

Abstract

In addition to its well-known roles as an electrophile and general acid, the side chain of histidine often serves as a hydrogen bond (H-bond) acceptor. These H-bonds provide a convenient pH-dependent switch for local structure and functional motifs. In hundreds of instances, a histidine caps the N-terminus of α- and 310-helices by forming a backbone NH···Nδ1 H-bond. To characterize the resilience and dynamics of the histidine cap, we measured the trans H-bond scalar coupling constant, 2hJNN, in several forms of Group 1 truncated hemoglobins and cytochrome b5. The set of 19 measured 2hJNN values were between 4.0 and 5.4 Hz, generally smaller than in nucleic acids (∼6–10 Hz) and indicative of longer, weaker bonds in the studied proteins. A positive linear correlation between 2hJNN and the difference in imidazole ring 15N chemical shift (Δ15N = |δ15Nδ1 – δ15Nε2|) was found to be consistent with variable H-bond length and variable cap population related to the ionization of histidine in the capping and noncapping states. The relative ease of 2hJNN detection suggests that this parameter can become part of the standard arsenal for describing histidines in helix caps and other key structural and catalytic elements involving NH···N H-bonds. The combined nucleic acid and protein data extend the utility of 2hJNN as a sensitive marker of local structural, dynamic, and thermodynamic properties in biomolecules.
The importance of hydrogen bonds (H-bonds) in the folding and structural organization of biomacromolecules has been recognized for decades. “Hydrogen Bonding in Biological Structures”1 presents an excellent historical perspective extending to the late 1980s. Since then, nuclear magnetic resonance (NMR) spectroscopy has emerged as an essential tool in the characterization of hydrogen bonding in both proteins and nucleic acids. Indeed, numerous NMR observables are available for assessing the presence, strength, and other fundamental characteristics of H-bonds. They include chemical shifts, fractionation factors, temperature coefficients, and hydrogen exchange rates. In favorable instances, these indirect manifestations can be complemented with direct detection of the H-bonding interaction. The parameter of interest in this work is the scalar coupling that connects NMR-active nuclei across the H-bond.2−5 H-Bond scalar couplings (HBCs) have the advantage of identifying donor and acceptor atoms and are extraordinarily sensitive to geometric properties such as H-bond length.5−9 In addition, because the magnitude of an HBC is affected by time and ensemble averaging, HBCs can also inform on the local dynamics and energetics of H-bonds within biomacromolecules.8−10
The predominant H-bond of proteins is the backbone–backbone N–H···O=C′ sustaining regular secondary structure. The 15N–13C three-bond (3hJNC′) and 1H–13C two-bond (2hJHC′) couplings associated with these and similar interactions have been measured in several proteins under various conditions.2,8,11−15 These couplings are invariably small (<1 Hz), which has focused application to diamagnetic proteins of fewer than 100 residues because of their favorable T1 and T2 relaxation times. In contrast, N–H···N H-bonds are relatively rare but exhibit 15N–15N two-bond couplings (2hJNN) between 2 and 11 Hz,16−18 an experimentally accessible range for proteins with less-than-optimal relaxation properties.
Protein N–H···N H-bonds frequently involve a histidine as the acceptor and another histidine or a backbone amide as the donor. A perusal of X-ray structures suggests an average of one bond of the latter type in every three deposited structures and fewer yet of the side chain to side chain type. However, such rarity does not imply that protein N–H···N H-bonds are insignificant; participation in active sites and critical elements of structure, for example, N-terminal caps of helices,19 along with unique ionization and tautomeric properties make histidines and their H-bonds especially interesting to study.
In its helix N-capping role, the histidine uses the Nδ1 atom as an H-bond acceptor and adopts the Nε2-H tautomeric state. Two capping configurations predominate, which we denote as i-to-i+3 and i-to-i–2, where i is the donor amide (Figure 1). Recently, we have reported the detection of i-to-i+3 N–H···Nδ1 H-bonds within the α-helix N-caps of ankyrin repeat (AR) proteins.18 The consensus AR N-cap has the sequence TXXH and is unusual in that the histidine Nδ1 atom acts as a bifurcated H-bond acceptor to both threonine amide and hydroxyl hydrogens (Figure 1B) in an arrangement that conditions inter-repeat packing and the geometry of the first turn of helix. In the study presented here, we expanded the AR protein experiments to a greater set of histidine N-caps and chose those caps in heme proteins (truncated hemoglobins and cytochrome b5) so that changes in heme placement, oxidation state, ligation state, or post-translational modification (PTM) could be exploited as perturbations to the N–H···Nδ1 bond.
Figure 1.
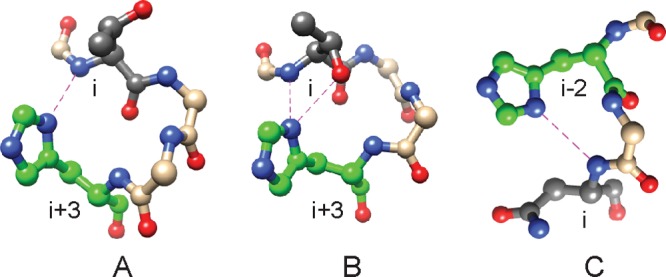
Helix capping by histidine. (A) i-to-i+3 N–H···Nδ1 H-bond in Synechococcus GlbN (PDB entry 4MAX).20 (B) Bifurcated i-to-i+3 N–H···Nδ1···H–Oγ interaction in the consensus AR (PDB entry 2BKG).21 (C) i-to-i–2 N–H···Nδ1 H-bond in rat mitochondrial cytochrome b5 (PDB entry 4HIL).
Truncated hemoglobins (TrHbs) share a basic architecture composed of seven helices labeled A–C and E–H by analogy to the canonical globins.22 In one group of TrHbs (TrHb1s), α-helix G is initiated by a strong start signal19 of the i-to-i+3 variety. The motif uses a histidine at the i+3 position in hundreds of TrHb1s, including those from the unicellular eukaryotes Chlamydomonas eugametos (CtrHb) and Chlamydomonas reinhardtii (THB1), and from the cyanobacteria Synechocystis sp. PCC 6803 (Synechocystis GlbN) and Synechococcus sp. PCC 7002 (Synechococcus GlbN). These homologous TrHb1s (∼40–50% identical) differ in the sequence of the cap and the residues immediately preceding and following it. The TrHb1 cap is close to the heme group and on the same side as the “proximal” histidine (Figure S1A), a conserved ligand to the iron in the ferric (oxidized) and ferrous (reduced) states.
THB1 and both GlbNs are “hexacoordinate” hemoglobins. They ligate the heme iron with the proximal histidine and a distal lysine (THB1)23 or histidine (GlbNs).24,25 Exogenous ligands such as O2 can displace the distal residue and force a conformational rearrangement bringing a tyrosine from the B helix (Tyr B10) and glutamine(s) from the E helix into the distal heme pocket.20,23,26 The B and E helices affected by the ligand replacement are remote from the helix-capping H-bond (Figure S1A). Comparing HBCs with and without exogenous ligand therefore offers an opportunity to assess long-range coupling between the distal and proximal sides of the heme.
In addition to hexacoordination, the two GlbNs have the ability to react with the heme group (Figure 2). The irreversible post-translational modification (PTM) involves His117, a noncoordinating histidine located on the H helix near the heme 2-vinyl group.27 Heme reduction in the absence of oxygen causes spontaneous formation of the His117 Nε2–2-Cα heme linkage (Figure 2B)28,29 and generates “GlbN-A”. Recent work has demonstrated that analogous histidine–heme modifications can be engineered at the 4-vinyl group with the Leu79His replacement.30 In the absence of the native (2-vinyl) cross-link, the 4-vinyl-reacted protein is denoted as GlbN-B (Figure 2C), whereas the doubly cross-linked material is termed GlbN-AB (Figure 2D). The “B” cross-link was also implanted into CtrHb with the Leu75His replacement (CtrHb-B).31 Panels C and D of Figure 2 emphasize the proximity of the engineered modification to the α-helix N-cap. For our purposes, the artificial linkages serve to probe the response of the G helix N-cap to a nearby structural perturbation.
Figure 2.

Heme b and its modifications in TrHb1s. (A) Heme b. (B) Native cross-link in wild-type Synechococcus and Synechocystis GlbN (GlbN-A). (C) Cross-link engineered in GlbN by placing a histidine at position 79 (GlbN-B). (D) Double cross-link engineered in GlbN (GlbN-AB). Amino acid labeling corresponds to that of Synechocystis GlbN.
The i-to-i–2 motif often has a proline at the i–1 position and forms a tight turn adequate for capping 310-helices. This second type of N-capping N–H···Nδ1 H-bond is found in the electron transfer protein cytochrome b5, where it initiates the C-terminal helix of the soluble heme domain. The H-bond is formed between His80 Nδ1 and Asp82 N–H and is more than 20 Å from the heme binding pocket (Figure S1B). The cap is preserved in the apoprotein,32 although an ∼1% capless population can be detected in the NMR data.32,33Figure 1C shows the structure of the 310 histidine N-cap.
The goals of this work were 2-fold. First, we wanted to explore the use of 2hJNN as a semiquantitative reporter of stress in helix caps formed with a histidine side chain. The HBC values collected on TrHb1s and cytochrome b5 in different states would inform on long-range transmission of perturbation. Second, we sought to add to the HBCs acquired on the helix caps of AR proteins18 and compare the NMR properties of protein N–H···N H-bonds with those of nucleic acids.3,34,35 These goals required the accurate measurement of 2hJNN HBCs in multiple proteins. As will be demonstrated, the data set uncovers trends in NMR parameters and extends the utility of 2hJNN HBCs for biophysical characterization in proteins. It also provides insights into the distinctive properties of HBCs involving an acceptor histidine.
Materials and Methods
Protein Expression and Purification
Overexpression and purification of uniformly 15N-labeled recombinant ferric TrHb1s and cytochrome b5 were achieved as described previously.23,25,36,37 A summary of the published procedures is provided in the Supporting Information. Covalently modified hemoglobins (GlbN-A, GlbN-B, GlbN-AB, and CtrHb-B) were produced by reduction of the ferric or cyanomet holoprotein with a 2–5-fold molar excess of dithionite (DT, >85%, Alfa-Aesar) for at least 15 min, followed by oxidation and purification.30
NMR Sample Preparation
Lyophilized ferric recombinant protein was dissolved in NMR buffer [0–125 mM sodium/potassium phosphate (pH 7.0–7.5) and 10% D2O]. Cyanide-bound ferric hemoglobins were prepared by addition of a 2–5-fold molar excess of KCN (J. T. Baker). Ferrous cytochrome b5 was produced by DT reduction of an ∼3 mM ferric protein (1.5-fold molar excess of DT to protein) sample. To prevent evaporation or oxidation, protein samples were transferred to Shigemi tubes and sealed with Parafilm M prior to the collection of NMR data. Protein concentrations ranged from 500 μM to 5 mM, although the concentrations of most samples were from ∼1.0 to 2.5 mM. Under the chosen conditions, each protein is monomeric. Specific NMR sample conditions are listed in Table S1.
