

Abstract
Purpose
To identify a melanoma miRNA expression signature that is predictive of outcome and then evaluate its potential to improve risk stratification when added to the standard of care staging criteria.
Experimental design
Total RNA was extracted from 59 formalin-fixed paraffin embedded (FFPE) melanoma metastases and hybridized to miRNA arrays containing 911 probes. We then correlated miRNA expression with post-recurrence survival and other clinicopathological criteria.
Results
We identified a signature of 18 miRNAs whose overexpression was significantly correlated with longer survival, defined as more than 18 months post-recurrence survival. Subsequent cross-validation showed that a small subset of these miRNAs can predict post-recurrence survival in metastatic melanoma with an estimated accuracy of 80.2% [95% CI: 79.8%, 80.6%]. In contrast to standard of care staging criteria, this six-miRNA signature significantly stratified stage III patients into “better” and “worse” prognostic categories, and a multivariate Cox regression analysis revealed the signature to be an independent predictor of survival. Furthermore, we demonstrated that most miRNAs from the signature also showed differential expression between patients with “better” and “worse prognosis” in the corresponding paired primary melanoma.
Conclusion
MiRNA signatures have potential as clinically relevant biomarkers of prognosis in metastatic melanoma. Our data suggest that molecularly-based models of risk assessment can improve the standard staging criteria and support the incorporation of miRNAs into such models.
Keywords: microRNA, melanoma, metastasis, prognosis, post-recurrence survival
INTRODUCTION
The treatment of metastatic melanoma remains a daunting clinical challenge, and the introduction of molecularly targeted therapies has failed to make a significant impact on overall survival. In the absence of effective therapy for stage IV melanoma, the early identification of patients at highest risk for the development of aggressive disease is critical. Thickness remains the single most important predictor of survival in localized melanoma, but the morphologically-based staging system only partly explains the variability in the natural history of melanoma. With advances in our understanding of melanoma tumorigenesis, there has been heightened interest in the utility of molecular markers as evidenced by the addition of mitotic index to the 2009 AJCC staging criteria (1). Several immunohistochemistry-based biomarkers such as Ki-67, and MMP-2 are promising in terms of their prognostic potential, but they are limited by inter-observer variability and lack of standardization (2–4). Thus, none of these markers have yet been integrated into common clinical practice.
MicroRNAs (miRNAs) are endogenous non-coding small RNAs that negatively regulate expression of their target genes at the posttranscriptional level via translational repression and/or cleavage (5). Recent data support the role of dysregulated miRNAs as oncogenes or tumor suppressors based on their ability to impact cell-cycle regulators and mediators of apoptosis that contribute to the development of the malignant phenotype (6–8). MiRNAs possess several features that make them attractive candidates for new prognostic biomarkers. First, they are upstream regulators that can simultaneously target large numbers of protein-coding genes and multiple cancer pathways. Second, miRNAs are the direct functional product of the corresponding gene. This is in contrast to mRNAs that need to be translated and often post-translationally modified to exert their function. Third, the stability of miRNAs in archival formalin-fixed paraffin embedded (FFPE) tissues allows them to be extracted from the initial biopsy specimen and quantified by standardized methods such as RT-PCR at any point during the patient’s clinical course (9). One of the primary limitations of mRNA-based gene expression profiling is the requirement for fresh frozen tissue, the acquisition of which is both labor intensive and time sensitive. Additionally, concerns have been raised about the possibility of compromising the accuracy of the staging when part of a frozen section from a thin melanoma is sampled for research purposes (10).
To assess the clinical significance of melanoma miRNA signatures, we first performed miRNA expression profiling on a cohort of human metastatic melanoma specimens with annotated clinical follow-up. An initial signature of miRNAs strongly associated with post-recurrence survival was identified and then further refined using supervised learning methods to a six miRNA set predictive of survival. The miRNA signature added prognostic value to the standard of care staging criteria in terms of the risk stratification of stage III patients. Notably, specific components of the prognostic signature in the metastatic tissue exhibited the same pattern of differential expression in the matched pair primary melanoma, suggesting that the miRNA signature may have early prognostic potential. Our data support the addition of molecularly-based models of risk assessment to the standard staging criteria and suggest that miRNA signatures hold promise as robust, clinically useful biomarkers with the ability to identify high risk patients at both early and late stages.
MATERIALS AND METHODS
Clinical Specimens
Primary and metastatic melanoma specimens were collected at the time of surgery from patients enrolled from 2002 to 2009. Congenital nevi were obtained from biopsies taken from patients without a history of skin cancer. Informed consent was obtained from all patients and approval acquired by the institutional review board (IRB) of NYU School of Medicine (#10362, #08-598). Patients enrolled in the IMCG are prospectively followed up every 3 months. Clinical status at last date of follow-up is recorded as “Alive, no melanoma,” “Alive with melanoma,” “Died with melanoma,” “Died, no melanoma,” or “Died, cause unknown.” All patients with a follow-up less than 20 months were deceased. When a patient is determined to be deceased, the patient’s history and last clinical status is reviewed with the medical oncology investigator (AP) to determine if melanoma was the cause of death. All tumors were classified according to the American Joint Committee on Cancer (AJCC) staging system. Only metastatic samples with tumor content >80% were included in the study. All congenital nevi and primary melanoma tissue were sectioned on Leica PEN Membrane Slides and macro- or microdissected.
RNA extraction
Total RNA was extracted as previously described (11). Briefly, ten sections of 10 µm of FFPE tissues were deparaffinized with xylene, washed in ethanol, and digested with proteinase K. RNA was extracted with acid phenol:chloroform followed by ethanol precipitation and DNAse digestion, or using the Qiagen miRNeasy FFPE kit. Total RNA quantity and quality were evaluated using a Nanodrop ND-1000 (Thermo Scientific) with an inclusion criteria of A260/A280 ≥ 1.8.
miRNA microarray expression profiling and data pre-processing
MiRNA microarrays were prepared as described previously (11). Each RNA sample (3.5 µg) was labeled by ligation of an RNA-linker, p-rCrU-Cy/dye to the miRNA 3' end. Hybridization and washing of the microarray slides were performed as previously described (11). Arrays were scanned using an Agilent Microarray Scanner Bundle G2565BA and analyzed using SpotReader software (Niles Scientific), to generate raw intensity data. Triplicate spots were combined into one signal by taking the logarithmic mean of the reliable spots. Quantile normalization was applied to make arrays comparable to one another (12). MiRNAs with low variance across samples (i.e. coefficient of variation < 1% on the log-scale) were filtered out, leaving 610 miRNAs for analysis.
