Grieshaber SS, Dooley CA, Fischer ER, Mead DJ, Carabeo RA, Hackstadt T. 2004. A chlamydial type III translocated protein is tyrosine phosphorylated at the site of entry and associated with recruitment of actin. Proc Natl Acad Sci U S A 101:10166–10171. doi: 10.1073/pnas.0402829101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lane BJ, Mutchler C, Al Khodor S, Grieshaber SS, Carabeo RA. 2008. Chlamydial entry involves TARP binding of guanine nucleotide exchange factors. PLoS Pathog 4:e1000014. doi: 10.1371/journal.ppat.1000014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Grieshaber S, Grieshaber N, Hackstadt T. 2003. Chlamydia trachomatis uses host cell dynein to traffic to the microtube organizing center in a p50 dynamitin independent process. J Cell Biol 116:3793–3802. doi: 10.1242/jcs.00695. [DOI] [PubMed] [Google Scholar]
- 12.Hackstadt T, Scidmore MA, Rockey DD. 1995. Lipid metabolism in Chlamydia trachomatis-infected cells: directed trafficking of Golgi-derived sphingolipids to the chlamydial inclusion. Proc Natl Acad Sci U S A 92:4877–4881. doi: 10.1073/pnas.92.11.4877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Carabeo RA, Mead DJ, Hackstadt T. 2003. Golgi-dependent transport of cholesterol to the Chlamydia trachomatis inclusion. Proc Natl Acad Sci U S A 100:6771–6776. doi: 10.1073/pnas.1131289100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kumar Y, Valdivia RH. 2008. Actin and intermediate filaments stabilize the Chlamydia trachomatis vacuole by forming dynamic structural scaffolds. Cell Host Microbe 4:159–169. doi: 10.1016/j.chom.2008.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Scidmore M. 2008. Chlamydia weave a protective cloak spun of actin and intermediate filaments. Cell Host Microbe 4:93–95. doi: 10.1016/j.chom.2008.07.006. [DOI] [PubMed] [Google Scholar]
- 16.Snavely EA, Kokes M, Dunn JD, Saka HA, Nguyen BD, Bastidas RJ, McCafferty DG, Valdivia RH. 2014. Reassessing the role of the secreted protease CPAF in Chlamydia trachomatis infection through genetic approaches. Pathog Dis 71:336–351. doi: 10.1111/2049-632X.12179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hybiske K, Stephens RS. 2007. Mechanisms of host cell exit by the intracellular bacterium Chlamydia. Proc Natl Acad Sci U S A 104:11430–11435. doi: 10.1073/pnas.0703218104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gong S, Yang Z, Lei L, Shen L, Zhong G. 2013. Characterization of Chlamydia trachomatis plasmid-encoded open reading frames. J Bacteriol 195:3819–3826. doi: 10.1128/JB.00511-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Johnson KA, Lee JK, Chen AL, Tan M, Sutterlin C. 2015. Induction and inhibition of CPAF activity during analysis of Chlamydia-infected cells. Pathog Dis 73:1–8. doi: 10.1093/femspd/ftv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bavoil PM, Hsia R. 1998. Type III secretion in Chlamydia: a case of deja vu? Mol Microbiol 28:860–862. doi: 10.1046/j.1365-2958.1998.00861.x. [DOI] [PubMed] [Google Scholar]
- 21.Belland RJ, Zhong G, Crane DD, Hogan D, Sturdevant D, Sharma J, Beatty WL, Caldwell HD. 2003. Genomic transcriptional profiling of the developmental cycle of Chlamydia trachomatis. Proc Natl Acad Sci U S A 100:8478–8483. doi: 10.1073/pnas.1331135100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fields KA, Fischer ER, Mead DJ, Hackstadt T. 2005. Analysis of putative Chlamydia trachomatis chaperones Scc2 and Scc3 and their use in the identification of type III secretion substrates. J Bacteriol 187:6466–6478. doi: 10.1128/JB.187.18.6466-6478.