NMR Data Acquisition
NMR spectroscopy was conducted using 600.13 MHz Bruker Avance or 600.53 MHz Avance-II spectrometers, each equipped with a cryogenic probe. 1H–15N HSQC, histidine-selective 1H–15N long-range (LR) HMQC,38 soft 1H–(N)–15N COSY,39 and quantitative 2hJNN constant-time spin–echo (CTSE) difference 1D/2D HSQC spectra39 were acquired as detailed elsewhere.18 A typical 2hJNN CTSE series consisted of 12–20 1D or 2D spectra using different τ modulation periods and two or three duplicate values. For a representative 1–2 mM protein sample, a single 2hJNN CTSE 1D spectrum was acquired with 512–1024 transients, a constant-time relaxation delay of ∼150 ms, and a recycle delay of ∼1.0 s and therefore required ∼10–20 min per data point. All ferric proteins in this study are paramagnetic (S = 1/2). Unless otherwise noted, NMR data were collected at 313 K to improve H-bond detection. 1H chemical shifts were referenced with respect to the water line (4.58 ppm at 313 K and 4.76 ppm at 298 K); 15N chemical shifts were referenced indirectly using the Ξ ratio.40
NMR Data Processing, Analysis, and Curve Fitting
NMR data were processed with NMRPipe41 or Topspin 2.1 (Bruker BioSpin). Spectra were analyzed using Sparky 3.42 For 2hJNN modulation data, 1D peak intensities were obtained using the deconvolution (mixed Lorentzian/Gaussian) routine of Topspin 2.1. 2D peak volumes were calculated by peak integration (Sparky 3). Peak intensities were tabulated and plotted as a function of the 2hJNN modulation time, τ. The fit to the data was performed with Kaleidagraph (Synergy Software) and the equation I(τ) = A cos(πJτ) to extract the initial amplitude (A) and J coupling magnitude (|2hJNN|).43 For some 2hJNN modulation curves, peak heights were substituted for intensities to yield better signal-to-noise ratios. However, the choice of heights or intensities did not significantly affect the best fit 2hJNN values. In most instances, the fitting error for 2hJNN was well below 0.1 Hz. The experimental error was based on peak intensities (or heights) in duplicate spectra collected at the beginning and end of an experimental series. The combined error estimate reported in Table 1 was typically <5%. The 2hJNN modulation curves plotted in Figure S3 have been normalized by their individually fitted amplitudes to facilitate comparison of peaks with different intensities.
Table 1. 2hJNN Values, Amide Donor 1H Chemical Shifts, and Histidine 15Nδ1 and 15Nε2 Chemical Shifts Observed for N–H···Nδ1 Helix-Capping H-Bondsa.
| labelb | protein | distalc | |2hJNN| (Hz) | 1H (ppm) | δ15Nε2 (ppm) | δ15Nδ1 (ppm) |
|---|---|---|---|---|---|---|
| CtrHb, Md,e | CN | 5.3 ± 0.2 | 11.41 | ndf | 254.9 | |
| A | CtrHb, md | CN | 5.2 ± 0.2 | 11.39 | 166.6 | 254.9 |
| B | CtrHb, M | CN | 5.0 ± 0.1 | 11.17 | 166.5 | 255.1 |
| C | CtrHb-B | CN | 4.0 ± 0.3 | 10.45 | 168.6 | 249.6 |
| D | THB1, Mg | CN | 4.8 ± 0.1 | 11.18 | 166.4 | 256.6 |
| E | THB1, mg | CN | 4.6 ± 0.3 | 11.28 | 166.9 | 256.9 |
| F | Syn7002h GlbN | His | 4.7 ± 0.2 | 10.77 | 166.9 | 256.4 |
| G | Syn7002 GlbN-A | His | 4.8 ± 0.2 | 10.97 | 167.3 | 256.6 |
| H | Syn6803i GlbNg | CN | 5.0 ± 0.2 | 11.34 | 165.9 | 257.7 |
| I | Syn6803 GlbN-Ag | CN | 4.8 ± 0.2 | 11.40 | 166.1 | 258.0 |
| J | Syn6803 GlbN | His | 4.9 ± 0.2 | 11.14 | 165.8 | 257.4 |
| K | Syn6803 GlbN-A | His | 4.9 ± 0.2 | 11.28 | 166.0 | 257.5 |
| L | Syn6803 GlbN-B | His | 4.5 ± 0.2 | 10.93 | 166.9 | 255.3 |
| M | Syn6803 GlbN-AB | His | 4.3 ± 0.2 | 10.87 | 167.6 | 254.5 |
| N | Syn6803 GlbN-B | CN | 5.4 ± 0.1 | 11.40 | 166.0 | 256.9 |
| O | Syn6803 GlbN-AB | CN | 5.0 ± 0.1 | 11.29 | 166.4 | 257.1 |
| P | ferric cyt b5 | His | 5.0 ± 0.1 | 10.88 | 165.0 | 250.5 |
| Q | ferrous cyt b5 | His | 5.0 ± 0.1 | 10.94 | 165.3 | 250.6 |
| R | apocyt b5j | − | 5.3 ± 0.1 | 11.15 | 165.0 | 250.3 |
Measured at 313 K, 10% D2O, and pH 7.0–7.2 on proteins containing ferric heme iron unless otherwise noted.
Distal ligand to the heme iron.
M, major heme isomer; m, minor heme isomer.
Measured at 283 K.
Not determined.
Measured at pH 7.3–7.5.
Syn7002, Synechococcus sp. PCC 7002.
Syn6803, Synechocystis sp. PCC 6803.
Measured at 298 K.
Simulation of the pH Response
The influence of N–H···Nδ1 H-bonding on the histidine ionization equilibrium, observed 2hJNN couplings, and imidazole 15N chemical shifts was explored using simulations performed with Scilab 5.4.1 (Scilab Entreprises S.A.S.). The thermodynamic model assumed the existence of four states: (1) a capped configuration in which the N–H···Nδ1 hydrogen bond is intact, coupling is active [2hJNN = 2hJNN(capped)], and the histidine adopts the Nε2-H neutral tautomer, (2) an open state in which the hydrogen bond is broken (2hJNN = 0) and the histidine occurs as the Nε2-H neutral tautomer, (3) an open state in which the hydrogen bond is broken (2hJNN = 0) and the histidine occurs as the Nδ1-H neutral tautomer, and (4) an open imidazolium form (2hJNN = 0). The cis state of the cytochrome Xxx–Pro bond was not detected, and the Xxx–Pro cis–trans equilibrium was not considered in the modeling.
The input parameters included the microscopic pKa for the capped state and both open states. For the open state, we chose the values of Nα-acetyl-histidine methylamide to eliminate terminal charge and intramolecular H-bonding effects.44 As determined by Tanokura,45 the imidazolium form dissociates to the Nε2H tautomer (open, Nε2H) with a pKa of 6.53 and to the Nδ1H tautomer (open, Nδ1H) with a pKa of 6.92. The Hill coefficients (nH) characterizing proton uptake46 were all held at unity. The fractional populations of the four forms (capped, Nε2H; open, Nε2H; open, Nδ1H; and open, +) were evaluated as a function of pH using eqs 1–5.
| 1 |
| 2 |
| 3 |
| 4 |
| 5 |
where Q is the partition function using the open protonated state as the reference. The predicted histidine 15N chemical shifts were calculated assuming fast exchange on the 15N chemical shift time scale using eqs 6 and 7:
| 6 |
| 7 |
The coefficients in eqs 6 and 7 correspond to the limiting 15N chemical shift values of Vila (neutral open tautomers)47 and Pelton and co-workers (imidazolium);38 the limiting 15N chemical shift values for the “capped, Nε2H” state were chosen to encompass the experimentally determined heme protein 15N chemical shifts. We note that the open state pKas were determined at 310 K.45 No correction was applied to these values when considering experimental data at 313 K. Open state pKas at 298 K were adjusted assuming a histidine enthalpy of ionization of +30 kJ/mol.48
Database Analysis
The structures of proteins determined by X-ray crystallography with better than 1.5 Å resolution were collected from the Protein Data Bank (July 30, 2015) and culled at 70% identity. Each of the 3253 structures was examined for the presence of amide–histidine N–Nδ1 pairs having an interatomic distance of <3.2 Å. Hydrogens were ignored, and a small number of structures having short N–Cδ2 distances were not assessed for the plausibility of alternative histidine χ2 angles. A representative set of helix caps (one chain per structure) was extracted from the data to obtain inter-nitrogen distance distributions for the two types of caps. The histogram in Figure 11 was optimized for bin size with a published method.49 In addition, 77 myoglobin structures (resolution better than 1.5 Å) containing the His24–His119 pair were collected and the Nε2–Nε2 distances measured for comparison with the N-capping H-bonds.
Figure 11.
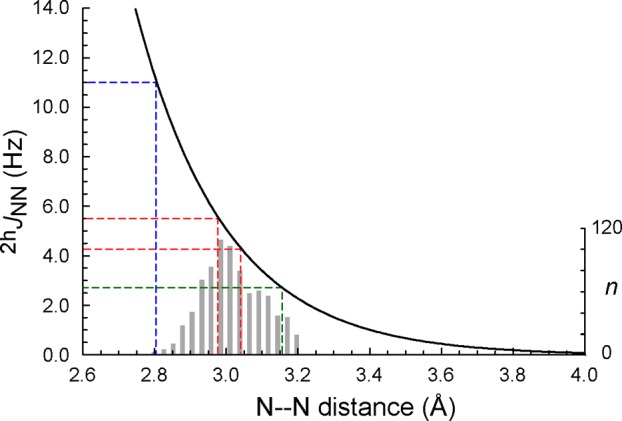
Dependence of 2hJNN(capped) on inter-nitrogen distance as approximated by the Fermi contact contribution to coupling.7 The equation is 2hJNN (Hz) = 795579 exp(−3.9868 d), where d is the inter-nitrogen distance in angstroms.7 The red dashed lines bracket the values measured in this work. The green dashed line indicates the 2hJNN(capped) predicted for the solvent-exposed cap of AR proteins. The blue dashed line indicates the value reported for the His24–His119 pair of apomyoglobin.17 The optimized histogram49 (right ordinate) is for 803 nonredundant i-to-i–2 and i-to-i+3 distances of <3.2 Å obtained from the PDB survey.