Statistical analysis
Significance Analysis of Microarrays (SAM), implemented in the Bioconductor package samr(†) was used to identify miRNAs significantly associated with post-recurrence survival using time from recurrence to death (or censored) as the outcome variable (13, 14). SAM computes the Cox regression coefficient for each miRNA and uses a permutation procedure to estimate the False Discovery Rates (FDR) and to select differentially expressed miRNAs while controlling for multiple comparisons using FDR (15). One thousand permutations of the data were used to estimate the FDRs and to select differentially expressed miRNAs. Additionally, the patients were dichotomized into two groups: a “longer survival” group (those who survived 18 months or more from the date of resection of the metastatic tumor, n = 36) and a “shorter survival” group (patients who survived less than 18 months, n = 23). A two-sample nonparametric comparison was used in SAM to identify miRNAs that are differentially expressed between these two groups. The significant gene lists resulting from the two types of analyses (survival and two-sample comparison) were then compared.
Construction of a ‘predictor biomarker’ based on miRNA expression
In order to develop a miRNA signature of post-recurrence survival, we used the following classification methods: nearest shrunken centroids (implemented in the Bioconductor package pamr (16)), classification trees (implemented in rpart (17)), Random Forests (implemented in randomForest (18)), AdaBoost with classification trees (19) as well as principal component logistic regression, using longer/shorter survival groups defined earlier as the outcome variable. The classification methods were used with and without pre-selection of input variables (i.e. miRNAs) based on an FDR criterion (i.e. FDR < 5%), whenever applicable. The prediction accuracy of the classification algorithms was estimated using 1000 cross-validation randomizations.
Comparison of the miRNA predictor with clinical predictors
We used the method of pre-validation (PV) (20, 21) to compare the prediction accuracy of the miRNA signature to that of TNM stage, site of metastasis, page at recurrence and other clinical and demographic variables. PV outputs a prediction for each patient based on a classifier (e.g. nearest shrunken centroids) that is estimated without using that patient’s data, thus reducing the bias that might arise from re-use of the data. The PV miRNA predictor was compared to other predictors of survival in a multivariate Cox regression analysis of post-recurrence survival. The Kaplan-Meier method was used to estimate the post-recurrence survival function (22). The log-rank test was used to compare the survival distribution between groups (23). All analyses were performed using the R language for statistical computing and the Bioconductor software (13, 24). Heatmap and hierarchical clustering analyses were done using Prism 4 software v4.0 (GraphPad Software, Inc. La Jolla, CA).
Bioinformatics analysis
DAVID bioinformatics resource (††) was used to conduct KEGG pathway analysis on predicted targets (according to TargetScan) of the miRNAs from the predictive signature. Most frequently represented pathways are assigned a p value calculated with a modified version of a Fisher-exact test (p-value cutoff of less than 0.1) showing significance of the association as compared to a random list using the human genome as a background.
Real-time PCR
Quantitative PCR (qRT-PCR) analysis of mir-126, mir-145, mir-143, mir-497, mir-150, mir-155, miR-342-3pand mir-455-3p was performed by using miRNA-specific TaqMan MicroRNA Assay Kit (Applied Biosystems), and an Applied Biosystems 7500 Sequence detection system. RNU44 small nuclear RNA was used for normalization of input RNA/cDNA levels. Each measurement was performed in triplicate and no-template controls were included for each assay.
RESULTS
miRNA signature distinguishes metastatic melanoma patients with worse prognosis
A miRNA expression profile of 911 miRNAs in 59 metastatic melanoma patients was obtained. These 59 patients were followed clinically for a median of 21 months (1–69 months range) after excision of the metastatic lesion. Clinicopathologic features of the 59 melanoma patients included in the study are presented in Table 1. A complete description of the stage at initial presentation, the stage at the time of tissue collection, and the metastatic site for each sample utilized is provided in Supplementary Table 1. The median survival of the entire cohort from the time of excision to date of last follow-up or death was 20 months. We evaluated the association between tumor miRNA expression profiles and post-recurrence survival based on the interval from metastasectomy to date of last follow-up or death. Using post-recurrence survival as the outcome variable in SAM, we identified 18 significant miRNAs (using an FDR of 5%), for which higher expression was associated with longer survival (Figure 1A and Table 2). Similar results were obtained using a two-sample nonparametric comparison to discriminate between patients with ‘longer survival’ (those who survived 18 months or more from the date of resection of the metastatic tumor, n=36) and ‘shorter survival’ (patients who survived less than 18 months, n=23). We chose 18 months as the threshold for longer/shorter survival based on the median survival of our own cohort (21 months), which is consistent with previous studies (25).
Table 1. Metastatic Patient Characteristics (n=59).
Clinical characteristics of the metastatic melanoma cohort.
| Variables | n (%) |
|---|---|
| Age (y) | |
| Mean (±SD) | 59.6±17.2 |
| Median | 59 |
| Sex | |
| Male | 38 (64.4) |
| Female | 21 (35.6) |
| Stage | |
| IIIB | 16 (27.1) |
| IIIC | 20 (33.9) |
| IV | 23 (39.0) |
| Anatomic Location | |
| Soft Tissue / Skin | 15 (25.4) |
| Regional Lymph Node | 26 (44.1) |
| Visceral | 7 (11.9) |
| Brain | 11 (18.6) |
| Treatment pre-surgery | |
| Treated | 40 (67.8) |
| Not treated | 19 (32.2) |
Figure 1.
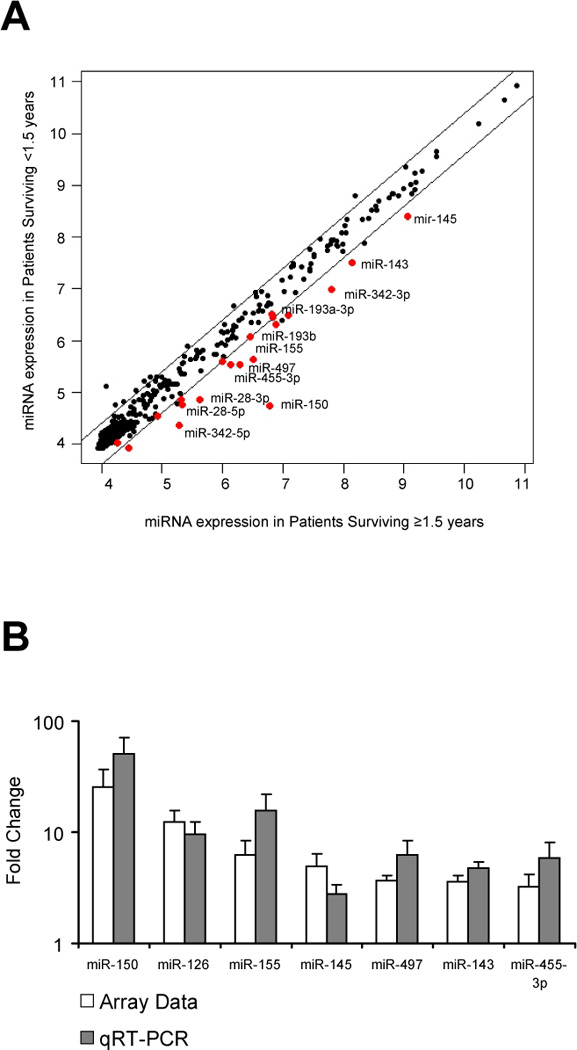
Identification of miRNAs differentially expressed in melanoma patients with longer post-recurrence survival (≥ 1.5 years). A. Mean normalized fluorescence for each microRNA (black dots) indicating expression levels as measured by microarray in patients surviving less than 1.5 years post-surgical resection compared to patients surviving 1.5 years or longer. MiRNAs significantly elevated in the longer survival group are indicated as red dots (FDR < 5%). Diagonal lines demarcate 1.5-fold difference levels in expression between the two groups. B. Real time PCR validation of microarray data. Expression levels of miRNAs were measured by real time RT-PCR in a subset on 10 samples, including 5 patients who survived at least 1.5 years after their recurrence and 5 patients who died in less than 1.5 years post-recurrence. Cycle threshold (Ct) values for each miRNA were normalized versus the housekeeping small RNA RNU44 (ΔCt) and represented as 2−ΔCt. The x axis shows 7 individual miRNA species examined both by microarray and real-time PCR; the y axis represents the mean value of the ratio of expression of the 7 miRNA species in microarray and real-time PCR (qRT-PCR).