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Johnson CM, Fisher DJ. 2013. Site-specific, insertional inactivation of incA in Chlamydia trachomatis using a group II intron. PLoS One 8:e83989. doi: 10.1371/journal.pone.0083989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kari L, Goheen MM, Randall LB, Taylor LD, Carlson JH, Whitmire WM, Virok D, Rajaram K, Endresz V, McClarty G, Nelson DE, Caldwell HD. 2011. Generation of targeted Chlamydia trachomatis null mutants. Proc Natl Acad Sci U S A 108:7189–7193. doi: 10.1073/pnas.1102229108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nguyen BD, Valdivia RH. 2012. Virulence determinants in the obligate intracellular pathogen Chlamydia trachomatis revealed by forward genetic approaches. Proc Natl Acad Sci U S A 109:1263–1268. doi: 10.1073/pnas.1117884109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Todd WJ, Caldwell HD. 1985. The interaction of Chlamydia trachomatis with host cells: ultrastructural studies of the mechanism of release of a biovar II strain from HeLa 229 cells. J Infect Dis 151:1037–1044. doi: 10.1093/infdis/151.6.1037. [DOI] [PubMed] [Google Scholar]
- 27.Nelson DE, Crane DD, Taylor LD, Dorward DW, Goheen MM, Caldwell HD. 2006. Inhibition of Chlamydiae by primary alcohols correlates with the strain-specific complement of plasticity zone phospholipase D genes. Infect Immun 74:73–80. doi: 10.1128/IAI.74.1.73-80.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jeffrey BM, Suchland RJ, Quinn KL, Davidson JR, Stamm WE, Rockey DD. 2010. Genome sequencing of recent clinical Chlamydia trachomatis strains identifies loci associated with tissue tropism and regions of apparent recombination. Infect Immun 78:2544–2553. doi: 10.1128/IAI.01324-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lutter E, Barger A, Nair V, Hackstadt T. 2013. Chlamydia trachomatis inclusion membrane protein CT228 recruits elements of the myosin phosphatase pathway to regulate release mechanisms. Cell Rep 3:1921–1931. doi: 10.1016/j.celrep.2013.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhong G, Fan P, Ji H, Dong F, Huang Y. 2001. Identification of a chlamydial protease-like activity factor responsible for the degradation of host transcription factors. J Exp Med 193:935–942. doi: 10.1084/jem.193.8.935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hou S, Lei L, Yang Z, Qi M, Liu Q, Zhong G. 2013. Chlamydia trachomatis outer membrane complex protein B (OmcB) is processed by the protease CPAF. J Bacteriol 195:951–957. doi: 10.1128/JB.02087-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Caldwell HD, Kromhout J, Schachter J. 1981. Purification and partial characterization of the major outer membrane protein of Chlamydia trachomatis. Infect Immun 31:1161–1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Carlson JH, Whitmire WM, Crane DD, Wicke L, Virtaneva K, Sturdevant DE, Kupko JJ, Porcella SF, Martinez-Orengo N, Heinzen RA, Kari L, Caldwell HD. 2008. The Chlamydia trachomatis plasmid is a transcriptional regulator of chromosomal genes and a virulence factor. Infect Immun 76:2273–2283. doi: 10.1128/IAI.00102-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen AL, Johnson KA, Lee JK, Sütterlin C, Tan M. 2012. CPAF: a chlamydial protease in search of an authentic substrate. PLoS Pathog 8:e1002842. doi: 10.1371/journal.ppat.1002842. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Plaque-forming efficiencies of C. muridarum and C. trachomatis serovar D. (A) PFU on L929 cell monolayers infected with a multiplicity of infection (MOI) of 0.0002 of C. muridarum P+, C. muridarum P−, or C. muridarum pBRCm. (B) PFU on L929 cell monolayers infected with an MOI of 0.0002 of C. trachomatis DP+ or DP−. The data shown represent the averages of three independent experiments. Download
A schematic showing the ORF and promoter locations for pgp3 and pgp4. Dashed lines indicate the deleted region for each construct. The vertical lines indicate the location of nonsense mutations, with the transversion mutation shown. Download