Results
N–H···N H-Bonding in Cyanomet CtrHb
We first present the data collected with one of the globins, namely CtrHb, in the ferric state with cyanide as the distal ligand [cyanomet CtrHb, or CtrHb-CN (PDB entry 1DLY)].50 The putative i-to-i+3 interaction in CtrHb involves Ser76 and His79 and is a convenient subject for illustrating the spectroscopic manifestations of the capping H-bonds along with the utility of solution phase characterization. The nuclei of interest are the backbone amide 15N and 1H nuclei and the histidine 15Nδ1, 1Hε1, 15Nε2, and 1Hδ2 nuclei. These are the targets of J-correlated experiments designed to establish unambiguously the existence of the N–H···Nδ1 H-bond.
The amide 1H–15N HSQC spectrum of CtrHb-CN shows two sets of cross-peaks (Figure 3A) corresponding to two heme orientations in the heme pocket. The major and minor isomers, related by an ∼180° rotation of the heme about its α–γ meso axis, are in slow exchange on the chemical shift time scale and occur in a 7:3 ratio in this protein.31 Sequence specific assignments identify the amide proton signals of Ser76 at downfield shifts of ∼11.2 ppm (major isomer) and ∼11.4 ppm (minor isomer). Notably, high-resolution HSQC data show that both Ser76 cross-peaks appear as 5 Hz doublets in the 15N dimension (Figure S2A,B), a fine structure that is absent from all other 15N–1H cross-peaks.
Figure 3.

Identification of the N–H···Nδ1 H-bond in CtrHb-CN. (A) 1H–15N HSQC (downfield amide 1H region). The minor heme isomer is denoted with a prime. (B) 1H–15N LR-HMQC (upfield 15N region). (C) 1H–15N LR-HMQC (downfield 15N region). (D) 1H–(N)–15Nδ1 COSY. (E) 1H-coupled 1Hε1–(Nδ1)–15N–(H) LR-COSY. (F) Ser76 N–H···Nδ1 His79 helix-capping scheme in C. eugametos CtrHb-CN. Correlated nuclei determined by the NMR data shown in panels D and E are highlighted with red and blue circles, respectively.
Histidine-selective 1H–15N LR-HMQC spectra obtained with CtrHb-CN were analyzed in previous work.31 They show that major His79 and minor His79 are deprotonated at neutral pH, adopt the Nε2H tautomer, and have Nδ1 signals at ∼255 ppm (Figure 3B,C and Table 1). Backbone amide 1H–15N HSQC spectra acquired with 15N decoupling centered at the His79 Nδ1 frequency during the evolution period contain collapsed Ser76 doublets (Figure S2C). This observation is in agreement with a ∼ 5 Hz 2hJNN caused by the Ser76–His79 H-bond. In addition, a weak cross peak between His79 15Nδ1 and the amide proton of Ser76 (major isomer) is detected in the high sensitivity 1H–15N LR-HMQC data (Figure S2D,E). Because these J-correlated nuclei are separated by 14 covalent bonds, the observed connectivity must arise via the one-bond trans HBC, 1hJHN.
To document further the presence of the N–H···Nδ1 H-bond, a soft 1H–(N)–15N COSY experiment18,39 was used. Unlike the LR-HMQC, the HNN-COSY pulse sequence utilizes the two-bond homonuclear HBC 2hJNN for N–H···N H-bond identification. This approach is advantageous because the 2hJNN coupling is often considerably larger than the 1hJHN coupling4 and therefore leads to a gain in sensitivity. Figure 3D illustrates the results of such an experiment. Two peaks are present in the spectrum and identify Ser76 amide 1H and His79 15Nδ1 within the N–H···N H-bonds (major and minor isomers). In agreement, the 1H-coupled 1H–(N)–15N–(H) LR COSY data51 shown in Figure 3E also display two 2hJNN-mediated peaks and correlate His79 1Hε1 with Ser76 15N, the latter split by its directly attached proton (|1JNH| ∼ 90 Hz). The NMR connectivity pattern shown in Figure 3 is unequivocal evidence of the N–H···Nδ1 H-bond between Ser76 and His79 in CtrHb-CN.
To determine if the 2hJNN coupling constants involving a histidine ring may be practical reporters for H-bond properties, we measured the magnitude of the 2hJNN couplings using a high-precision quantitative CTSE difference HSQC experiment.43 With this method, amide NH groups within N–H···Nδ1 H-bonds undergo 2hJNN modulation according to the timing (τ) of a histidine-selective 15Nδ1 inversion pulse. Repeating the experiment for different values of τ yields a modulation curve from which the magnitude of 2hJNN can be accurately extracted. Figure 4A presents the downfield region of 1D data collected for CtrHb-CN. The intensities of the resolved Ser76 NH protons were plotted as a function of the 2hJNN modulation time τ and fit according to the relationship I(τ) = A cos(πJτ).43
Figure 4.

Measurement of the 2hJNN magnitude in CtrHb-CN. (A) Stack plot showing the resolved amide proton resonances of Ser76 (major and minor isomers) as a function of the 2hJNN modulation time, τ. (B) Normalized Ser76 NH peak intensities (major isomer, black circles) as a function of τ. The horizontal black line indicates zero intensity, and the black dashed vertical line corresponds to the null time for 2hJNN = 5 Hz (100 ms). Simulated 2hJNN curves (black lines) between 4 and 6 Hz in 0.2 Hz increments are included for comparison.
The data in Figure 4B yield a well-defined value (|2hJNN| = 5.0 ± 0.1 Hz, major isomer) for the Ser76–His79 N–H···Nδ1 interaction, in agreement with the directly observed splitting (Figure S2A,B). The minor heme isomer has the same coupling constant within error [|2hJNN| = 5.2 ± 0.2 Hz (Figure S3A)]. Decreasing the temperature from 313 to 283 K yielded a relatively constant |2hJNN| [5.3 ± 0.2 Hz, major isomer (Figure S3S)]. The magnitude of the CtrHb-CN 2hJNN couplings is within the range expected from the few previously reported instances of amide N–H···N His H-bonds (∼2–6 Hz).16,18
N–H···N H-Bonding in THB1-CN and Synechocystis GlbN-CN
We collected HSQC, LR-HMQC, HNN-COSY, and 2hJNN CTSE difference spectra to characterize the N–H···N H-bonds in THB1-CN (Asn87–His90) and Synechocystis GlbN-CN (Asn80–His83). These proteins yielded HNN-COSY signals (Figure 5) and 2hJNN values (Table 1 and Figure S3D,E,H) similar to those of CtrHb-CN, in a suggestion that sequence context and the identity of the amide donor (Ser in CtrHb and Asn in Synechocystis GlbN and THB1) have minimal effects on the helix-capping H-bond. GlbN-CN with native heme PTM (GlbN-A-CN) yielded a 2hJNN value [4.8 ± 0.2 Hz (Figure S3I)] close to that in its unmodified form. Thus, the native His117–2-Cα cross-link appears to have little influence on the N–H···Nδ1 H-bond in the cyanomet complexes.
Figure 5.
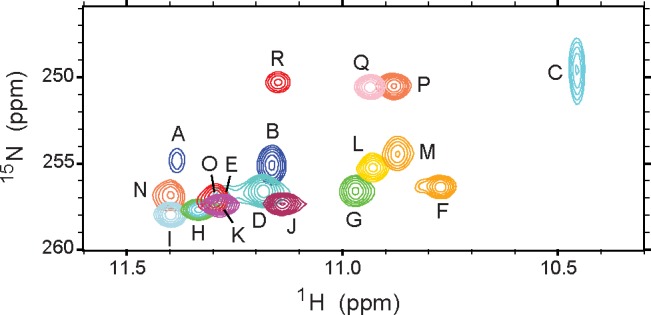
Overlay of HNN-COSY spectra. Data collected on hemoglobins and holocytochrome b5 at 313 K and apocytochrome b5 at 298 K (peak R). Each cross-peak corresponds to the amide 1H and His 15Nδ1 of an N–H···Nδ1 H-bond, correlated by 2hJNN. The peak labels are defined in the first column of Table 1.
N–H···N H-Bonding in Bis-histidine Synechocystis and Synechococcus GlbNs
To examine how binding of cyanide to the distal heme site and the consequent conformational rearrangement alter the N–H···N N-cap, we prepared Synechococcus and Synechocystis GlbNs with and without PTM in their ferric bis-histidine state. All four species yielded observable HNN-COSY cross-peaks (Figure 5) and 2hJNN values of 4.7–4.9 Hz (Table 1 and Figure S3F,G,J,K). These results indicate insensitivity to the distal ligand and associated structural differences in the wild-type GlbNs, regardless of heme modification status. In the case of Synechococcus GlbN, which contains a TXXH motif as in the previously studied AR proteins,18 no evidence of bifurcated N–H···Nδ1···H–Oγ H-bonding was observed. In addition, the relatively constant magnitude of 2hJNN detected for wild-type TrHb1s from C. eugametos, C. reinhardtii, Synechococcus, and Synechocystis (4.6–5.2 Hz) was greater than the 2hJNN values obtained for the bifurcated H-bonds of ARs (1.8–4.1 Hz),18 in support of a shorter N···Nδ1 distance in the former proteins.
Influence of Engineered His-Heme PTM on the Helix-Capping N–H···N H-Bond
Non-native heme modifications can be introduced into Synechocystis GlbN as depicted in panels C and D of Figure 2. Synechocystis GlbN-B and GlbN-AB exhibit bis-histidine coordination as in the wild-type protein.30Figure 5 shows the correlations between Asn80 backbone amide 1H and His83 15Nδ1 and therefore the presence of the helix-capping N–H···N H-bonds. However, the data demonstrate systematic upfield shifts for Asn80 1H and His83 15Nδ1 nuclei relative to GlbN-A. Measurement of the 2hJNN couplings (Table 1 and Figure S3L,M) also suggests H-bond lengthening (or weakening) relative to the bis-histidine wild-type reference.