Table 2. MicroRNAs significantly associated with post-recurrence survival.
MicroRNAs significantly associated with post-recurrence survival. Eighteen miRNAs found overexpressed in metastatic tissues of patients with longer survival (≥1.5 years) compared to that of patients with shorter survival (<1.5 years). Fold change, false discovery rate (FDR) and Cox regression coefficient (d), chromosome location and host genes (when pertinent) are indicated.
| miRNA | Score (d) | FDR (%) | Fold Change | Chromosome Location |
Host Gene |
|---|---|---|---|---|---|
| hsa-miR-150 | −3.98 | 0 | 9.74 | 19q 13.33 | Intergenic |
| hsa-miR-455-3p | −3.52 | 0 | 1.77 | 9q32 | COL27A1 |
| hsa-miR-145 | −3.06 | 0 | 1.92 | 5q 33.1 | Intergenic |
| hsa-miR-342-3p | −2.91 | 0 | 2.56 | 14q 32.2 | EVL |
| hsa-miR-497 | −2.87 | 0 | 1.72 | 17p 13.1 | AC027763.1 |
| hsa-miR-155 | −2.77 | 0 | 2.17 | 21q 21.3 | Intergenic |
| hsa-miR-342-5p | −2.66 | 0 | 3.9 | 14q 32.2 | EVL |
| hsa-miR-143 | −2.61 | 0 | 1.79 | 5q 33.1 | Intergenic |
| hsa-miR-193a-3p | −2.43 | 0 | 1.34 | 17q 11.2 | Intergenic |
| hsa-miR-146b-5p | −2.38 | 0 | 1.59 | 10q 24.32 | Intergenic |
| hsa-miR-28-3p | −2.32 | 0 | 1.6 | 3q 28 | LPP-201 |
| hsa-miR-10b | −2.14 | 0 | 1.61 | 2q 31.1 | HOXD3 |
| hsa-miR-193b | −2.08 | 0 | 1.3 | 16 p3.12 | Intergenic |
| hsa-miR-28-5p | −1.87 | 3 | 1.51 | 3q 28 | LPP-201 |
| hsa-miR-142-5p | −1.86 | 3 | 2.69 | 17q 22 | Intergenic |
| hsa-miR-143* | −1.73 | 4.86 | 1.36 | 5q 33.1 | Intergenic |
| hsa-miR-126 | −1.73 | 4.86 | 1.83 | 9q34.3 | EGFL7 |
| hsa-miR-214 | −1.72 | 4.86 | 1.26 | 1q 24.3 | DNM3 |
Of the 59 patients included in the study, 19 (32%) had treatment prior to surgery (55% chemotherapy, 35% immunotherapy, 45% radiation). When we excluded patients with prior treatment from the analyses, 10 of the 18 miRNAs were still found to be significantly associated with post-recurrence survival based on the remaining 40 treatment-naïve patients using SAM and the FDR cutoff of 5%.
Validation of microarray data by real time RT-PCR
To validate the data obtained using the microarray platform, we quantified the expression of 7 miRNAs (miR-150, miR-126, miR-155, miR-145, miR-497, miR-143 and miR-455-3p) in 10 specimens using real time PCR designed to detect mature miRNAs. The data were normalized to the endogenous control small nuclear RNA RNU44. Real time PCR showed that all 7 selected miRNAs display higher expression levels in the 5 cases with longer survival compared to the 5 cases with worse prognosis (Figure 1B and Supplementary Table 2). Six of the 7 miRNA gene chip expression assays were significantly correlated with the RT-PCR results demonstrating an average Pearson correlation coefficient of 0.82 (range 0.73–0.97; Supplementary Table 2).
A small set of miRNAs predicts post-recurrence survival
In order to develop a miRNA signature of post-recurrence survival, we used supervised learning methods (see Methods section). All miRNAs as well as significant subsets of miRNAs (i.e. FDR < 5%) were used as input variables in the classification algorithms. The prediction accuracy of the classification algorithms was estimated using cross-validation. Provided in Supplementary Table 3 are training error rates as well as estimated true error rates of the classifiers along with 95% confidence intervals obtained from 1000 cross-validation randomizations. The method of nearest shrunken centroids performed best with an estimated average prediction accuracy of 80.2% (95% confidence interval from 79.8% to 80.6%).
Based on 1000 cross-validation randomizations, the miRNA signatures obtained by the method of nearest shrunken centroids were most often comprised of the following six miRNAs: mir-150, mir-342-3p, mir-455-3p, mir-145, mir-155 and mir-497. The median number of miRNAs used by the nearest shrunken centroids method was 2 (range 1–10). The number of miRNAs used by the classification trees was 1 and 2. These findings suggest that a small number of miRNAs might be sufficient to accurately predict post-recurrence survival.
miRNA signature is an independent predictor of post-recurrence survival in metastatic melanoma
We next used the method of pre-validation (PV) to derive a miRNA predictor of post-recurrence survival. The PV miRNA predictor assigned each patient to a “better prognosis” (n = 43) or a “worse prognosis” (n = 16) group using the nearest shrunken centroids classifier. The median survival of patients in “better” and “worse” prognostic groups were 880 days (95% CI: [653, NA]) and 189 days (95% CI: [111, 438]) respectively. The Kaplan-Meier estimated survival curves of the “better prognosis” and “worse prognosis” groups predicted by the PV miRNA signature are shown in Figure 2C. The PV miRNA predictor was able to segregate patients into “better prognosis” and “worse prognosis” groups with significance comparable to that of stage at recurrence (log-rank p<0.0001, Figure 2A) and the site of recurrence (log-rank p=0.0026, Figure 2B). Among the cohort of patients with stage III melanoma, the AJCC designation of IIIB versus IIIC was unable to significantly risk stratify the patients based on post-recurrence survival (log-rank p=0.48, Suppl. Figure 1A). The PV miRNA predictor, however, was able to significantly separate stage IIIB and IIIC patients into better and worse prognosis groups based on post-recurrence survival (log-rank p=0.0046) (Suppl. Figure 1B).
Figure 2.
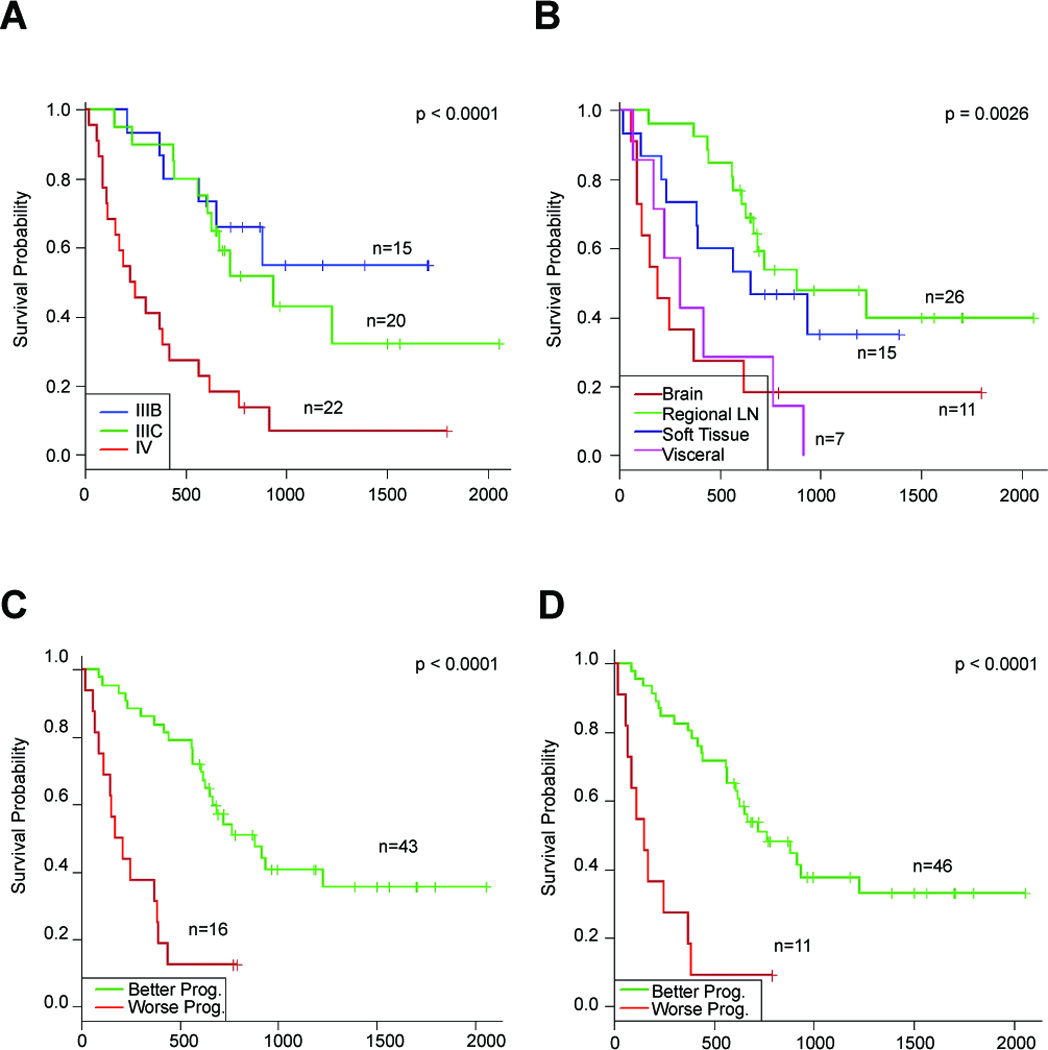
Kaplan-Meier estimates of post-recurrence survival stratified by (A) stage at recurrence, (B) site of metastasis, (C) “better prognosis” and “worse prognosis” groups predicted by the pre-validated miRNA signature, and (D) “Better prognosis” and “worse prognosis” groups predicted by the optimal predictor of post-recurrence survival that combines the pre-validated miRNA signature and stage.
We next used a multivariate Cox proportional hazard regression analysis to compare the PV miRNA signature to standard clinicopathological factors including age, sex, stage, site of metastasis, recurrence number and time to first recurrence in their ability to predict post-recurrence survival. The miRNA predictor was significantly associated with a longer post-recurrence survival in metastatic patients (HR 3.42; 95%CI: [1.49–7.86]; p=0.0038) with only a designation of stage IV exhibiting a comparable predictive value (HR 5.69; 95% CI: [1.41, 22.9]; p=0.0144) (Table 3A). When the PV miRNA signature and stage were included in the model, other variables such as age, sex, or time to first recurrence, were not significant. We did find, however, that select miRNAs from the signature were related to stage and site of metastasis (Suppl. Table 4). Specifically, all six miRNAs were significantly associated with stage at recurrence (Suppl. Table 4 and Suppl. Figure 1C) and all except for mir-342-3p were associated with site of metastasis (Suppl. Table 4). In order to further optimize the prediction model, we removed all variables except stage and the miRNA predictor. A new Cox hazard regression model inclusive of only stage and the signature showed that both a designation of stage IV (HR 3.76; p = 0.0072) and the PV miRNA signature (HR 3.16; p = 0.0029) were significant predictors of post-recurrence survival (Table 3B). Finally, based on this Cox model, we derived an optimal predictor of post-recurrence survival that combined the PV miRNA and AJCC stage variables and segregated the patients into “better prognosis” and “worse prognosis” groups using the median hazard ratio as the cutoff point. The Kaplan-Meier estimates of survival functions of these two groups are shown in Figure 2D (p<0.0001).
Table 3. Cox proportional hazards modeling (CPHM) to predict post-recurrence survival in metastatic melanoma based on clinical, pathological and miRNA expression predictors.
Hazard ratios and confidence interval for various predictors based on Cox proportional hazards models of post-recurrence survival. Hazard ratios and 95% Confidence Intervals (CIs) for predictors obtained using (A) the Cox proportional hazards model or (B) the optimal Cox proportional hazards model for predicting post-recurrence survival.
| A. Hazard ratios and 95% Confidence Intervals (CIs) for predictors obtained using CPHM for predicting post-recurrence survival | |||
|---|---|---|---|
| Predictor | Hazard Ratio |
95% CI | p-value |
| PV miRNA | 3.42 | [1.49, 7.86] | 0.0038 |
| Stage IIIC(*) | 2.09 | [0.61, 7.09] | 0.24 |
| Stage IV(*) | 5.69 | [1.41, 22.9] | 0.0144 |
| Regional LN(†) | 1.57 | [0.36, 6.77] | 0.55 |
| Soft tissue/skin(†) | 2.38 | [0.68, 8.29] | 0.17 |
| Visceral(†) | 1.88 | [0.59, 5.99] | 0.28 |
| Recurrence number | 1.03 | [0.89, 1.19] | 0.67 |
| Time to 1st recurrence | 1 | [1.00, 1.00] | 0.79 |
| B. Hazard ratios and 95% CI for predictors in the optimal Cox CPHM for predicting post-recurrence survival | |||
|---|---|---|---|
| Predictor | Hazard Ratio |
95% CI | p-value |
| PV miRNA | 3.16 | [1.48, 6.72] | 0.0029 |
| Stage IIIC(§) | 1.32 | [0.48, 3.57] | 0.59 |
| Stage IV(§) | 3.76 | [1.43, 9.88] | 0.0072 |
Baseline: stage IIIB
Baseline: brain mets
Baseline: stage IIIB
These findings demonstrate the potential of specific miRNAs to provide a molecular classification model predictive of melanoma patient survival that can enhance the current morphologically-based staging criteria.