Upon replacement of the distal histidine with cyanide in GlbN-B and GlbN-AB, the H-bonds persist (Figure 5). In contrast to observations in the bis-histidine state, Asn80 1H and His83 15Nδ1 resonances have large downfield shifts. Furthermore, a significant (∼16–20%) increase in the 2hJNN coupling constants is measured in both GlbN-AB-CN (+0.7 Hz; 2hJNN = 5.0 Hz) and GlbN-B-CN (+0.9 Hz; 2hJNN = 5.4 Hz) (Table 1 and Figure S3O,N). This is presumably due to H-bond relaxation relative to the strained bis-histidine complexes. Thus, formation of the engineered linkage to the FG turn, adjacent to the helix N-cap, couples distal cyanide binding to the proximal side of the heme pocket in a manner not observed in the wild-type proteins.
A non-native histidine–heme covalent modification analogous to that in Synechocystis GlbN-B can be engineered in CtrHb.31 HNN-COSY (Figure 5) and 2hJNN modulation experiments performed with CtrHb-B-CN (Figure S3C) demonstrate that the N–H···Nδ1 H-bond is considerably perturbed (2hJNN = 4.0 Hz) relative to its wild-type reference [2hJNN = 5.0 Hz (Table 1)]. These results suggest that the formation of the engineered cross-link to the FG turn in CtrHb-B-CN generates strain in the adjacent helix-capping H-bond. The ∼20% decrease in 2hJNN observed for CtrHb-B-CN compared to wild-type CtrHb-CN is in contrast to that of the GlbN-B-CN and GlbN-CN pair [8% increase by cross-linking (Table 1)]. The opposite effect of the engineered cross-link in CtrHb-CN and GlbN-CN on the G helix N-cap implicates a difference in the conformational plasticity of neighboring elements of structure. It is worth noting that the F-helix/FG loop sequence preceding the cap in CtrHb (VPHL) is shorter by two residues than in GlbN (VENHGL).
N–H···N H-Bonding in the His-Pro-Asp Motif of Cytochrome b5
To study the i-to-i–2 motif and its response to iron redox changes and heme binding, we prepared the ferric, ferrous, and apoprotein states of cytochrome b5. As in the hemoglobins, the N–H donor proton resonates downfield from its random shift (HSQC data on the ferric form shown in Figure S4A). Also in agreement with previous work,36 histidine-selective LR-HMQC spectra confirm that His80 is neutral and that its Nδ1 atom is deprotonated (Figure S4B). HNN-COSY data recorded for the ferric protein (Figure S4C), the ferrous protein, and the apoprotein (Figure 5) support the presence of the N–H···Nδ1 H-bond (Figure 1C) in all three forms of the cytochrome. Measurement of 2hJNN returned values of 5.0, 5.0, and 5.3 Hz for the ferric form (313 K), ferrous form (313 K), and apoprotein form (298 K, for stability reasons), respectively (Table 1 and Figure S3P–R). Thus, the His-Pro-Asp N–H···N H-bond of cytochrome b5 depends little on the iron redox state or the presence of the heme. Additionally, the cytochrome b5i-to-i–2 H-bond appears to be similar to those detected within the TrHb1 i-to-i+3 motifs.
Discussion
N–H···N H-Bonding, 2hJNN, and Histidine pKa
The 2hJNN data obtained with the hemoglobins and cytochrome suggest that this parameter can register long-range influences and be a reliable indicator of the state of an N-capping H-bond. However, to interpret the 2hJNN values, it is essential to consider that the observables may be ensemble averages over the H-bonded state having a limiting 2hJNN equal to 2hJNN(capped) and one or more nonbonded states having a 2hJNN equal to 0. We will assume that nonbonded states expose the histidine to solvent, in which case there are three possible open species: the imidazolium (protonated) state, the Nε2-H tautomer (imidazolium pKa ∼ 6.53), and the Nδ1-H tautomer (imidazolium pKa ∼ 6.92).44,45,52 The two tautomers partition in an ∼5:2 ratio dictated by the pKa values. The set of relevant equilibria is presented in Figure 6. Although the details of this scheme are expected to differ from protein to protein and conditions to conditions, the thermodynamic analysis provides a framework with which to scrutinize the significance of the 2hJNN measurements.
Figure 6.

Scheme used to assess the effect of fast exchange averaging on histidine–amide H-bonding and histidine 15N chemical shifts. The histidine can adopt an H-bonded state in which 2hJNN is active. The open states are characterized by a 2hJNN of 0 and partition according to sample pH and microscopic pKa values. Limiting 15Nδ1 and 15Nε2 chemical shifts38,47 are listed in parentheses. The limiting 15N chemical shifts for the H-bonded state are estimates based on globins with large Δ15N values and low apparent histidine pKa values.
To perform the simulation shown in Figure 7A, the microscopic pKa of the histidine in the H-bonded state was set to 4, 5, or 6. For each pKa value, the fractional population of all four states was calculated as a function of pH using eqs 1–5. As the histidine pKa in the capped state approaches that of the open state, the maximal attainable fraction of the H-bonded form drops below 1. Under such conditions, the observed 2hJNN value does not reach 2hJNN(capped) and the observed histidine chemical shifts do not correspond to the pure state limiting chemical shifts. Capping histidine pKas have been published32,36,53 or were estimated in this work (Figures S6–S8 and Table S2); they suggest that in most cases, the pKa is sufficiently low (<4.5) that a direct measurement of 2hJNN(capped) is possible. Open/closed averaging may be influential for the solvent-exposed cap of AR proteins,18 CtrHb-B-CN, and cytochrome b5, and in those instances, extrapolation of the observed 2hJNN is necessary to estimate 2hJNN(capped). In this framework, 2hJNN(capped) is a parameter determined primarily by bond length,54 and Kclose (Figure 6) is a measure of bond strength directly related to the free energy of H-bond formation, ΔGf°.55
Figure 7.
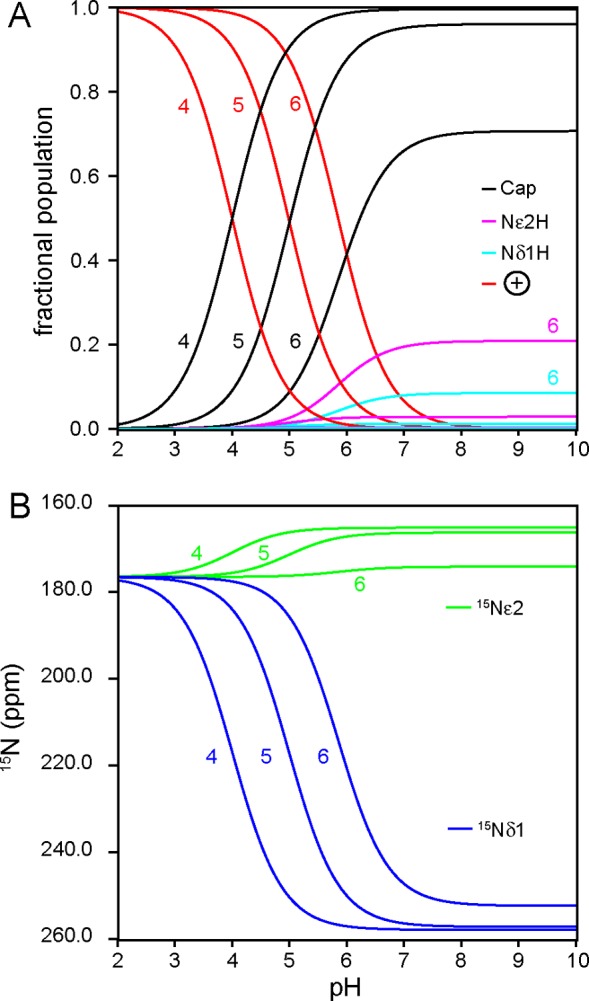
Effect of capping histidine pKa on (A) microstate fractional populations and (B) histidine 15Nδ1 and 15Nε2 chemical shifts. Fast exchange averaging among imidazolium (red trace), capped Nε2-H (black trace), and open Nδ1-H and Nε2-H histidine tautomers (cyan and magenta traces, respectively) was assumed. In panel B, the limiting 15N chemical shift values were as defined in Figure 6. In both simulations, the capping histidine pKa was set to 4, 5, or 6 as indicated on the traces. As the capping histidine pKa nears the model compound values, a significant proportion of open states persists at neutral and higher pH, which reduces the maximal observable Δ15N.
For the i-to-i+3 N–H···N N-cap of consensus AR proteins, we found that buried H-bonds have larger observed couplings (2hJNN ∼ 4–5 Hz) than H-bonds exposed to solvent (2hJNN ∼ 2 Hz). We also note that H-bonds with larger 2hJNN values are exhibited by histidines having a depressed ionization constant (pKa < 3), whereas those with smaller values have an only moderately depressed pKa (∼5.7). Assuming a macroscopic pKa of 5.7 (Figure S6), a simulation similar to that shown in Figure 7 leads to a closed state population of 77% at the pH of the AR 2hJNN measurement (6.6). The model indicates that increasing the pH by one unit leads to a closed state population of 86% and an increase in 2hJNN (+0.23 Hz), in good agreement with pH-dependent NMR data (Figure S6). Extrapolation to a vanishing population of open states predicts a 2hJNN(capped) of ∼2.7 Hz for the solvent-exposed H-bond, which can be meaningfully compared to the 2hJNN(capped) of buried H-bonds [where 2hJNN ∼ 2hJNN(capped) ∼ 4 Hz].
The same analysis was applied to CtrHb-CN. NMR data demonstrate that His79 in WT CtrHb-CN remains neutral until global acid unfolding, suggesting a pKa of <3.5 (Figure S7A,B). In contrast, His79 in L75H CtrHb-B-CN undergoes partial protonation at pH 5.5 in support of a pKa near 5 (Figure S7C,D). Thus, at pH 7.2, the wild-type N–H···Nδ1 H-bond is formed ∼100% of the time and the L75H variant H-bond only ∼95% of the time. A hypothetical fully formed H-bond in L75H CtrHb-B-CN would therefore give rise to a 2hJNN(capped) value of ∼4.2 Hz, lower than the wild-type value of 5.0 Hz. The parameters used in the simulations are listed in Table S3.
In both examples, the low 2hJNN(capped) and low observed 2hJNN values are likely manifestations of H-bond lengthening and concomitant increased sampling of open states, respectively. The contribution from open/closed averaging is also possible in cytochrome b5. When the estimated apoprotein pKas of the H-bonded (∼5.0)36 and open (∼6.9)32 states were used in the simulation, 2hJNN(capped) was evaluated at ∼5.5 Hz. In general, the modeling can be adjusted to account for deviations of the open state pKa from the fully exposed residue and guide the choice of experimental conditions under which a satisfactory estimate of 2hJNN(capped) can be obtained.