Select miRNAs retain predictive capacity in primary matched pairs
We next sought to investigate if the miRNA prognostic signature found in metastatic melanoma tissue could also be detected in the corresponding primary tumor. We utilized real-time PCR to assess expression levels of the 6 miRNAs that most frequently comprise the prediction signature (and an endogenous control, RNU44) in a subset of 20 matched primary-metastatic cases with <1.5 year post-recurrence survival (n=10) and ≥1.5 year post-recurrence survival (n=10). The clinicopathological features of these primary specimens are summarized in Supplementary Table 5. In order to establish the basal expression level of the 6 miRNAs in the ‘normal’ melanocytic lineage, we analyzed their expression in congenital nevi (n=5) and compared it to that of the 20 aforementioned matched pairs. Primary and metastatic expressions were positively correlated for all six miRNAs, although the strength of the correlation reached statistical significance only for miR-145 (Spearman’s rho = 0.54, p = 0.05). Five of the six microRNAs had higher average expression in the primary tumor of patients with longer survival (consistent with what we observed in the metastatic samples), although these differences did not reach statistical significance (Figure 3). Additionally, miR-497, -155, -150, -342-3p and -145 were significantly overexpressed in metastatic melanoma compared to nevi (p-values < 0.02) and miR-150 and miR-155 were also significantly elevated in primary melanoma compared to nevi (p-values < 0.01). MiR-455-3p was significantly down-regulated in patients with shorter survival in both primary and metastatic samples compared to nevi (p = 0.0027 and 0.14, respectively). These results suggest that miRNAs may be useful markers of prognosis prior to the development of metastases.
Figure 3.
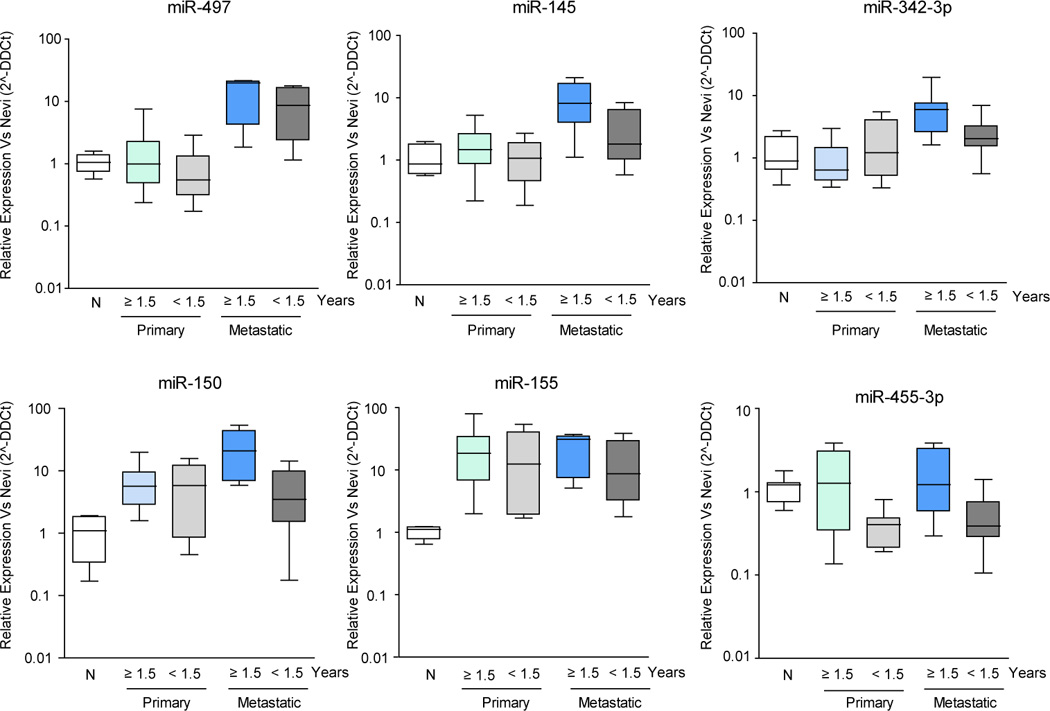
Expression of predictor miRNAs in paired primary-metastatic melanomas relative to their levels in congenital nevi. Box-plots in logarithmic scale represent the expression of the 6 miRNAs most commonly included in the ‘predictor’ set, measured by quantitative PCR in paired primary-metastatic samples, relative to their respective levels in congenital nevi. Data was normalized to small nuclear RNA RNU44. The "box" part contains the middle 50% of the data, the line in the box indicates the median value, and the whiskers indicate the minimum and maximum data values.
DISCUSSION
In this study we identified a miRNA signature in metastatic melanoma tissue that was predictive of post-recurrence survival such that patients with higher expression levels of the signature elements had longer survival. The miRNA signature was able to significantly stratify stage III patients into better and worse prognosis groups based on survival probability better than the standard classification of IIIB and IIIC. Some miRNAs from the signature recapitulated the differential expression between prognostic categories in the matched pair primary tissue suggesting that miRNA signatures may play a role in prognosis at early stages.
It is generally recognized that there are limitations in the current staging system, and it is expected that the addition of molecularly-based prognostic risk stratification will ultimately result in optimized, personalized cancer treatment. A clinically useful biomarker must be reflective of melanoma biology but more important, it should impact patient care. To our knowledge, no genome-wide miRNA studies defining signatures associated with survival or prognosis have been reported in cutaneous melanoma to date. One recent study, in which only 16 miRNAs were analyzed, found that elevated miR-15b was associated with worse recurrence free survival and overall survival in primary melanoma (26). Our report, which focuses on metastatic melanoma, identifies a multimarker signature of miRNAs correlated with outcome. A number of studies have described miRNA alterations associated with melanoma progression (7, 27–31). While these studies offer new insight into the role of miRNAs in melanoma pathogenesis, they do not address the prognostic significance of those findings.
Our miRNA signature was able to enhance the predictive potential of conventional staging for stage III patients, allowing for a significant risk stratification based on post-recurrence survival that was not attainable using the AJCC designation of IIIB or IIIC. The optimal model was one that incorporated both the miRNA signature and stage at recurrence. Primary treatment for stage III patients typically includes surgical resection of the lymph nodes, but the options for post-surgical adjuvant therapy are varied and include observation, clinical trials, radiation to the nodal basin, or interferon alpha therapy. As demonstrated in our patient cohort, the conventional staging system offers little guidance as to which type of adjuvant therapy (if any) is indicated for stage III patients based on an appropriate assessment of risk. The addition of the miRNA signature to staging, however, was able to provide clear risk stratification based on survival. Thus, one could imagine a clinical scenario in which patients with a worse prognosis could be targeted for interferon therapy or entered into a clinical trial, whereas those with better prognosis could be observed and spared the morbidity of further therapy.
Although our miRNA signature was developed from an expression profile of tissues from stage III and IV patients, an ideal prognostic marker is one that can also risk-stratify early in the disease course. We assessed the expression pattern of our predictive miRNA signature in 20 matched pair cases to assess if the differential expression of the 6 select miRNAs and their association with survival was maintained in the primary melanoma. Five of the 6 miRNAs in the signature demonstrated the same expression and association with survival trends in the primary and metastatic tissue. The difference did not reach statistical significance, however, possibly because of the low sample size of the matched pair cohort. Further studies in a larger set of primary tissues are needed to adequately assess the utility of miRNAs in predicting recurrence at early melanoma stages.
An association between any of the 6 miRNAs from the predictive signature and melanoma outcome has not been previously reported, but several of these miRNAs have been identified as being relevant to tumorigenesis and prognosis in other malignancies (Supplementary Table 6). miR-155 has been shown to play an oncogenic role in both hematopoietic malignancies (32) and solid cancers including breast, cervical, and clear renal cell carcinoma (33–35). In our cohort, however, increased expression of miR-155 in metastatic tissue was associated with longer survival. The same result was also noted in previous studies of pancreatic cancer (36) inviting speculation regarding the nature of miR-155s oncogenic effect. These differences may be attributable to tumor-specific or host-specific effects given the well-established link between miR-155 and the regulation of immune function (37). It has been demonstrated that expression of miR-155 increases upon activation of immune cells by TLRs, cytokines and other antigens (38). Thus it is possible that although immune function is initially stimulated in early tumor development, the response and corresponding levels of miR-155 expression eventually decrease with the development of an increasingly malignant phenotype. Nonetheless, a recent study has shown antiproliferative and pro-apoptotic activities of miR-155 in melanoma cell lines (39), implying that miRNA-155 could be a direct negative regulator of melanoma cell proliferation and survival.
Our results demonstrate that higher expression of miR-145 in metastatic tissue is associated with longer survival. This finding is consistent with previous reports in colon, lung, breast, and prostate cancer suggesting a tumor suppressor role for miR-145 (33). Higher expression of miR-145 in tissue from patients with longer survival might be interpreted as an attempt to impair tumor progression as it has previously been shown that p53 transcriptionally induces miR-145 to repress c-Myc (40). It is possible that elevated levels of miR-145 serve as a functional read-out of preserved p53 activity and increased efficiency of DNA damage repair or pro-apoptotic mechanisms. On the other hand, miR-497 has been found suppressed in colon, breast, prostate, ovarian, gastric and lung cancer (41, 42). Similarly, miR-342-3p expression is reduced in human colorectal cancer (along with the hosting gene EVL) by means of CpG island methylation (43), and its restoration induces an apoptotic response. In a comparative genomic hybridization (CGH) study on DNA copy number abnormalities of genomic regions containing known miRNA genes, mir-342 was found downregulated in a minority of melanoma cell lines (8/45) (44). MiR-150 has been involved in the maturation of B-cells, has been found upregulated in lymphocytic leukemias, and it targets oncogenes such as c-Myb (45) and the receptor P2X7 (46). Interestingly, it has also been shown to be downregulated in Chronic Myeloid Leukemia (CML) by BCR-ABL1(47). Further functional analyses in melanoma would be required to establish whether these miRNA alterations represent ‘passenger’ defects reflective of prognosis, or whether they actively participate in tumor progression.
We also identified miR-455-3p as downregulated in both primary and metastatic melanoma compared to nevi in patients with shorter survival. The mature, processed form of the miRNA (miR-455) has also been shown to be downregulated in primary melanoma cell lines compared to normal melanocytes (48). Transcription factor PAX6, a putative target of miR-455, has been shown to play a critical role in the self-renewal and differentiation of neural stem cells (49). Melanocytes are derivatives of the neural crest, and comparative genomic studies have shown an association between overexpression of genes such as NEDD9 (neural precursor cell expressed, developmentally downregulated) and increased invasive potential (50). Thus, it is possible that the loss of miR-455 and its subsequent impact on PAX6 expression may disrupt the normal progression of melanogenesis resulting in an immature melanocyte with increased migratory capacity and enhanced metastatic potential. Again, further mechanistic studies would be needed to further explore the relationship between miR-455-3p, PAX6, cell differentiation, and metastatic potential.
Although we recognize that it is not possible to draw firm conclusions regarding the mechanism of action of the prognostic miRNAs identified in the signature, our preliminary pathway analyses revealed that the putative targets of the miRNAs in the signature converge upon common pathways known to be altered in melanoma (e.g., Wnt, MAPK, and TGFβ) and other cancers (CLL, colorectal, endometrial and pancreatic cancers) (Suppl. Table 7). Because miRNAs are able to simultaneously modulate multiple genes from different pathways, it is plausible that they might play a role in the complex process of melanoma progression and metastasis. While not definitive, this analysis suggests that the set of miRNAs identified in our study not only have prognostic capacity but may also be reflective of the underlying biology. Further supporting this possibility, recent reports suggest that genomic regions frequently altered in melanoma are enriched for miRNA genes (44). Many of the miRNAs from our signature are located in genomic regions previously reported as altered in melanoma such as loss of 9q32 (miR-455-3p) in melanoma cell lines, gain of the 5q locus (miR-145) in acral melanoma, and gain of 21q (miR-155) in uveal melanoma (Supplementary Table 8).
In conclusion, our results demonstrate the potential of miRNAs as clinically useful markers of prognosis in metastatic melanoma patients. A 6 miRNA signature was able to improve risk stratification for stage III patients suggesting that miRNAs may serve as a useful molecular adjunct to the current morphologic staging system in identifying high risk patients who might benefit from adjuvant therapy. Differential expression of most miRNAs from the predictor signature was also observed in the matched pair primary tissue suggesting that the miRNA signature may also play a role in prognosis of early lesions. Further studies in a larger cohort of primary melanoma are needed to better define the role of the signature in predicting the development of aggressive disease.
Supplementary Material
STATEMENT OF TRANSLATIONAL RELEVANCE.