Fast Exchange Averaging and 2hJNN–Chemical Shift Correlations
Histidine nitrogen shifts have been the subject of multiple studies. 14N NMR studies of azoles56 and extension to 15N NMR of free histidine in solution,44 model imidazoles,57 and histidine within the catalytic triad of α-lytic protease58 define the ring nitrogens as one of two types: the pyrrole α-type, which is protonated and has an upfield 15N shift (167.5 ppm in imidazole and 176.5 ppm in imidazolium),38 and the pyridine β-type, which is deprotonated and resonates downfield (15N = 249.5 ppm).38 The canonical limiting values for neutral histidine were recently revised with density functional theory (DFT) calculations,47 which return shifts of 167.5 ppm (Nε2) and 261.5 ppm (Nδ1) for the Nε2-H tautomer and shifts of 183.5 ppm (Nδ1) and 266.5 ppm (Nε2) for the Nδ1-H tautomer (Figure 6). According to the revised values, the theoretical maximal separation between the Nδ1 and Nε2 signals (Δ15N = |δ15Nδ1 – δ15Nε2|) is 94 ppm for the Nε2-H tautomer and 83 ppm for the Nδ1-H tautomer.
Under fast exchange on the chemical shift time scale, the 15N chemical shifts of the neutral ring reflect the ∼5:2 Nε2-H:Nδ1-H ratio, and Δ15N is smaller than for the pure tautomeric states. As the pH is lowered, protonation of the ring occurs, which changes the β-nitrogen to an α-like nitrogen and also reduces Δ15N. The N-capping interaction favors the Nε2-H tautomer and contributes primarily to a large Δ15N, but secondarily, H-bonding tends to shift the acceptor Nδ1 β-type nitrogen upfield of its maximal value,59 thereby leading to a small reduction in Δ15N. These competing influences and local context typically render histidine 15N chemical shifts difficult to interpret.
A plot of 2hJNN versus Δ15N is shown in Figure 8 for the globins, cytochrome b5, and AR proteins. The observed 15N separations are below the DFT-derived maximum value of 94 ppm.
Figure 8.
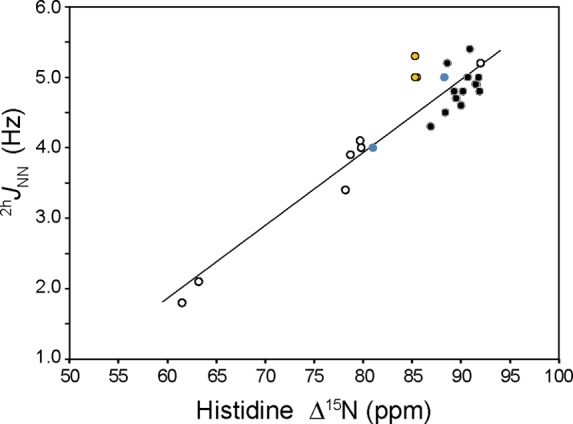
Plot of 2hJNN vs histidine Δ15N for globins (black fill), cytochrome b5 (orange fill), and AR proteins (○). Data for WT CtrHb-CN (for the major isomer, 2hJNN = 5.0 Hz and Δ15N = 88.3 ppm) and L75H CtrHb-B-CN (2hJNN = 4.0 Hz, and Δ15N = 81 ppm) are colored blue. Linear regression of the full data set returns a slope of 0.10 Hz/ppm and an x-intercept (Δ15N for 2hJNN = 0 Hz) of 43.3 ppm (r2 = 0.88). Linear regression of the AR protein data alone yields a slope of 0.11 Hz/ppm and an x-intercept of 45.4 ppm (r2 = 0.98), whereas the heme protein data alone (omitting apo cytochrome b5) return a slope of 0.06 Hz/ppm and an x-intercept of 10.5 ppm (r2 = 0.29).
Although significant scatter is present in the heme protein data, the composite Δ15N values are positively correlated to 2hJNN such that larger couplings are associated with larger Δ15N. This behavior is characterized by large variations in the acceptor (15Nδ1) chemical shift [larger coupling, larger shift (Table 1)]. The other ring nitrogen (15Nε2H) displays modest variations that are negatively correlated to 2hJNN (larger coupling, smaller shift). The AR protein data span the greatest range of 2hJNN (1.8–5.2 Hz), Δ15N (61.5–92.0 ppm), and pKa (<3.0–5.7) values; in addition, their 2hJNN versus Δ15N correlation is the strongest. In contrast, the heme protein data cluster around similar 2hJNN (4.0–5.4 Hz), Δ15N (81.0–91.9 ppm), and pKa (<4.0–5.0) values and show only a weak positive correlation between 2hJNN and Δ15N.
The positive correlation between the histidine Nδ1 chemical shift and 2hJNN is noteworthy. For a pyridine β-type acceptor that is H-bonded 100% of the time, as H-bond length decreases, we expect the magnitude of 2hJNN to increase and the 15Nδ1 shift to decrease (i.e., move upfield) as the nitrogen nucleus assumes greater α character.9 The reverse trend is observed. To determine if open/closed averaging may be responsible, we simulated the pH profile of the 15N chemical shifts as a function of histidine pKa predicted by the model shown in Figure 6. The imidazole 15N shifts, calculated with eqs 6 and 7, are presented in Figure 7B. Interestingly, the model predicts a linear relationship between Δ15N and the fractional population of the capped form; thus, a linear correlation between Δ15N and the magnitude of 2hJNN may also be expected. Assuming pH 7.2 and 2hJNN(capped) = 5 Hz, the model predicts a 0.09 Hz/ppm line with an x-intercept (Δ15N for 2hJNN = 0 Hz) of 37 ppm, values similar to those obtained from linear regression of the experimental data (Figure 8).
The 2hJNN values and chemical shifts for heme proteins (Table 1) and AR proteins18 were combined in a plot of 2hJNN coupling versus amide 1H chemical shifts (Figure 9). In this plot, the shifts were corrected for random coil values and the effect of nearest neighbors in the primary structure.60 Paramagnetic effects, though estimated to be small at the cap site (∼0.2 ppm), are likely to contribute to the scatter of the heme protein data along with secondary and tertiary structure differences. Despite this referencing problem, a positive correlation emerges by which greater 1H deshielding accompanies greater 2hJNN coupling, as anticipated.61
Figure 9.
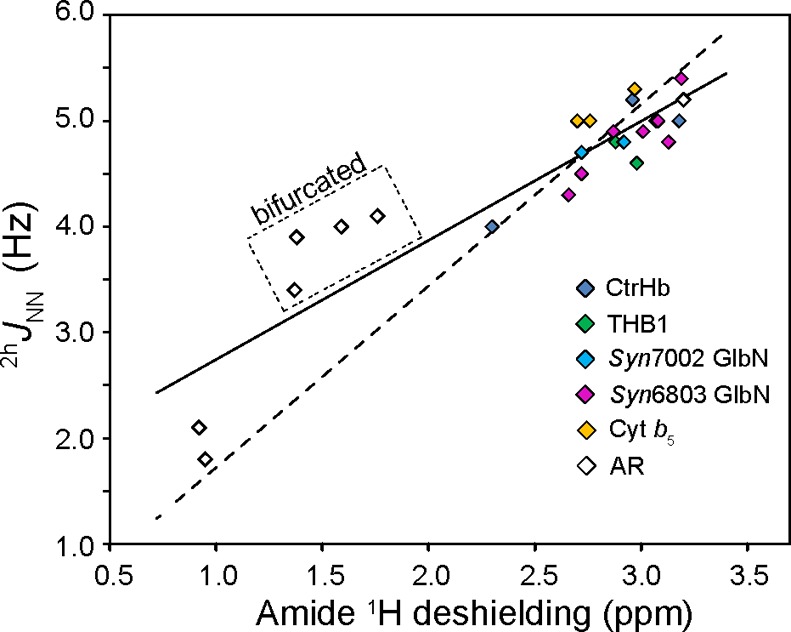
Plot of 2hJNN vs the amide 1H-corrected chemical shift in heme proteins (fill colors correspond to Figure S5) and AR proteins (◇).18 Linear regression of the entire data set gives a slope of 1.13 Hz/ppm and a y-intercept of 1.62 Hz [r2 = 0.84 (—)]. The non-zero y-intercept is attributed to the lack of data at low J values. Fixing the y-intercept to zero yields a best fit slope of 1.72 Hz/ppm [r2 = 0.59 (−–−)]. Linear regression of the heme protein data alone returns a slope of 1.13 Hz/ppm and a y-intercept of 1.56 Hz [r2 = 0.55 (not shown)].
The chemical shift trend in Figure 9 is reminiscent of the trend observed for Hoogsteen and Watson–Crick base pairs in triplex DNA34 and prompts a comparison of the new protein data to published nucleic acid information. In nucleic acids, 2hJNN has been especially useful for inspecting the diversity of H-bond geometries. For example, ultra-high-resolution crystallographic structures of model U-A and G-C base pairs support the possibility that the former have slightly shorter N–N H-bonding distances (Δd ∼ 0.05 Å).62,63 In agreement, the central imino N1–H···N3 H-bond within an RNA G-C base pair typically displays 2hJNN values between 5 and 7 Hz, on average smaller than those of the imino N3–H···N1 H-bond found in RNA (DNA) U-A (T-A) pairs (6–8 Hz).3,4,64 Similarly, cooperative networks of amino N2–H···N7 H-bonds found in G-quadruplex DNA display 2hJNN values of 6–8 Hz.35 Hoogsteen C-G base pairs involving protonated cytidine contain short, electrostatically stabilized H-bonds (N3+–H···N7) and display the largest 2hJNN values yet measured in a biomacromolecule (10–11 Hz).34 On the other hand, A-A mismatches, which have amino N6–H···N7-type H-bonds, tend to display low 2hJNN values (2–3 Hz), in support of their nonideal geometry.39 In addition, a positive correlation between the imino donor 1H chemical shift and the magnitude of 2hJNN, which encompasses C+(N3)–G(N7), T(N3)–A(N1), T(N3)–A(N7), and G(N1)–C(N3) bond types within triplex DNA, has been reported.34 Similar observations have been made between the amide donor 1H shift and |3hJNC′| within N–H···O=C′ protein H-bonds.11
The parallel between protein and nucleic acid N–H···N data is apparent in a plot of 2hJNN couplings versus raw 1H chemical shifts (Figure 10). The different absolute magnitude of protein and nucleic acid 2hJNN values supports the idea that in solution, both Watson–Crick and Hoogsteen N–H···N H-bonds in the DNA triplex are shorter (and possibly stronger) than the helix-capping amide N–H···Nδ1 His H-bond in proteins. This observation may reflect the fact that the difference in pKa between the donor and acceptor groups in the nucleic acids is significantly smaller than that of an amide–histidine pair.7,65 The analysis of combined raw protein amide and nucleic acid imino 1H chemical shifts is particularly susceptible to the referencing problem mentioned above, but remarkably, the aggregate data describe a relatively constant slope and consistent linear behavior.