Emerging evidence suggests that microRNAs (miRNAs) play an important role in melanoma tumorigenesis, but the clinical implications of these findings have not been well defined. Here we demonstrate that a specific signature of a small set of miRNAs is predictive of post-recurrence survival in a cohort of metastatic melanoma patients. We further explored the prognostic relevance of miRNAs and found that the addition of the miRNA signature to the standard of care staging criteria improved risk stratification for stage III patients. Furthermore, we found that most miRNAs from the metastatic signature also showed differential expression between patients with “better” and “worse prognosis” in the corresponding primary melanoma. These data suggest that miRNA signatures may be clinically useful prognostic biomarkers for both early and late stage melanoma, and thus may have the potential to influence clinical management by identifying high risk patients most likely to benefit from adjuvant therapy and/or heightened surveillance. Further studies of the clinical utility of miRNA-based monitoring assays are warranted.
Acknowledgements
We thank Rosetta Genomics (Rehovot, Israel) for the miRNA array data, the NYU Genomics Facility for the use of the high-throughput qPCR ABI 7900 SDS equipment, and the NYUCI Histopathology Core for tissue sectioning and LCM use.
Grant support: This work was funded by a Cancer Center Support Grant (NIH/NCI 2 P30 CA16087-23), the Elsa U. Pardee Foundation and the ConCerN Foundation. M.F.S. has been supported by an Alfonso Martin-Escudero fellowship.
REFERENCES
- 1.Balch CM, Gershenwald JE, Soong SJ, et al. Final Version of 2009 AJCC Melanoma Staging and Classification. J Clin Oncol. 2009 doi: 10.1200/JCO.2009.23.4799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Berger AJ, Camp RL, Divito KA, Kluger HM, Halaban R, Rimm DL. Automated quantitative analysis of HDM2 expression in malignant melanoma shows association with early-stage disease and improved outcome. Cancer Res. 2004;64:8767–8772. doi: 10.1158/0008-5472.CAN-04-1384. [DOI] [PubMed] [Google Scholar]
- 3.O'Reilly KE, Rojo F, She QB, et al. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res. 2006;66:1500–1508. doi: 10.1158/0008-5472.CAN-05-2925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yancovitz M, Yoon J, Mikhail M, et al. Detection of mutant BRAF alleles in the plasma of patients with metastatic melanoma. J Mol Diagn. 2007;9:178–183. doi: 10.2353/jmoldx.2007.060135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Meister G, Landthaler M, Dorsett Y, Tuschl T. Sequence-specific inhibition of microRNA- and siRNA-induced RNA silencing. Rna. 2004;10:544–550. doi: 10.1261/rna.5235104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ma L, Teruya-Feldstein J, Weinberg RA. Tumour invasion and metastasis initiated by microRNA-10b in breast cancer. Nature. 2007;449:682–688. doi: 10.1038/nature06174. [DOI] [PubMed] [Google Scholar]
- 7.Segura MF, Hanniford D, Menendez S, et al. Aberrant miR-182 expression promotes melanoma metastasis by repressing FOXO3 and microphthalmia-associated transcription factor. Proc Natl Acad Sci U S A. 2009;106:1814–1819. doi: 10.1073/pnas.0808263106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tavazoie SF, Alarcon C, Oskarsson T, et al. Endogenous human microRNAs that suppress breast cancer metastasis. Nature. 2008;451:147–152. doi: 10.1038/nature06487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Glud M, Klausen M, Gniadecki R, et al. MicroRNA expression in melanocytic nevi: the usefulness of formalin-fixed, paraffin-embedded material for miRNA microarray profiling. J Invest Dermatol. 2009;129:1219–1224. doi: 10.1038/jid.2008.347. [DOI] [PubMed] [Google Scholar]
- 10.Florell SR, Meyer LJ, Boucher KM, et al. Increased melanocytic nevi and nevus density in a G-34T CDKN2A/p16 melanoma-prone pedigree. J Invest Dermatol. 2008;128:2122–2125. doi: 10.1038/jid.2008.51. [DOI] [PubMed] [Google Scholar]
- 11.Rosenfeld N, Aharonov R, Meiri E, et al. MicroRNAs accurately identify cancer tissue origin. Nat Biotechnol. 2008;26:462–469. doi: 10.1038/nbt1392. [DOI] [PubMed] [Google Scholar]
- 12.Bolstad BM, Irizarry RA, Astrand M, Speed TP. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics. 2003;19:185–193. doi: 10.1093/bioinformatics/19.2.185. [DOI] [PubMed] [Google Scholar]
- 13.Bioconductor. Bioconductor: Open software development for computational biology and bioinformatics. 2007 doi: 10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tusher VG, Tibshirani R, Chu G. Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci U S A. 2001;98:5116–5121. doi: 10.1073/pnas.091062498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Benjamini Y, Drai D, Elmer G, Kafkafi N, Golani I. Controlling the false discovery rate in behavior genetics research. Behav Brain Res. 2001;125:279–284. doi: 10.1016/s0166-4328(01)00297-2. [DOI] [PubMed] [Google Scholar]
- 16.Tibshirani R, Hastie T, Narasimhan B, Chu G. Diagnosis of multiple cancer types by shrunken centroids of gene expression. Proc Natl Acad Sci U S A. 2002;99:6567–6572. doi: 10.1073/pnas.082099299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Breiman L, Friedman JM, Olshen R, Stone C. Classification and Regression Trees. New York: Wadsworth; 1984. [Google Scholar]
- 18.Breiman L. Random Forests. Machine Learning. 2001;45:5–32. [Google Scholar]
- 19.Freund Y, Schapire R. Experiments with a new boosting algorithm. Machine Learning: Proceedings of the Thirteenth International Conference, Morgan Kauffman, San Francisco. 1996:148–156. [Google Scholar]
- 20.Tibshirani RJ, Efron B. Pre-validation and inference in microarrays. Stat Appl Genet Mol Biol. 2002;1 doi: 10.2202/1544-6115.1000. Article1. [DOI] [PubMed] [Google Scholar]
- 21.Hofling B, Tibshirani RJ. A Study of Pre-validation. Annals of Applied Statistics. 2008;2 [Google Scholar]
- 22.Kaplan EL, Meier P. Nonparametric estimation from incomplete observations. J Amer Statist Assoc. 1958;53:457–481. [Google Scholar]
- 23.Mantel N. Evaluation of survival data and two new rank order statistics arising in its consideration. Cancer Chemother Rep. 1966;50:163–170. [PubMed] [Google Scholar]
- 24.Team RDC. R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria ISBN 3-900051-07-0. 2008 [Google Scholar]