Figure 10.

Plot of 2hJNN vs donor 1H chemical shift for globin and cytochrome b5 (this work), AR protein,18 and nucleic acid34 N–H···N H-bonds. A linear regression of the combined data returns a slope of 1.13 Hz/ppm and an x-intercept (1H chemical shift for 2hJNN = 0) of 6.83 ppm (r2 = 0.94).
2hJNN and Internuclear Distance
As per Figure 7, capping histidines with a pKa near or below 4 and open state pKas close to model compound values are expected to have a neutral-pH 2hJNN equal to 2hJNN(capped) and be directly related to the geometry of the H-bond. Del Bene and co-workers have demonstrated that the coupling originates principally from the Fermi contact interaction and that, accounting for differences in equilibrium bond length, the coupling is not particularly sensitive to the hybridization or charge state of the donor and acceptor groups.6,7,54 The inter-nitrogen distance is the main determinant of the coupling magnitude, with angular dependence becoming steep only at large deviations from ideal geometry. The distance dependence is captured by an exponential decay7 that can be used to rationalize our results. Figure 11 places the estimated 2hJNN(capped) in this context. The range covered by the hemoglobin and cytochrome values is framed by red dotted lines; the corresponding N–N atomic separations are between ∼2.98 and 3.04 Å. The lowest predicted 2hJNN(capped) of 2.7 Hz is associated with a separation of 3.16 Å.
The accuracy of J-derived distances can be cross-validated with a survey of N-capping characteristics in available protein crystallographic structures. For the i-to-i–2 H-bonds with an intervening Pro, the mean inter-nitrogen distance is 3.00 ± 0.08 Å (n = 298). For the i-to-i+3 H-bonds, the distribution has a similar mean of 3.00 ± 0.09 Å (n = 70). These values are in good agreement with the predicted distances obtained from our solution 2hJNN data. Interestingly, the His–His Nε2-H···Nε2 bond in apomyglobin has a significantly larger 2hJNN (∼11 Hz).17 Inspection of multiple holomyoglobin structures reveals a short inter-nitrogen distance [2.77 ± 0.10 Å (n = 77)]. The histidine pair has the same properties in apo- and holomyoglobin (buried, low acceptor pKa),66 and it is therefore reasonable to expect that the H-bond characteristics are maintained. Again using the Fermi contact curve of Figure 11,7 a remarkable correspondence between the measured J and N–N distance is obtained (blue dashed line).
It is difficult to associate a free energy with the variation in 2hJNN detected here, in part because of uncertainty in the determination of Kclose in Figure 6. However, the work of Del Bene suggests that in the relevant range of distances, a 1 Hz difference in 2hJNN(capped) corresponds to several kilojoules per mole in binding energy.54 In agreement, the 2 unit increase in histidine pKa for CtrHb-B compared to that of CtrHb (Figure S7) corresponds to a 100-fold change in Kclose (assuming a common model pKa for the open states) and gives rise to a 0.8 Hz difference in 2hJNN(capped). Thus, qualitatively, the perturbations caused by heme modification do appear to have a significant local effect on H-bond geometry and, consequently, on the open/closed equilibrium of the helix cap. A full quantitative analysis would require knowledge of the pH dependence of protein stability and measurement of the histidine “open” state pKas.
Conclusion
In this work, we demonstrated that helix-capping N–H···N H-bonds can be routinely detected in 15N-labeled proteins using hydrogen bond scalar coupling experiments. Direct assignment of H-bonding nuclei was achieved by tailoring HNN-COSY and CTSE difference experiments for protein amide 15N–1H and histidine 15Nδ1 nuclei. Evaluation of the 2hJNN coupling constants, along with knowledge of the histidine pKa, provides a convenient metric for the length and relative strength of N–H···N H-bonds. In addition, a linear correlation between 2hJNN couplings and donor 1H chemical shift emerges as observed for N–H···N H-bonds in Watson–Crick and Hoogsteen DNA base pairs. The absolute magnitude of the protein couplings tends to be smaller than those in nucleic acids and hints that in solution, DNA/RNA N–H···N H-bond lengths are likely shorter (∼2.8–2.9 Å) than those in the studied proteins (∼3.0–3.1 Å). Other varieties of N–H···N H-bonds, for example, those using tryptophan as a donor or deprotonated lysine as an acceptor, are feasible as well. Indeed, the LR-HNN-COSY experiments51 (Figure 3E) could be used to overcome instances in which rapid hydrogen exchange precludes detection of the shared proton.
The open/closed capping model (Figure 6) suggests that longer, weaker H-bonds will lead to decreased 2hJNN (and decreased amide 1H and imidazole Δ15N chemical shifts) through two primary mechanisms: (1) increased N–N distance leading to decreased n → σ* donation and weaker Fermi contact interaction and (2) enhanced sampling of the open non-H-bonded states (each with 2hJNN = 0). Thus, only for strong H-bonds [∼100% closed, where 2hJNN = 2hJNN(capped)] can the relative magnitude of 2hJNN be interpreted purely in terms of H-bond geometry. For weaker H-bonds, the expected distance and angular dependencies are obscured; specifically, open/closed averaging will always lead to a decrease in the observed 2hJNN value and therefore a population-dependent overestimate of H-bond length.
We envision that 2hJNN measurements can be routinely extended to resolve persistent questions of enzyme mechanism. Perturbation of individual H-bonds could be probed in the presence or absence of inhibitors, within a series of homologous enzymes or variants, and under different environmental conditions. The sensitivity of 2hJNN to bonding geometry and time-averaged population could reveal information not otherwise accessible by other solution methods or inspection of crystallographic structures. Several proteins for which NMR data have been published can be used as examples. (1) His95, in the active site of triosephosphate isomerase (PDB entry 1TIM),67 has a depressed pKa that allows this residue to be an efficient electrophile over a broad pH range.68 H-Bond formation with the backbone amide of Glu97 is thought to be responsible at least in part for the low pKa.68,69 (2) Likewise, His187, in the active site of uracil DNA glycosylase, functions in the neutral state, and its low pKa is attributed to a capping interaction.70,71 (3) In E2 ligases, the histidine of the conserved His-Pro-Asn motif has been proposed to play a catalytic role, but there is also support for a strictly structural role (histidine locked in the cap position).72 (4) Tautomeric state switching in the dual-histidine motif of peptidyl prolyl isomerase has been proposed as an integral feature of the allosteric mechanism.73 In each of these instances, measurements of 2hJNN HBCs would settle controversies or provide a robust indicator of H-bond presence and strength in the solution state.
Acknowledgments
The authors thank George Rose and Christopher Falzone for helpful discussions and critical reading of the manuscript. All NMR experiments were conducted in the Johns Hopkins University Biomolecular NMR Center.
Glossary
Abbreviations
- 1D
one-dimensional
- 2D
two-dimensional
- AR
ankyrin repeat
- CtrHb
heme domain of C. eugametos LI637 hemoglobin
- CtrHb-B
CtrHb with heme covalently attached to the FG turn
- CTSE
constant-time spin–echo
- DT
sodium dithionite
- GlbN
Synechocystis or Synechococcus hemoglobin
- GlbN-A
GlbN with heme covalently attached to the H helix
- GlbN-AB
GlbN with heme covalently attached to the FG turn and H helix
- GlbN-B
GlbN with heme covalently attached to the FG turn
- H-bond
hydrogen bond
- HBC
hydrogen bond scalar coupling
- nJxy
scalar coupling between nuclei x and y separated by n bonds
- 2hJNN
two-bond N–N HBC
- LR
long-range
- Mb
myoglobin
- PDB
Protein Data Bank
- PTM
post-translational modification
- THB1
hemoglobin 1 from C. reinhardtii
- TrHb1
Group 1 truncated hemoglobin.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.biochem.5b01002.
Details of protein preparation, NMR experiment conditions, simulation details, additional spectra, J modulation curves, and pH titrations (three tables and eight figures) (PDF)
This work was supported by National Science Foundation Grant MCB-1330488 to J.T.J.L. and National Institutes of Health Grant T32 GM008403 for L.Q.
The authors declare no competing financial interest.
Supplementary Material
References
- Jeffrey G. A., and Saenger W. (1994) Hydrogen Bonding in Biological Structures, Springer, New York. [Google Scholar]
- Cornilescu G.; Hu J. S.; Bax A. (1999) Identification of the hydrogen bonding network in a protein by scalar couplings. J. Am. Chem. Soc. 121, 2949–2950 10.1021/ja9902221. [DOI] [Google Scholar]
- Dingley A. J.; Grzesiek S. (1998) Direct observation of hydrogen bonds in nucleic acid base pairs by internucleotide 2JNN couplings. J. Am. Chem. Soc. 120, 8293–8297 10.1021/ja981513x. [DOI] [Google Scholar]
- Pervushin K.; Ono A.; Fernandez C.; Szyperski T.; Kainosho M.; Wüthrich K. (1998) NMR scalar couplings across Watson-Crick base pair hydrogen bonds in DNA observed by transverse relaxation optimized spectroscopy. Proc. Natl. Acad. Sci. U. S. A. 95, 14147–14151 10.1073/pnas.95.24.14147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dingley A. J.; Cordier F.; Grzesiek S. (2001) An introduction to hydrogen bond scalar couplings. Concepts Magn. Reson. 13, 103–127. [DOI] [Google Scholar]
- Del Bene J. E.; Perera S. A.; Bartlett R. J. (2001) 15N,15N spin-spin coupling constants across N-H-N and N-H+-N hydrogen bonds: can coupling constants provide reliable estimates of N-N distances in biomolecules?. Magn. Reson. Chem. 39, S109–S114 10.1002/mrc.913. [DOI] [Google Scholar]
- Del Bene J. E.; Elguero J. (2006) Systematic ab initio study of 15N-15N and 15N-1H spin-spin coupling constants across N-H+-N hydrogen bonds: Predicting N-N and N-H coupling constants and relating them to hydrogen bond type. J. Phys. Chem. A 110, 7496–7502 10.1021/jp0613642. [DOI] [PubMed] [Google Scholar]
- Barfield M.; Dingley A. J.; Feigon J.; Grzesiek S. (2001) A DFT study of the interresidue dependencies of scalar J-coupling and magnetic shielding in the hydrogen-bonding regions of a DNA triplex. J. Am. Chem. Soc. 123, 4014–4022 10.1021/ja003781c. [DOI] [PubMed] [Google Scholar]
- Jaravine V. A.; Alexandrescu A. T.; Grzesiek S. (2001) Observation of the closing of individual hydrogen bonds during TFE-induced helix formation in a peptide. Protein Sci. 10, 943–950 10.1110/ps.48501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luy B.; Marino J. P. (2000) Direct evidence for Watson-Crick base pairs in a dynamic region of RNA structure. J. Am. Chem. Soc. 122, 8095–8096 10.1021/ja0000910. [DOI] [Google Scholar]
- Cordier F.; Grzesiek S. (1999) Direct observation of hydrogen bonds in proteins by interresidue 3hJNC′ scalar couplings. J. Am. Chem. Soc. 121, 1601–1602 10.1021/ja983945d. [DOI] [PubMed] [Google Scholar]
- Cordier F.; Grzesiek S. (2004) Quantitative comparison of the hydrogen bond network of A-state and native ubiquitin by hydrogen bond scalar couplings. Biochemistry 43, 11295–11301 10.1021/bi049314f. [DOI] [PubMed] [Google Scholar]
- Liu A. Z.; Hu W. D.; Majumdar A.; Rosen M. K.; Patel D. J. (2000) NMR detection of side chain-side chain hydrogen bonding interactions in 13C/15N-labeled proteins. J. Biomol. NMR 17, 305–310 10.1023/A:1008390813387. [DOI] [PubMed] [Google Scholar]
- Cordier F.; Wang C.; Grzesiek S.; Nicholson L. K. (2000) Ligand-induced strain in hydrogen bonds of the c-Src SH3 domain detected by NMR. J. Mol. Biol. 304, 497–505 10.1006/jmbi.2000.4274. [DOI] [PubMed] [Google Scholar]
- Cordier F.; Rogowski M.; Grzesiek S.; Bax A. (1999) Observation of through-hydrogen-bond 2hJHC′ in a perdeuterated protein. J. Magn. Reson. 140, 510–512 10.1006/jmre.1999.1899. [DOI] [PubMed] [Google Scholar]
- Eletsky A.; Heinz T.; Moreira O.; Kienhofer A.; Hilvert D.; Pervushin K. (2002) Direct NMR observation and DFT calculations of a hydrogen bond at the active site of a 44 kDa enzyme. J. Biomol. NMR 24, 31–39 10.1023/A:1020697627485. [DOI] [PubMed] [Google Scholar]
- Hennig M.; Geierstanger B. H. (1999) Direct detection of a histidine-histidine side chain hydrogen bond important for folding of apomyoglobin. J. Am. Chem. Soc. 121, 5123–5126 10.1021/ja990340o. [DOI] [Google Scholar]
- Preimesberger M. R.; Majumdar A.; Aksel T.; Sforza K.; Lectka T.; Barrick D.; Lecomte J. T. J. (2015) Direct NMR detection of bifurcated hydrogen bonding in the α-helix N-caps of ankyrin repeat proteins. J. Am. Chem. Soc. 137, 1008–1011 10.1021/ja510784g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aurora R.; Rose G. D. (1998) Helix capping. Protein Sci. 7, 21–38 10.1002/pro.5560070103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenke B. B.; Lecomte J. T. J.; Heroux A.; Schlessman J. L. (2014) The 2/2 hemoglobin from the cyanobacterium Synechococcus sp. PCC 7002 with covalently attached heme: comparison of X-ray and NMR structures. Proteins: Struct., Funct., Genet. 82, 528–534 10.1002/prot.24409. [DOI] [PubMed] [Google Scholar]
- Binz H. K.; Kohl A.; Pluckthun A.; Grutter M. G. (2006) Crystal structure of a consensus-designed ankyrin repeat protein: implications for stability. Proteins: Struct., Funct., Genet. 65, 280–284 10.1002/prot.20930. [DOI] [PubMed] [Google Scholar]
- Perutz M. F. (1979) Regulation of oxygen affinity of hemoglobin: influence of structure of the globin on the heme iron. Annu. Rev. Biochem. 48, 327–386 10.1146/annurev.bi.48.070179.001551. [DOI] [PubMed] [Google Scholar]
- Johnson E. A.; Rice S. L.; Preimesberger M. R.; Nye D. B.; Gilevicius L.; Wenke B. B.; Brown J. M.; Witman G. B.; Lecomte J. T. J. (2014) Characterization of THB1, a Chlamydomonas reinhardtii truncated hemoglobin: linkage to nitrogen metabolism and identification of lysine as the distal heme ligand. Biochemistry 53, 4573–4589 10.1021/bi5005206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couture M.; Das T. K.; Savard P. Y.; Ouellet Y.; Wittenberg J. B.; Wittenberg B. A.; Rousseau D. L.; Guertin M. (2000) Structural investigations of the hemoglobin of the cyanobacterium Synechocystis PCC 6803 reveal a unique distal heme pocket. Eur. J. Biochem. 267, 4770–4780 10.1046/j.1432-1327.2000.01531.x. [DOI] [PubMed] [Google Scholar]
- Scott N. L.; Falzone C. J.; Vuletich D. A.; Zhao J.; Bryant D. A.; Lecomte J. T. J. (2002) The hemoglobin of the cyanobacterium Synechococcus sp. PCC 7002: Evidence for hexacoordination and covalent adduct formation in the ferric recombinant protein. Biochemistry 41, 6902–6910 10.1021/bi025609m. [DOI] [PubMed] [Google Scholar]
- Trent J. T. 3rd; Kundu S.; Hoy J. A.; Hargrove M. S. (2004) Crystallographic analysis of Synechocystis cyanoglobin reveals the structural changes accompanying ligand binding in a hexacoordinate hemoglobin. J. Mol. Biol. 341, 1097–1108 10.1016/j.jmb.2004.05.070. [DOI] [PubMed] [Google Scholar]
- Vu B. C.; Jones A. D.; Lecomte J. T. J. (2002) Novel histidine-heme covalent linkage in a hemoglobin. J. Am. Chem. Soc. 124, 8544–8545 10.1021/ja026569c. [DOI] [PubMed] [Google Scholar]
- Nothnagel H. J.; Preimesberger M. R.; Pond M. P.; Winer B. Y.; Adney E. M.; Lecomte J. T. J. (2011) Chemical reactivity of Synechococcus sp. PCC 7002 and Synechocystis sp. PCC 6803 hemoglobins: covalent heme attachment and bishistidine coordination. JBIC, J. Biol. Inorg. Chem. 16, 539–552 10.1007/s00775-011-0754-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preimesberger M. R.; Pond M. P.; Majumdar A.; Lecomte J. T. J. (2012) Electron self-exchange and self-amplified posttranslational modification in the hemoglobins from Synechocystis sp. PCC 6803 and Synechococcus sp. PCC 7002. JBIC, J. Biol. Inorg. Chem. 17, 599–609 10.1007/s00775-012-0880-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preimesberger M. R.; Wenke B. B.; Gilevicius L.; Pond M. P.; Lecomte J. T. J. (2013) Facile heme vinyl posttranslational modification in a hemoglobin. Biochemistry 52, 3478–3488 10.1021/bi400289e. [DOI] [PubMed] [Google Scholar]
- Rice S. L.; Preimesberger M. R.; Johnson E. A.; Lecomte J. T. J. (2014) Introduction of a covalent histidine-heme linkage in a hemoglobin: A promising tool for heme protein engineering. J. Inorg. Biochem. 141, 198–207 10.1016/j.jinorgbio.2014.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lecomte J. T. J.; Moore C. D. (1991) Helix formation in apocytochrome b5: the role of a neutral histidine at the N-cap position. J. Am. Chem. Soc. 113, 9663–9665 10.1021/ja00025a037. [DOI] [Google Scholar]
- Davis R. B. Jr.; Lecomte J. T. J. (2006) A dynamic N-capping motif in cytochrome b5: evidence for a pH-controlled conformational switch. Proteins: Struct., Funct., Genet. 63, 336–348 10.1002/prot.20759. [DOI] [PubMed] [Google Scholar]
- Dingley A. J.; Masse J. E.; Peterson R. D.; Barfield M.; Feigon J.; Grzesiek S. (1999) Internucleotide scalar couplings across hydrogen bonds in Watson-Crick and Hoogsteen base pairs of a DNA triplex. J. Am. Chem. Soc. 121, 6019–6027 10.1021/ja9908321. [DOI] [Google Scholar]
- Dingley A. J.; Peterson R. D.; Grzesiek S.; Feigon J. (2005) Characterization of the cation and temperature dependence of DNA quadruplex hydrogen bond properties using high-resolution NMR. J. Am. Chem. Soc. 127, 14466–14472 10.1021/ja0540369. [DOI] [PubMed] [Google Scholar]
- Moore C. D.; al-Misky O. N.; Lecomte J. T. J. (1991) Similarities in structure between holocytochrome b5 and apocytochrome b5: NMR studies of the histidine residues. Biochemistry 30, 8357–8365 10.1021/bi00098a012. [DOI] [PubMed] [Google Scholar]