- 25.Francken AB, Accortt NA, Shaw HM, et al. Prognosis and determinants of outcome following locoregional or distant recurrence in patients with cutaneous melanoma. Ann Surg Oncol. 2008;15:1476–1484. doi: 10.1245/s10434-007-9717-9. [DOI] [PubMed] [Google Scholar]
- 26.Satzger I, Mattern A, Kuettler U, et al. MicroRNA-15b represents an independent prognostic parameter and is correlated with tumor cell proliferation and apoptosis in malignant melanoma. Int J Cancer. 2009 doi: 10.1002/ijc.24960. [DOI] [PubMed] [Google Scholar]
- 27.Yan D, Zhou X, Chen X, et al. MicroRNA-34a inhibits uveal melanoma cell proliferation and migration through downregulation of c-Met. Invest Ophthalmol Vis Sci. 2009;50:1559–1565. doi: 10.1167/iovs.08-2681. [DOI] [PubMed] [Google Scholar]
- 28.Muller DW, Bosserhoff AK. Integrin beta 3 expression is regulated by let-7a miRNA in malignant melanoma. Oncogene. 2008;27:6698–6706. doi: 10.1038/onc.2008.282. [DOI] [PubMed] [Google Scholar]
- 29.Igoucheva O, Alexeev V. MicroRNA-dependent regulation of cKit in cutaneous melanoma. Biochem Biophys Res Commun. 2009;379:790–794. doi: 10.1016/j.bbrc.2008.12.152. [DOI] [PubMed] [Google Scholar]
- 30.Felicetti F, Errico MC, Bottero L, et al. The promyelocytic leukemia zinc finger-microRNA-221/-222 pathway controls melanoma progression through multiple oncogenic mechanisms. Cancer Res. 2008;68:2745–2754. doi: 10.1158/0008-5472.CAN-07-2538. [DOI] [PubMed] [Google Scholar]
- 31.Bemis LT, Chen R, Amato CM, et al. MicroRNA-137 targets microphthalmia-associated transcription factor in melanoma cell lines. Cancer Res. 2008;68:1362–1368. doi: 10.1158/0008-5472.CAN-07-2912. [DOI] [PubMed] [Google Scholar]
- 32.Eis PS, Tam W, Sun L, et al. Accumulation of miR-155 and BIC RNA in human B cell lymphomas. Proc Natl Acad Sci U S A. 2005;102:3627–3632. doi: 10.1073/pnas.0500613102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Iorio MV, Ferracin M, Liu CG, et al. MicroRNA gene expression deregulation in human breast cancer. Cancer Res. 2005;65:7065–7070. doi: 10.1158/0008-5472.CAN-05-1783. [DOI] [PubMed] [Google Scholar]
- 34.Jung M, Mollenkopf HJ, Grimm C, et al. MicroRNA profiling of clear cell renal cell cancer identifies a robust signature to define renal malignancy. J Cell Mol Med. 2009 doi: 10.1111/j.1582-4934.2009.00705.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang X, Tang S, Le SY, et al. Aberrant expression of oncogenic and tumor-suppressive microRNAs in cervical cancer is required for cancer cell growth. PLoS ONE. 2008;3:e2557. doi: 10.1371/journal.pone.0002557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Roldo C, Missiaglia E, Hagan JP, et al. MicroRNA expression abnormalities in pancreatic endocrine and acinar tumors are associated with distinctive pathologic features and clinical behavior. J Clin Oncol. 2006;24:4677–4684. doi: 10.1200/JCO.2005.05.5194. [DOI] [PubMed] [Google Scholar]
- 37.O'Connell RM, Chaudhuri AA, Rao DS, Baltimore D. Inositol phosphatase SHIP1 is a primary target of miR-155. Proc Natl Acad Sci U S A. 2009;106:7113–7118. doi: 10.1073/pnas.0902636106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Thai TH, Calado DP, Casola S, et al. Regulation of the germinal center response by microRNA-155. Science. 2007;316:604–608. doi: 10.1126/science.1141229. [DOI] [PubMed] [Google Scholar]
- 39.Levati L, Alvino E, Pagani E, et al. Altered expression of selected microRNAs in melanoma: antiproliferative and proapoptotic activity of miRNA-155. Int J Oncol. 2009;35:393–400. [PubMed] [Google Scholar]
- 40.Sachdeva M, Zhu S, Wu F, et al. p53 represses c-Myc through induction of the tumor suppressor miR-145. Proc Natl Acad Sci U S A. 2009;106:3207–3212. doi: 10.1073/pnas.0808042106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Guo J, Miao Y, Xiao B, et al. Differential expression of microRNA species in human gastric cancer versus non-tumorous tissues. J Gastroenterol Hepatol. 2009;24:652–657. doi: 10.1111/j.1440-1746.2008.05666.x. [DOI] [PubMed] [Google Scholar]
- 42.Yan LX, Huang XF, Shao Q, et al. MicroRNA miR-21 overexpression in human breast cancer is associated with advanced clinical stage, lymph node metastasis and patient poor prognosis. Rna. 2008;14:2348–2360. doi: 10.1261/rna.1034808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Garzon R, Pichiorri F, Palumbo T, et al. MicroRNA gene expression during retinoic acid-induced differentiation of human acute promyelocytic leukemia. Oncogene. 2007;26:4148–4157. doi: 10.1038/sj.onc.1210186. [DOI] [PubMed] [Google Scholar]
- 44.Zhang L, Huang J, Yang N, et al. microRNAs exhibit high frequency genomic alterations in human cancer. Proc Natl Acad Sci U S A. 2006;103:9136–9141. doi: 10.1073/pnas.0508889103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lin YC, Kuo MW, Yu J, et al. c-Myb is an evolutionary conserved miR-150 target and miR-150/c-Myb interaction is important for embryonic development. Mol Biol Evol. 2008;25:2189–2198. doi: 10.1093/molbev/msn165. [DOI] [PubMed] [Google Scholar]
- 46.Zhou L, Qi X, Potashkin JA, Abdul-Karim FW, Gorodeski GI. MicroRNAs miR-186 and miR-150 down-regulate expression of the pro-apoptotic purinergic P2X7 receptor by activation of instability sites at the 3'-untranslated region of the gene that decrease steady-state levels of the transcript. J Biol Chem. 2008;283:28274–28286. doi: 10.1074/jbc.M802663200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Agirre X, Jimenez-Velasco A, San Jose-Eneriz E, et al. Down-regulation of hsa-miR-10a in chronic myeloid leukemia CD34+ cells increases USF2-mediated cell growth. Mol Cancer Res. 2008;6:1830–1840. doi: 10.1158/1541-7786.MCR-08-0167. [DOI] [PubMed] [Google Scholar]
- 48.Mueller DW, Rehli M, Bosserhoff AK. miRNA expression profiling in melanocytes and melanoma cell lines reveals miRNAs associated with formation and progression of malignant melanoma. J Invest Dermatol. 2009;129:1740–1751. doi: 10.1038/jid.2008.452. [DOI] [PubMed] [Google Scholar]
- 49.Sansom SN, Griffiths DS, Faedo A, et al. The level of the transcription factor Pax6 is essential for controlling the balance between neural stem cell self-renewal and neurogenesis. PLoS Genet. 2009;5:e1000511. doi: 10.1371/journal.pgen.1000511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kim M, Gans JD, Nogueira C, et al. Comparative oncogenomics identifies NEDD9 as a melanoma metastasis gene. Cell. 2006;125:1269–1281. doi: 10.1016/j.cell.2006.06.008. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.