- Scott N. L.; Lecomte J. T. J. (2000) Cloning, expression, purification, and preliminary characterization of a putative hemoglobin from the cyanobacterium Synechocystis sp. PCC 6803. Protein Sci. 9, 587–597 10.1110/ps.9.3.587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelton J. G.; Torchia D. A.; Meadow N. D.; Roseman S. (1993) Tautomeric states of the active-site histidines of phosphorylated and unphosphorylated IIIGlc, a signal-transducing protein from Escherichia coli, using two-dimensional heteronuclear NMR techniques. Protein Sci. 2, 543–558 10.1002/pro.5560020406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majumdar A.; Kettani A.; Skripkin E. (1999) Observation and measurement of internucleotide 2JNN coupling constants between 15N nuclei with widely separated chemical shifts. J. Biomol. NMR 14, 67–70 10.1023/A:1008335502416. [DOI] [PubMed] [Google Scholar]
- Wishart D. S.; Bigam C. G.; Yao J.; Abildgaard F.; Dyson H. J.; Oldfield E.; Markley J. L.; Sykes B. D. (1995) 1H, 13C and 15N chemical shift referencing in biomolecular NMR. J. Biomol. NMR 6, 135–140 10.1007/BF00211777. [DOI] [PubMed] [Google Scholar]
- Delaglio F.; Grzesiek S.; Vuister G. W.; Zhu G.; Pfeifer J.; Bax A. (1995) NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- Goddard T. D., and Kneller D. G. (2006) SPARKY 3, University of California, San Francisco. [Google Scholar]
- Bax A.; Vuister G. W.; Grzesiek S.; Delaglio F.; Wang A. C.; Tschudin R.; Zhu G. (1994) Measurement of homo- and heteronuclear J couplings from quantitative J correlation. Methods Enzymol. 239, 79–105 10.1016/S0076-6879(94)39004-5. [DOI] [PubMed] [Google Scholar]
- Blomberg F.; Maurer W.; Rueterjans H. (1977) Nuclear magnetic resonance investigation of 15N-labeled histidine in aqueous solution. J. Am. Chem. Soc. 99, 8149–8159 10.1021/ja00467a005. [DOI] [PubMed] [Google Scholar]
- Tanokura M. (1983) 1H-NMR study on the tautomerism of the imidazole ring of histidine residues. I. Microscopic pK values and molar ratios of tautomers in histidine-containing peptides. Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol. 742, 576–585 10.1016/0167-4838(83)90276-5. [DOI] [PubMed] [Google Scholar]
- Roux-Fromy M. (1982) On the Hill plot of NMR data for titration of proteins residues. Biophys. Struct. Mech. 8, 289–306 10.1007/BF00537207. [DOI] [PubMed] [Google Scholar]
- Vila J. A. (2012) Limiting values of the 15N chemical shift of the imidazole ring of histidine at high pH. J. Phys. Chem. B 116, 6665–6669 10.1021/jp211196r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boschcov P.; Seidel W.; Muradian J.; Tominaga M.; Paiva A. C. M.; Juliano L. (1983) Ionization constants and thermodynamic parameters of histidine and derivatives. Bioorg. Chem. 12, 34–44 10.1016/0045-2068(83)90005-6. [DOI] [Google Scholar]
- Shimazaki H.; Shinomoto S. (2007) A method for selecting the bin size of a time histogram. Neural Comput. 19, 1503–1527 10.1162/neco.2007.19.6.1503. [DOI] [PubMed] [Google Scholar]
- Pesce A.; Couture M.; Dewilde S.; Guertin M.; Yamauchi K.; Ascenzi P.; Moens L.; Bolognesi M. (2000) A novel two-over-two α-helical sandwich fold is characteristic of the truncated hemoglobin family. EMBO J. 19, 2424–2434 10.1093/emboj/19.11.2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majumdar A.; Kettani A.; Skripkin E.; Patel D. J. (1999) Observation of internucleotide NH···N hydrogen bonds in the absence of directly detectable protons. J. Biomol. NMR 15, 207–211 10.1023/A:1008357304708. [DOI] [PubMed] [Google Scholar]
- Reynolds W. F.; Peat I. R.; Freedman M. H.; Lyerla J. R. Jr. (1973) Determination of the tautomeric form of the imidazole ring of L-histidine in basic solution by carbon-13 magnetic resonance spectroscopy. J. Am. Chem. Soc. 95, 328–331 10.1021/ja00783a006. [DOI] [PubMed] [Google Scholar]
- Vu B. C.; Vuletich D. A.; Kuriakose S. A.; Falzone C. J.; Lecomte J. T. J. (2004) Characterization of the heme-histidine cross-link in cyanobacterial hemoglobins from Synechocystis sp. PCC 6803 and Synechococcus sp. PCC 7002. JBIC, J. Biol. Inorg. Chem. 9, 183–194 10.1007/s00775-003-0512-1. [DOI] [PubMed] [Google Scholar]
- Del Bene J. E.; Bartlett R. J. (2000) N-N spin-spin coupling constants [2hJ(15N-15N)] across N-H···N hydrogen bonds in neutral complexes: To what extent does the bonding at the nitrogens influence 2hJN-N?. J. Am. Chem. Soc. 122, 10480–10481 10.1021/ja002735+. [DOI] [Google Scholar]
- Sigala P. A.; Ruben E. A.; Liu C. W.; Piccoli P. M.; Hohenstein E. G.; Martinez T. J.; Schultz A. J.; Herschlag D. (2015) Determination of hydrogen bond structure in water versus aprotic environments to test the relationship between length and stability. J. Am. Chem. Soc. 137, 5730–5740 10.1021/ja512980h. [DOI] [PubMed] [Google Scholar]
- Witanowski M.; Webb G. A.; Stefaniak L.; Januszewski H.; Grabowski Z. (1972) Nitrogen-14 nuclear magnetic resonance of azoles and their benzo derivatives. Tetrahedron 28, 637–653 10.1016/0040-4020(72)84027-4. [DOI] [Google Scholar]
- Roberts J. D.; Chun Y.; Flanagan C.; Birdseye T. R. (1982) A nitrogen-15 nuclear magnetic resonance study of the acid-base and tautomeric equilibria of 4-substituted imidazoles and its relevance to the catalytic mechanism of α-lytic protease. J. Am. Chem. Soc. 104, 3945–3949 10.1021/ja00378a027. [DOI] [Google Scholar]
- Bachovchin W. W.; Roberts J. D. (1978) Nitrogen-15 nuclear magnetic resonance spectroscopy. State of histidine in catalytic triad of α-lytic protease. Implications for charge-relay mechanism of peptide-bond cleavage by serine proteases. J. Am. Chem. Soc. 100, 8041–8047 10.1021/ja00494a001. [DOI] [Google Scholar]
- Bachovchin W. W. (1986) 15N NMR spectroscopy of hydrogen-bonding interactions in the active site of serine proteases: evidence for a moving histidine mechanism. Biochemistry 25, 7751–7759 10.1021/bi00371a070. [DOI] [PubMed] [Google Scholar]
- Schwarzinger S.; Kroon G. J.; Foss T. R.; Chung J.; Wright P. E.; Dyson H. J. (2001) Sequence-dependent correction of random coil NMR chemical shifts. J. Am. Chem. Soc. 123, 2970–2978 10.1021/ja003760i. [DOI] [PubMed] [Google Scholar]
- Benedict H.; Shenderovich I. G.; Malkina O. L.; Malkin V. G.; Denisov G. S.; Golubev N. S.; Limbach H. H. (2000) Nuclear scalar spin-spin couplings and geometries of hydrogen bonds. J. Am. Chem. Soc. 122, 1979–1988 10.1021/ja9907461. [DOI] [Google Scholar]
- Rosenberg J. M.; Seeman N. C.; Day R. O.; Rich A. (1976) RNA double-helical fragments at atomic resolution. II. The crystal structure of sodium guanylyl-3′,5′-cytidine nonahydrate. J. Mol. Biol. 104, 145–167 10.1016/0022-2836(76)90006-1. [DOI] [PubMed] [Google Scholar]
- Seeman N. C.; Rosenberg J. M.; Suddath F. L.; Kim J. J.; Rich A. (1976) RNA double-helical fragments at atomic resolution. I. The crystal and molecular structure of sodium adenylyl-3′,5′-uridine hexahydrate. J. Mol. Biol. 104, 109–144 10.1016/0022-2836(76)90005-X. [DOI] [PubMed] [Google Scholar]
- Alkorta I.; Elguero J.; Denisov G. S. (2008) A review with comprehensive data on experimental indirect scalar NMR spin-spin coupling constants across hydrogen bonds. Magn. Reson. Chem. 46, 599–624 10.1002/mrc.2209. [DOI] [PubMed] [Google Scholar]
- Bloomfield V. A., Crothers D. M., and Tinoco I. (2000) Nucleic Acids: Structures, Properties, and Functions, University Science Books, Sausalito, CA. [Google Scholar]
- Cocco M. J.; Kao Y.-H.; Phillips A. T.; Lecomte J. T. J. (1992) Structural comparison of apomyoglobin and metaquomyoglobin: pH titration of histidines by NMR spectroscopy. Biochemistry 31, 6481–6491 10.1021/bi00143a018. [DOI] [PubMed] [Google Scholar]
- Banner D. W.; Bloomer A.; Petsko G. A.; Phillips D. C.; Wilson I. A. (1976) Atomic coordinates for triose phosphate isomerase from chicken muscle. Biochem. Biophys. Res. Commun. 72, 146–155 10.1016/0006-291X(76)90972-4. [DOI] [PubMed] [Google Scholar]
- Lodi P. J.; Knowles J. R. (1991) Neutral imidazole is the electrophile in the reaction catalyzed by triosephosphate isomerase: structural origins and catalytic implications. Biochemistry 30, 6948–6956 10.1021/bi00242a020. [DOI] [PubMed] [Google Scholar]
- Lodi P. J.; Knowles J. R. (1993) Direct evidence for the exploitation of an α-helix in the catalytic mechanism of triosephosphate isomerase. Biochemistry 32, 4338–4343 10.1021/bi00067a024. [DOI] [PubMed] [Google Scholar]
- Drohat A. C.; Xiao G.; Tordova M.; Jagadeesh J.; Pankiewicz K. W.; Watanabe K. A.; Gilliland G. L.; Stivers J. T. (1999) Heteronuclear NMR and crystallographic studies of wild-type and H187Q Escherichia coli uracil DNA glycosylase: electrophilic catalysis of uracil expulsion by a neutral histidine 187. Biochemistry 38, 11876–11886 10.1021/bi9910880. [DOI] [PubMed] [Google Scholar]
- Drohat A. C.; Jagadeesh J.; Ferguson E.; Stivers J. T. (1999) Role of electrophilic and general base catalysis in the mechanism of Escherichia coli uracil DNA glycosylase. Biochemistry 38, 11866–11875 10.1021/bi9910878. [DOI] [PubMed] [Google Scholar]
- Cook B. W.; Shaw G. S. (2012) Architecture of the catalytic HPN motif is conserved in all E2 conjugating enzymes. Biochem. J. 445, 167–174 10.1042/BJ20120504. [DOI] [PubMed] [Google Scholar]
- Wang J.; Tochio N.; Kawasaki R.; Tamari Y.; Xu N.; Uewaki J. I.; Utsunomiya-Tate N.; Tate S. I. (2015) Allosteric breakage of the hydrogen bond within the dual-histidine motif in the active site of human Pin1 PPIase. Biochemistry 54, 5242–5253 10.1021/acs.biochem.5b00606. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.